
Electrocatalytic hydrogenation of lignin monomer to methoxy-cyclohexanes with high faradaic efficiency†
Miao
Wang‡
ab,
Tao
Peng‡
ab,
Chenxin
Yang
a,
Baiyao
Liang
a,
Henan
Chen
a,
Mohan
Kumar
ab,
Yun
Zhang
 a and
Wei
Zhao
*a
a and
Wei
Zhao
*a
aInstitute for Advanced Study, Shenzhen University, Shenzhen 518060, China. E-mail: weizhao@szu.edu.cn
bCollege of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
First published on 1st December 2021
Abstract
Developing efficient renewable electrocatalytic processes in chemical manufacturing is of commercial interest, especially from biomass-derived feedstock. Selective electrocatalytic hydrogenation (ECH) of biomass-derived lignin monomers to high-value oxygen-functional compounds is promising towards achieving this goal. However, ECH has to date lacked the satisfied selectivity to upgrade lignin monomers to high-value oxygenated chemicals due to the reduction of vulnerable −OCH3 that exists in most lignin monomers. Herein we report carbon-felt supported ternary RhPtRu catalysts with a record faradaic efficiency (FE) of 62.8% and selectivity of 91.2% to methoxy-cyclohexanes (2-methoxy-cyclohexanol and 2-methoxy-cyclohexanone) from guaiacol, via a strong inhibition effect on the cleavage of the methoxy group, representing the best performance compared to previous reports. We further conducted a brief TEA to demonstrate a profitable ECH of guaiacol to high-value methoxy-cyclohexanes using our designed RhPtRu ternary catalysts.
Today, chemical manufacture commonly undergo energy-intensive processes with high temperatures and pressures, which leads to enormous greenhouse gas (e.g., CO2) emissions into the environment.1–5 The valorization of lignocellulosic biomass has the potential to reduce the overdependence on fossil feedstock in the production of chemicals.6–15 Each year, humans throw away more than 40 million tons of inedible plant materials such as wheat stems, corn stove and wood shavings.16 It is hugely appealing to convert these discarded plants into high value-added chemicals. Additionally, renewable electricity is another pathway in response to the overdependence upon fossil energy. 67% of electricity resources by the middle of this century will be provided by renewable energy sources (e.g., solar photovoltaic, wind, and hydro).17,18 Renewable electricity consumption, as well as energy innovation, plays a key role in mitigating greenhouse-gas emissions. Thus, electricity-driven processes (e.g., electrocatalysis) for the valorization of lignocellulosic biomass are promising ways to fulfil the goal of carbon neutrality and mitigate the ever-increasing energy crisis.
