
Clusterin increases mitochondrial respiratory chain complex I activity and protects against hexavalent chromium-induced cytotoxicity in L-02 hepatocytes
Yuanyuan
Xiao
,
Ming
Zeng
,
Lirong
Yin
 ,
Na
Li
and
Fang
Xiao
*
,
Na
Li
and
Fang
Xiao
*
Department of Health Toxicology, Xiangya School of Public Health, Central South University, Changsha 410078, PR China. E-mail: fangxiao@csu.edu.cn; Tel: +86 -731-84487130
First published on 15th November 2018
Abstract
Previous evidence revealed significant elevated liver cancer mortality in the areas where water was contaminated with hexavalent chromium [Cr(VI)], which highlighted that we should pay more attention to Cr(VI)-induced cytotoxicity in hepatocytes. We found that Clusterin (CLU) was up-regulated in Cr(VI)-exposed L-02 hepatocytes, but the role CLU played in Cr(VI)-induced cytotoxicity has never been explored. In the present study, we demonstrate Cr(VI) targeted mitochondrial respiratory chain complex I (MRCC I) activity and induced reactive oxygen species (ROS) accumulation, which caused mitochondrial damage that was characterized by the increase of permeability transition pore (PTP) open rate, the collapse of mitochondrial membrane potential (MMP), and the release of apoptosis-inducing factor (AIF) and Cytochrome C (Cyt C) from mitochondria to cytoplasm, which then induced cell viability loss and increased aspartate transaminase (AST)/alanine transaminase (ALT) leakage. We reveal that Cr(VI) may regulate CLU expression through the ROS-ataxia telangiectasia mutant (ATM)–insulin-like growth factor 1 (IGF-1) axis, and CLU expression was positively correlated to MRCC I activity. We further confirmed that CLU may regulate MRCC I activity via modulating its subunit nicotinamide adenine dinucleotide dehydrogenase (ubiquinone) Fe–S protein 3 (NDUFS3) expression. By the establishment of CLU over-expression cells, we found that over-expression of CLU alleviated Cr(VI)-induced MRCC I inhibition and further rescued cell viability loss and reduced AST and ALT leakage. Thus, we reached the conclusion that the CLU-induced increase of MRCC I activity protected against Cr(VI)-induced cytotoxicity. The present research will provide new experimental evidence for thoroughly clarifying the cytotoxicity and the carcinogenic mechanism of Cr(VI).
Introduction
The genotoxic, mutagenic, and cytotoxic effects of heavy metal, such as hexavalent chromium [Cr(VI)], exposure have long been a hot topic of toxicology research. Cr(VI) and its compounds are widely used in numerous industrial processes including chrome plating, stainless steel manufacturing, and leather tanning.1 Chromium (Cr) can be absorbed through the gastrointestinal and respiratory tracts and the skin. An ecological mortality study conducted in Greece revealed significant elevated liver cancer mortality in the area where water was contaminated with Cr(VI),2 which highlighted that we should pay more attention to Cr(VI)-induced cytotoxicity in the hepatocytes.Several previous studies have demonstrated that reactive oxygen species (ROS) accumulation and oxidative stress play a key role in the cytotoxicity and carcinogenicity of heavy metals such as Cr(VI).3,4 The mitochondrial respiratory chain, which is also known as the electron transport chain (ETC), releases a considerable amount of superoxide during normal oxidative metabolism.5 Mitochondria produce ATP by oxidative phosphorylation via the mitochondrial respiratory chain, which consists of five multi-subunit mitochondrial respiratory chain complexes (MRCCs) (I–V) composed of 75 nuclear DNA-encoded and 13 mitochondrial DNA encoded proteins.6 MRCC I (nicotinamide adenine dinucleotide (NADH): ubiquinone oxidoreductase) is the first and the largest protein complex of the ETC, and it has an essential role in maintaining mitochondrial function and integrity. MRCC I consists of at least 43 subunits and accepts electrons from NADH and transports them to co-factor ubiquinone via several iron–sulphur clusters.7 The known sites of ROS production in the mitochondrial ETC are MRCC I and III.8
Clusterin (CLU), also known as apolipoprotein J, is a soluble 80 kDa heterodimeric glycoprotein that is abundant in physiological fluids.9 There are two major transcript isoforms of CLU due to alternative splicing, which consequently yield two protein isoforms with distinct functions, anti-apoptotic secreted CLU (sCLU) and pro-apoptotic nuclear CLU (nCLU).10 sCLU is the main form of CLU under normal physiological conditions, and in this paper, CLU refers to sCLU. As a stress-induced chaperone, CLU blocks protein misfolding and aggregation in a manner similar to small heat shock proteins (HSPs) (sHsps),11 inhibits apoptosis by interacting with activated Bax and protects the cells from ER stress-induced apoptosis through a physical interaction with 78 kDa glucose-regulated protein (GRP78).12
We found that CLU was up-regulated in Cr(VI)-exposed L-02 hepatocytes, but the role of CLU played in Cr(VI)-induced cytotoxicity has never been explored. To the best of our knowledge, this is the first research study that links the anti-apoptosis function of CLU with MRCC I, which will provide new experimental evidence for thoroughly clarifying Cr(VI)-induced cytotoxicity. CLU is over-expressed in various cancer tissues and plays an important role in chemotherapy resistance and metastatic spread, thus we think it may help the hepatocytes to escape apoptosis and then undergo malignant transformation after Cr(VI) exposure.
