
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring an anomaly: the synthesis of 7,7′-diazaindirubin through a 7-azaindoxyl intermediate†
James A. Shriver *,
Katelyn R. Wang,
Andrew C. Patterson,
James R. DeYoung and
Richard J. Lipsius
*,
Katelyn R. Wang,
Andrew C. Patterson,
James R. DeYoung and
Richard J. Lipsius
Central College, 812 University St., Pella, IA 50219, USA. E-mail: shriverj@central.edu
First published on 6th October 2020
Abstract
Two independent methods generating 7-azaindoxyl as an intermediate verify that 7,7′-diazaindirubin is formed exclusively over 7,7′-diazaindigo. This contrasts with long-standing knowledge related to the reactivity of indoxyl, which proceeds via a radical-initiated homodimerization process, leading to indigo. A series of experiments confirms 7-azaindoxyl as an intermediate with results suggesting a condensation pathway followed by oxidation.
Introduction
Indirubin (1), a side product from commercial indigo (2) production (Fig. 1), is emerging as a promising pharmaceutical platform. Indirubin derivatives have been studied for their anti-cancer properties,1 potential to treat diabetes,2 and anti-inflammatory response.3 Modern understanding began when the crystal structures, illustrating the interaction between cyclin dependent kinase-2 with both indirubin-3′-oxime and indirubin-5-sulfonate, were elucidated in 1999.4 Ultimately, this has led to promising leads inter alia in the treatment of gliomas,5 MLL leukemia cells,6 and pancreatic cancer for indirubin derivatives,7 and melanoma and non-small cell lung cancer cells for 7,7′-diazaindirubin derivatives.8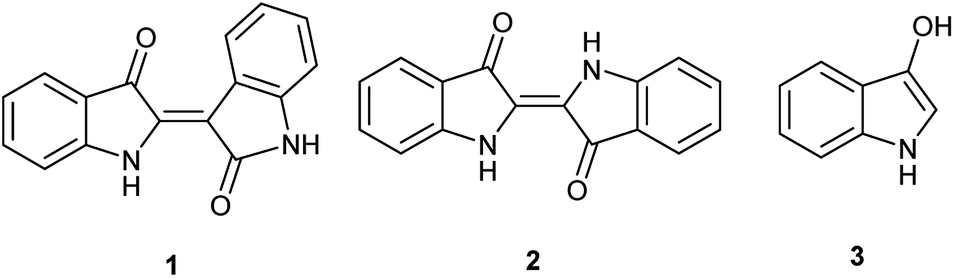 | ||
| Fig. 1 Chemical structures for indirubin (1), indigo (2) and indoxyl (3). | ||
Indigo has been used and independently developed as a dye in many cultures,9 dated as early as 6000 years ago at an archaeological site in Peru.10 Traditionally, precursors are derived from natural sources that can vary with geographic location.11 These compounds give rise to indoxyl (3-hydroxyindole, 3, Fig. 1), a reactive intermediate that spontaneously converts to indigo in air with selectivity over indirubin. This has been confirmed through numerous studies of indoxyl-generating compounds under oxidative conditions, and it proceeds through a free radical-based mechanism. For example, the action by oxygen under basic conditions suggests a free radical process after generation of indoxyl.12 Moreover, convincing evidence13 by Berlin and co-workers supports this observation more broadly with other oxidants and includes the possibility of formation under neutral and acidic conditions with the appropriate oxidant.
In contrast, the synthesis of indirubin is thought to occur due to formation of small amounts of isatin, 4, by oxidation of 3.14 Subsequent condensation occurs between indoxyl and isatin as leveraged in the traditional, and still dominant preparative method for indirubin introduced by Baeyer in the 1880s and illustrated in Scheme 1.15 Contemporaneously, there are a limited number of new preparations for indirubin through classical synthetic methods other than those which employ the classical Baeyer approach. In one example, 3-formyl indigo is reacted with Oxone® to form indirubin in a 6% yield as a biproduct during the synthesis of isatoic anhydrides.16 Though limited in its assessment, 4 was reacted with 2-oxoindole with PCl5 to form 5′ and 6′-substituted indirubins.17 A more promising protocol was accomplished through the reductive coupling of isatin using potassium borohydride18 where yields ranging from 63%–91% were seen for an array of electron-rich indirubins.