Realising this capability will surely benefit from the selective hydrogenation of lignin-derived substances to high-value methoxy chemicals that are key intermediates in the production of pharmaceuticals, pesticides, perfumes and natural products.19–24 Currently, thermocatalytic hydrogenation is widely applied to upgrade lignin monomers to the needed chemicals under elevated temperatures of 100–500 °C and H2 pressures (1–200 bar)25 powered by fossil fuel systems, leading to substantial CO2 emissions. In addition, high-pressure H2 is associated with safety hazards and increased capital costs. Electrocatalytic hydrogenation (ECH), potentially coupling with renewable electricity and replacing H2 gas with protons generated on the electrode surface through water splitting, offers a promising route to convert lignin monomers to high-value methoxy organic chemicals under mild conditions (e.g., room temperature and ambient pressure). However, most of the present studies have to date focused on deoxygenating lignin monomers to bulk chemicals such as phenol, benzene, or alkane derivatives thereby reducing the intrinsically high-value methoxy groups, but the low market value of these bulk chemicals inhibits a profitable process for an emerging technology such as ECH.26
We sought therefore to convert lignin monomers to high-value methoxy-cyclohexanes. At present, the main bottleneck for the ECH of lignin monomers to high-value methoxy-cyclohexanes is the thermodynamically favored scission of the methoxy group (–OCH3) in an electron-rich environment due to the lower dissociation energy of –OCH3, and thus achieving high FE and selectivity toward target products with abundant methoxy groups is also rendered challenging due to the competing reaction (e.g., HER).27–31
In this work, we developed ternary carbon-felt supported alloy electrocatalysts, i.e., RhPtRu, via a facile, one-step electro-deposition strategy (see the ESI†) to achieve superior FE and selectivity to high-value methoxy-cyclohexanes (2-methoxy-cyclohexanol and 2-methoxy-cyclohexanone) from the lignin monomer guaiacol. Fig. 1 schematically shows the configuration of the two-chambered H-cell utilized here, in which the ECH process of lignin monomers occurs on the cathode, while the OER occurs on the anode. The two compartments are separated by a bipolar membrane (BPM) with a characteristic function of producing protons and hydroxyl ions at the cathode and anode, respectively.32–36 The BPM is capable of offering a relatively stable chemical environment for electrochemical reaction enabled by an effective inhibition of product crossover.37,38
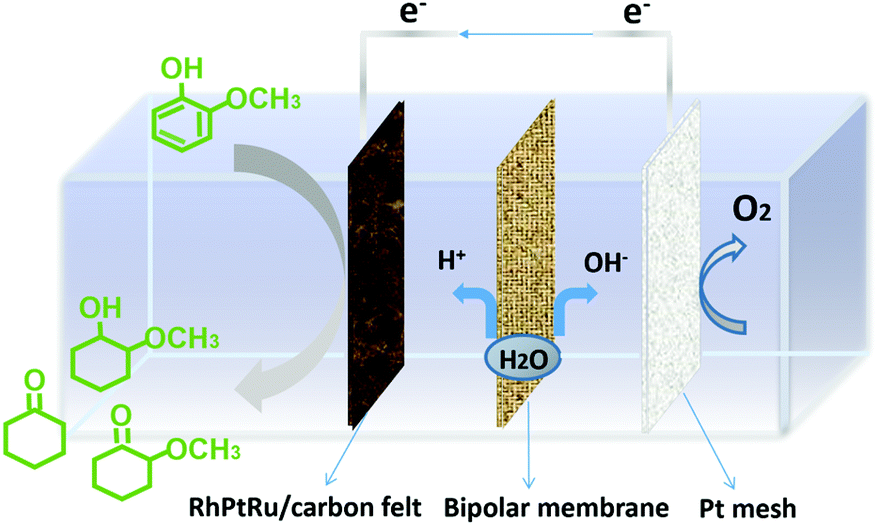 | ||
| Fig. 1 Schematic illustration of the ECH of representative lignin monomers in a H-cell system separated by a bipolar membrane. | ||
