
In situ synthesis of pyridinium-based ionic porous organic polymers with hydroxide anions and pyridinyl radicals for halogen-free catalytic fixation of atmospheric CO2†
Ke
Liu
,
Zixuan
Xu
,
He
Huang
,
Yadong
Zhang
,
Yan
Liu
,
Zhiheng
Qiu
,
Minman
Tong
 ,
Zhouyang
Long
and
Guojian
Chen
*
,
Zhouyang
Long
and
Guojian
Chen
*
School of Chemistry and Materials Science, Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou, 221116, China. E-mail: gjchen@jsnu.edu.cn
First published on 23rd November 2021
Abstract
Pyridinium-based ionic porous organic polymers (Py-iPOPs) were constructed from newly designed acetonitrile-functionalized ionic liquids and multi-carbaldehyde monomers by base-catalyzed Knoevenagel condensation. The obtained Py-iPOPs having in situ formed hydroxide anions and pyridinyl radicals were regarded as efficient metal-free and halogen-free heterogeneous catalysts for atmospheric CO2 fixation under mild conditions.
Carbon dioxide (CO2) is one of the major greenhouse gases responsible for climate change and environmental issues. At present, many efforts are being made to achieve the carbon emissions peak and carbon neutrality all over the world. Meanwhile, CO2 is also regarded as an abundant C1 resource, which can be converted into diverse fuels and value-added chemicals by various catalytic reaction processes.1,2 In this context, catalytic conversion of CO2 and epoxides into useful cyclic carbonates is an attractive, atom economic and energy-saving non-redox route to achieve CO2 utilization and promote carbon neutrality.3,4 Towards the catalytic CO2 fixation, most of the efficient catalysts largely depend on the catalytic active sites related to nucleophilic halogen anions cooperating with various electrophilic metal sites or hydrogen bond donors, which have afforded excellent catalytic performances.4 However, from the perspective of green chemistry, metal-free and halogen-free catalysts are desirable and environmentally friendly, so as to obtain high-quality cyclic carbonates excluding metal or halogen residues.5,6 Indeed, pioneering works have been carried out in CO2 conversion using a variety of halogen-free homogeneous catalysts including N-organic bases, imidazolium carboxylates (NHC-CO2), salophens and ionic liquids (ILs) with hydroxide, bicarbonate and amino acid anions.5,6 Considering the facile recovery of catalysts and easy separation of products, efficient metal-free and halogen-free heterogeneous catalysts are highly desired, however, only a few examples have been reported,7,8 but unfortunately, they often require relatively harsh conditions to achieve the satisfactory catalytic activities. Therefore, it is an important and challenging task to develop efficient halogen-free heterogeneous catalysts for the conversion of atmospheric CO2 under mild conditions.
Porous organic polymers (POPs) are regarded as promising heterogeneous catalysts due to their fascinating characteristics such as abundant porosity, good stability and diverse catalytic functionality.9 In the past decade, metal sites and/or halogen anions were introduced into POPs to get various metal-based POPs and imidazolium or pyridinium-based ionic POPs for catalytic CO2 conversion.10–16 Recently, N-heterocyclic carbenes (NHCs) or NHC-CO2 adduct porous polymers have been reported as metal-free and halogen-free heterogeneous catalysts for CO2 conversion, but high reaction temperatures are still required in catalytic processes.7,17 Also, the syntheses of these porous polymeric catalysts undergo relatively complex procedures related to two or three steps by post-treatments.7,17 Besides, a series of porous poly(ionic liquid)s with hydroxide, amino acid and azolide anions were prepared as efficient heterogeneous catalysts for CO2 fixation under metal-/halogen-free conditions.18–20 Unfortunately, two or three steps are also needed to obtain the targeted halogen-free poly(ionic liquid)s by post-treating with an excess amount of NaOH aqueous solution or continuously neutralizing with various amino acids or azolides. Inspired by these valuable works, we aim to design task-specific ionic POPs with halogen-free anions in one-pot and one-step, which would be regarded as highly efficient heterogeneous catalysts for CO2 conversion under ambient conditions.
Herein, we report a succinct one-pot strategy to construct a new series of pyridinium-based ionic porous organic polymers (denoted Py-iPOPs) with in situ formed hydroxide anions (OH−) and pyridinyl radicals towards halogen-free catalytic fixation of atmospheric CO2. As shown in Scheme 1, this elaborate design relies on base-catalyzed Knoevenagel condensation reaction between a newly designed acetonitrile-functionalized pyridinium ionic liquid (IL-ACN, Scheme S1†) and benzene-1,3,5-tricarbaldehyde (TFB), which was inspired by the eye-catching sp2-carbon conjugated covalent organic frameworks (COFs) and ionic vinylene-linked COFs.21–23 To the best of our knowledge, the present Py-iPOPs are the first examples of sp2-carbon-linked ionic POPs using cyano-substituted activated methylene-based ionic linkers. It is noted that the typical low-cost inorganic base KOH not only acts as the base catalyst for achieving the Knoevenagel polycondensation but also can remove the Br− anions and provide OH− anions within Py-iPOP-1 by ion-exchange during the synthetic process. More interestingly, the stable pyridinyl radicals were in situ formed by one-electron reduction of IL-ACN during the KOH base-involved one-pot process. By virtue of the active OH− anions and pyridinyl radicals, the typical Py-iPOP-1 was regarded as a superior halogen-free heterogeneous catalyst for atmospheric CO2 fixation under mild conditions.
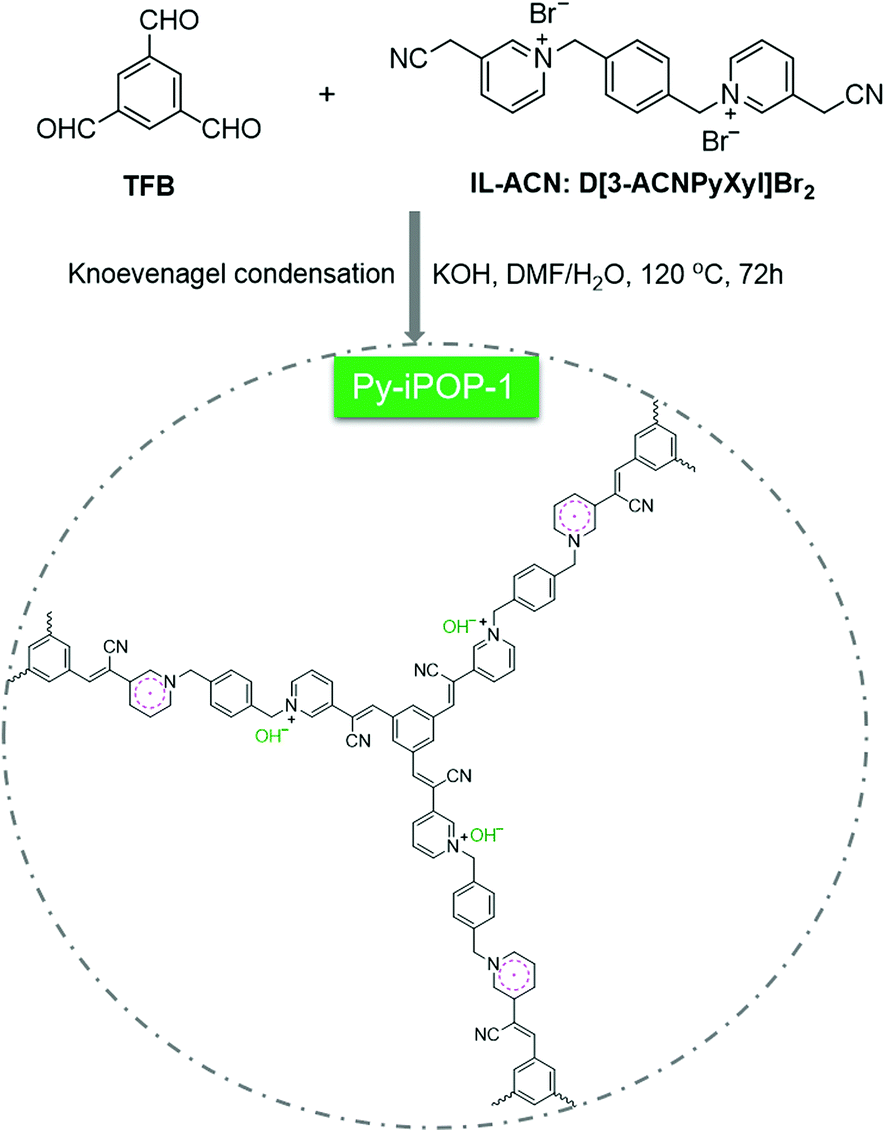 | ||
| Scheme 1 In situ synthesis of the pyridinium-based ionic porous organic polymer (Py-iPOP-1) with hydroxide anions and pyridinyl radicals by the KOH-catalyzed Knoevenagel condensation reaction. | ||
The one-pot synthesis of the typical cyano vinylene-linked Py-iPOP-1 was accomplished by KOH-catalyzed Knoevenagel condensation reaction between multi-aldehyde knotted TFB and cyano-substituted ionic linker IL-ACN in the mixture solvent of DMF/H2O (v/v: 5/1) at 120 °C for 72 h under solvothermal conditions. The key ionic linker IL-ACN was first synthesized by the quaternization reaction of 3-pyridineacetonitrile and α,α′-dibromo-p-xylene, which was confirmed by 1H NMR and 13C NMR spectra (Fig. S1†) plus elemental analysis (EA). Besides, the control polymers Py-iPOP-2 and Py-iPOP-3 were prepared using similar methods by replacing the KOH base catalyst with NaOH and Cs2CO3 (Schemes S2 and S3†). These Py-iPOPs were insoluble in common organic solvents such as tetrahydrofuran (THF), N,N-dimethylformamide (DMF) and dimethylsulfoxide (DMSO), indicative of their highly cross-linked networks.
The porosities of Py-iPOPs were investigated by N2 sorption measurements. As shown in Fig. S2A,† all the Py-iPOPs exhibit Type II isotherms with remarkable uptakes at high relative pressures, P/P0 = 0.80–0.99, indicating the presence of abundant large-sized mesoporosity within polymers. The Barrett–Joyner–Halenda (BJH) pore size distributions (Fig. S2B†) can reasonably demonstrate that Py-iPOPs have enriched large mesopores with the most probable pore sizes distributed from 23.5 to 33.6 nm. As listed in Table S1,† Py-iPOPs possess moderate Brunauer–Emmett–Teller (BET) surface areas ranging from 39 to 65 m2 g−1. Among them, Py-iPOP-1 that was prepared using KOH as the base catalyst not only has a good polymer yield (54%) but also possesses the highest BET surface area of 65 m2 g−1 and total pore volume of 0.41 cm3 g−1. The relatively low surface areas of these Py-iPOPs may be due to the rotation of methylene linkers and the filling of high contents of ionic sites paired with large counter-anions in the ionic networks.24,25 Indeed, all the polymers Py-iPOPs have high contents of pyridinium IL moieties (1.96–2.01 mmol g−1) calculated by the EA results (Table S1†). As depicted in the SEM images (Fig. S3†), the typical polymer Py-iPOP-1 shows the fluffy nano-morphology that was aggregated by many cross-linked nanoparticles. Moreover, the abundant mesoporosity of Py-iPOP-1 should originate from the nanocavities between the interconnected polymeric nanoparticles. X-ray diffraction (XRD) patterns (Fig. S4†) confirm that Py-iPOP-1 lost most of the sharp Bragg diffraction peaks that can be assigned to crystalline monomers TFB and IL-ACN, indicating the successful formation of semi-crystalline highly cross-linked polymers. The XRD pattern of Py-iPOP-1 exhibits two obvious broad diffraction peaks at 11.5° and 18.4° with corresponding d-spacing values of 0.75 and 0.48 nm, indicating a certain regular arrangement of knots and ionic linkers within the polymer.
The chemical structure of Py-iPOP-1 was confirmed by the Fourier transform infrared (FTIR) spectra and solid-state 13C cross-polarization/magic angle spinning nuclear magnetic resonance (CP/MAS NMR). In the FTIR spectra (Fig. S5†), for the monomer TFB, the obvious peak at 1696 cm−1 was attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of aldehyde groups,23 but it had completely disappeared in the formed Py-iPOP-1, indicating a high degree of polymerization by fully consuming the aldehyde groups. The emergence of two new signals at 1603 cm−1 and 1019 cm−1 is attributed to the stretching vibration of the CC bond and the bending vibration of the –CC–H bond.23,26 Compared with the sharp peak (CN+) at 1633 cm−1 for pyridinium-based IL-ACN, the relatively broad peak centered at 1648 cm−1 should be assigned to the stretching vibration of CN+ originating from the incorporated pyridinium moieties.27,28 These results indicate the successful synthesis of the pyridinium-based vinylene-linked Py-iPOP-1. Besides, the stretching vibration of cyano groups at 2242 cm−1 for IL-ACN was red-shifted to 2188 cm−1 for Py-iPOP-1, indicating the new location of the cyano side groups in the sp2-carbon conjugated part of Py-iPOP-1.21,26 The broad bands at 3446 and 3397 cm−1 should be attributed to the adsorbed water in IL-ACN and Py-iPOP-1, and originated from the chemical adsorption of water from the synthetic system or moist air due to their IL-based hydrophilic moieties.24–31 The existence of stably adsorbed water was further verified by thermogravimetric analysis (TGA) of the vacuum-dried sample Py-iPOP-1. As shown in Fig. S6,† the initial loss of weight (6.5 wt%) below 200 °C was mainly due to the removal of the adsorbed water. The TGA curve also indicates that Py-iPOP-1 has good thermal stability up to 250 °C. Although it is hard to distinguish OH− anions from H-bonded water, the obvious red-shift at 3397 cm−1 from 3446 cm−1 and a small adsorbed peak (O–H) at 1386 cm−1 could indicate the existence of OH− anions in Py-iPOP-1.18,24 In the 13C CP/MAS NMR spectrum (Fig. S7†), the broad peak centered at 163.5 ppm is ascribed to the sp2-carbon atoms on the pyridinium ring.23,27,33 The obvious peaks located at 137.9, 125.4 and 40.6 ppm are attributed to carbon atoms of the CC linkages, aromatic phenyl groups and methylene linkers within Py-iPOP-1, respectively.28,32,33 The obvious peak at 116.9 ppm that is assigned to cyano groups in the IL-ACN linker (Fig. S1B†) was changed to a weak peak at 107.8 ppm, indicating the formation of CC linkages with cyano side groups in the polymeric skeleton of Py-iPOP-1.23,34
O stretching vibration of aldehyde groups,23 but it had completely disappeared in the formed Py-iPOP-1, indicating a high degree of polymerization by fully consuming the aldehyde groups. The emergence of two new signals at 1603 cm−1 and 1019 cm−1 is attributed to the stretching vibration of the CC bond and the bending vibration of the –CC–H bond.23,26 Compared with the sharp peak (CN+) at 1633 cm−1 for pyridinium-based IL-ACN, the relatively broad peak centered at 1648 cm−1 should be assigned to the stretching vibration of CN+ originating from the incorporated pyridinium moieties.27,28 These results indicate the successful synthesis of the pyridinium-based vinylene-linked Py-iPOP-1. Besides, the stretching vibration of cyano groups at 2242 cm−1 for IL-ACN was red-shifted to 2188 cm−1 for Py-iPOP-1, indicating the new location of the cyano side groups in the sp2-carbon conjugated part of Py-iPOP-1.21,26 The broad bands at 3446 and 3397 cm−1 should be attributed to the adsorbed water in IL-ACN and Py-iPOP-1, and originated from the chemical adsorption of water from the synthetic system or moist air due to their IL-based hydrophilic moieties.24–31 The existence of stably adsorbed water was further verified by thermogravimetric analysis (TGA) of the vacuum-dried sample Py-iPOP-1. As shown in Fig. S6,† the initial loss of weight (6.5 wt%) below 200 °C was mainly due to the removal of the adsorbed water. The TGA curve also indicates that Py-iPOP-1 has good thermal stability up to 250 °C. Although it is hard to distinguish OH− anions from H-bonded water, the obvious red-shift at 3397 cm−1 from 3446 cm−1 and a small adsorbed peak (O–H) at 1386 cm−1 could indicate the existence of OH− anions in Py-iPOP-1.18,24 In the 13C CP/MAS NMR spectrum (Fig. S7†), the broad peak centered at 163.5 ppm is ascribed to the sp2-carbon atoms on the pyridinium ring.23,27,33 The obvious peaks located at 137.9, 125.4 and 40.6 ppm are attributed to carbon atoms of the CC linkages, aromatic phenyl groups and methylene linkers within Py-iPOP-1, respectively.28,32,33 The obvious peak at 116.9 ppm that is assigned to cyano groups in the IL-ACN linker (Fig. S1B†) was changed to a weak peak at 107.8 ppm, indicating the formation of CC linkages with cyano side groups in the polymeric skeleton of Py-iPOP-1.23,34
The chemical states and elemental compositions of Py-iPOP-1 were further confirmed by X-ray photoelectron spectroscopy (XPS). The full XPS survey spectrum (Fig. S8A†) shows the dominant elemental compositions of C (79.25 at%), N (10.73 at%) and O (9.93 at%) that are represented by the atomic concentration (at%). To our delight, a very small amount of residual Br− anions (ca. 0.09 at%) within Py-iPOP-1 (Fig. S8B†) could be negligible, confirming that Br− anions were almost completely replaced by OH− anions via the ion-exchange process. The presence of OH− anions is reflected by a divided OH− peak at 531.4 eV in the high-resolution O 1s XPS spectrum of Py-iPOP-1 (Fig. 1A).19,35 The chemically absorbed H2O is also detected by the signal at 532.5 eV,24,29 in accordance with the results in FTIR and TGA. In the C 1s spectrum (Fig. 1B), three deconvoluted peaks with binding energy (BE) values at 284.3, 284.8 and 286.0 eV for Py-iPOP-1 are ascribed to carbons of cyano side groups, aromatic rings and vinylene/methylene linkers, C–N and CN bonds in the pyridinium rings.28,36 To our surprise, three electronic states for N atoms are recognized from the divided peaks from the N 1s spectra (Fig. 1C) for Py-iPOP-1. The main peak that appeared at 399.5 eV originated from the inherent cyano groups (–CN) in Py-iPOP-1.26,32 In particular, the divided peak with a higher BE value at 401.0 eV is assigned to the N atoms of cationic Py-N+ moieties, while another peak with a lower BE value at 398.7 eV should be attributed to the N atoms in the reduced pyridinyl radicals (Py˙–N).28,33,37 The molar ratio of Py-N+ to Py˙–N was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.53 based on their atomic concentrations as shown in Fig. 1C implying that about 60% of pyridinium cationic N atoms were in situ reduced to pyridinyl radical-based neutral N atoms during the strong base KOH-catalyzed Knoevenagel condensation reaction. Furthermore, the electron paramagnetic resonance (EPR) spectrum of Py-iPOP-1 (Fig. 1D) exhibits a characteristic sharp signal with a g value of 2.003, manifesting the successful formation of pyridinyl radicals, similar to the common viologen organic radicals.28,37 In fact, a series of stable pyridinyl radicals were discovered in the last century by one-electron reduction of 1-alkylpyridinium ions using chemical, electrochemical and photochemical reduction procedures.38 To the best of our knowledge, the present Py-iPOP-1 should be regarded as the first example of pyridinyl radical-based porous organic materials. The cationic radical Py-iPOP-1 was formed by the in situ reduction of dicationic IL-ACN under the KOH-involving alkaline conditions without using extra reducing agents such as Na2S2O4. Besides, another two cationic radical porous polymers, Py-iPOP-2 with OH− anions and Py-iPOP-3 with CO32− anions, were also obtained using NaOH and Cs2CO3 as the base catalysts, which were fully confirmed by their XPS spectra (Fig. S9 and S10†). Indeed, our recent works have provided some clues to prepare viologen or phenanthroline cationic radicals within porous polymers under alkaline K2CO3-involving synthetic conditions.28,37
:1.53 based on their atomic concentrations as shown in Fig. 1C implying that about 60% of pyridinium cationic N atoms were in situ reduced to pyridinyl radical-based neutral N atoms during the strong base KOH-catalyzed Knoevenagel condensation reaction. Furthermore, the electron paramagnetic resonance (EPR) spectrum of Py-iPOP-1 (Fig. 1D) exhibits a characteristic sharp signal with a g value of 2.003, manifesting the successful formation of pyridinyl radicals, similar to the common viologen organic radicals.28,37 In fact, a series of stable pyridinyl radicals were discovered in the last century by one-electron reduction of 1-alkylpyridinium ions using chemical, electrochemical and photochemical reduction procedures.38 To the best of our knowledge, the present Py-iPOP-1 should be regarded as the first example of pyridinyl radical-based porous organic materials. The cationic radical Py-iPOP-1 was formed by the in situ reduction of dicationic IL-ACN under the KOH-involving alkaline conditions without using extra reducing agents such as Na2S2O4. Besides, another two cationic radical porous polymers, Py-iPOP-2 with OH− anions and Py-iPOP-3 with CO32− anions, were also obtained using NaOH and Cs2CO3 as the base catalysts, which were fully confirmed by their XPS spectra (Fig. S9 and S10†). Indeed, our recent works have provided some clues to prepare viologen or phenanthroline cationic radicals within porous polymers under alkaline K2CO3-involving synthetic conditions.28,37
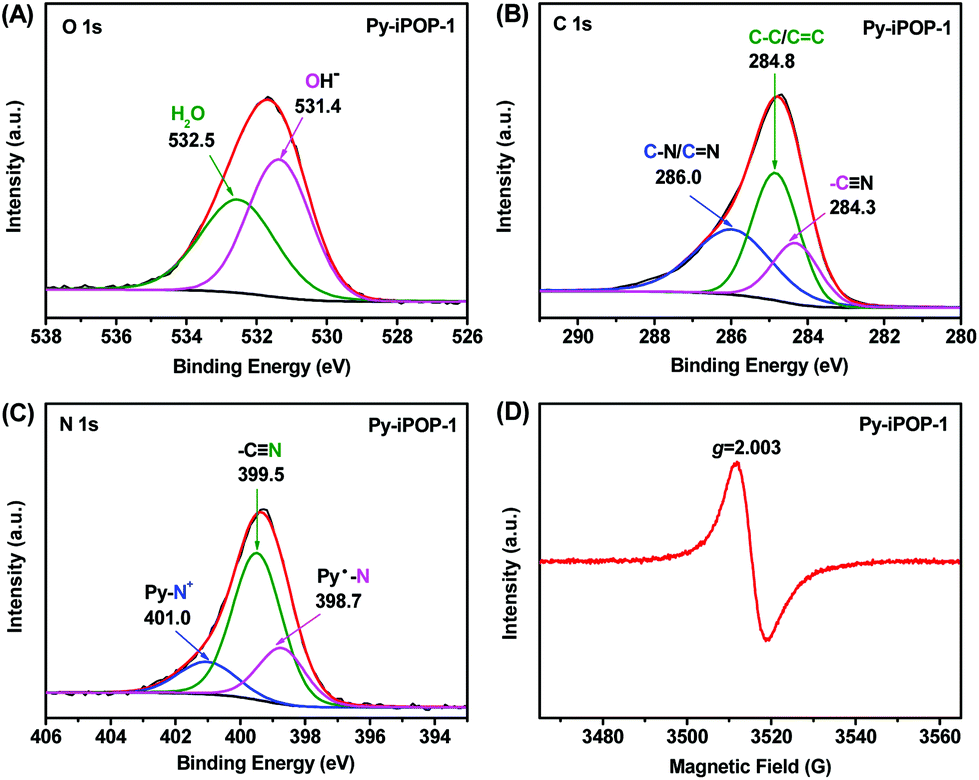 | ||
| Fig. 1 XPS spectra of Py-iPOP-1: (A) O 1s, (B) C 1s and (C) N 1s. (D) EPR spectrum of Py-iPOP-1 at room temperature. | ||
By virtue of basic hydroxide anions and active pyridinyl radicals within polymers, Py-iPOPs were employed as metal-free and halogen-free heterogeneous catalysts for the conversion of CO2 into cyclic carbonates under mild conditions. First, the catalytic performances of a series of Py-iPOP-1, Py-iPOP-2 and Py-iPOP-3 were carried out in the cycloaddition of CO2 with a model substrate glycidol in the absence of solvents and co-catalysts under atmospheric pressure (0.1 MPa) using CO2 balloons (Table 1). At a low temperature of 40 °C for 72 h, these catalysts Py-iPOPs afforded moderate yields of 68%–80% for the product glycerol carbonate. Among them, OH− anion-paired Py-iPOP-1 gave a much higher yield of 80% than Py-iPOP-2 (68%) also with the OH− anion (Table 1, entries 1 and 2), which may be due to the higher N and Py-IL contents based on the EA and XPS results (seen in Table S1, Fig. S8A and S9A†), and also close to the larger BET surface area (65 m2 g−1) of Py-iPOP-1. Besides, Py-iPOP-1 also provides a bit better yield than Py-iPOP-3 (73%) with CO32− anions (Table 1, entry 3), which should be attributed to their same catalytically active bicarbonate anions (HCO3−).39 It was reported that regardless of OH− anions or CO32− anions with H2O, they can smoothly react with CO2 to produce the same nucleophilic HCO3− anion with a high leaving ability, which was regarded as the true catalytic active site for the activation and conversion of CO2 and epoxides into cyclic carbonates.18,39–41 The higher basicity of OH− anions than that of CO32− anions makes for the easy formation of the HCO3− active anions without an activation barrier,18,39 giving rise to the better catalytic activity of Py-iPOP-1. The presence of a small amount of inbuilt H-bonded water in Py-iPOPs can promote the conversion of CO32− anions to HCO3− anions during the reaction, which was indicated by the reaction of K2CO3·1.5H2O with CO2 to produce KHCO3.40 By optimizing the reaction conditions, the optimal metal-free and halogen-free heterogeneous catalyst Py-iPOP-1 afforded a very high yield of 99% with a high selectivity of 99% at 60 °C for 48 h (Table 1, entry 4), which was not only comparable to the yield of 99% (Table 1, entry 6) over the control homogeneous catalyst bromide anion-dominated IL-ACN, but also surpassed the corresponding yields (82%, 85%, 56% and 70%, Table 1, entries 7–10) over various inorganic bases KOH, KHCO3, K2CO3 and K2CO3·(1.5H2O). The better catalytic activity of K2CO3·(1.5H2O) than that of K2CO3 indicates the important role of crystal water for enhancing the formation of KHCO3 when reacting with CO2.40 It is worth noting that different inorganic bases and IL-ACN have different basicity in terms of their pH values in the aqueous solution in the order KOH > K2CO3 > KHCO3 > IL-ACN. However, the obtained catalytic activities (Table 1, entries 6–9) are not well consistent with their basicity, which are in agreement with the nucleophilicity of different anions in the order of Br− > HCO3− > OH− > CO32−.5,6 Therefore, a good nucleophilic anion with a high leaving ability is the key catalytic active site for CO2 conversion.39 Very interestingly, the above inorganic bases exhibited good catalytic performances in the CO2 cycloaddition with glycidol, which were different from the results of absent catalytic activities using KOH and KHCO3 for the cycloaddition reactions between CO2 and 1,2-epoxyhexane or styrene oxide in previous works.39,41 The above control results suggest that the true catalytic roles of OH− anions and CO32− anions in both inorganic bases and Py-iPOPs should be attributed to the in situ formed HCO3− anions. Certainly, the better catalytic behavior of Py-iPOP-1 than those of KOH and KHCO3 also indicates that pyridinium cations and pyridinyl radicals could play key roles in catalysis.39,41 In fact, pioneering works have revealed that pyridine or pyridinyl radicals can react with CO2 to yield the carbamate radical intermediate [Py+-CO2−],42,43 which might promote the catalytic efficiency in this work.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | T (°C) | t (h) | Yieldc (%) | Selectivityc (%) |
| a Reaction conditions: glycidol (2 mmol), CO2 balloon (0.1 MPa), the catalyst (40 mg, 4 mol% based on the Py-IL content), temperature (T = 30–60 °C), time (t = 48–96 h). b The dosages of IL-ACN, KOH, K2CO3, KHCO3 and K2CO3·(1.5H2O) were 20 mg, 4.5 mg, 5.6 mg, 8.0 mg and 6.6 mg based on the Py-IL content (2.01 mmol g−1) in Py-iPOP-1. c Yield and selectivity of the product 4-(hydroxymethyl)-1,3-dioxolan-2-one were determined from the 1H NMR spectra. | |||||
| 1 | Py-iPOP-1 | 40 | 72 | 80 | 99 |
| 2 | Py-iPOP-2 | 40 | 72 | 68 | 99 |
| 3 | Py-iPOP-3 | 40 | 72 | 73 | 99 |
| 4 | Py-iPOP-1 | 60 | 48 | 99 | 99 |
| 5 | Py-iPOP-1 | 30 | 96 | 72 | 99 |
| 6 | IL-ACNb | 60 | 48 | 99 | 99 |
| 7 | KOHb | 60 | 48 | 82 | 99 |
| 8 | KHCO3b | 60 | 48 | 85 | 99 |
| 9 | K2CO3b | 60 | 48 | 56 | 99 |
| 10 | K2CO3·(1.5H2O) | 60 | 48 | 70 | 99 |
The excellent substrate compatibility was also investigated by Py-iPOP-1 in the CO2 cycloaddition with various epoxides. As shown in Fig. 2, the small-sized epoxides 1a–1d (glycidol, epichlorohydrin, epibromohydrin and 1,2-epoxybutane) could be smoothly converted to corresponding cyclic carbonates (2a–2d) in high yields of 99% under mild conditions (60 °C for 48 h) and even obtain considerable yields of 80–99% at near room temperatures (30–40 °C). For the commonly used substrate epichlorohydrin, the catalytic performance of Py-iPOP-1 that was carried out under atmospheric and mild conditions could exceed the catalytic behaviors over many previously reported metal-/halogen-free heterogeneous catalysts (seen Table S2† for details),7,8,17–19 and even compared favourably with many halogen anion-dominated porous ionic polymers.10,12,13,24,27–29 For some large-sized inert substrates (1e–1h) including 1,2-epoxyhexane, 1,2-epoxyoctane, glycidyl phenyl ether and allyl glycidyl ether, high yields of 88–96% could be also achieved by increasing temperatures to 80–120 °C. In addition, a five-cycle assessment in the CO2 conversion with glycidol demonstrated that the robust heterogeneous catalyst Py-iPOP-1 afforded good reusability without loss of catalytic activities (Fig. S19†), owing to the well-preserved chemical structures and catalytic active sites of the recovered catalyst (seen FTIR in Fig. S20† and XPS in Fig. S21†).
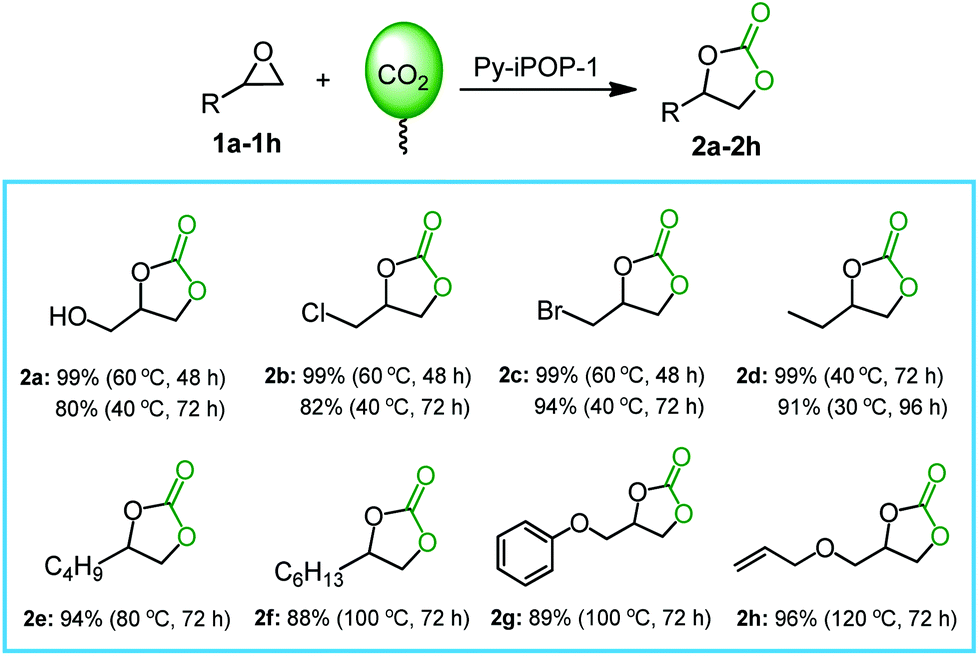 | ||
| Fig. 2 The substrate scope for CO2 cycloaddition with epoxides catalyzed by Py-iPOP-1. Reaction conditions: substrate (2 mmol), the catalyst (40 mg for 1a–1e, 50 mg for 1f–1h), CO2 balloon (0.1 MPa), 30–120 °C, 48–96 h. The optimal yields of all the products were determined by 1H NMR analyses of crude products and the detailed spectra and data were provided in the Fig. S11–S18.† | ||
Briefly, the well-designed polymer Py-iPOP-1 behaves as a remarkable metal-halogen-free heterogeneous catalyst for CO2 conversion, which can be attributed to the enriched active sites including OH− anions and pyridinyl radicals. It is worth pointing out that the basic polymer Py-iPOP-1 with OH− anions exhibits considerable CO2 adsorption capacities (i.e., 0.82 and 0.55 mmol g−1 at 273 and 298 K, 0.1 MPa, Fig. S22†), which can contribute to the enrichment of CO2 for the subsequent reaction, and also make for sufficient interaction between the CO2 molecules, activated epoxides and catalytic active sites.27,28 Besides, the fluffy nano-morphology and abundant mesoporosity can be beneficial for the sufficient exposure and high dispersion of catalytic active sites, which enhance the mass transfer of epoxides to the catalyst and thus improve the catalytic efficiency in solvent-free multi-phase reaction systems.28,44
Based on our experimental results and previous reports, a plausible catalytic mechanism for the halogen-free synthesis of cyclic carbonates by reacting CO2 with epoxides using Py-iPOP-1 is described in Scheme S4. First, the basic anions OH− within Py-iPOP-1 directly reacted with CO2 to produce HCO3− anions with a good nucleophilicity and leaving ability18,39 while [Py+-CO2−] adducts derived from active pyridinyl radicals and CO2 could be converted to the corresponding bicarbonate salt [Py+]-[HCO3−] in the presence of a small amount of absorbed water within Py-iPOP-1.41,45,46 Then, the epoxide was together activated by hydrogen-bonding interactions of the O atom from epoxide with the α-proton of pyridinium cations and free –OH groups of the absorbed water molecule, thus promoting the activation and ring opening of the epoxide via the C–O bond polarization.14–16,24,29 Meanwhile, the formed nucleophilic HCO3− anions attacked the β-carbon atom of the epoxide with less steric hindrance, leading to the formation of the oxygen ion intermediate.18,39,46 In particular, the increased temperatures can accelerate this rate-determining step for the epoxide ring opening by hydrogen-bonding interactions and a nucleophilic attack.46–49 Successively, the inserted CO2 was coupled with the negative O atom of the intermediate by a nucleophilic attack and the carbonate link was formed.39,46Lastly, the final product cyclic carbonate was obtained by the intramolecular ring closure of the carbonate link and release of HCO3− anions.18,46 The catalyst Py-iPOP-1 can be recovered by the reversible conversion of the immediate catalyst with HCO3− anions with the desorption of CO2. In a word, the basic microenvironment of Py-iPOP-1 with OH− anions and pyridinyl radicals can be beneficial for the facile formation of nucleophilic HCO3− anions and enhancement of the simultaneous CO2 capture and catalytic conversion. For the catalyst Py-iPOP-3, CO32− anions can also be converted to the active HCO3− anions when reacting with CO2 and a small amount of absorbed water, which obeys a similar bicarbonate anion-involved catalytic mechanism to Py-iPOP-1.
In summary, we reported a new series of vinylene-linked pyridinium-based ionic radical porous organic polymers with hydroxide anions or carbonate anions, which were directly constructed by one-pot Knoevenagel polycondensation. By virtue of active OH− anions and pyridinyl radicals, the typical heterogeneous catalyst Py-iPOP-1 exhibited impressive metal-free and halogen-free catalytic performance in the conversion of CO2 into cyclic carbonates under mild conditions. This work carves out a general way to construct more kinds of task-specific ionic radical porous organic polymers for exploiting more diverse applications such as other types of metal-free heterogeneous catalytic reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21603089), the Natural Science Foundation of Jiangsu Province (BK20160209), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (16KJB150014), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX20_2354) and the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP).Notes and references
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- S. Subramanian, Y. Song, D. Kim and C. T. Yavuz, ACS Energy Lett., 2020, 5, 1689–1700 CrossRef CAS.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- F. Zhang, Y. Wang, X. Zhang, X. Zhang, H. Liu and B. Han, Green Chem. Eng., 2020, 1, 82–93 CrossRef.
- D.–H. Lan, N. Fan, Y. Wang, X. Gao, P. Zhang, L. Chen, C.–T. Au and S.–F. Yin, Chin. J. Catal., 2016, 37, 826–845 CrossRef CAS.
- S. N. Talapaneni, O. Buyukcakir, S. H. Je, S. Srinivasan, Y. Seo, K. Polychronopoulou and A. Coskun, Chem. Mater., 2015, 27, 6818–6826 CrossRef CAS.
- S. Subramanian, J. Park, J. Byun, Y. Jung and C. T. Yavuz, ACS Appl. Mater. Interfaces, 2018, 10, 9478–9484 CrossRef CAS.
- K. Huang, J.-Y. Zhang, F. Liu and S. Dai, ACS Catal., 2018, 8, 9079–9102 CrossRef CAS.
- R. Luo, X. Liu, M. Chen, B. Liu and Y. Fang, ChemSusChem, 2020, 13, 3945–3966 CrossRef CAS.
- R. Luo, M. Chen, X. Liu, W. Xu, J. Li, B. Liu and Y. Fang, J. Mater. Chem. A, 2020, 8, 18408–18424 RSC.
- S. Subramanian, J. Oppenheim, D. Kim, T. S. Nguyen, W. M. H. Silo, B. Kim, W. A. Goddard III and C. T. Yavuz, Chem, 2019, 5, 3232–3242 CAS.
- Y. Zhang, K. Liu, L. Wu, H. Huang, Z. Xu, Z. Long, M. Tong, Y. Gu, Z. Qin and G. Chen, Dalton Trans., 2021, 50, 11878–11888 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS.
- C. Liu, L. Shi, J. Zhang and J. Sun, Chem. Eng. J., 2022, 427, 131633 CrossRef CAS.
- Y. Zhao, H. Huang, H. Zhu and C. Zhong, Microporous Mesoporous Mater., 2022, 329, 111526 CrossRef CAS.
- X. Wang, Q. Dong, Z. Xu, Y. Wu, D. Gao, Y. Xu, C. Ye, Y. Wen, A. Liu, Z. Long and G. Chen, Chem. Eng. J., 2021, 403, 126460 CrossRef CAS.
- Z. Guo, X. Cai, J. Xie, X. Wang, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 12812–12821 CrossRef CAS.
- Y. Zhou, W. Zhang, L. Ma, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9387–9398 CrossRef CAS.
- Y. He, X. Li, W. Cai, H. Lu, J. Ding, H. Li, H. Wan and G. Guan, ACS Sustainable Chem. Eng., 2021, 9, 7074–7085 CrossRef CAS.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS.
- S. Xu, M. Richter and X. Feng, ACC Mater. Res., 2021, 2, 252–265 CrossRef CAS.
- F. Meng, S. Bi, Z. Sun, B. Jiang, D. Wu, J.-S. Chen and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 13614–13620 CrossRef CAS.
- Y. Zhang, G. Chen, L. Wu, K. Liu, H. Zhong, Z. Long, M. Tong, Z. Yang and S. Dai, Chem. Commun., 2020, 56, 3309–3312 RSC.
- S. Hou, M. Meng, D. Liu and P. Zhang, ChemSusChem, 2021, 14, 3059–3063 CrossRef CAS PubMed.
- R. Bu, L. Zhang, X.-Y. Liu, S.-L. Yang, G. Li and E.-Q. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 26431–26440 CrossRef CAS PubMed.
- G. Chen, X. Huang, Y. Zhang, M. Sun, J. Shen, R. Huang, M. Tong, Z. Long and X. Wang, Chem. Commun., 2018, 54, 12174–12177 RSC.
- Y. Zhang, K. Liu, L. Wu, H. Zhong, N. Luo, Y. Zhu, M. Tong, Z. Long and G. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16907–16916 CrossRef CAS.
- Y. Zhang, K. Zhang, L. Wu, K. Liu, R. Huang, Z. Long, M. Tong and G. Chen, RSC Adv., 2020, 10, 3606–3614 RSC.
- J. Byun, H. A. Patel, D. Thirion and C. T. Yavuz, Polymer, 2017, 126, 308–313 CrossRef CAS.
- Y. Byun, S. H. Je, S. N. Talapaneni and A. Coskun, Chem. – Eur. J., 2019, 25, 10262–10283 CrossRef CAS.
- J. Xu, Y. He, S. Bi, M. Wang, P. Yang, D. Wu, J. Wang and F. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12065–12069 CrossRef CAS.
- J. Lin, S. Bi, Z. Fan, Z. Fu, Z. Meng, Z. Hou and F. Zhang, Polym. Chem., 2021, 12, 1661–1667 RSC.
- Y. Wei, W. Chen, X. Zhao, S. Ding, S. Han and L. Chen, Polym. Chem., 2016, 7, 3983–3988 RSC.
- L. Chen, J. Chang, Y. Zhang, Z. Gao, D. Wu, F. Xu, Y. Guo and K. Jiang, Chem. Commun., 2019, 55, 3406–3409 RSC.
- M. Zhang, M. Lu, Z.-L. Lang, J. Liu, M. Liu, J.-N. Chang, L.-Y. Li, L.-J. Shang, M. Wang, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 6500–6506 CrossRef CAS.
- G. Chen, Y. Zhang, K. Liu, X. Liu, L. Wu, H. Zhong, X. Dang, M. Tong and Z. Long, J. Mater. Chem. A, 2021, 9, 7556–7565 RSC.
- E. M. Kosower, Stable pyridinyl radicals, Prep. Org. Chem., 1983, 117–162 CAS.
- T. Ema, K. Fukuhara, T. Sakai, M. Ohbo, F.-Q. Bai and J.-y. Hasegaw, Catal. Sci. Technol., 2015, 5, 2314–2321 RSC.
- C. Zhao, X. Chen and C. Zhao, Ind. Eng. Chem. Res., 2010, 49, 12212–12216 CrossRef CAS.
- J. Liu, G. Yang, Y. Liu, D. Wu, X. Hu and Z. Zhang, Green Chem., 2019, 21, 3834–3838 RSC.
- M. Z. Kamrath, R. A. Relph and M. A. Johnson, J. Am. Chem. Soc., 2010, 132, 15508–15511 CrossRef CAS.
- Y. Yan, E. L. Zeitler, J. Gu, Y. Hu and A. B. Bocarsly, J. Am. Chem. Soc., 2013, 135, 14020–14023 CrossRef CAS PubMed.
- G. Chen, Y. Zhang, J. Xu, X. Liu, K. Liu, M. Tong and Z. Long, Chem. Eng. J., 2020, 381, 122765 CrossRef CAS.
- H. Zhou, W. Chen, J.-H. Liu, W.-Z. Zhang and X.-B. Lu, Green Chem., 2020, 22, 7832–7838 RSC.
- K. R. Roshan, R. A. Palissery, A. C. Kathalikkattil, R. Babu, G. Mathai, H.-S. Leed and D.-W. Park, Catal. Sci. Technol., 2016, 6, 3997–4004 RSC.
- M. Liu, L. Liang, X. Li, X. Gao and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- H. Zhong, Y. Su, X. Chen, X. Li and R. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef CAS PubMed.
- F. Zhang, S. Bulut, X. Shen, M. Dong, Y. Wang, X. Cheng, H. Liu and B. Han, Green Chem., 2021, 23, 1147–1153 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03465k |
| This journal is © The Royal Society of Chemistry 2022 |