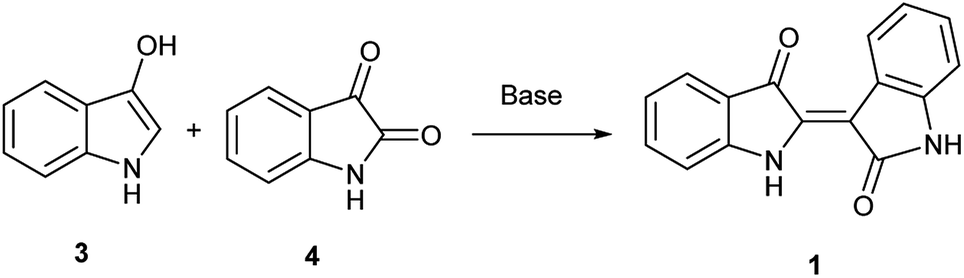 | ||
| Scheme 1 Original Baeyer synthesis of 1. | ||
More recently, methods employing biological or enzyme-promoted pathways have been explored to generate indirubins and other indigoid species. Insight into this category of reaction can be gleaned by a look at traditional biological synthesis of indigo, which is nearly always accompanied by indirubin. An elaborated example of this is seen through the enzymatic hydrolysis of indican (indoxyl-β-D-glucoside) isolated from Polygonum tincturum.19 A tissue culture where Polygonum tincturum was fed with an array of indoles to produce indigo and indirubin without isolation.20 Additionally, Maugard identified isatan C during an early (June) harvest of woad, an indigo-producing plant, and discerned that it promoted increased formation of indirubin through an isatin precursor.21 A novel approach using Cytochrome P450 enzymes was highlighted in work by Guengerich which also noted an array of oxidation products beyond indoxyl including oxindole and dioxindole.22 Further elaboration of this process with Cytochrome P450 mutant enzymes formed an array of synthetic indirubin and indigo compounds.23 Recently, it was shown that cysteine can shift the selectivity of a flavin-containing monooxygenase from indigo to indirubin.24
Herein, an anomaly to the innate selectivity towards indigo under indoxyl-generating conditions is explored for 7-azaindole, 5, which exclusively produces 7,7′-diazaindirubin, 6. In this instance, the intermediate is specifically 7-azaindoxyl, 7 (Fig. 2). This preference was observed for two independent approaches, both of which show significant promise as viable platforms for the low-cost production of a wide array of indirubin derivatives.
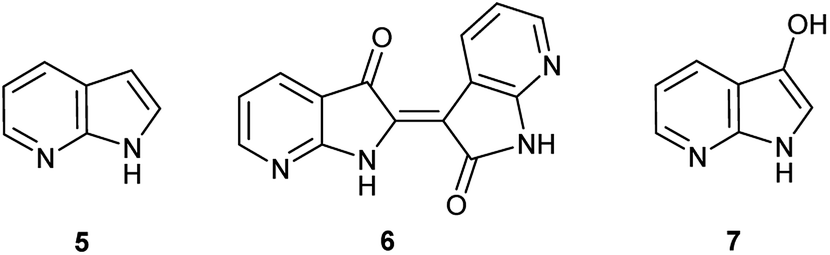 | ||
| Fig. 2 Chemical structures for 7-azaindole (5), 7,7′diazaindirubin (6), and 7-azaindoxyl (7). | ||
Results
To initiate indirubin synthesis, a protocol developed by Yamamoto and co-workers was employed. Their work produced indigo from indole in up to 82% yield with a molybdenum catalyst in the presence of cumene peroxide.25 Utilizing a scaled-down variant of this procedure with increased catalyst load, this method converted 5 to 6 with no evidence of the corresponding indigo (Scheme 2).‡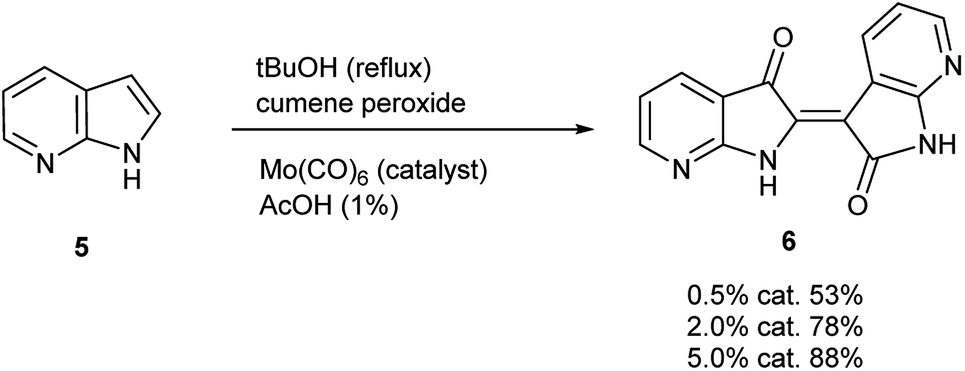 | ||
| Scheme 2 Molybdenum-catalyzed synthesis of 7,7′-diazaindirubin (6). | ||
The next task was to confirm that the corresponding indoxyl was produced as an intermediate during the reaction. Germane to this goal, the original publication by Yamamoto et al. verified the presence of indoxyl through LCMS during the synthesis of 2. To independently test for the presence of an indoxyl intermediate under these conditions, 4 was employed as a trapping agent for indoxyl, harkening back to the original process developed by Baeyer shown in Scheme 1.15 For indole (8), indirubin (1) was produced preferentially over 2 in an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. For 5, 7′-azaindirubin (9) was formed exclusively under standard reaction conditions (Scheme 3).
:1 ratio. For 5, 7′-azaindirubin (9) was formed exclusively under standard reaction conditions (Scheme 3).
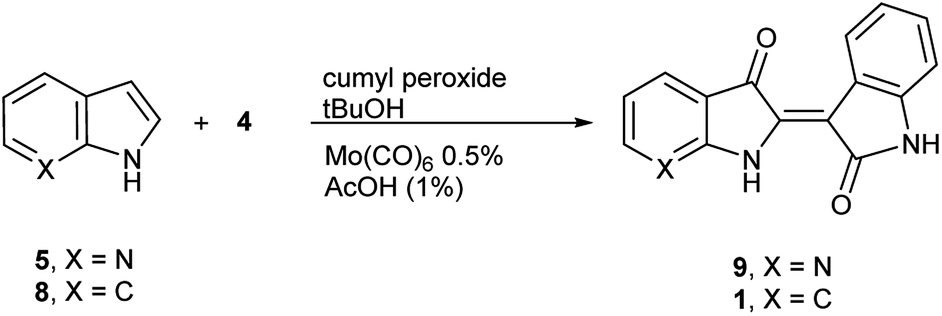 | ||
| Scheme 3 Trapping of indoxyl intermediates for molybdenum-catalysed procedure. | ||
The protocol from Scheme 2 was applied to selected indoles to help assess the process. Broadly, the results illustrate that electron-deficient indoles are more amenable towards indirubin synthesis than relatively electron rich systems. Notably, 6-nitroindole, indole-5-carbonitrile and indole-5-carboxylic acid gave rise to appreciable quantities of the corresponding indirubin, while the parent system and 5-methoxyindole formed only indigo products. These results are summarized in Table 1, showing the percent recovery and relative abundance of indirubin and indigo, which have the same molecular weight, as assessed by 1H NMR integration.
:indigo with 2% Mo(CO)6 catalyst
The second method to generate 6 was accomplished through the synthesis of 7-azaindoxyl acetate (17) and subsequent solvolysis of the acetate group. Synthesis of 17 was performed by oxidation of 5 with (diacetoxyiodo)benzene (DIB) using a modified protocol at larger scale (20 mmol), detailed in the ESI.†26 While saponification of indoxyl acetate is well known to produce indigo, mimicking reaction conditions under neutral-to-acidic conditions is more problematic. Ultimately, acid-catalysed transesterification§ at 150 °C gave rise to 6 as the sole product as shown in Scheme 3. In contrast, saponification of 17 leads to the corresponding indigo.8
As a condensation pathway was suspected, a Lewis acid was used to facilitate indirubin formation. It has been established that rare earth triflates as Lewis acids are helpful in facilitating condensation reactions, including aldol condensations.27 The initial screening of indoles shown in Table 1 have generally produced a mixture of indigo and indirubin derivatives for electron deficient systems. The example of 6-nitroindole produced a preferred amount of 11 compared to 10 in a 2:9 ratio of 10:11 (Fig. 3). When 2% Yb(OTf)3 was added to the reaction, a 3:1 ratio of 10:11 was observed by NMR (Fig. 4).¶ It is important to note that Yb(OTf)3 inhibited total recovery of product(s) for all examples explored, likely due to a deleterious effect on the molybdenum catalyst.
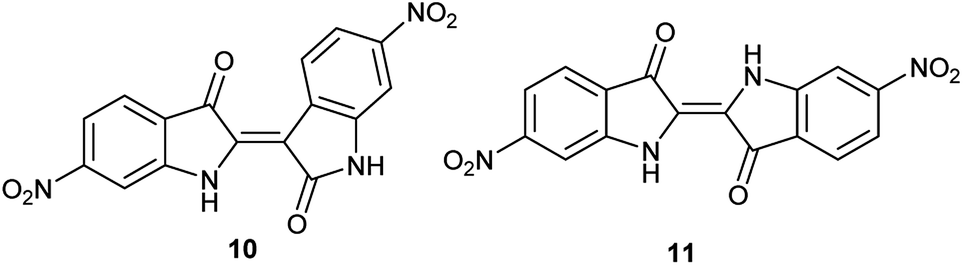 | ||
| Fig. 3 Structures of 6,6′-dinitroindirubin (10) and 6,6′-dinitroindigo (11). | ||
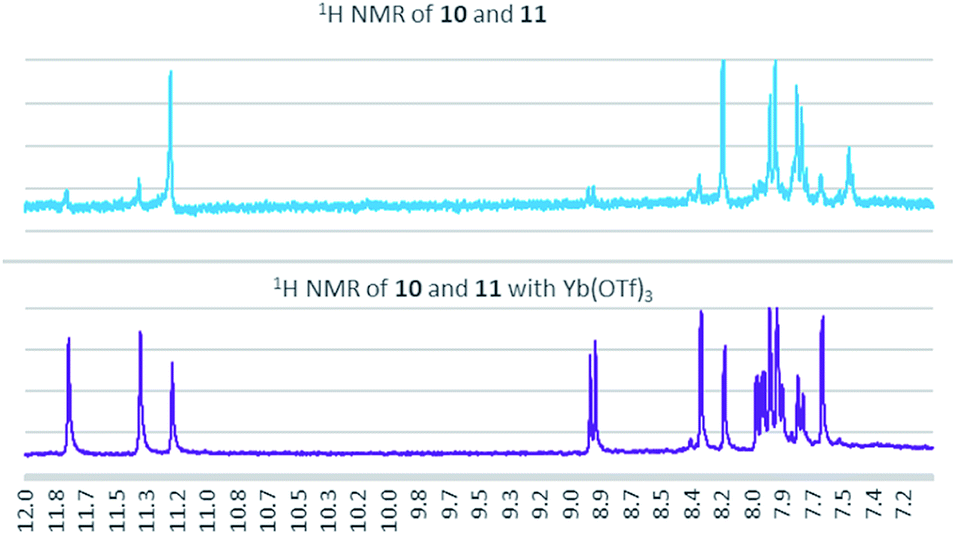 | ||
| Fig. 4 1H NMR for the formation of 10 and 11 with and without 2% Yb(OTf)3. | ||
Discussion
The collective results supply insight into the likely mechanism for this reaction. The reaction in Scheme 2 illustrated an unambiguous switch in selectivity from indigo to indirubin when utilizing 5 instead of indole as the reactant. It was initially assumed that this observation was due to the electron deficient character of 5, which was subsequently supported in the evaluation shown in Table 1 for other substituted indoles. As this reaction was already known to proceed through an indoxyl intermediate from prior research, this strongly suggests that the ability of the 3-position in indoxyl to behave as an electrophile is enhanced. To verify the results from Yamamoto for the parent system and to confirm that an indoxyl intermediate is also present for 5, a trapping experiment with 4 was run as shown in Scheme 3. This result helps substantiate that an alternate mechanism, with initial oxidation at the 2-position, is not in play.Given the complexity of the system from Scheme 2, it was important to independently verify this result, allowing removal of peroxide and the molybdenum catalyst as variables. Scheme 4 illustrates this alternate approach, which lead to the same result under acidic (but not basic) conditions. Moreover, the fact that peroxide was not necessary to promote this transformation strongly suggests that the reaction is proceeding through condensation pathway to generate 7 and is followed by oxidation with air to form 6. This was further affirmed by the addition of a rare earth triflate, which promoted the formation of indirubin over indigo for 6-nitroindole. A mechanistic pathway is illustrated in Scheme 5, which hypothesizes the intermolecular aldol condensation between the keto and enol forms of 7 followed by oxidation with air (or peroxide).
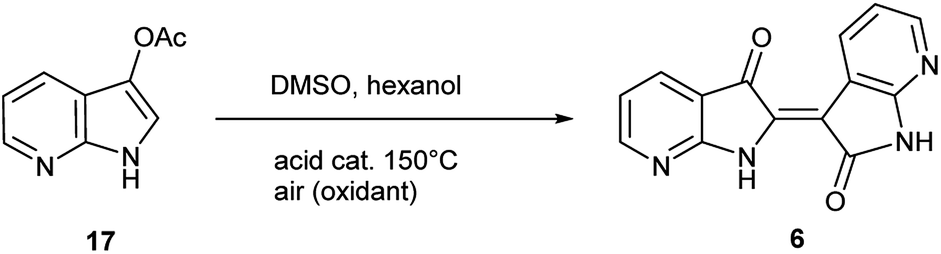 | ||
| Scheme 4 Acid-catalysed transesterification on 17 leading to 6. | ||
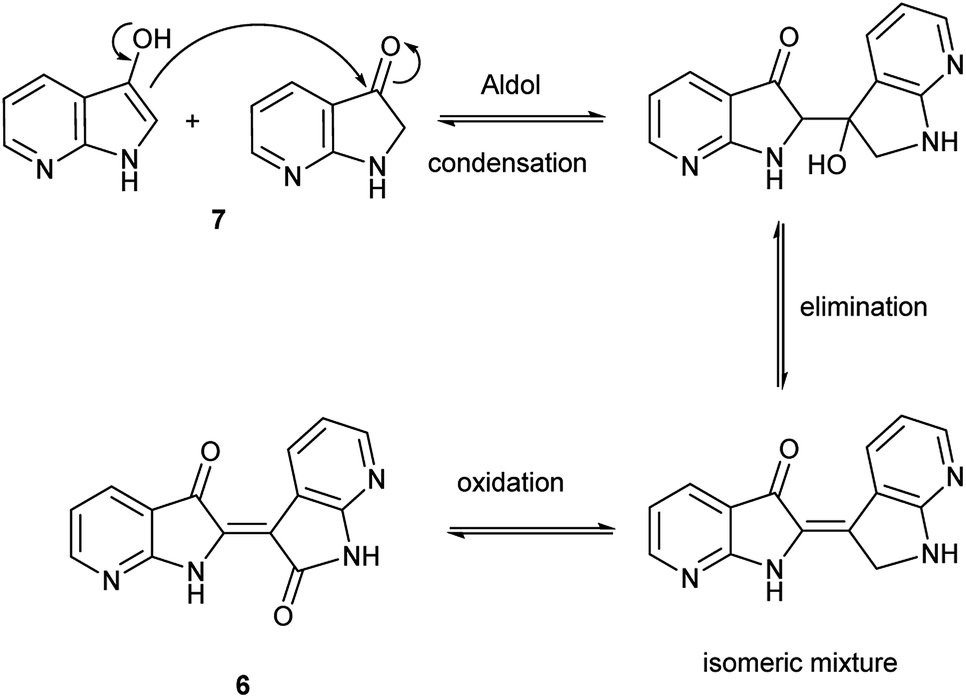 | ||
| Scheme 5 Proposed mechanism for the synthesis of 6 from 7. | ||
Conclusions
In summary, the anomalous behaviour where certain indoxyl compounds form indirubin, illustrated in detail for 7, is likely due to a previously unidentified condensation pathway shown in Scheme 4. This is a striking result given the ubiquitous nature of indigo and the extent of research around it. Moreover, there is significant opportunity for the scope to include, at the least, an array of electron deficient indoles. This is particularly true given the indirubin-enhancing effect seen for Yb(OTf)3. In addition to possible methodology development, future studies will explore the extent of keto–enol tautomerization and its role in promoting this anomalous pathway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like the thank Central College and the Chemistry Department for support of this research. In addition, we would like to thank the following for summer funding for undergraduates: The Moore Family Foundation (Andrew Patterson), the Arthur J. Bosch Endowment for Student Research (Katelyn Wang) and the John Wesselink Endowment (Katelyn Wang). Katelyn Wang will continue this research during the 2020–21 academic year as a recipient of a Goldwater Fellowship.Notes and references
- (a) Y. Wang, P. M. Hoi, J. Yuet-Wa Chan and M.-Y. Simon, Anti-Cancer Agents Med. Chem., 2014, 9, 1213 CrossRef; (b) G. Y. Wu, J. Z. Liu, F. D. Fang and J. Zuo, Sci. Sin., Ser. B, 1982, 10, 1071 Search PubMed.
- (a) S. Sharma and R. Taliyan, Biochem. Biophys. Res. Commun., 2014, 452, 1009 CrossRef CAS; (b) T. Konno, K. Sasaki, K. Kobayashi and T. Murata, Mol. Med. Rep., 2000, 21, 1552 Search PubMed.
- W. Gao, Y. Guo, C. Wang, Y. Lin, L. Yu, T. Sheng, Z. Wu and Y. Gong, Acta Histochem., 2016, 118, 606 CrossRef CAS.
- (a) R. Hoessel, S. LeClerc, J. A. Endicott, M. E. Noble, A. Lawrie, P. Tunnah, M. Leost, E. Damiens, D. Marie, D. Marko, E. Niederberger, W. Tang, G. Eisenbrand and L. Meijer, Nat. Cell Biol., 1999, 1, 60 CrossRef CAS; (b) T. G. Davies, P. Tunnah, L. Meijer, D. Marko, G. Eisenbrand, J. A. Endicott and M. E. M. Noble, Structure, 2001, 9, 389 CrossRef CAS.
- S. Kotliarova, S. Pastorino, L. C. Kovell, Y. Kotliarov, H. Song, W. Zhang, R. Bailey, D. Maric, J. C. Zenklusen, J. Lee and H. A. Fine, Cancer Res., 2008, 68, 6643 CrossRef CAS.
- Z. Wang, K. S. Smith, M. Murphy, O. Piloto, T. C. Somervialle and M. L. Cleary, Nature, 2008, 455, 1205 CrossRef CAS.
- B. Marchand, I. Tremblay, S. Cagnol and M. J. Boucher, Carcinogenesis, 2012, 33, 529 CrossRef CAS.
- X. Cheng, K.-H. Merz, S. Vatter, J. Christ and S. Wölfl, Bioorg. Med. Chem., 2014, 22, 247 CrossRef CAS.
- Indirubin, the Red Shade of Indigo, ed. L. Meijer, N. Guyard, L. A. Skaltsounis and G. Eisenbrand, Life in Progress, Station Biologique, Roscoff, France, 2006 Search PubMed.
- J. C. Splitstoser, T. D. Dillehay, J. Wouters and A. Claro, Sci. Adv., 2016, 2, 1 Search PubMed.
- K. G. Gilbert, D. J. Hill, C. Crespo, A. Mas, M. Lewis, B. Rudolph and D. T. Cooke, Phytochem. Anal., 2000, 11, 18 CrossRef CAS.
- G. A. Russell and G. Kaupp, J. Am. Chem. Soc., 1969, 91, 3851 CrossRef CAS.
- A. N. Zelentskii, G. V. Fomin, N. V. Kutafina and A. A. Berlin, Russ. Chem. Bull., 1970, 19, 1105 CrossRef.
- For a review: A. Medvedev, O. Buneeva and V. Glover, Biologics, 2007, 1, 151 CAS.
- (a) A. Baeyer, Ber. Dtsch. Chem. Ges., 1882, 15, 50 CrossRef; (b) F. Sánchez-Viesca and R. Gómez, Am. J. Chem., 2018, 8, 85 Search PubMed.
- A. C. Nelson, E. S. Kalinowski, T. L. Jacobson and P. Grundt, Tetrahedron Lett., 2013, 54, 6804 CrossRef CAS.
- H. Saito, K. Tabata, S. Hanada, Y. Kanda, T. Suzuki and S. Miyairi, Bioorg. Med. Chem. Lett., 2011, 21, 5370 CrossRef CAS.
- C. Wang, J. Yan, M. Du, J. A. Burlison, C. Li, Y. Sun, D. Zhao and J. Liu, Tetrahedron, 2017, 73, 2780 CrossRef CAS.
- T. Maugard, E. Enaud, A. d. L. Sayette, P. Choisy and M. D. LeGoy, Biotechnol. Prog., 2002, 18, 1104 CrossRef CAS.
- J.-Y. Shim, Y.-J. Chang and S.-U. Kim, Biotechnol. Lett., 1998, 20, 1139 CrossRef CAS.
- E. E. Maugard, P. Choisy and M. D. Legoy, Phytochem, 2001, 58, 897 CrossRef.
- E. M. J. Gillam, L. M. Notley, H. Cai, J. J. DeVoss and F. Guengerich, Biochem, 2000, 39, 13817 CrossRef CAS.
- (a) F. P. Guengerich, J. L. Sorrells, S. Schmitt, J. A. KRauser, P. Aryal and L. Meijer, J. Med. Chem., 2004, 47, 3236 CrossRef CAS; (b) Z.-L. Wu, P. Aryal, O. Lozach, L. Meijer and F. P. Guengerich, Chem. Biodiversity, 2005, 2, 51 CrossRef CAS.
- J. Kim, J. Lee, P.-G. Lee, E.-J. Lee, W. Kroutil and B.-g. Kim, ACS Catal., 2019, 9, 9539 CrossRef CAS.
- (a) Y. Inoue, Y. Yamamoto, H. Suzuki and U. Takaki, New Dev. in Selective Oxidation II, Proceedings of the Second World Congress and Fourth European Workshop Meeting Studies in Surface Science and Catalysis, 1994, p. 615 Search PubMed; (b) Y. Yamamoto, Y. Inoue, U. Takaki and H. Suzuki, Bull. Chem. Soc. Jpn., 2011, 84, 82 CrossRef CAS.
- K. Lie, P. Wen, J. Lie and G. Huang, Synthesis, 2010, 21, 3623 Search PubMed.
- (a) S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Chem. Rev., 2002, 102, 2227 CrossRef CAS; (b) T. Mukaiyama, K. Naraska and T. Banno, Chem. Lett., 1973, 2, 1011 CrossRef; (c) A. Kumar, I. Ahmad and M. S. Rao, Can. J. Chem., 2008, 86, 899 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07144g |
| ‡ While initially scaled at 5 mmol, the reaction was performed once (non-optimized) at a 100 mmol scale to afford 5.66 g (43% yield) of 6. |
| § The reaction was performed for 24 h with either trichloroacetic acid (14%) or Yb(OTf)3 (34%) to yield solely 6 without further optimization. |
| ¶ Due to low solubility, the 1H NMR was taken with 2000 scans at 300 MHz on the crude precipitate after filtration. |
| This journal is © The Royal Society of Chemistry 2020 |