The morphology of the as-obtained carbon-felt supported RhPtRu sample was first investigated by SEM. The SEM images with three different magnifications represent the formation of hierarchical one-dimensional (1D) architecture, with interconnected carbon fibers, and attached catalyst particles on the surface of the fibers (Fig. 2a–c). This kind of carbon fiber provides an open space for more contacts between the catalyst and electrolyte and supplies a sufficient number of available active sites on the three-phase-interface as well as enhances the kinetics of charge transfer during the ECH process.39–42
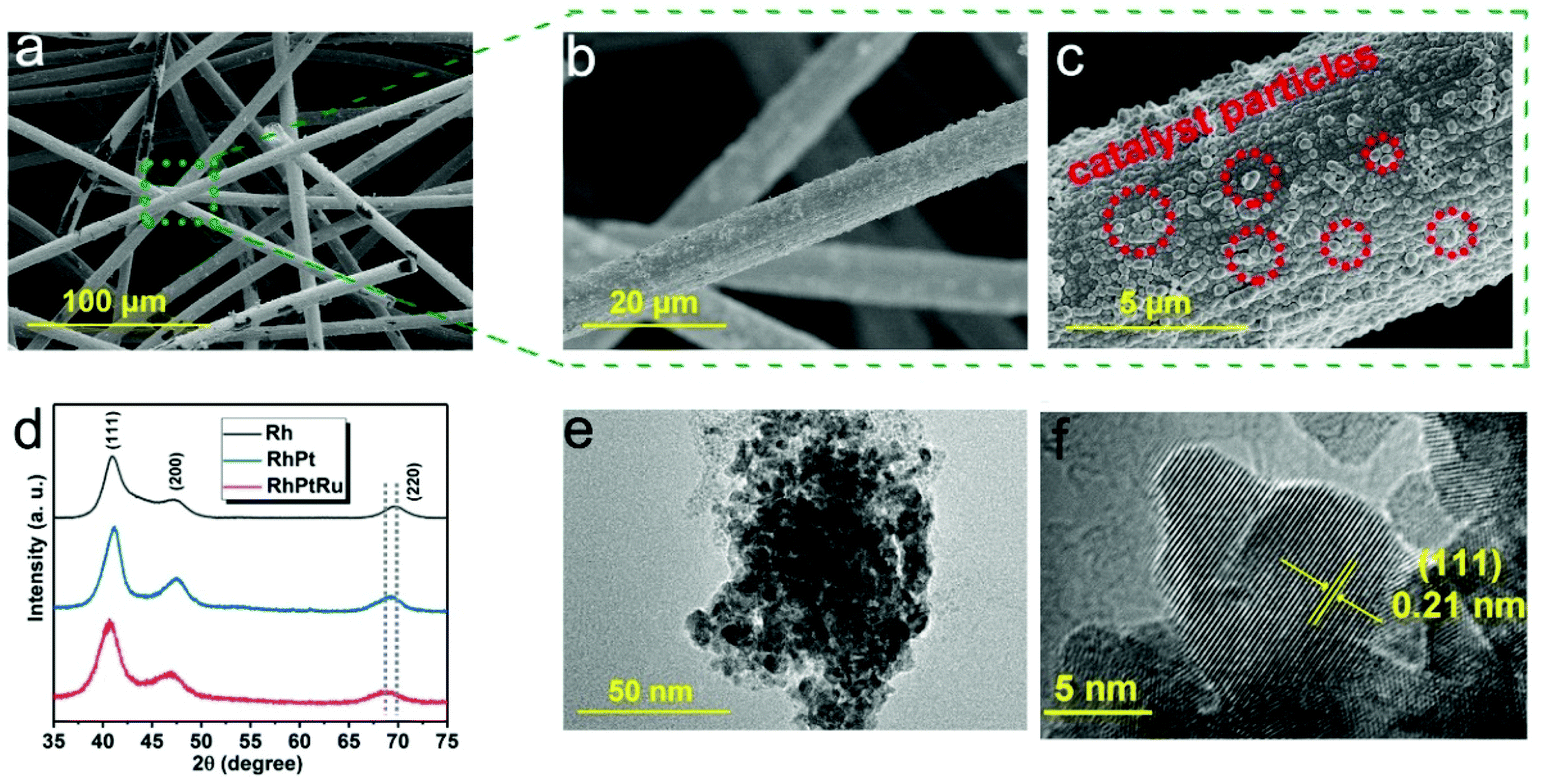 | ||
| Fig. 2 (a–c) SEM images of the RhPtRu alloy on carbon felt. (d) XRD patterns of Rh, RhPt, and RhPtRu samples. (e) TEM image of the RhPtRu alloy. (f) HRTEM image of the RhPtRu alloy. | ||
The Bragg peaks for the as-obtained samples in the X-ray diffraction pattern can be well indexed by a cubic structure with the space group of Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (Fig. 2d). Specifically, the diffraction peaks at around 41°, 47°, and 69° correspond to the (111), (200), and (220) planes of a face-centered cubic structure, respectively.43 No other impurity phase was observed in the pattern. The XRD peaks of RhPt and RhPtRu shift to lower 2θ values compared to pure Rh, indicating the formation of RhPt and RhPtRu alloys.44
m (Fig. 2d). Specifically, the diffraction peaks at around 41°, 47°, and 69° correspond to the (111), (200), and (220) planes of a face-centered cubic structure, respectively.43 No other impurity phase was observed in the pattern. The XRD peaks of RhPt and RhPtRu shift to lower 2θ values compared to pure Rh, indicating the formation of RhPt and RhPtRu alloys.44
The formation of RhPtRu alloys is further confirmed by XPS spectra, wherein Rh 3d and Pt 4f peaks slightly shift to a higher binding energy and Ru 3p peaks shift to a lower binding energy due to alloying (Fig. S1†).45 The high-resolution XPS spectra in Fig. S1a† show that the Rh 3d spectrum is well fitted with spin–orbit doublets (∼307.5 eV and ∼312.2 eV) assigned to Rh 3d5/2 and Rh 3d3/2.46,47 The XPS spectrum of Pt 4f core level (Fig. S1b†) shows the spin–orbit doublet peaks of 4f 7/2 and 4f 5/2 at around 71.6 eV and 74.9 eV, respectively.30,46,48 Compared with the binding energy of Pt, that of the Pt 4f level in RhPt and RhPtRu is slightly positively shifted to a higher value. In addition, Fig. S1c† displays the Ru 3p spectrum, where Ru 3p3/2 and Ru 3p1/2 are located at 462.0 eV and 484.1 eV, respectively.49 The XPS analysis shows an atomic Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt:Ru ratio of 44:15:41 (Fig. S3†) on the surface of the optimized RhPtRu catalysts that has the most selective hydrogenation activity.
:Pt:Ru ratio of 44:15:41 (Fig. S3†) on the surface of the optimized RhPtRu catalysts that has the most selective hydrogenation activity.
The TEM images further confirm a metallic nanocluster morphology (Fig. 2e). The HRTEM image shows a crystal lattice with a d-spacing of 0.21 nm, matching with the (111) Miller index of the exposed facet of the RhPtRu crystal (Fig. 2f). The elemental distribution of the RhPtRu alloy was further investigated by scanning TEM-energy dispersive X-ray (STEM-EDX) analysis. The results indicate that the elements Rh, Pt, and Ru are uniformly distributed in the sample (Fig S2†).
The ECH of guaiacol was conducted using a two-chambered H-cell with 0.2 M HClO4 solution as both catholyte and anolyte. The FE toward 2-methoxy-cyclohexanol and 2-methoxy-cyclohexanone using monometallic Rh catalyst is significantly increased due to an incorporation of heteroatoms (i.e., Pt and Ru), and the designed ternary RhPtRu catalysts achieve an FE of 47.9% toward 2-methoxy-cyclohexanol and an FE of 14.9% to 2-methoxy-cyclohexanone, representing a total FE of 62.8% to methoxy-cyclohexanes (Fig. 3a). This outstanding performance obtained here is possibly due to a synergistic effect between the ternary metallic elements. Rh and Pt show high adsorption energies of guaiacol and thus a high coverage of guaiacol on the catalyst surface,50 facilitating the ECH of guaiacol. Linear sweep voltammetry plots indicate that alloying Pt and Rh with Ru suppresses the competing HER reaction (Fig. S4†) since Ru shows lower HER activity compared to Pt and Rh.51 The suppressed HER leads to the increased ECH of guaiacol. Partial current densities toward methoxy-cyclohexanes on RhPtRu catalysts show the largest value (32.4 mA cm−2) (Fig. S6†) compared to all other catalysts screened in this work (e.g., Rh, Pt, Ru, RhPt, RhRu, PtRu). Fig. 3b presents the product distribution of guaiacol electrocatalytic hydrogenation over RhPtRu catalysts under different reaction duration. The pie chart with the mole fraction of various products using RhPtRu catalysts (Fig. 3c) suggests that 2-methoxy-cyclohexanol and 2-methoxy-cyclohexanone as major products occupy 62.04% and 29.11%, respectively, while the cyclohexanone as a by-product derived from the overhydrogenation of the aromatic ring accounts for only 8.85%. We then further investigated the durability of the RhPtRu catalysts using chronoamperometry measurements at a constant current density of 50 mA cm−2 (Fig. 3e).
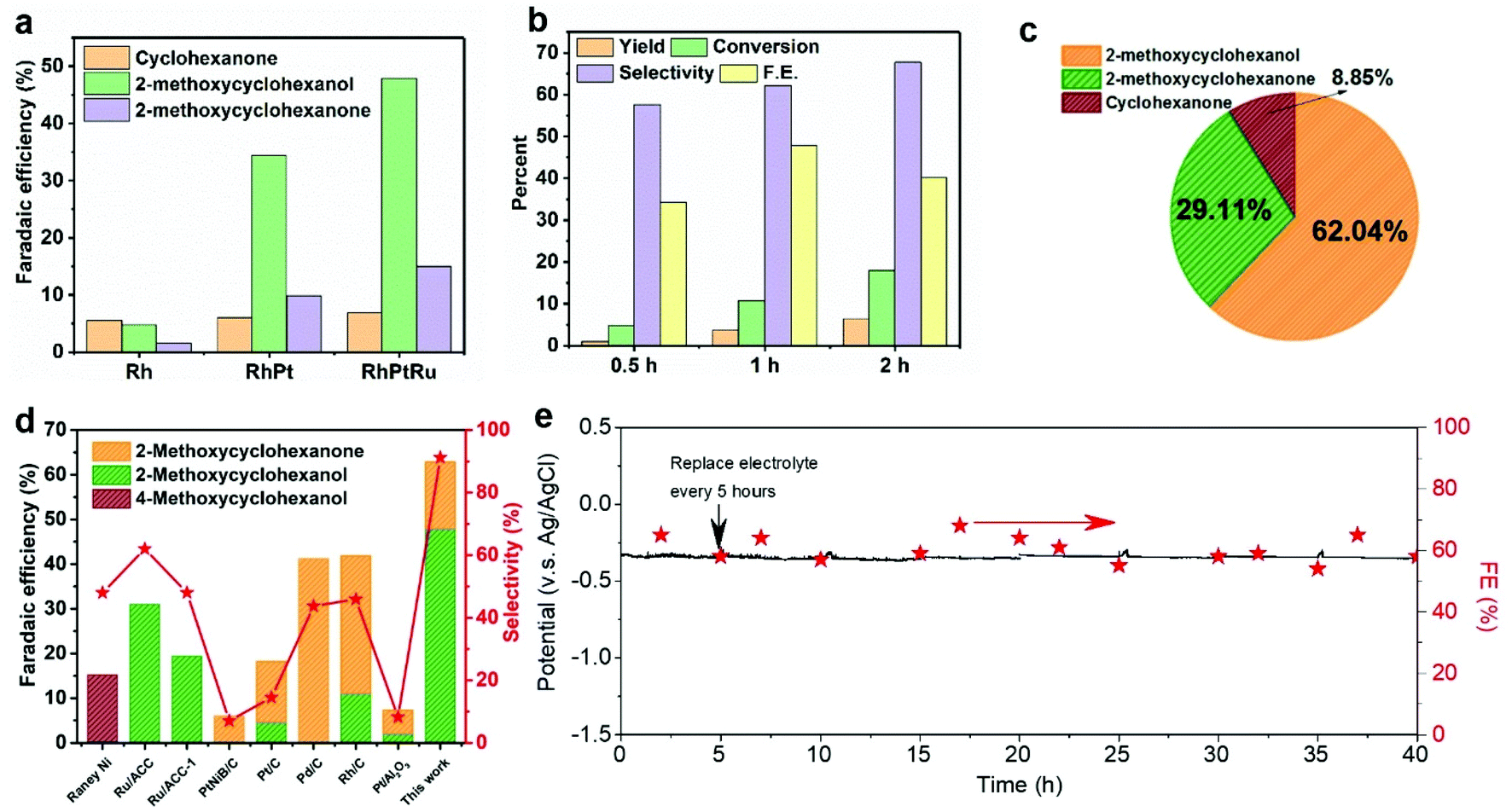 | ||
| Fig. 3 (a) Faradaic efficiency (FE) toward various hydrogenated products from guaiacol using Rh, RhPt, and RhPtRu catalysts at 50 mA cm−2 for a 1-hour electrocatalytic reaction. (b) Yield, conversion, selectivity, and FE towards 2-methoxycyclohexanol obtained by upgrading guaiacol using a RhPtRu catalyst at 50 mA cm−2 for up to 2 h. (c) Pie chart with the molar ratio of various hydrogenated products over the RhPtRu catalyst at 50 mA cm−2 after a 1-h electrocatalytic reaction. (d) Comparison of this work with representative literature that targeted the preservation of oxygenated functional groups (OFGs, −OCH3) via ECH. Right-Y axis represents the selectivity of methoxylated products over various catalysts. (e) The ECH of guaiacol on RhPtRu catalysts at an applied current density of 50 mA cm−2 using a H-cell system with 50 mL of catholyte for 40 h, showing the potential and FE to the target methoxylated products. Note that the catholyte was replaced every 5 h. | ||
As discussed above, a ternary RhPtRu catalyst was successfully obtained in this work via an electro-deposition route onto a carbon felt that displays a good electrochemical activity and superior selectivity toward methoxylated products when utilized as a cathodic electrocatalyst that are remarkably optimized than from previous reports in the ECH of guaiacol. The electrocatalytic performances in comparison with the literature are shown in Fig. 3d and Table S1.† Specifically, the RhPtRu catalyst in this work has achieved the highest FE (47.9%) for 2-methoxy-cyclohexanol to date, and more excitingly, 91.2% ultra-high selectivity toward methoxylated products (i.e., 2-methoxy-cyclohexanol and 2-methoxy-cyclohexanone) has been obtained simultaneously here.
The XRD (Fig. S7†) analysis indicates that the crystal structure of RhPtRu catalysts remains stable during 1 h of ECH. We further examined the stability of the ternary RhPtRu by conducting the ECH of guaiacol for 40 h. Our catalyst maintains a potential of ∼−0.35 V vs. Ag/AgCl (saturated with KCl) and FE of ∼60% to the target methoxylated products at an applied current density of 50 mA cm−2 over the 40-hour ECH (Fig. 3e). TEM, STEM with EDX mapping (Fig. S8†), and XPS (Fig. S9†) indicate that our RhPtRu maintains its morphological and compositional features over the 40-hour ECH.
We conducted a technoeconomic analysis (TEA) to evaluate the profitability to produce methoxy-cyclohexanes using renewable electricity powered ECH (details of TEA refer to the supplementary text and Fig. 4). The TEA indicates that the plant gate levelized cost of methoxy-cyclohexanes is 63.5 $ kg−1 in a H-cell system is profitable at the applied current density of 50 mA cm−2 (Fig. 4b), indicating a profitable ECH of guaiacol to methoxy-cyclohexanes having a market price of 431 $ kg−1. Overall, the breakdown analysis of the plant gate levelized cost indicates that the capital cost of the electrolyte, electrolyzer, membrane, and noble metal catalysts is the main factor affecting the total cost. The high FE of 62.8 and selectivity of 92% of our designed RhPtRu catalysts significantly decreases the electricity cost to 0.93 $ kg−1.
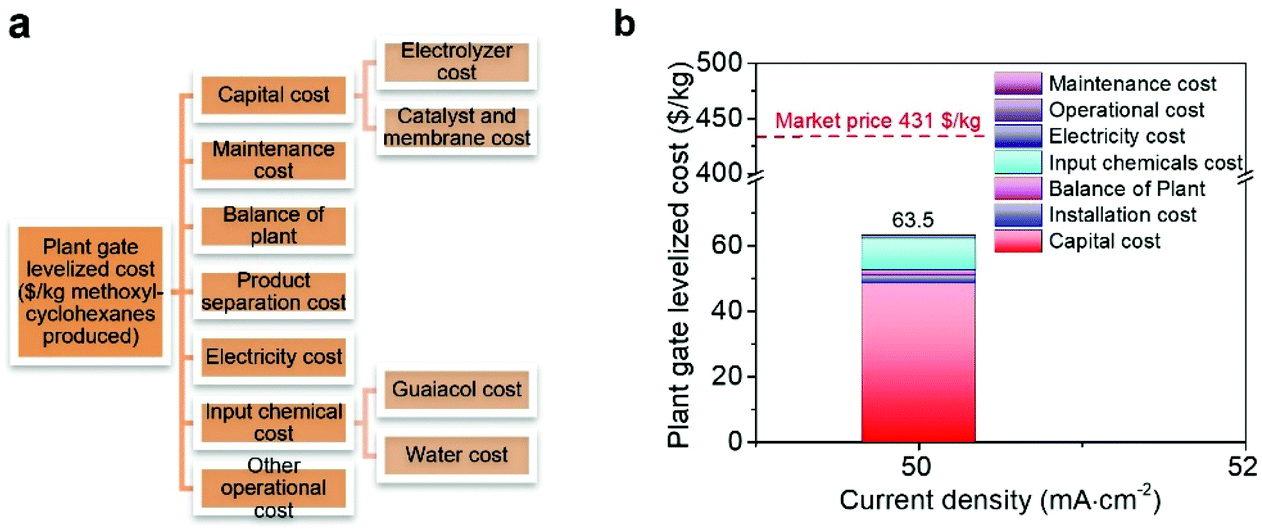 | ||
| Fig. 4 (a) TEA model. (b) Breakdown of cost of methoxy-cyclohexanes from electrocatalytic hydrogenation of guaiacol at a current density of 50 mA cm−2 on RhPtRu catalysts. | ||
Conclusions
In summary, valorization of lignin-derived monomers through ECH offers a promising pathway for the selective production of high value-added chemical products using renewable electricity. Herein, FE of 62.8% and high selectivity of 91.2% toward high-value methoxy-cyclohexanes were achieved through the selective hydrogenation of guaiacol using the designed carbon felt-supported RhPtRu catalysts. A brief TEA indicates our designed RhPtRu ternary catalysts enable a profitable ECH of guaiacol to high value methoxy-cyclohexanes.Author contributions
M. W., T. P. and Y. Z. conducted the electrochemical experiments and XRD, and drafted the manuscript. T. P. carried out the TEA study, stability test, SEM, HRTEM, STEM with EDX mapping, and XPS. T. P. wrote and revised the manuscript. C. Y., B. L., and H. C. assisted in GC calibration and electrode preparation. W. Z. supervised this project.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the support from the Natural Science Foundation of China (21972096) and the Shenzhen Science and Technology Program (JCYJ20190808150615285).Notes and references
- Y. Sun, K. Xu, Z. Wei, H. Li, T. Zhang, X. Li, W. Cai, J. Ma, H. J. Fan and Y. Li, Adv. Mater., 2018, 30, 1802121 CrossRef.
- Y. Pan, H. Ren, H. Du, F. Cao, Y. Jiang, H. Du and D. Chu, J. Mater. Chem. A, 2018, 6, 22497–22502 RSC.
- M. Ledendecker, G. Clavel, M. Antonietti and M. Shalom, Adv. Funct. Mater., 2015, 25, 393–399 CrossRef CAS.
- S. Sultan, J. N. Tiwari, A. N. Singh, S. Zhumagali, M. Ha, C. W. Myung, P. Thangavel and K. S. Kim, Adv. Energy Mater., 2019, 9, 1900624 CrossRef.
- T. Ouyang, Y.-Q. Ye, C.-Y. Wu, K. Xiao and Z.-Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 4923–4928 CrossRef CAS.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS PubMed.
- W. Liu, W. You, W. Sun, W. Yang, A. Korde, Y. Gong and Y. Deng, Nat. Energy, 2020, 5, 759–767 CrossRef CAS.
- X. Cui, A.-E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Nat. Commun., 2016, 7, 11326 CrossRef.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Heroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- D. J. Xiao, E. D. Chant, A. D. Frankhouser, Y. Chen, A. Yau, N. M. Washton and M. W. Kanan, Nat. Chem., 2019, 11, 940–947 CrossRef CAS.
- C. J. Bondue, F. Calle-Vallejo, M. C. Figueiredo and M. T. M. Koper, Nat. Catal., 2019, 2, 243–250 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- K. Sanderson, Nature, 2011, 474, S12–S14 CrossRef CAS PubMed.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, Green Chem., 2020, 22, 7233–7264 RSC.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- J. Fu, Z. Ren, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2018, 564, 395–399 CrossRef CAS.
- E. J. Corey and X. M. Cheng, The logic of chemical synthesis, John Wiley & Sons, New York, 1989, p. 436 Search PubMed.
- J. Le Bras, D. Chatterjee and J. Muzart, Tetrahedron Lett., 2005, 46, 4741–4743 CrossRef CAS.
- T. Felicetti, R. Cannalire, D. Pietrella, G. Latacz, A. Lubelska, G. Manfroni, M. L. Barreca, S. Massari, O. Tabarrini, K. Kieć-Kononowicz, B. D. Schindler, G. W. Kaatz, V. Cecchetti and S. Sabatini, J. Med. Chem., 2018, 61, 7827–7848 CrossRef CAS PubMed.
- G. Valdameri, C. Gauthier, R. Terreux, R. Kachadourian, B. J. Day, S. M. B. Winnischofer, M. E. M. Rocha, V. Frachet, X. Ronot, A. Di Pietro and A. Boumendjel, J. Med. Chem., 2012, 55, 3193–3200 CrossRef CAS PubMed.
- Hiv drugs global market report 2020–30: Covid-19 implications and growth, Vol, The Business Research Company, 2020, pp. 1–200 Search PubMed.