Material and methods
Reagents
Roswell Park Memorial Institute (RPMI)-1640 medium and fetal bovine serum (FBS) were purchased from Gibco (Gaithersburg, MD, USA). Potassium dichromate (K2Cr2O7) was obtained from Changsha chemical reagents company (changsha, China). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), MitoTEMPO, glutamate (Glu), malate (Mal), and rotenone (ROT) were obtained from Sigma (St Louis, MO, USA). Apoptosis-inducing factor (AIF) (B-9) (sc-55519), Cytochrome C (Cyt C) (A-8) (sc-13156), CLU-α (B-5) (sc-5289), ataxia telangiectasia mutant (ATM) (1A1) (sc-73615), and nicotinamide adenine dinucleotide dehydrogenase (ubiquinone) Fe–S protein 3 (NDUFS3) (D-4) (sc-374282) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Insulin-like growth factor 1 (IGF-1) (20215-1-AP) and β-actin (60008-1-Ig) were obtained from Proteintech (Wuhan, China). Cleaved Caspase-9 (AF5240) and Cleaved Caspase-3 (AF7022) were purchased from Affinity Biosciences (Shanghai, China). All other chemicals and solvents were of analytical grade or the best pharmaceutical grade.Cell lines
The immortalized human cell line L-02 hepatocyte and highly metastatic human hepatocellular carcinoma (HCC) cell line HCCLM3 were purchased from China Center for Type Culture Collection of Wuhan University. Another HCC HepG2 line was obtained from Type Culture Collection of Chinese Academy of Sciences, Shanghai, China.Establishment of CLU over-expression and knockdown cells
The cDNA encoding CLU (pDONR223-CLU) (Changsha Yingrun Biotechnology Co., Ltd, Changsha, China) was amplified by reverse transcription polymerase chain reaction (RT-PCR). CLU was cloned into pLVX-IRES-puro vector (Clontech, Mountain View, CA) and expression vectors of CLU were generated. Lentiviral carrying CLU cDNA was generated and harvested as described previously.13 pYr-LV-CLU small hairpin RNA (shRNA), a sCLU-RNA interference (RNAi) lentiviral vector, was constructed (Changsha Yingrun Biotechnology Co., Ltd, Changsha, China). A GFP-lentiviral vector (pYr-LV-GFP) was used as a negative control. Lentiviral carrying CLU shRNA or scrambled shRNA was used to infect target cells.Cell viability detection
The hepatocytes were seeded at 104 cells per well in a 96-well plate for MTT assay. The chemicals of indicated final concentrations were added to the well. Then, 10 μl of 5 mg ml−1 MTT solution was added into each well for an additional incubation of 4 h. The cells were exposed to 150 μl of dimethyl sulfoxide (DMSO). The absorbance was read on an ELISA reader Versamax (Molecular Devices, Sunnyvale, CA, USA) at 570 nm.Alanine aminotransaminase (ALT) and aspartate aminotransaminase (AST) level measurement
ALT and AST levels in the supernatants of hepatocytes were detected using the commercial kits (Jiancheng Institute of Biological Products, Nanjing, China) as described previously.14Mitochondria isolation
The treated L-02 hepatocytes were collected (300 g for 5 min) and then washed twice with ice-cold PBS and re-suspended with five volumes of buffer A (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, and 0.1 mM phenylmethylsulfonyl fluoride, pH 7.5). The cells were left on ice for 2 min, homogenized with a syringe, and then centrifuged twice at 1500g for 15 min at 4 °C. The supernatant obtained was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 15 min at 4 °C. The resulting obtained mitochondrial pellets were re-suspended in buffer A. The protein concentrations of the mitochondrial suspensions were determined using the Coomassie blue protein-binding method, as explained by Bradford.15
000g for 15 min at 4 °C. The resulting obtained mitochondrial pellets were re-suspended in buffer A. The protein concentrations of the mitochondrial suspensions were determined using the Coomassie blue protein-binding method, as explained by Bradford.15
Permeability transition pore (PTP) open rate determination
The opening of the PTP was determined using a mitochondrial PTP detection kit obtained from GenMed Scientifics Inc. (Shanghai, China) according to the protocol. Briefly, the temperature of the enzyme-labeled instrument was set to 25 °C, and the wavelength was 540 nm. A 96-well plate was placed into the enzyme-labeled instrument to measure the blank value; readings were taken every 30 s for 10 min and then set back to zero. The mitochondrial suspension (70 μl) was added to the well, and then GENMED buffer (Reagent A) (170 μl) was added to the suspension. The plate was read using the enzyme-labeled instrument and the absorbance (A) was recorded. PTP open rate (%) = (A of the control group − A of the treatment group) × 100%/A of the control group.Mitochondrial membrane potential (MMP) determination
The cationic fluorescent dye Rhodamine 123 (Rh123) was used for the estimation of MMP. The hepatocytes were stained with 2 μg ml−1 Rh123 for 30 min in the dark. The fluorescence was monitored using a fluorescence spectrophotometer at the excitation and emission wavelengths of 495 nm and 535 nm, respectively.Western blotting
Protein from whole cell lysates was extracted using an EpiQuik Whole Cell Extraction Kit (Epigentek, Farmingdale, NY, USA), and protein from the cytosolic fraction was extracted using a Nuclear/Cytosol Fractionation Kit (Biovision, Danvers, MA, USA). Protein (50 μg) extracted from the whole cell lysates (Cleaved Caspase-9, Cleaved Caspase-3, CLU, ATM, IGF-1, NDUFS3, and β-actin) or from the cytosolic fraction (AIF, Cyt C, and β-actin) was separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a polyvinylidene difluoride (PVDF) membrane for western blotting analysis. Primary antibodies were used. The membranes were washed and incubated with secondary antibodies. Immunoreactive proteins were detected by enhanced chemiluminescence (ECL).ROS level detection
The intracellular ROS level was measured using hydroethidine (HE) (Molecular Probes, Inc., Eugene, Ore). HE is a non-fluorescent compound that can diffuse through cell membranes and then can be rapidly oxidized to ethidium under the action of O2˙−. Briefly, the cells were treated with 2 μM HE for 15 min at 37 °C, and then they were analyzed by flow cytometry. ROS level was quantified using the mean intensity of fluorescence (MIF).MRCC I activity detection
The activity of mitochondrial respiratory chain complex (MRCC) I was determined with a Mito Complex I activity assay kit (GenMed Scientifics Inc., Shanghai, China) according to the protocol.Quantitative real-time PCR (qPCR)
The total cellular RNA of the hepatocytes was isolated using the RNeasy Mini Kit (QIAGEN, Hilden, Germany). For cDNA synthesis, a total of 5 μg of RNA was reverse transcribed to cDNA using the PrimeScript RT reagents kit (Takara, Dalian, China) according to the manufacturer's instructions. qPCR analysis was performed using SYBR®Premix Taq™ (Takara, Dalian, China) with the 7900HT Fast Real-Time PCR System (Applied Biosystems, Inc., Foster City, CA, USA), and each PCR reaction was repeated at least three times. ACTB was used as a control. The data were analyzed by the 2−ΔΔCt method. PCR protocol: initial denaturation for 2 min at 95 °C, 35 cycles of 95 °C for 10 s, 60 °C for 5 s, and 72 °C for 12 s. Sequences for qPCR primers: CLU, 5′-GAGCAGCTGAACGAGCAGTTT-3′ (forward), 5′-CTTCGCCTTGCGTGAGGT-3′ (reverse); ACTB, 5′-ATCATGTTTGAGACCTTCAACA-3′ (forward), 5′-CATCTCTTGCTCGAAGTCCA-3′ (reverse).Statistical analyses
All data are expressed as mean ± standard deviation (SD) of at least 3 independent experiments. Statistical significance for differences among groups was calculated by one-way analysis of variance (ANOVA). All statistical analyses were performed using SPSS ver.19.0 software. p < 0.05 was considered statistically significant.Results
Cr(VI)-Induced cytotoxicity was associated with the inhibition of MRCC I activity
The cytotoxic effect of Cr(VI) on L-02 hepatocytes was evaluated using the MTT method. Treatment with Cr(VI) (0, 5, 10, 15 μM) for 24 h inhibited cell growth in a dose-dependent manner (Fig. 1A). The inhibition rate was about 40% at 15 μM. Cr(VI) treatment also induced a remarkable elevation in both serum ALT and AST levels when compared with the normal level (Fig. 1B), because the damaged hepatocytes released both ALT and AST into the extracellular space. We also demonstrated that Cr(VI) exposure caused mitochondrial damage, which was characterized by the change of mitochondrial permeability transition. As shown in Fig. 1C, Cr(VI) treatment obviously increased PTP opening rate. Since the MMP is a highly sensitive indicator of the mitochondrial permeability transition, the effect of Cr(VI) on MMP was measured by Rh123 staining. As shown in Fig. 1D, Cr(VI) significantly decreased the MMP in a dose-dependent manner. MMP and fluorescence intensity reported in our experiment are negatively correlated because quenching of Rh123 was proportional to the potential, and the increase in fluorescence means depolarization. Our results showed that Cr(VI) significantly caused mitochondrial damage such as the opening of PTP and the collapse of the MMP. These events could lead to the release of AIF and Cyt C from mitochondria into the cytoplasm and further result in the initiation of apoptosis. As shown in Fig. 1E, treatment with different concentrations of Cr(VI) induced a significant increase of AIF, Cyt C, Caspase-9, and Caspase-3 protein levels.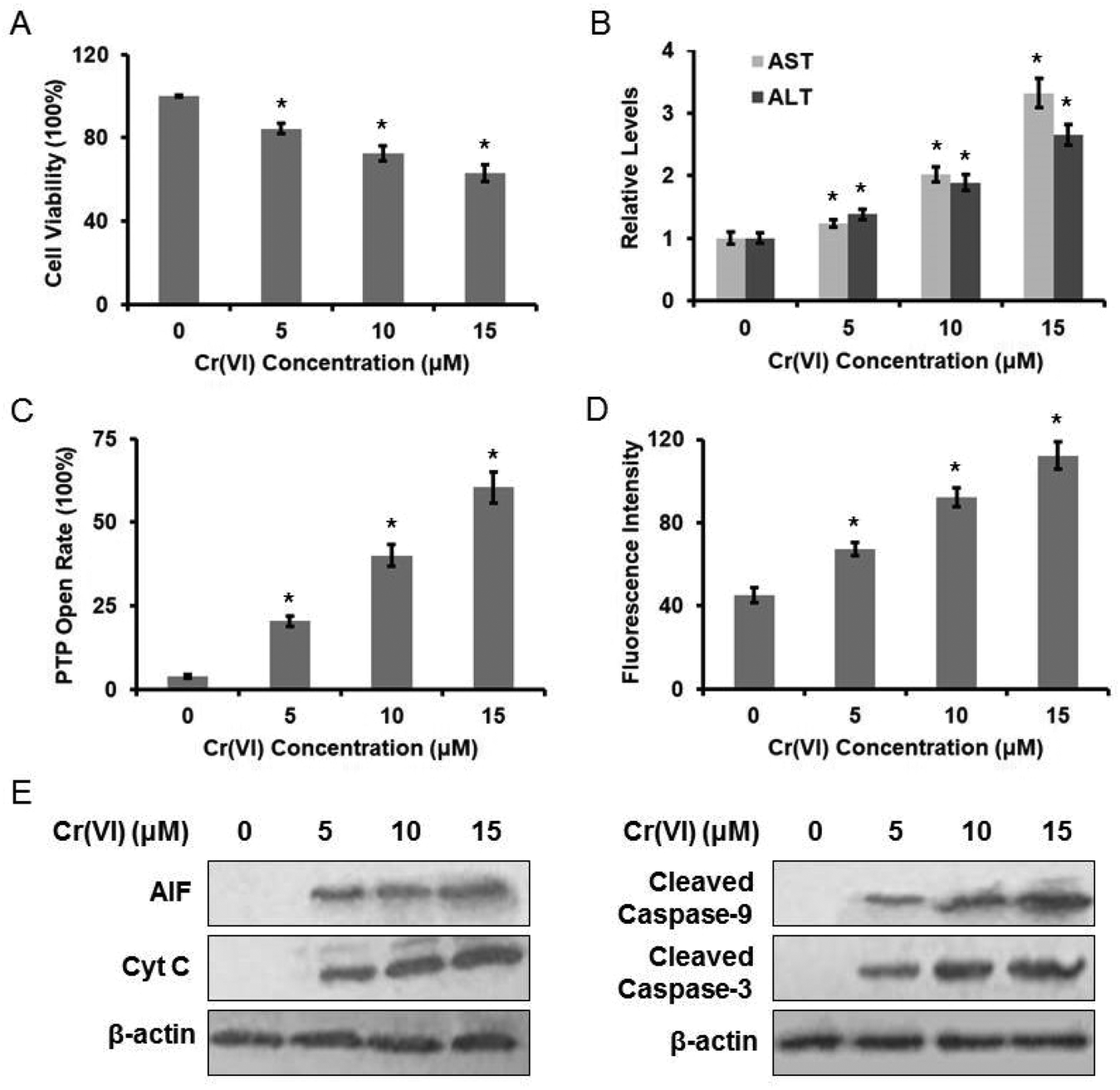 | ||
| Fig. 1 Cr(VI)-Induced cytotoxicity in L-02 hepatocytes. L-02 hepatocytes were exposed to different concentrations of Cr(VI) (0, 5, 10, and 15 μM) for 24 h. (A) Cell viability was determined by MTT assay. (B) AST and ALT levels were examined using the commercial kits. (C) PTP opening rate was determined using the commercial kit. (D) MMP was assayed with the cationic fluorescent dye Rh123. (E) The protein levels of AIF, Cyt C, cleaved Caspase-9 and cleaved Caspase-3 were examined using western blotting. *p < 0.05, compared with control. | ||
We previously demonstrated that Cr(VI) targeted MRCC I to induce ROS-dependent Caspase-3 activation.16 As shown in Fig. 2A, different concentrations of Cr(VI) induced significant ROS formation in the hepatocytes in a dose-dependent manner. We also confirmed that Cr(VI) treatment caused the inhibition of MRCC I activity (Fig. 2B), which indicated the dysfunction of ETC. Treatment with the MRCC I substrates 10 mM glutamate/10 mM malate (Glu/Mal) for 10 min prior to Cr(VI) exposure restored the viability of the hepatocytes, indicating that Cr(VI) exerted a cytotoxic effect on cell viability through MRCC I (Fig. 2C). The application of Glu/Mal also alleviated the Cr(VI)-induced increase of AST and ALT levels (Fig. 2D). To better elucidate the role of MRCC I played in Cr(VI)-induced cytotoxicity, we also used a MRCC I inhibitor rotenone (ROT), whose inhibitory effect on MRCC I is shown in Fig. 2E. ROT treatment (5 μM, 24 h) significantly enhanced the Cr(VI)-induced increase of ROS formation (Fig. 2F), aggravated the Cr(VI)-induced inhibition of cell viability (Fig. 2G), and augmented the Cr(VI)-induced increase of AST and ALT leakage (Fig. 2H).
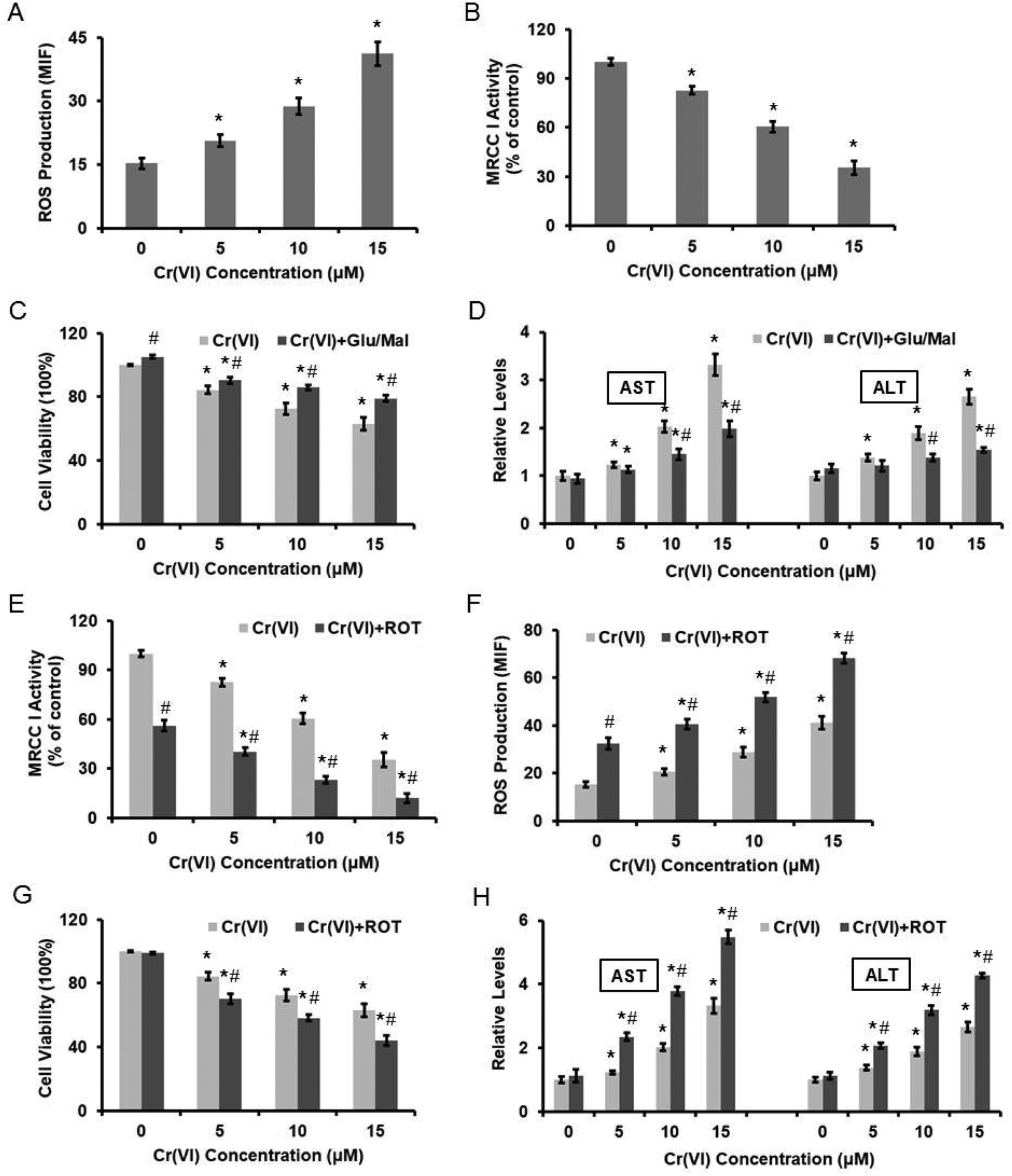 | ||
| Fig. 2 Cr(VI)-Induced cytotoxicity was associated with the inhibition of MRCC I activity. L-02 hepatocytes were exposed to different concentrations of Cr(VI) (0, 5, 10, and 15 μM) for 24 h. (A) ROS level was measured using HE and was quantified by the MIF. (B) MRCC I activity was determined using the commercial kit. L-02 hepatocytes were treated with the MRCC I substrates 10 mM Glu/10 mM Mal for 10 min prior to Cr(VI) exposure. (C) Cell viability. (D) AST and ALT levels. ROT (5 μM, 24 h) was used to treat L-02 hepatocytes together with Cr(VI). (E) MRCC I activity. (F) ROS level. (G) Cell viability. (H) AST and ALT levels. *p < 0.05, compared with control. #p < 0.05, compared with Cr(VI) alone treatment. | ||
CLU was up-regulated in Cr(VI)-exposed L-02 hepatocytes
It was reported that CLU serum levels correlated positively with workers’ exposure to heavy metals and to Cr blood and urine concentrations, and CLU was a sensitive biomarker of the organismal oxidative stress caused by ROS accumulation.17 We then examined CLU expression in Cr(VI)-exposed L-02 hepatocytes and found that Cr(VI) up-regulated both mRNA (Fig. 3A) and protein (Fig. 3B) levels of CLU in a dose-dependent manner. Since CLU has long been implicated in protecting cells from apoptosis,18 we speculated that the up-regulation of CLU after Cr(VI) exposure may also have an anti-apoptosis function.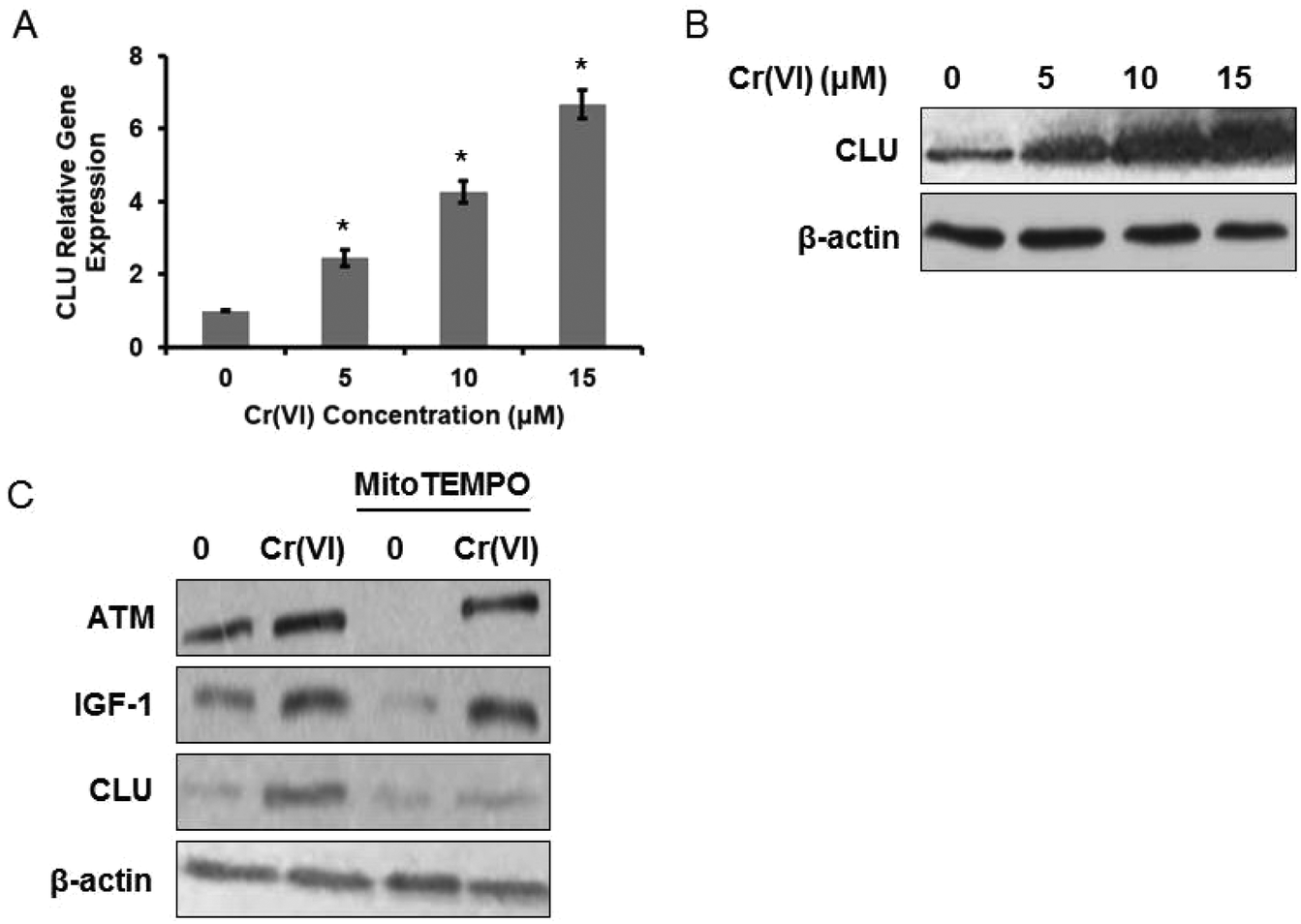 | ||
| Fig. 3 CLU was up-regulated in Cr(VI)-exposed L-02 hepatocytes. L-02 hepatocytes were exposed to different concentrations of Cr(VI) (0, 5, 10, and 15 μM) for 24 h. (A) CLU mRNA expression was determined using qPCR. The data were analyzed by the 2−ΔΔCt method. (B) CLU protein level was examined using western blotting. The hepatocytes were exposed to 15 μM Cr(VI) for 24 h with or without MitoTEMPO pretreatment (10 μM for 1 h). (C) The protein expression levels of ATM, IGF-1, and CLU were determined. *p < 0.05, compared with control. | ||
We next explored the mechanism associated with the Cr(VI)-induced increase of CLU. Heavy metal-mediated formation of ROS causes various modifications to DNA bases (such as 8-oxo-dG),19 ataxia telangiectasia mutant (ATM) kinase is a key protein mediating cellular responses to DNA damage, and ATM-dependent IGF-1 induction has been confirmed to regulate CLU expression after DNA damage.20 Based on previous evidence, we speculated that Cr(VI) may regulate CLU expression through the ROS-ATM-IGF-1 axis. As shown in Fig. 3C, Cr(VI) exposure up-regulated the protein expression levels of ATM, IGF-1 and CLU. Pretreatment with MitoTEMPO (10 μM, 1 h), a mitochondria-targeted antioxidant, alleviated the Cr(VI)-induced increase of these protein levels.
CLU may regulate MRCC I activity
Since we confirmed that MRCC I inhibition-related ROS formation accounted for Cr(VI)-induced cytotoxicity, we speculated that CLU may protected against Cr(VI)-induced cytotoxicity through modulation of MRCC I activity. We first wanted to demonstrate if CLU expression was correlated to MRCC I activity. CLU has been confirmed to play a critical role in cancer metastasis,21 and a positive correlation between CLU expression and Gleason pattern was observed in prostate cancer. In addition, CLU overexpression promoted invasion and metastasis in HCC.22 We chose two HCC cell lines, HepG2 (low metastatic potential) and HCCLM3 (high metastatic potential), to confirm our hypothesis. We examined CLU expression in L-02 hepatocytes and the two HCC cell lines, and we found that HepG2 showed a similar CLU expression level while HCCLM3 showed a significantly higher CLU expression level compared with that of L-02 hepatocytes (Fig. 4A). MRCC I activity shown in Fig. 4B revealed a similar change. MRCC I activity of HCCLM3 was about 2.87 times higher than that of L-02 hepatocytes.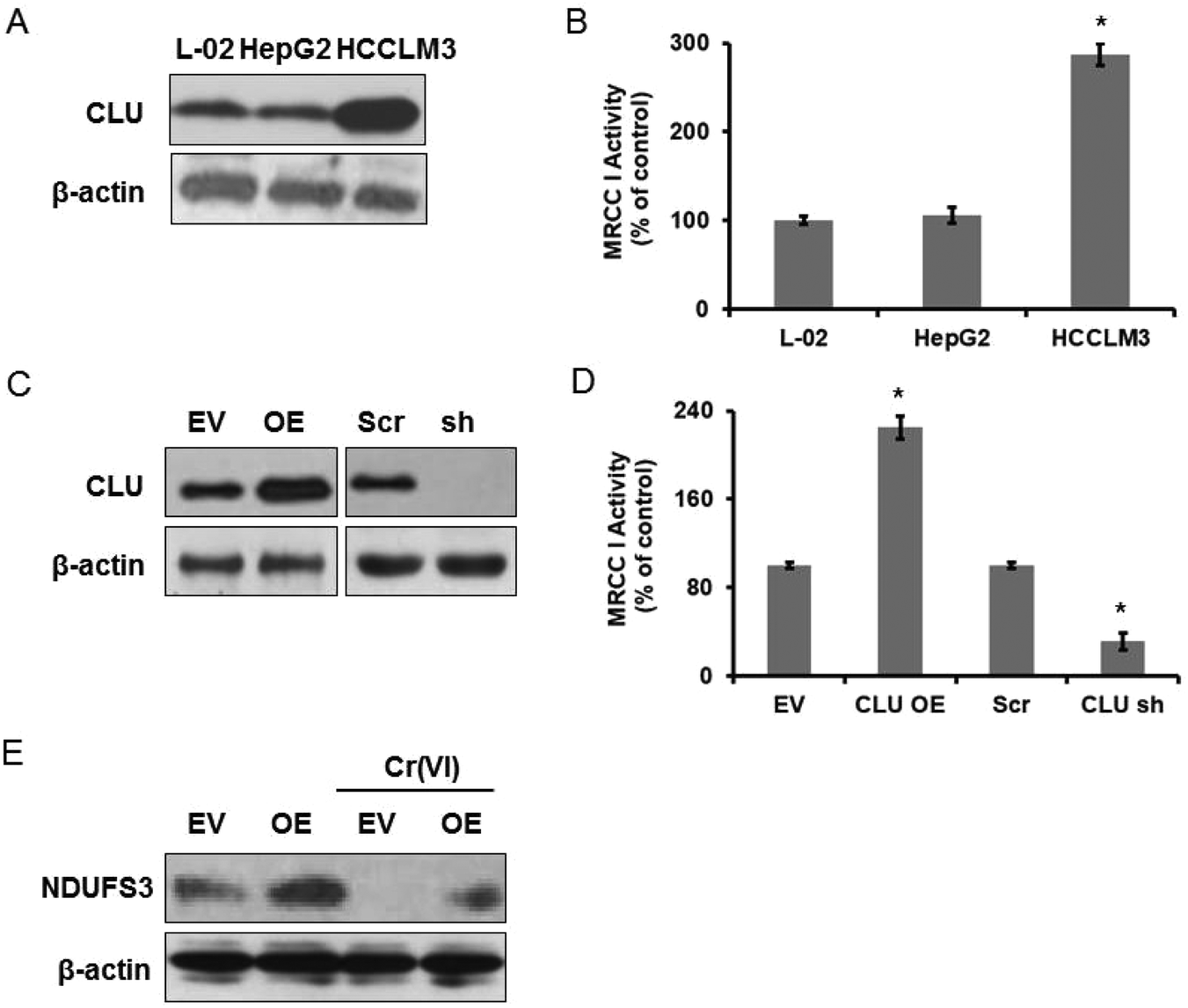 | ||
| Fig. 4 CLU may regulate MRCC I activity. Human L-02 hepatocytes and two HCC cell lines HepG2 and HCCLM3 were used. (A) CLU protein level was examined using western blotting. (B) MRCC I activity was determined using the commercial kit. The hepatocytes were transfected either with CLU-cDNA (EV-transfected cells served as a control) or with CLU shRNA (Scr-transfected cells served as a control). (C) CLU protein level. (D) MRCC I activity. (E) NDUFS3 protein level was examined using western blotting. *p < 0.05, compared with control. | ||
To better elucidate CLU function, we over-expressed and knocked down CLU in L-02 hepatocytes. The western blotting result shown in Fig. 4C revealed increased CLU expression in the hepatocytes transfected with CLU-cDNA (CLU OE). Empty vector (EV)-transfected cells served as a control. CLU expression was dramatically decreased in the hepatocytes transfected with CLU shRNA. Cells transfected with scrambled (Scr) sequence served as a control. We then determined MRCC I activity and found that CLU OE cells showed higher activity and CLU sh cells showed lower activity compared with that of the relative control cells (Fig. 4D). The above results confirmed that CLU expression was positively correlated to MRCC I activity.
Mitochondrial protein NDUFS3 is a core component of MRCC I in the respiratory chain of the mitochondrial matrix for MRCC I assembly and activity.23 NDUFS3 encodes one of the iron–sulfur protein components of MRCC I, and mutations or inhibition of this gene have been confirmed to be associated with MRCC I deficiency. We examined NDUFS3 protein expression and the result is shown in Fig. 4E. CLU OE cells showed higher expression of NDUFS3, compared with the EV cells, and alleviated Cr(VI)-induced inhibition of NDUFS3 expression. Therefore, we thought CLU may regulate MRCC I activity via modulating NDUFS3 expression, but the exact regulatory mechanism remained to be explored.
CLU-induced increase of MRCC I activity protected against Cr(VI)-induced cytotoxicity
We next examined the effect of CLU on Cr(VI)-induced cytotoxicity. Over-expression of CLU alleviated the inhibition of MRCC I activity (Fig. 5A), the accumulation of intracellular ROS level (Fig. 5B), the increase of PTP opening rate (Fig. 5C), and the collapse of MMP (Fig. 5D) after exposure of different concentrations of Cr(VI). Furthermore, over-expression of CLU reduced the Cr(VI)-induced release of AIF and Cyt C from mitochondria to cytoplasm and inhibited the increase of cleaved Caspase-9 and cleaved Caspase-3 expression levels (Fig. 5E). CLU also rescued Cr(VI)-induced loss of cell viability (Fig. 5F) and reduced the Cr(VI)-induced increase of AST and ALT leakage to extracellular space (Fig. 5G). These results together suggested that the CLU-induced increase of MRCC I activity protected against Cr(VI)-induced cytotoxicity.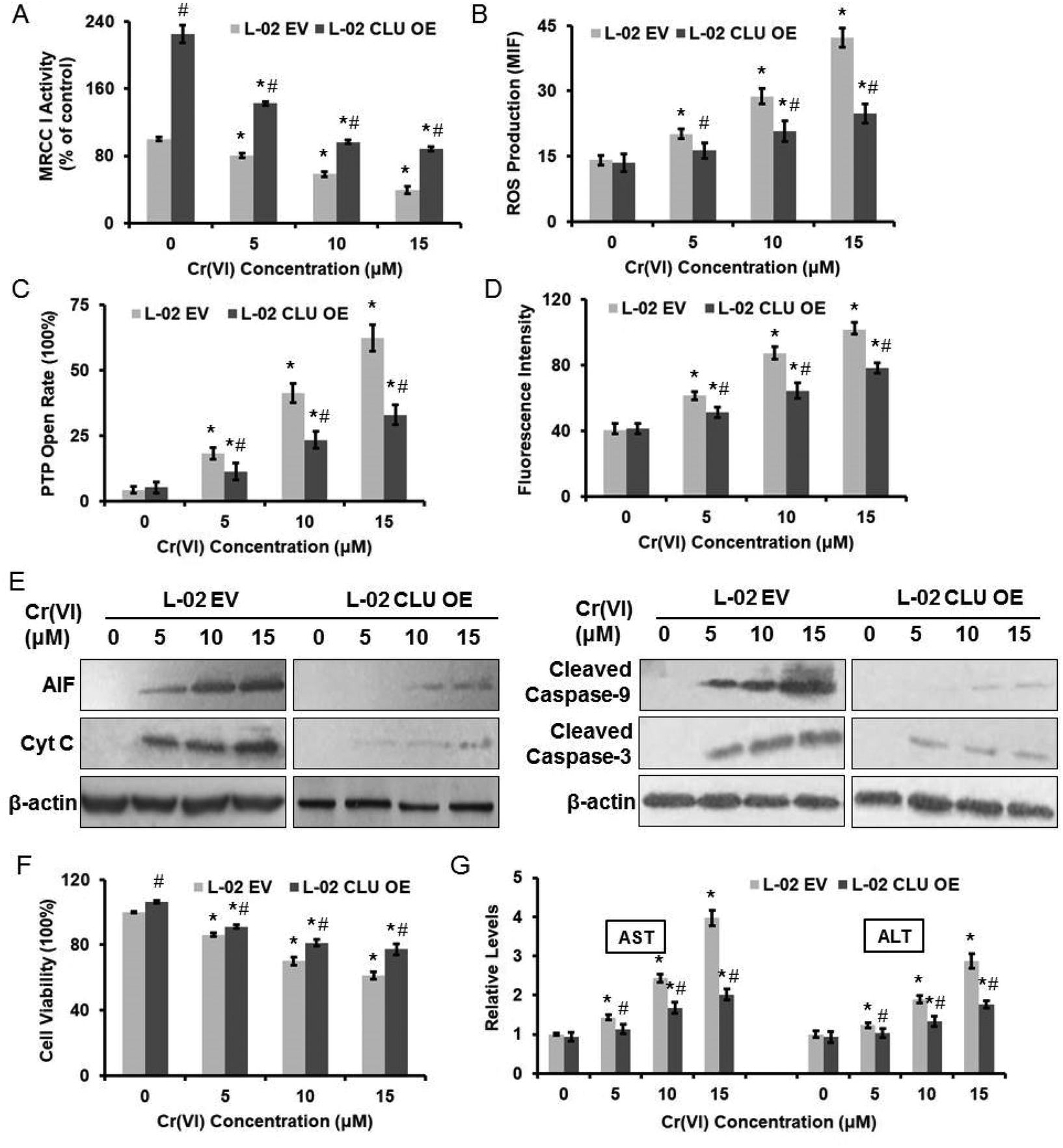 | ||
| Fig. 5 CLU-induced increase of MRCC I activity protected against Cr(VI)-induced cytotoxicity. L-02 EV and OE cells were exposed to different concentrations of Cr(VI) (0, 5, 10, and 15 μM) for 24 h. (A) MRCC I activity was determined using the commercial kit. (B) ROS level was measured using HE. (C) PTP opening rate was determined using the commercial kit. (D) MMP was assayed with the cationic fluorescent dye Rh123. (E) The protein expression levels of AIF, Cyt C, cleaved Caspase-9 and cleaved Caspase-3 were examined using western blotting. (F) Cell viability was determined by MTT assay. (G) AST and ALT levels were examined using the commercial kits. *p < 0.05, compared with control. #p < 0.05, compared with L-02 EV hepatocytes. | ||
Discussion
We demonstrated in the present study that Cr(VI) targeted MRCC I and induced ROS accumulation, caused mitochondrial damage that was characterized by the increase of PTP opening rate, the collapse of MMP, and the release of AIF and Cyt C from mitochondria to cytoplasm, and then induced cell viability loss and increased AST/ALT leakage. The PTP is known as a voltage-dependent, nonselective high-conductance inner mitochondrial membrane channel that allows solutes up to 1500 Da to pass freely in and out of mitochondria. The PTP opens under conditions of mitochondrial damage; the opening is greatly enhanced by ATP depletion and oxidative stress. MMP is a key parameter that represents mitochondrial function. The collapse of MMP can imply the increase of PTP opening, which results in cellular dysfunction and even cell death.MRCC I, the critical entry point of mitochondrial ETC, is also one of the main sites of ROS production.24 The electron donor NADH from the TCA cycle can be oxidized by MRCC I and then initiates electron transport. ROT is a phytogenic toxin that can bind to MRCC I and increase superoxide release into the mitochondrial matrix.25 To better elucidate the role of MRCC I played in Cr(VI)-induced cytotoxicity, we used MRCC I inhibitor ROT and its substrates Glu/Mal to treat the hepatocytes. We found that ROT treatment significantly enhanced the Cr(VI)-induced increase of ROS formation, aggravated the inhibition of cell viability, and augmented the increase of AST and ALT leakage, while Glu/Mal treatment showed the opposite results. We confirmed that the inhibition of MRCC I activity played a key role in Cr(VI)-induced cytotoxicity.
We found that Cr(VI)-induced cytotoxicity was accompanied with a high expression of CLU, which interacts with a broad spectrum of molecules and regulates several functions including cell proliferation/survival.26 An accumulation of CLU has been reported in the injured organs of various diseases.27,28 Over-expression of CLU has been identified in various types of malignant tumors, which correlates with advanced tumor stage, poor prognosis, and chemotherapy resistance.29 Furthermore, the immunohistochemistry revealed that CLU staining is mainly cytoplasmic and only occasionally in the nucleus in malignant tumor tissue,30 suggesting that elevated CLU, especially sCLU, disrupts the balance of apoptosis regulation and leads to malignant transformation and tumor progression. Evidence also suggested that CLU was an independent predictive factor for prognosis of HCC and it facilitated metastasis through eukaryotic translation initiation factor 3 subunit I (EIF3I)/Akt/matrix metalloproteinase 13 (MMP13) signaling.31 We confirmed that HCCLM3 with high metastatic potential showed high CLU expression as well as high MRCC I activity, while HepG2 with low metastatic potential showed relatively low CLU expression as well as low MRCC I activity, indicating that CLU expression was correlated to MRCC I activity. Cyt C, which is released upon the increase of PTP opening, contributes to the activation of Apoptosome.32 Apoptosome further activates Caspase-9 and results in Caspase-3 activation.33 We established CLU OE and sh cell lines and obtained similar results. Over-expression of CLU alleviated Cr(VI)-induced MRCC I inhibition, ROS accumulation, PTP opening, and MMP collapse. Furthermore, over-expression of CLU reduced Cr(VI)-induced AIF and Cyt C release, inhibited Caspase-3 activation, and further rescued cell viability loss, and reduced AST and ALT leakage. Thus, we reached the conclusion that CLU-induced increase of MRCC I activity protected against Cr(VI)-induced cytotoxicity.
The model of our working hypothesis is shown in Fig. 6. Cr(VI) inhibited MRCC I, led to intracellular ROS accumulation and then caused cytotoxicity. The Cr(VI)-mediated burst generation of ROS induced a DNA damage response, which activated ATM kinase and its down-stream executor IGF-1, and resulted in up-regulation of CLU. NDUFS3, one of the precursor subunits in the 45-subunit MRCC I, is known to play a vital role in the proper assembly of intact, functional MRCC I. The reduced expression of NDUFS3 may inhibit the normal function of MRCC I, lead to decreased MRCC I activity, and further induce mitochondrial dysfunction.6 Cr(VI)-Induced high expression of CLU may activate the MRCC I subunit NDUFS3, which increased MRCC I activity, inhibited ROS production, and further alleviated cytotoxicity. Thus, we think that Cr(VI)-induced high expression of CLU protected against cytotoxicity via increasing MRCC I activity. Since CLU plays an important role in the occurrence, development and metastasis of malignant tumors, we think it may help the hepatocytes to escape apoptosis and then undergo malignant transformation after Cr(VI) exposure. It was also reported that NDUFS3 expression was significantly higher in invasive carcinoma tissues when compared with normal tissues.34 If NDUFS3 has a functional role in regulating metabolic features of transformed cells, CLU-induced up-regulation of NDUFS3 may also contribute to the carcinogenesis of Cr(VI). Although we demonstrated that CLU may regulate MRCC I activity via modulating NDUFS3 expression, the exact regulatory mechanism remains to be explored and further research work is also needed, because MRCC I is a giant complex with 44 subunits, and its function is very complicated and not well demonstrated.
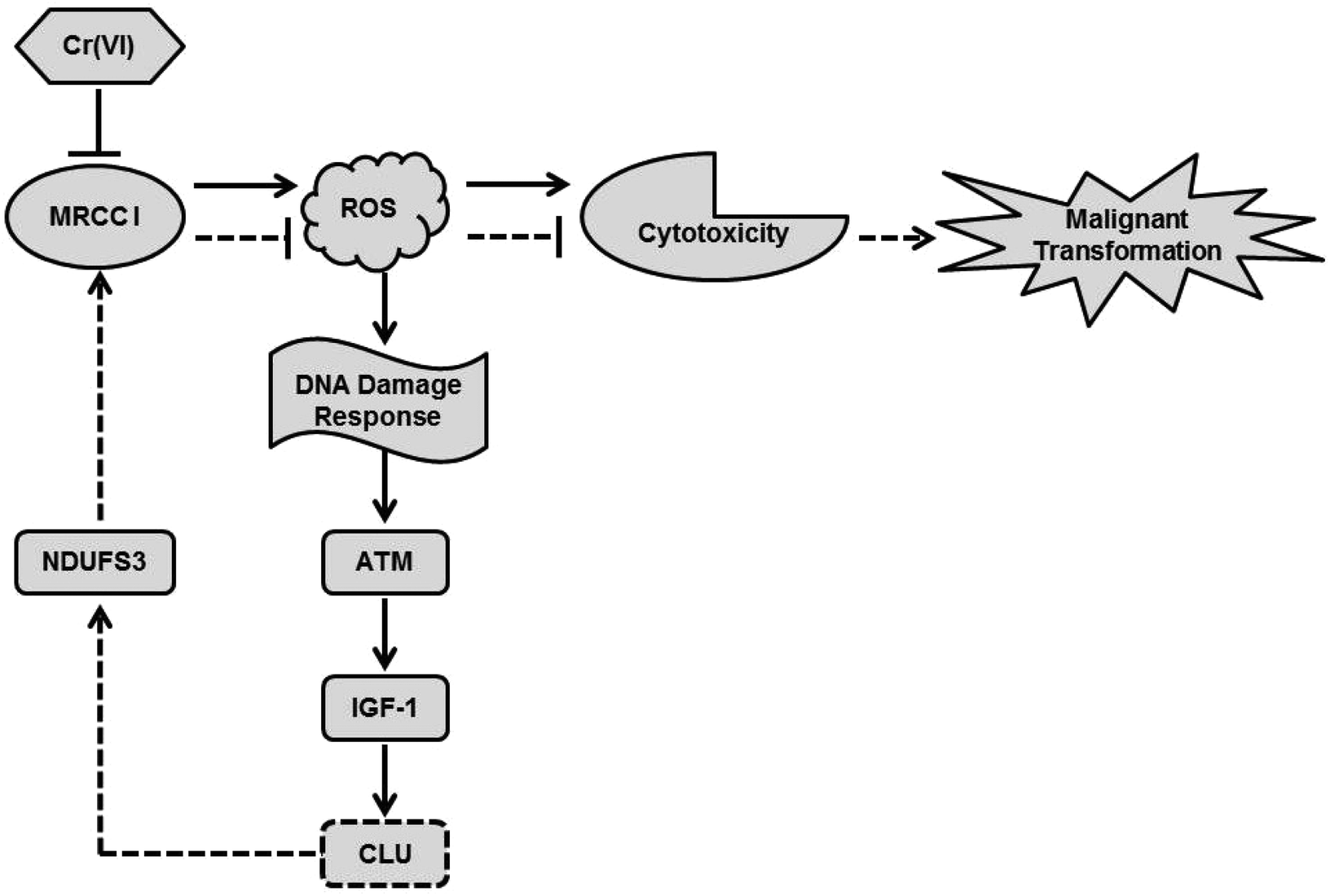 | ||
| Fig. 6 The model of our working hypothesis. Cr(VI) inhibited MRCC I, led to intracellular ROS accumulation and then caused cytotoxicity. ROS could induce a DNA damage response, which activated ATM kinase and its down-stream executor IGF-1, and resulted in up-regulation of CLU. High expression of CLU may activate the MRCC I subunit NDUFS3, which increased MRCC I activity, inhibited ROS production, and further alleviated cytotoxicity. CLU may help the hepatocytes to escape apoptosis and then undergo malignant transformation after Cr(VI) exposure. | ||
Conclusions
In conclusion, our findings unravel a novel mechanism wherein CLU increased MRCC I activity and protected against Cr(VI)-induced cytotoxicity in L-02 hepatocytes, which will provide new experimental evidence for thoroughly clarifying the toxic effects of Cr(VI). Cr(VI)-Induced high expression of CLU may also help the hepatocytes to escape apoptosis and then undergo malignant transformation after Cr(VI) exposure.Author contributions
F.X. designed the experiments, supervised the project and wrote the paper. Y.Y.X., L.R.Y., and N.L. performed the experiments and analyzed the data.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank all of the individuals in this laboratory for their valuable ideas and suggestions. This research was financially supported by the National Natural Science Foundation of China (No. 81773478).References
- M. Costa and C. B. Klein, Crit. Rev. Toxicol., 2006, 36, 155 CrossRef CAS PubMed.
- A. Linos, A. Petralias, C. A. Christophi, E. Christoforidou, P. Kouroutou, M. Stoltidis, A. Veloudaki, E. Tzala, K. C. Makris and M. R. Karagas, Environ. Health, 2011, 10, 50 CrossRef CAS PubMed.
- A. K. Patlolla, B. Constance, H. Diahanna and P. B. Tchounwou, Int. J. Environ. Res. Public Health, 2009, 6, 643–653 CrossRef CAS PubMed.
- A. Patlolla, C. Barnes, C. Yedjou, V. Velma and P. Tchounwou, Environ. Toxicol., 2010, 24, 66–73 CrossRef PubMed.
- D. B. Zorov, M. Juhaszova and S. J. Sollott, Physiol. Rev., 2014, 94, 909 CrossRef CAS PubMed.
- R. O. Vogel, C. E. Dieteren, V. D. H. Lp, P. H. Willems, J. A. Smeitink, W. J. Koopman and L. G. Nijtmans, J. Biol. Chem., 2007, 282, 7582–7590 CrossRef CAS PubMed.
- S. M. Budde, L. P. van denHeuvel and J. A. Smeitink, Mitochondrion, 2002, 2, 109–115 CrossRef CAS PubMed.
- D. M. Rhoads, A. L. Umbach, C. C. Subbaiah and J. N. Siedow, Plant Physiol., 2006, 141, 357–366 CrossRef CAS PubMed.
- I. P. Trougakos and E. S. Gonos, Int. J. Biochem. Cell Biol., 2002, 34, 1430–1448 CrossRef CAS PubMed.
- K. S. Leskov, D. Y. Klokov, J. Li, T. J. Kinsella and D. A. Boothman, J. Biol. Chem., 2003, 278, 11590–11600 CrossRef CAS PubMed.
- F. Loison, L. Debure, P. Nizard, G. P. Le, D. Michel and D. Y. Le, Biochem. J., 2006, 395, 223–231 CrossRef CAS PubMed.
- C. Wang, K. Jiang, D. Gao, X. Kang, C. Sun, Q. Zhang, Y. Li, L. Sun, S. Zhang and K. Guo, PLoS One, 2013, 8, e55981 CrossRef CAS PubMed.
- G. Liu, G. Yang, B. Chang, I. Mercadouribe, M. Huang, J. Zheng, R. C. Bast, S. H. Lin and J. Liu, J. Natl. Cancer Inst., 2010, 102, 812–827 CrossRef CAS PubMed.
- Q. Liang, Y. Zhang, M. Zeng, L. Guan, Y. Xiao and F. Xiao, Toxicol. Res., 2018, 7, 521–528 RSC.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- F. Xiao, Y. Li, L. Dai, Y. Deng, Y. Zou, P. Li, Y. Yang and C. Zhong, Int. J. Mol. Med., 2012, 30, 629–635 CrossRef CAS PubMed.
- E. C. Alexopoulos, X. Cominos, I. P. Trougakos, M. Lourda, E. S. Gonos and V. Makropoulos, Bioinorg. Chem. Appl., 2008, 2008, 420578 Search PubMed.
- L. E. French, A. Wohlwend, A. P. Sappino, J. Tschopp and J. A. Schifferli, J. Clin. Invest., 1994, 93, 877 CrossRef CAS PubMed.
- M. Valko, H. Morris and M. T. Cronin, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS PubMed.
- E. M. Goetz, B. Shankar, Y. Zou, J. C. Morales, X. Luo, S. Araki, R. Bachoo, L. D. Mayo and D. A. Boothman, Oncogene, 2011, 30, 3745–3754 CrossRef CAS PubMed.
- C. Wang, K. Jiang, X. Kang, D. Gao, C. Sun, Y. Li, L. Sun, S. Zhang, X. Liu and W. Wu, Int. J. Biochem. Cell Biol., 2012, 44, 2308–2320 CrossRef CAS PubMed.
- S. H. Lau, J. D. Sham, C. H. Tzang, D. Tang, N. Ma, L. Hu, Y. Wang, J. M. Wen, G. Xiao and W. M. Zhang, Oncogene, 2006, 25, 1242–1250 CrossRef CAS PubMed.
- D. Martinvalet, D. M. Dykxhoorn, R. Ferrini and J. Lieberman, Cell, 2008, 133, 681–692 CrossRef CAS PubMed.
- D. C. Wallace, Novartis Foundation symposium, 2001.
- J. F. Turrens, J. Physiol., 2010, 552, 335–344 CrossRef PubMed.
- M. R. Wilson and S. B. Easterbrook-Smith, Biochim. Biophys. Acta, 1992, 1159, 319 CrossRef CAS.
- J. R. Silkensen, G. B. Schwochau and M. E. Rosenberg, Biochem. Cell Biol., 1994, 72, 483–488 CrossRef CAS PubMed.
- L. Bertram and R. E. Tanzi, Nat. Rev. Neurol., 2010, 6, 11–13 CrossRef CAS PubMed.
- J. Ischia and A. I. So, Nat. Rev. Urol., 2013, 10, 386–395 CrossRef CAS PubMed.
- D. D. Miller, A. Emley, S. Yang, J. E. Richards, J. E. Lee, A. Deng, M. P. Hoang and M. Mahalingam, Mod. Pathol., 2012, 25, 505–515 CrossRef CAS PubMed.
- C. Wang, G. Jin, H. Jin, N. Wang, Q. Luo, Y. Zhang, D. Gao, K. Jiang, D. Gu and Q. Shen, Oncotarget, 2015, 6, 2903–2916 Search PubMed.
- X. Huang, D. Zhai and Y. Huang, Mol. Cell. Biochem., 2001, 224, 1–7 CrossRef CAS PubMed.
- C. C. Wu and S. B. Bratton, Mol. Cell. Oncol., 2017, 4, e1281865 CrossRef PubMed.
- S. Suhane, D. Berel and V. K. Ramanujan, Biochem. Biophys. Res. Commun., 2011, 412, 590–595 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |