
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRestriction of the conrotatory motion in photo-induced 6π electrocyclic reaction: formation of the excited state of the closed-ring isomer in the cyclization†
Tatsuhiro Nagasaka a,
Hikaru Sotomea,
Soichiro Morikawaa,
Lucas Martinez Uriarteb,
Michel Sliwab,
Tsuyoshi Kawaic and
Hiroshi Miyasaka*a
a,
Hikaru Sotomea,
Soichiro Morikawaa,
Lucas Martinez Uriarteb,
Michel Sliwab,
Tsuyoshi Kawaic and
Hiroshi Miyasaka*a
aDivision of Frontier Materials Science and Center for Advanced Interdisciplinary Research, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan
bUniv. Lille, CNRS, UMR 8516, LASIR, Laboratoire de Spectrochimie Infrarouge et Raman, Lille 59000, France
cGraduate School of Science and Technology, Division of Materials Science, Nara Institute of Science and Technology, Ikoma, Nara, 630-0192, Japan. E-mail: miyasaka@chem.es.osaka-u.ac.jp
First published on 27th May 2020
Abstract
The electrocyclic reaction dynamics of a photochromic dithiazolylarylene derivative, 2,3-dithiazolylbenzothiophene (DTA) was investigated by using time-resolved transient absorption and fluorescence spectroscopies. The closed-ring isomer of DTA undergoes cycloreversion through the conical intersection mediating the potential energy surfaces of the excited and ground states, which is in agreement with the Woodward–Hoffmann rules for the electrocyclic reactions of 6π electron systems. On the other hand, a large portion of the open-ring isomer undergoes cyclization along the distinct reaction scheme, in which the cyclization takes place in the excited state manifold leading to the formation of the excited state of the closed-ring isomer. The suppression of the geometrical motion of DTA due to the intramolecular interaction could open a new efficient reaction pathway resulting in the formation of the electronically excited state of the product.
Introduction
Electrocyclic reactions1,2 in the excited state take important roles in various photo-functions of molecular systems. Photoisomerization of diarylethene derivatives3–5 is one of the electrocyclic reactions in 6π electron systems, where the cyclization in the open-ring isomer and the cycloreversion in the closed-ring one take place upon photoexcitation. Owing to their excellent properties of high durability and thermal stability, a number of studies have been so far accumulated and various new photo-responses have been reported, such as photomechanical motion,6,7 fluorescent switching,8–10 reaction enhancement via multiphoton absorption11–16 and so forth.5Much attention has been also focused onto the elucidation of the photoisomerization mechanism to acquire principles for rational design of the advanced molecules based on important factors regulating the reaction properties such as yields and rate constants. On the reaction yields of the cyclization and cycloreversion, it was experimentally observed that the summation of both reaction yields was at most unity in various diarylethene derivatives.5 Ultrafast spectroscopy of reaction dynamics revealed that the product (closed- or open-ring isomer) in the ground state was produced with the same time constant of the decay of the reactant (open- or closed-ring isomer in the excited state).12–29 From these experimental results as well as theoretical investigations,30–35 it has been deduced that the cyclization and cycloreversion reactions take place on the 2A potential energy surface and the same conical intersection (CI) mediates the 2A and 1A (ground state) in both the cyclization and cycloreversion reactions as shown in Fig. 1a. That is, the cyclization and cycloreversion reaction yields are mainly regulated by the branching ration, χ, at CI.24
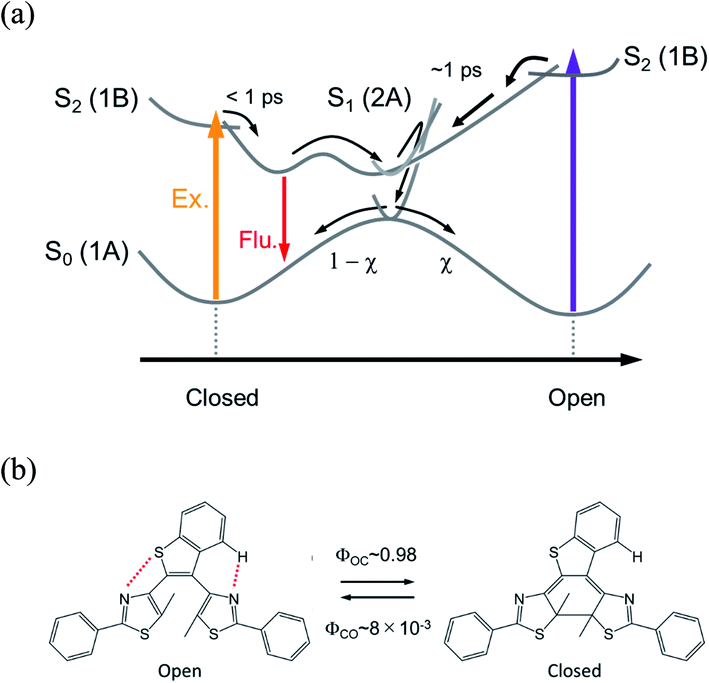 | ||
| Fig. 1 (a) Conventional electrocyclic reaction mechanism of diarylethene derivative. (b) Photochromic reaction of a dithiazolylarylene derivative, DTA, with quantum yields in n-hexane solution. | ||
Although the reaction mechanism in the 6π electron systems could be, in principle, interpreted in the framework of the Woodward–Hoffman rules1,2 followed by the Van der Lugt/Oosterhoff model,36 recent theoretical investigations reported the importance of multidimensional coordinate and the specific molecular geometry around CI29,34,35 that control the quantum yield. Multidimensional motions in molecules suggest various reaction coordinates could be extracted in the case where some specific geometrical motion is enhanced or suppressed during the reaction, leading to reaction pathways accompanied with new responses. Along this line, we have investigated the cyclization and cycloreversion dynamics of a terarylene derivative, 2,3-dithiazolylbenzothiophene (DTA), whose structure is similar to those of diarylethene derivatives as shown in Fig. 1b.37 In general, the open-ring isomer of diarylethene derivatives have two distinct conformers: one is reactive anti-parallel conformer and the other is non-reactive parallel conformer.4,5 Thus, the presence of the parallel conformer is one of the origins for decreasing the apparent cyclization reaction yield. In the case of DTA, however, the molecular geometry is fixed to the anti-parallel conformation by the intramolecular hydrogen bonding and S–N interaction, thereby leading to the cyclization reaction yield of 0.98 in n-hexane. This restricted geometry is partially released in polar solvents such as methanol, and increases the population of the parallel conformer, leading to the decrease of net cyclization reaction yield (0.54 in methanol).37
These intramolecular interactions could also restrict important geometrical movement for the cyclization, such as the conrotatory motion in the Woodward–Hoffmann framework and the deformation of the molecular symmetry. Our present study on this derivative actually shows that the cyclization reaction upon UV irradiation occurs through two pathways, the conventional one and also another one going through the excited state of the closed-ring isomer. In the following, we discuss this new reaction dynamics and mechanism that should allow the design of new efficient photochromic materials.
Experimental
Femtosecond transient absorption spectroscopy
The laser system with dual optical parametric amplifiers (OPAs) was used for transient absorption spectroscopy.12–14 A femtosecond laser pulse from a Ti:Sapphire oscillator (Tsunami, Spectra-Physics, 802 nm, 100 fs, 820 mW, 80 MHz) was guided into a regenerative amplifier at 1 kHz repetition rate (Spitfire, Spectra-Physics). The amplified light pulse was divided into two portions. One was guided to an OPA system (TOPAS-Prime, Light-Conversion) for generation of the excitation light centered at 330 nm (ca. 290 nJ) and 630 nm (ca. 68 nJ), which were respectively used for the measurements of the cyclization and cycloreversion reactions. The other 802 nm pulse was led to the other OPA (TOPAS-Prime, Light-Conversion) and converted into 1180 nm. This infrared pulse was focused into a 2 mm CaF2 plate for generation of the femtosecond white light continuum which was used as a probing pulse. A part of this white light was used as a reference beam for calibration of the intensity fluctuation of the probe pulse. Both of the white light pulses were monitored by two multichannel photodiode arrays (PMA-10, Hamamatsu) equipped with polychromators. The polarization of the excitation pulse was set at the magic angle with respect to that of the probe pulse. The group velocity dispersion of the white light probe pulse was evaluated on the basis of an optical Kerr signal of carbon tetrachloride, and used for correction of transient absorption spectra. 3000 probe pulses were collected for obtaining spectra at each delay time.The sample solution was set in a rotation cell with an optical length of 2 mm and absorbance of the solution at excitation wavelength is around 1. Continuous visible light from a Xenon lamp (LAX-103, Asahi Spectra, 400–700 nm) equipped with a mirror module (LAX-VIS 400–700, Asahi Spectra) was irradiated into the sample solution to keep the concentration of the open-ring isomer constant during the measurements of the cyclization reaction. On the other hand, for the measurements of the cycloreversion reaction, DTA was first converted to the closed-ring isomer upon UV irradiation before the measurement of the transient absorption spectra and used without cw irradiation because the change of the concentration was negligibly small due to the small cycloreversion reaction yield (0.8%).37
Steady-state spectroscopy and materials
Steady-state absorption and fluorescence spectra were respectively measured by a Hitachi U-3500 spectrophotometer and a JASCO FP 8300 fluorimeter. 2,3-Dithiazolylbenzothiophene (DTA) was synthesized and purified as reported previously.37 n-Hexane (Wako, infinity pure grade) was used without purification. The sample solution was purged with N2 gas. For the transient absorption spectroscopy, optical cell with 2 mm length was used. On the other hand, a quartz cell with a 1 cm optical length was employed for steady-state absorption and fluorescence measurements. A longpass filter (R60, HOYA Corporation, edge wavelength: 600 nm) was introduced in the detection path to block the scattering of the excitation beam. All the measurements were performed at 22 ± 2 °C.Results and discussion
Steady-state spectroscopy
Fig. 2 shows steady-state absorption spectra of the open-ring isomer of DTA, DTA(o), and the closed-ring one, DTA(c), together with the fluorescence spectrum of DTA(c). The open-ring isomer has absorption bands only in the UV region, while the closed-ring isomer has the absorption also in the visible region. This absorption band of DTA(c) is ascribable to the optically allowed transition from the 1A state to the 1B state on the basis of the large molar absorption coefficient as 9500 M−1cm−1 at 597 nm.37 Thus, the visible irradiation first pumps up DTA(c) in the ground state to the 1B state and it undergoes the cycloreversion process in the excited state. The very weak emission with the maximum at 800 nm was observed in the fluorescence spectrum of DTA(c). The Stokes-shift was ca. 3000 cm−1 from the absorption shoulder at 650 nm. This value is smaller than that of the closed-ring isomer of diarylethene derivatives (typically ca. 4500 cm−1 in nonpolar solutions),12,24 suggesting that the present DTA(c) undergoes relatively small geometrical rearrangement in the excited state compared to those of the usual diarylethene derivatives.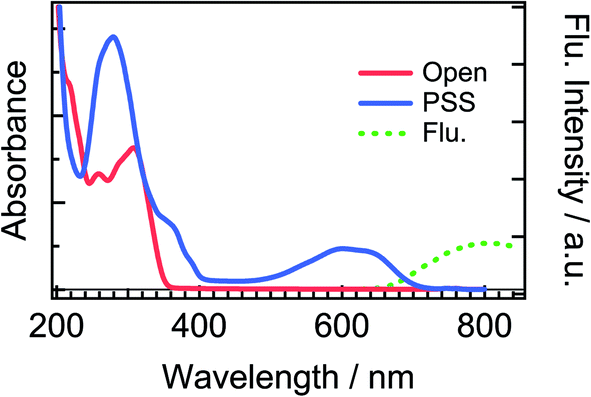 | ||
| Fig. 2 Steady-state absorption and fluorescence spectra of DTA in n-hexane solution. The solid red line is the absorption spectrum of the open-ring isomer, DTA(o), and the solid blue line is that in the photostationary state (PSS) upon irradiation at 330 nm. The dotted green line is the fluorescence spectrum of DTA(c) excited at 600 nm. The fluorescence quantum yield of DTA(c) in n-hexane solution was estimated to be <1 × 10−3, using cresyl violet (ΦFlu. = 0.54) in methanol solution as a reference sample.38 | ||
Dynamics of cycloreversion reaction
Fig. 3 shows time-resolved transient absorption spectra of DTA(c) in n-hexane solution, excited with a femtosecond 630 nm laser pulse. Immediately after the excitation, a negative signal appears in the wavelength region of 580–680 nm together with positive bands in the wavelength ranges of 400–580 nm and >680 nm. The former negative band is safely ascribed to the bleaching of the ground state, while the latter positive one is ascribable to the excited state absorption. Absorption bands with maxima at 820 and 920 nm evolve in sub-ps time region. At and after ca. 1 ps following the excitation, no remarkable spectral evolution was observed and all absorption bands decay until 100 ps. At 150 ps, the complete disappearance of the transient difference absorption spectra and recovery of the baseline signal are consistent with the small cycloreversion reaction yield (0.8%) of DTA(c).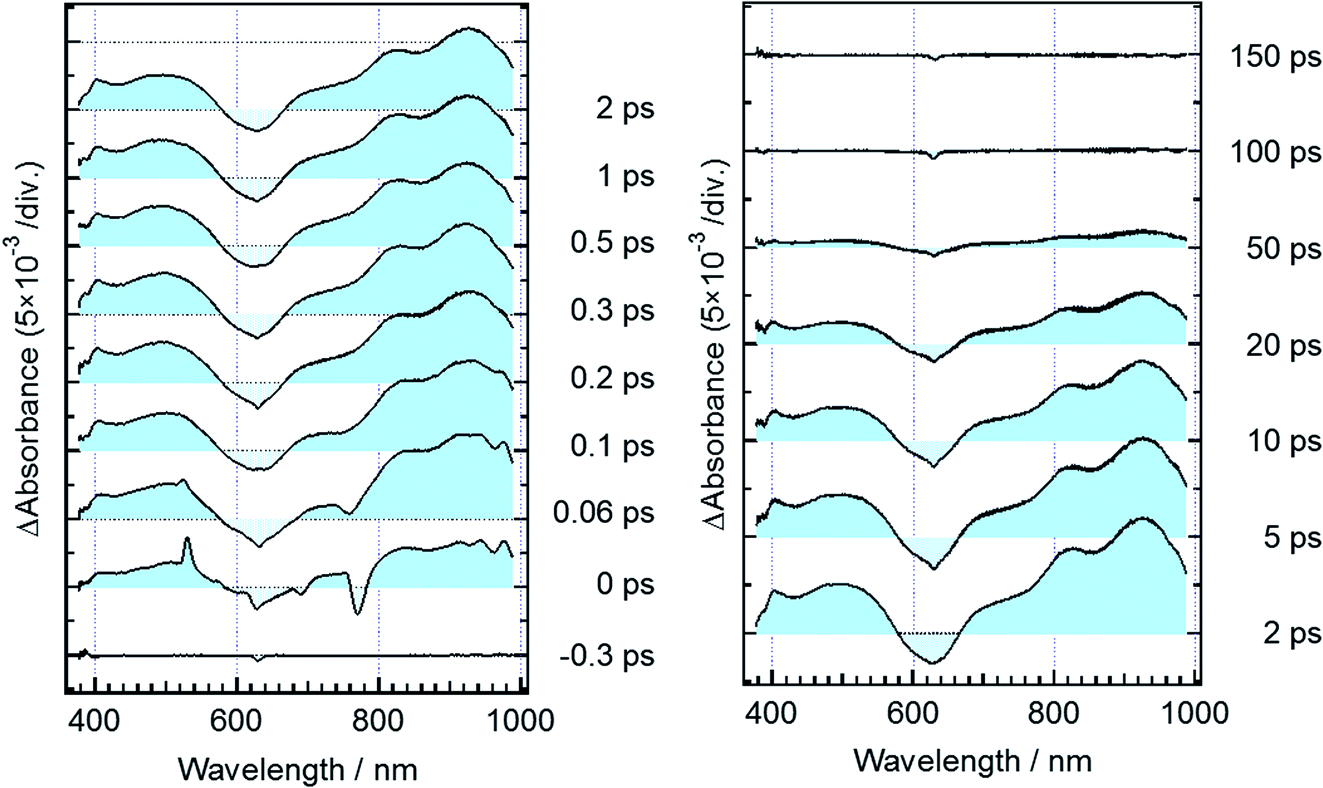 | ||
| Fig. 3 Transient absorption spectra of DTA(c) in n-hexane solution, excited with a femtosecond 630 nm laser pulse. Negative and positive signals around 780 and 530 nm in the spectrum at 0 ps originate from stimulated Raman scattering of the solvent. | ||
To quantitatively elucidate the reaction dynamics, we applied the global fitting analysis with a triple-exponential function to the transient absorption data of DTA(c). This analysis yields three time constants of 75.6 ± 0.3 fs, 1.7 ± 0.05 ps, and 25.4 ± 0.09 ps. The decay-associated spectra (DAS) of each component are shown in Fig. S1.† In Fig. 4, we show time profiles of transient absorption signals at 610, 925, and 985 nm as examples of the analysis. The time profile at 985 nm shows a subpicosecond decay, which corresponds to a positive band in the longer wavelength region than 840 nm in the first DAS (Fig. S1†). This ultrafast evolution is ascribable to the internal conversion from Franck–Condon state (1B) to the subsequent 2A state with different electronic symmetry, as have been generally observed in the previous reports on other 6π electron systems.12,15,16,22–24 The second time constant of 1.7 ps is probably due to the vibrational cooling and geometrical rearrangement in the 2A state, which is consistent with the relative large Stokes shift, compared with typical aromatic compounds. The third component (25.4 ps) is safely ascribable to the lifetime of the 2A state because this time constant is close to the fluorescence lifetime of DTA(c) (21 ps, Fig. S2†). Taken together with above discussion, DTA(c) in the Franck–Condon 1B excited state by the 630 nm absorption undergoes the internal conversion to the 2A state with the time constant < 100 fs and the subsequent 2A state decays with the time constant of 25 ps after the vibrational cooling accompanied with the geometrical rearrangement.
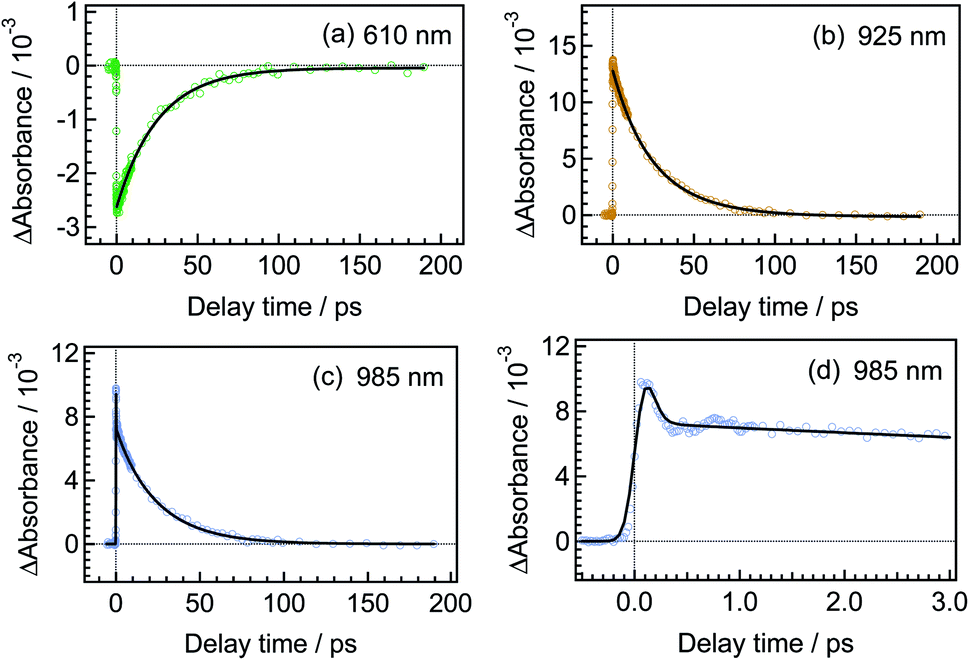 | ||
| Fig. 4 Temporal behaviors of transient absorbance of DTA(c) in n-hexane solution, excited with a femtosecond 630 nm laser pulse. The monitoring wavelengths are 610 nm for (a), 925 nm for (b) and 985 nm for (c). The time profile at 985 nm in the early time stage is shown in the panel (d). | ||
Dynamics of cyclization reaction
Fig. 5 shows transient absorption spectra of the open-ring isomer, DTA(o), in n-hexane solution excited with a femtosecond laser pulse at 330 nm. Immediately after the excitation, the absorption band with a maximum around 440 nm appears together with broad absorption bands in the entire wavelength region of the observation (400–985 nm). In the subpicosecond time region after the excitation, the absorption band at 440 nm decreases while 540 nm band increases concomitantly with the appearance of the absorption around 820 and 920 nm. The temporal evolution of the spectra indicates that the absorption band around 440 nm ascribable to the excited Franck–Condon state evolves in time into new bands around 540, 820 and 960 nm, which are due to the successor state of the former band. At and after ca. 2 ps following the excitation, an absorption band around 500–700 nm, which is due to the ground state absorption of DTA(c), appears concomitantly with the decrease of the absorption bands around 440, 820 and 930 nm.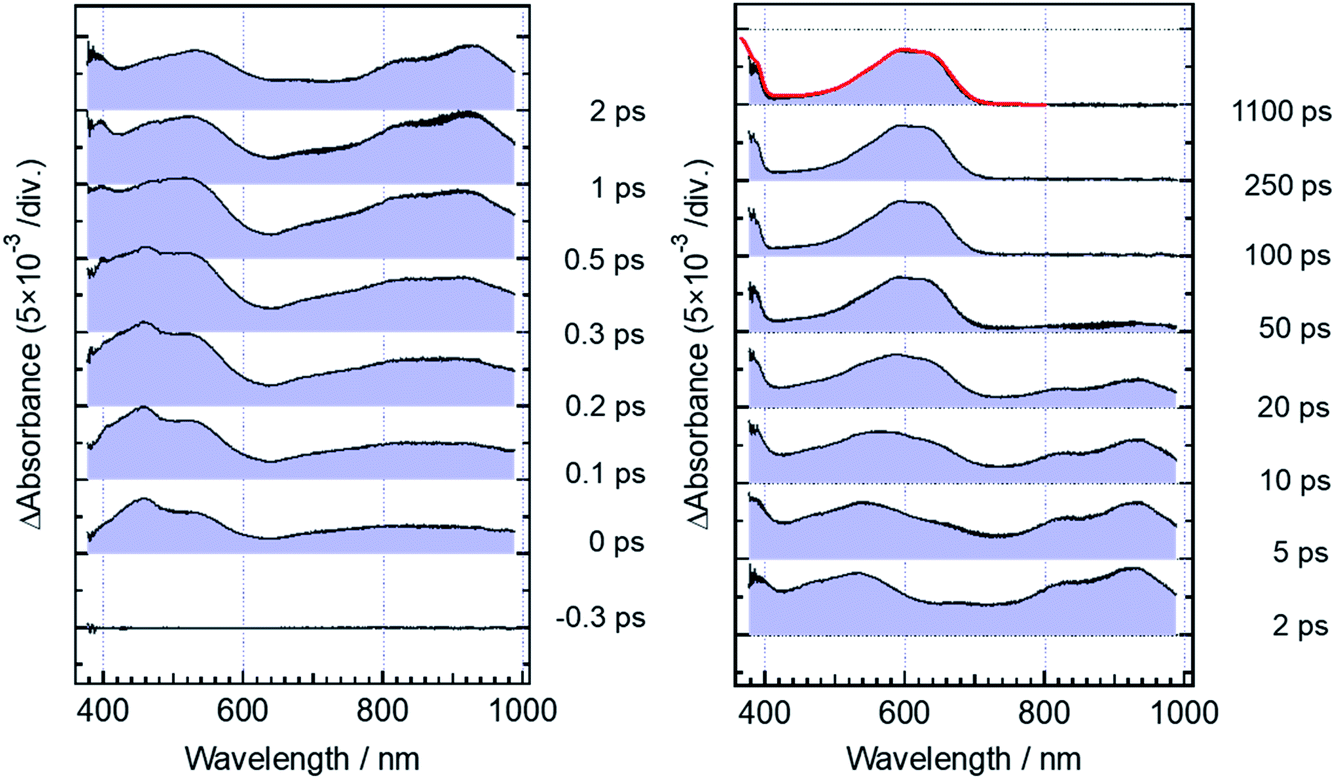 | ||
| Fig. 5 Transient absorption spectra of DTA(o) in n-hexane solution, excited with a femtosecond 330 nm laser pulse. The red curve shows the steady-state absorption spectrum of DTA(c). | ||
For the analysis of the dynamics of the cyclization reaction, we employed a model based on a weighted sum of three exponential decays in the time range of 0.15–3000 ps to reproduce the kinetic traces of transient absorption signals. Time constants retrieved by the global analysis were 380 ± 7 fs, 1.9 ± 0.03 ps and 24.0 ± 0.08 ps. In Fig. 6a and b, we show the results at 600 and 925 nm as examples of the analysis. The DAS of these three components are shown in Fig. 6c–e. The first DAS (380 fs) shows a negative band with a peak around 930 nm for the wavelength range > 800 nm, which corresponds to the rise of transient absorbance with this time constant, indicating that the excited Franck–Condon state interconverts into some new species characterized by an absorption maximum around 930 nm as explained in the previous section. The second DAS (1.9 ps) also shows a negative band in the wavelength range of 550–700 nm where DTA(c) in the ground state has an absorption band (steady-state spectra, Fig. 2). This result indicates that the time constant of 1.9 ps is ascribable to the formation of DTA(c). Actually, similar time constants in the range of 0.5 to a few ps were reported for the cyclization reaction of diarylethene derivatives.5,20,21 The third DAS shows the band shape quite similar to the transient absorption spectrum of the closed-ring isomer excited at 630 nm as was shown in Fig. 3. In addition, the time constant of the third DAS (24 ps) is almost the same with that the decay time constants of the fluorescence and the transient absorption of DTA(c). These results strongly suggest that the excited state of the closed-ring isomer is, at least in part, produced by the excitation of the open-ring isomer, DTA(o), by the UV light excitation of the open-ring isomer.
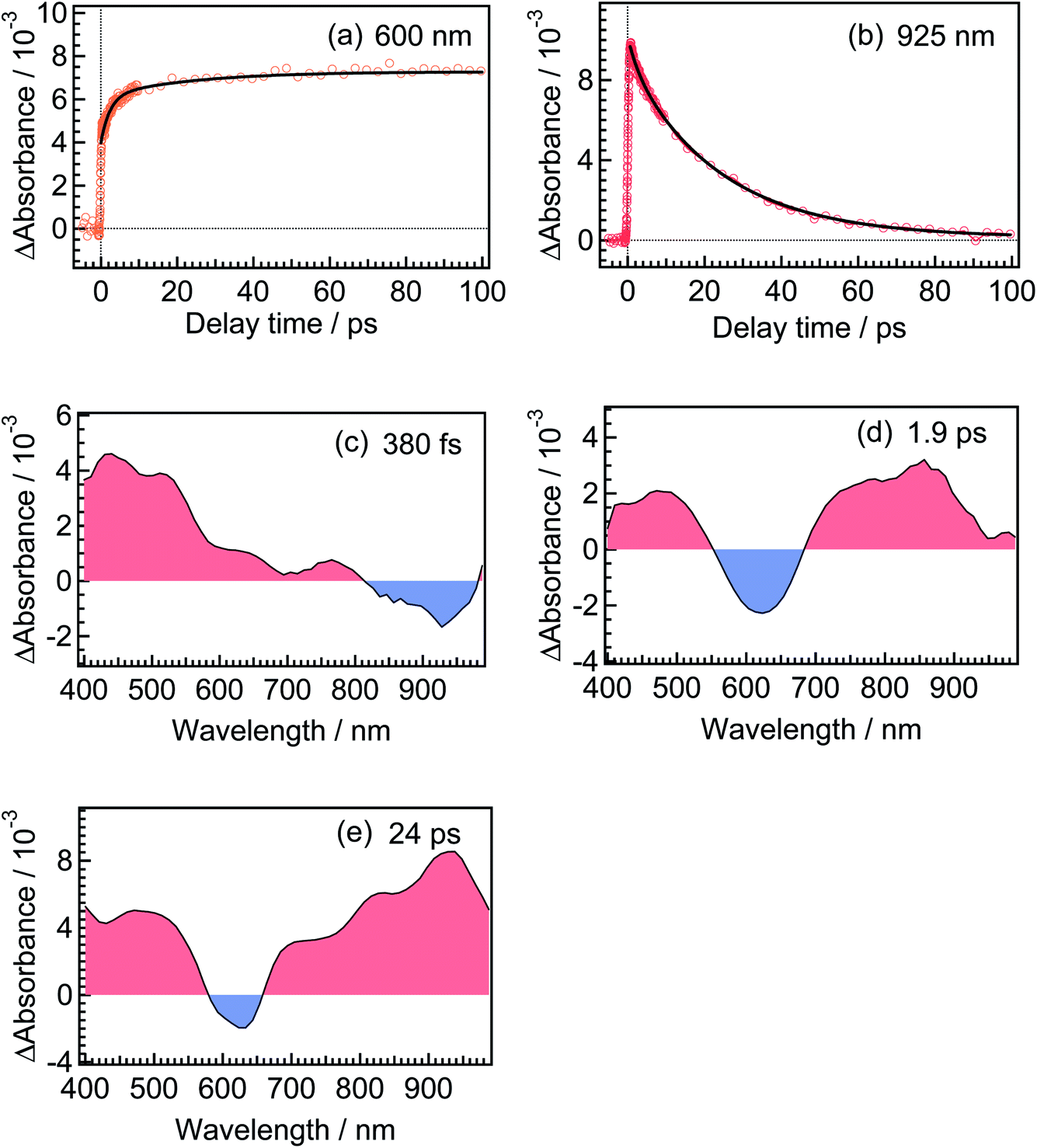 | ||
| Fig. 6 Temporal behaviors of transient absorbance of DTA(o) in n-hexane solution, excited with a femtosecond 330 nm laser pulse. The monitoring wavelengths were 600 and 925 nm for (a) and (b), respectively. Decay-associated spectra (DAS) obtained from the global analysis of the transient absorption spectra of DTA(o). The extracted time constants for DAS spectra were 380 fs for (c), 1.9 ps for (d) and 24 ps for (e). | ||
To more precisely elucidate the formation of the excited state of the closed-ring isomer in the cyclization reaction, we further analyzed the transient absorption spectra of DTA(c) and DTA(o) in the following manner. The transient absorbance of the excited state of DTA(c), ΔAc, is represented by eqn (1). Formation of the open-ring isomer is not included in this equation because the cycloreversion reaction yield is small (<0.8%).
| ΔAC(t) = εα1Cα1(t) + εα2Cα2(t) − εC{Cα1(t) + Cα2(t)} | (1) |
| ΔAC(t) ≈ (εα2 − εC)Cα2(t) | (2) |
The right side of eqn (2) corresponds to the difference spectrum of the species α2 with the 25 ps lifetime and the DTA(c) in the ground state.
On the other hand, the transient absorbance in the wavelength region ≥ 400 nm after the excitation of DTA(o) is represented by eqn (3),
| ΔAO(t) = εβ1Cβ1(t) + εβ2Cβ2(t) + εβ3Cβ3(t) + εCCC(t) | (3) |
| CC(∞)=Cβ1(t)+Cβ2(t)+Cβ3(t)+CC(t) | (4) |
| ΔAO(t) ≈ εβ3Cβ3(t) + εC{CC(∞) − Cβ3(t)} | (5) |
By subtracting εCCC(∞) from the both sides of eqn (5), we can derive eqn (6).
| ΔAO(t) − εCCC(∞) ≈ (εβ3 − εC)Cβ3(t) | (6) |
The right side of eqn (6) corresponds to the difference absorption spectrum between the species β3 with the 24 ps lifetime and the ground state of DTA(c).
The spectrum obtained by eqn (6) for β3 at 10 ps is shown in Fig. 7a, where the spectrum at 250 ps following the excitation of the open-ring isomer was used as the spectrum of εCCC(∞) and the transient spectrum at 10 ps following the excitation of DTA(c) is also shown. Although a slight deviation between the two spectra is observed in the 400–750 nm range where DTA(c) has absorption bands in the ground state, the spectral feature of these two spectra is similar to each other. This result strongly suggests that the species β3 in the cyclization dynamics appearing in the cyclization reaction is identical with α2, the excited state of the closed-ring isomer. It should be noted that some of the closed-ring isomer DTA(c) is produced with the time constant of 1.9 ps as was discussed in previous sections. This result indicates that some of the DTA(o) undergoes the cyclization reaction directly leading to the ground state of DTA(c). Accordingly, the slight deviation in Fig. 7a might be due to the vibrationally unrelaxed DTA(c) in the ground state produced via the channel with the 1.9 ps time constant. To confirm the contribution of the vibrationally hot DTA(c), we plotted the spectra at 25 ps, where this deviation becomes smaller. This result indicates that the deviation is mainly ascribable to the cooling process of DTA(c).
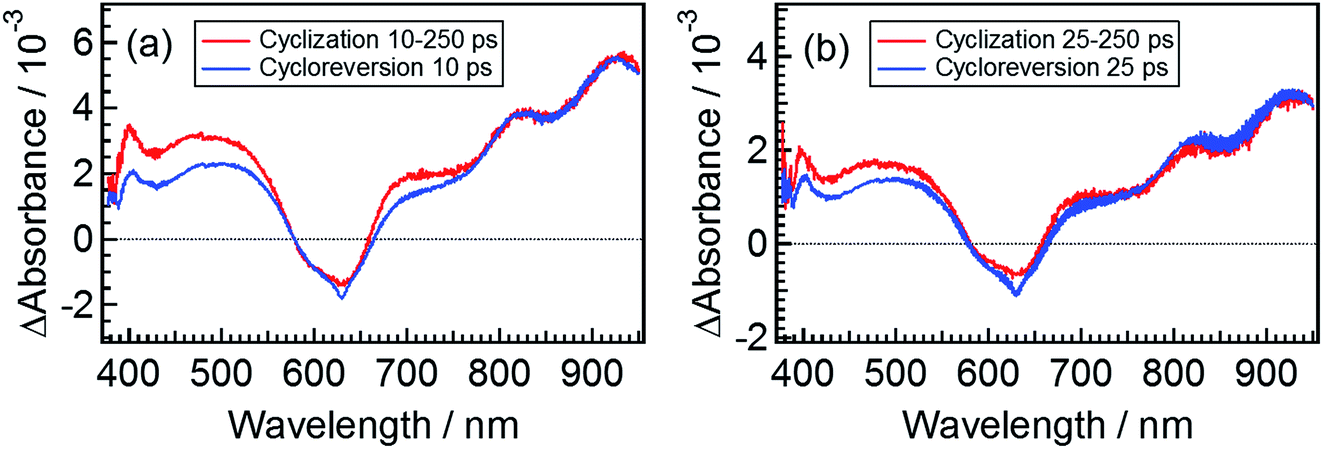 | ||
| Fig. 7 Extracted spectra of the intermediates appearing in the cyclization and cycloreversion reactions. The solid red lines are the transient spectrum of DTA(o) at 250 ps subtracted from the transient spectrum at (a) 10 ps and (b) 25 ps. The solid blue lines are the transient absorption spectra of DTA(c) at (a) 10 ps and (b) 25 ps. | ||
To more directly confirm the cyclization in the excited state manifold, we measured a fluorescence spectrum of DTA(o). Before this measurement, we prepared the fresh sample of DTA(o) and confirmed that no emission was observed by the excitation under the visible light irradiation to ensure no detectable closed-ring isomer in the sample solution.39 The fluorescence spectrum by the excitation of DTA(o) at 300 nm is shown in Fig. 8. In addition to the fluorescence from the trace of the parallel conformer (<ca. 2%) of DTA(o) in the wavelength range of 340–520 nm (ESI, Fig. S3†), a broad emission with the maximum at 800 nm was observed in the wavelength region > 680 nm, which is identical to the fluorescence spectrum of DTA(c), as was shown in Fig. 2. In addition, the fluorescence excitation spectrum monitored in the wavelength region (330–500 nm) was almost identical with the absorption spectrum of DTA(o) (ESI, Fig. S4†). This result directly indicates that the emission of the excited DTA(c) originates from the excitation of DTA(o) by UV irradiation and it shows unambiguously that the excited state of DTA(c) is produced after the excitation of DTA(o).
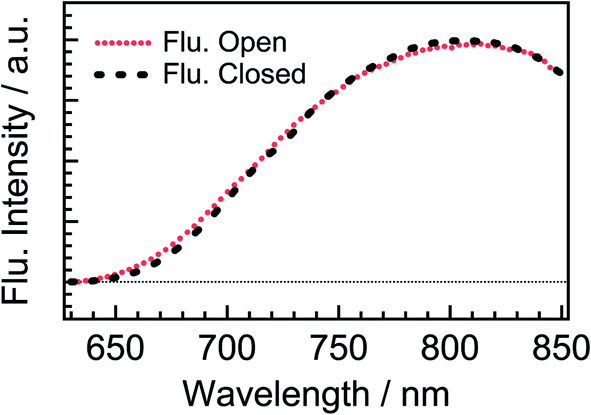 | ||
| Fig. 8 Steady-state fluorescence spectrum of DTA(o) in n-hexane solution (red dotted curve) excited at 300 nm. The fluorescence spectrum of DTA(c) in n-hexane solution (black dashed curve) is also shown for comparison. Both the spectra were normalized at 810 nm. | ||
As was shown in Fig. 5 and 6, some portion of the exited DTA(o) undergoes the cyclization reaction leading to the formation of the excited state of DTA(c). The portion of the formation of the excited state of the closed-ring isomer was quantitatively estimated on the basis of the results with the transient absorption spectroscopy of DTA(o). In this estimation, we evaluated the molar absorption coefficient of the excited state of DTA(c) in n-hexane and determined the branching ratio by comparing the amount of the excited state of DTA(c) and the ground state of DTA(c) produced by the excitation of DTA(o). The details of the calculation are shown in the ESI.† From this estimation, it was revealed that ca. 60% of the excited DTA(o) in the Franck–Condon state leads to the formation of DTA(c) in the excited state, while the 40% follows the reaction scheme via the 2A/1A conical intersection.
It is worth mentioning that the time constant of the formation of DTA(c) in the ground state, 1.9 ps, is different from that for DTA(c) in the excited state, 380 fs. This result strongly implies that the pathway leading to the formation of DTA(c) in the excited state is branched around the FC region. That is, 60% of the molecules undergo the cyclization through the unconventional pathway while the rest of the molecule traces the routes to the CI leading to the ground state. Although the detailed elucidation on the mechanism is underway, these results suggest that the suppression of the geometrical rearrangement takes an important role in the formation of the excited state of the closed-ring isomer. According to theoretical works on the 6π electrocyclic reactions,32,40,41 the PESs of the 2A and 1A (ground) states can be described along the two structural coordinates: one is the simple coordinate along the distance between the two reactive carbon atoms and the other is the coordinate out of the C2 symmetry toward the CI. The characteristic shape of the PESs is based on the fact that high-level calculations such as CASSCF discovered the symmetry-broken molecular geometry at the CI, and markedly different from that of the funnel type CI in trans–cis isomerization of stilbene derivatives. In the present DTA system, the molecular framework is fixed by the intramolecular non-covalent interactions. These restrictions probably inhibit the geometrical motion along the symmetry-broken coordinate, which may make it possible for the excited DTA(o) to circumvent the CI and reach DTA(c) in the excited state on the 2A PES. Actually, the restriction leading to the different reaction pathway have been reported in other photoinduced isomerization reactions.42,43
Conclusion
The reaction dynamics of DTA is summarized in Fig. 9. In the cycloreversion reaction, DTA(c) is pumped into the Franck–Condon state (1B state) and then converted into the subsequent 2A state within 100 fs. This 2A population is relaxed into the ground state of DTA(c) via the conical intersection with the time constant of 25 ps. On the other hand, two reaction pathways were observed in the cyclization process. One is through the conventional pathway via the 2A/1A conical intersection, which has been generally reported for 6π electron systems. In the present system, ca. 40% of the molecules excited to the Franck–Condon region evolve along the downward slope of the potential energy surface, and lead to the closed-ring isomer via the conical intersection with the time constant of 1.9 ps. On the other hand, the remaining ca. 60% of the molecules undergoes the cyclization process through the reaction pathway different from general 6π electron systems. In this newly found mechanism, the excited state of DTA(c) is produced from DTA(o) in the Franck–Condon state with the time constant of 380 fs, namely, the chemical bond is produced between the reactive carbon atoms within the excited state manifold, which is out of the framework of the Woodward–Hoffmann rules. Although the detailed investigation is underway for the clear elucidation of the mechanism, the restraint of the molecular geometry by the intramolecular interaction is playing an important role in this reaction pathway. In the photoinduced cyclization and cycloreversion reactions of general 6π electron systems, the same conical intersection has been predicted for both reactions. As a result, the summation of the cyclization and cycloreversion reaction yields is less than unity. On the other hand, the present result could lead to the efficient reversible switching system with the summation of both reaction yields beyond 1. Such results are important for application and the design of novel efficient photochromic 6π electron systems.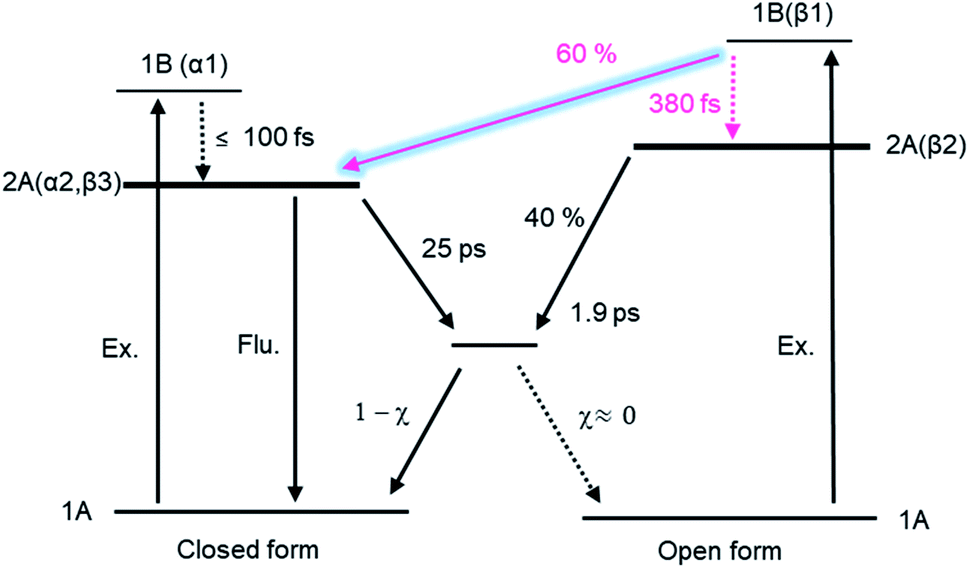 | ||
| Fig. 9 Electrocyclic reaction mechanism of DTA. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP26107002 and JP26107006 in Scientific Research on Innovative Areas “Photosynergetics” and JSPS KAKENHI Grant Number JP19J10328 for JSPS Research Fellow. The authors appreciate Mr Ryosuke Sei in Osaka University for his assistance in the steady-state spectroscopy, Dr Tetsuro Katayama in Tokushima University for his assistance in the femtosecond transient absorption spectroscopy, and Prof. Kenji Kamada in AIST for the fluorescence measurements. This work was also supported by the CNRS LIA “Nano-synergetics”.Notes and references
- R. B. Woodward and R. Hoffmann, Angew. Chem. Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS.
- R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Verlag Chemie, Weinheim, 1970 Search PubMed.
- M. Irie and M. Mohri, J. Org. Chem., 1988, 53, 803–808 CrossRef CAS.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- R. Nishimura, A. Fujimoto, N. Yasuda, M. Morimoto, T. Nagasaka, H. Sotome, S. Ito, H. Miyasaka, S. Yokojima, S. Nakamura, B. L. Feringa and K. Uchida, Angew. Chem. Int. Ed., 2019, 58, 13308–13312 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759–760 CrossRef CAS PubMed.
- K. Uno, H. Niikura, M. Morimoto, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 13558–13564 CrossRef CAS PubMed.
- M. Irie and M. Morimoto, Bull. Chem. Soc. Jpn., 2018, 91, 237–250 CrossRef CAS.
- Y. Yokoyama and K. Nakatani, Photon-Working Switches, Springer, Berlin, 2017 Search PubMed.
- H. Sotome, T. Nagasaka, K. Une, S. Morikawa, T. Katayama, S. Kobatake, M. Irie and H. Miyasaka, J. Am. Chem. Soc., 2017, 139, 17159–17167 CrossRef CAS PubMed.
- H. Sotome, T. Nagasaka, K. Une, C. Okui, Y. Ishibashi, K. Kamada, S. Kobatake, M. Irie and H. Miyasaka, J. Phys. Chem. Lett., 2017, 8, 3272–3276 CrossRef CAS PubMed.
- T. Nagasaka, T. Kunishi, H. Sotome, M. Koga, M. Morimoto, M. Irie and H. Miyasaka, Phys. Chem. Chem. Phys., 2018, 20, 19776–19783 RSC.
- C. L. Ward and C. G. Elles, J. Phys. Chem. Lett., 2012, 3, 2995–3000 CrossRef CAS PubMed.
- C. L. Ward and C. G. Elles, J. Phys. Chem. A, 2014, 118, 10011–10019 CrossRef CAS PubMed.
- D. Guillaumont, T. Kobayashi, K. Kanda, H. Miyasaka, K. Uchida, S. Kobatake, K. Shibata, S. Nakamura and M. Irie, J. Phys. Chem. A, 2002, 106, 7222–7227 CrossRef CAS.
- Y. Asano, A. Murakami, T. Kobayashi, A. Goldberg, D. Guillaumont, S. Yabushita, M. Irie and S. Nakamura, J. Am. Chem. Soc., 2004, 126, 12112–12120 CrossRef CAS PubMed.
- M. Boggio-Pasqua, M. Ravaglia, M. J. Bearpark, M. Garavelli and M. A. Robb, J. Phys. Chem. A, 2003, 107, 11139–11152 CrossRef CAS.
- P. R. Hania, R. Telesca, L. N. Lucas, A. Pugzlys, J. van Esch, B. L. Feringa, J. G. Snijders and K. Duppen, J. Phys. Chem. A, 2002, 106, 8498–8507 CrossRef CAS.
- H. Jean-Ruel, M. Gao, M. A. Kochman, C. Lu, L. C. Liu, R. R. Cooney, C. A. Morrison and R. J. D. Miller, J. Phys. Chem. B, 2013, 117, 15894–15902 CrossRef CAS PubMed.
- D. T. Valley, D. P. Hoffman and R. A. Mathies, Phys. Chem. Chem. Phys., 2015, 17, 9231–9240 RSC.
- T. Buckup, C. Sarter, H.-R. Volpp, A. Jäschke and M. Motzkus, J. Phys. Chem. Lett., 2015, 6, 4717–4721 CrossRef CAS PubMed.
- H. Sotome, K. Une, T. Nagasaka, S. Kobatake, M. Irie and H. Miyasaka, J. Chem. Phys., 2020, 152, 034301 CrossRef PubMed.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS PubMed.
- S. H. Pullen, N. A. Anderson, L. A. Walker II and R. J. Sension, J. Chem. Phys., 1998, 108, 556–563 CrossRef CAS.
- N. Kuthirummal, F. M. Rudakov, C. L. Evans and P. M. Weber, J. Chem. Phys., 2006, 125, 133307 CrossRef PubMed.
- S. Adachi, M. Sato and T. Suzuki, J. Phys. Chem. Lett., 2015, 6, 343–346 CrossRef CAS PubMed.
- K. Kosma, S. A. Trushin, W. Fuss and W. E. Schmid, Phys. Chem. Chem. Phys., 2009, 11, 172–181 RSC.
- A. Nenov, P. Kölle, M. A. Robb and R. de Vivie-Riedle, J. Org. Chem., 2010, 75, 123–129 CrossRef CAS PubMed.
- M. Garavelli, P. Celani, M. Fato, M. J. Bearpark, B. R. Smith, M. Olivucci and M. A. Robb, J. Phys. Chem., 1997, 101, 2023–2032 CrossRef CAS.
- M. Garavelli, C. S. Page, P. Celani, M. Olivucci, W. E. Schmid, S. A. Trushin and W. Fuss, J. Phys. Chem. A, 2001, 105, 4458–4469 CrossRef CAS.
- H. Tamura, S. Nanbu, T. Ishida and H. Nakamura, J. Chem. Phys., 2006, 124, 084313 CrossRef PubMed.
- S. Deb and P. M. Weber, Annu. Rev. Phys. Chem., 2011, 62, 19–39 CrossRef CAS PubMed.
- B. C. Arruda and R. J. Sension, Phys. Chem. Chem. Phys., 2014, 16, 4439–4455 RSC.
- W. T. A. M. Van der Lugt and L. J. Oosterhotf, J. Am. Chem. Soc., 1969, 91, 6042–6049 CrossRef CAS.
- S. Fukumoto, T. Nakashima and T. Kawai, Angew. Chem. Int. Ed., 2011, 50, 1565–1568 CrossRef CAS PubMed.
- D. Magde, J. H. Brannon, T. L. Cremers and J. Olmsted III, J. Phys. Chem., 1979, 83, 696–699 CrossRef CAS.
- The fluorescence measurements of DTA(o) was performed with special care about the accumulation of DTA(c), which is produced upon the UV irradiation of DTA(o) during the data acquisition. First, we measured the fluorescence spectrum of DTA(o) in n-hexane solution excited at 300 nm, and then measured that of the same solution upon the excitation at 600 nm, which is rigorously resonant with the visible absorption of DTA(c). If a significant amount of DTA(c) had been accumulated in the quartz cell, we would have detected the fluorescence of DTA(c). However, no emission was observed in this control experiment. This procedure ensured that the contribution of DTA(c) is negligibly small under the present condition.
- M. B. Pasqua, M. Ravaglia, M. J. Bearpark, M. Garavelli and M. A. Robb, J. Phys. Chem. A, 2003, 107, 11139–11152 CrossRef.
- D. M. Tapia, A. Perrier, M. J. Bearpark, M. A. Robb, B. Lasorne and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 18463–18471 RSC.
- S. Aloïse, M. Sliwa, Z. Pawlowska, J. Réhault, J. Dubois, O. Poizat, G. Buntinx, A. Perrier, F. Maurel, S. Yamaguchi and M. Takeshita, J. Am. Chem. Soc., 2010, 132, 7379–7390 CrossRef PubMed.
- C. J. Otolski, A. M. Raj, V. Ramamurthy and C. G. Elles, J. Phys. Chem. Lett., 2019, 10, 121–127 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03523h |
| This journal is © The Royal Society of Chemistry 2020 |