- X. Wang, S. Zhu, S. Wang, J. Wang, W. Fan and Y. Lv, Appl. Catal., A, 2018, 568, 231–241 CrossRef CAS.
- W. Lan and J. S. Luterbacher, ACS Cent. Sci., 2019, 5, 1642–1644 CrossRef CAS PubMed.
- C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron, D. J. Miller and J. E. Jackson, Green Chem., 2015, 17, 601–609 RSC.
- Z. Li, M. Garedew, C. H. Lam, J. E. Jackson, D. J. Miller and C. M. Saffron, Green Chem., 2012, 14, 2540–2549 RSC.
- M. Garedew, D. Young-Farhat, J. E. Jackson and C. M. Saffron, ACS Sustainable Chem. Eng., 2019, 7, 8375–8386 CrossRef CAS.
- Y. Zhou, Y. Gao, X. Zhong, W. Jiang, Y. Liang, P. Niu, M. Li, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1807651 CrossRef.
- W. Liu, W. You, Y. Gong and Y. Deng, Energy Environ. Sci., 2020, 13, 917–927 RSC.
- A. ter Heijne, H. V. M. Hamelers, V. de Wilde, R. A. Rozendal and C. J. N. Buisman, Environ. Sci. Technol., 2006, 40, 5200–5205 CrossRef CAS.
- M. D. Eisaman, K. Parajuly, A. Tuganov, C. Eldershaw, N. Chang and K. A. Littau, Energy Environ. Sci., 2012, 5, 7346–7352 RSC.
- M. Mandal, ChemElectroChem, 2021, 8, 1448–1450 CrossRef CAS.
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- S. Valluri and S. K. Kawatra, Fuel Process. Technol., 2021, 213, 106691 CrossRef CAS.
- Y. C. Li, Z. Yan, J. Hitt, R. Wycisk, P. N. Pintauro and T. E. Mallouk, Adv. Sustainable Syst., 2018, 2, 1700187 CrossRef.
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Science, 2020, 369, 1099–1103 CrossRef CAS PubMed.
- Y. Hou, T. Huang, Z. Wen, S. Mao, S. Cui and J. Chen, Adv. Energy Mater., 2014, 4, 1400337 CrossRef.
- X. Fan, Z. Peng, R. Ye, H. Zhou and X. Guo, ACS Nano, 2015, 9, 7407–7418 CrossRef CAS PubMed.
- J.-S. Li, S.-L. Li, Y.-J. Tang, M. Han, Z.-H. Dai, J.-C. Bao and Y.-Q. Lan, Chem. Commun., 2015, 51, 2710–2713 RSC.
- M. Wang, Y. Zhang, T. Peng, M. Kumar, Q.-T. Zhang and W. Zhao, Diam. Relat. Mater., 2021, 118, 108508 CrossRef CAS.
- L. Rao, Y.-X. Jiang, B.-W. Zhang, Y.-R. Cai and S.-G. Sun, Phys. Chem. Chem. Phys., 2014, 16, 13662–13671 RSC.
- K.-W. Park, J.-H. Choi, K.-S. Ahn and Y.-E. Sung, J. Phys. Chem. B, 2004, 108, 5989–5994 CrossRef CAS.
- Q. Wang, Y.-W. Zhou, Z. Jin, C. Chen, H. Li and W.-B. Cai, Catalysts, 2021, 11, 925 CrossRef CAS.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, I. Gueye, O. Seo, J. Kim, S. Hiroi, O. Sakata, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2020, 11, 12731–12736 RSC.
- H. Guo, Z. Fang, H. Li, D. Fernandez, G. Henkelman, S. M. Humphrey and G. Yu, ACS Nano, 2019, 13, 13225–13234 CrossRef CAS.
- P. Wang, Y. Zhang, R. Shi and Z. Wang, ACS Appl. Energy Mater., 2019, 2, 2515–2523 CrossRef CAS.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef.
- J. Akinola, I. Barth, B. R. Goldsmith and N. Singh, ACS Catal., 2020, 10, 4929–4941 CrossRef CAS.
- A. R. Zeradjanin, A. Vimalanandan, G. Polymeros, A. A. Topalov, K. J. J. Mayrhofer and M. Rohwerder, Phys. Chem. Chem. Phys., 2017, 19, 17019–17027 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03523a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |