
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation on voltage loss in organic triplet photovoltaic devices based on Ir complexes†
Yingzhi
Jin‡
a,
Jie
Xue‡
b,
Juan
Qiao
 *b and
Fengling
Zhang
*a
*b and
Fengling
Zhang
*a
aDepartment of Physics, Chemistry and Biology, Linköping University, Linköping SE-58183, Sweden. E-mail: fengling.zhang@liu.se
bKey Lab of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: qjuan@mail.tsinghua.edu.cn
First published on 30th October 2019
Abstract
Voltage losses in singlet material-based organic photovoltaic devices (OPVs) have been intensively studied, whereas, only a few investigations on triplet material-based OPVs (T-OPVs) are reported. To investigate the voltage loss in T-OPVs, two homoleptic iridium(III) complexes based on extended π-conjugated benzo[g]phthalazine ligands, Ir(Ftbpa)3 and Ir(FOtbpa)3, are synthesized as sole electron donors. T-OPVs are fabricated by mixing two donors with phenyl-C71-butyric acid methyl ester (PC71BM) as an electron acceptor. Insertion of oxygen-bridges as flexible inert δ-spacers in Ir(FOtbpa)3 has slightly elevated both the lowest unoccupied molecular orbital and the highest occupied molecular orbital levels compared to those of Ir(Ftbpa)3, which results in a lower charge transfer (CT) state energy (ECT) for Ir(FOtbpa)3-based devices. However, a higher Voc (0.88 V) is observed for Ir(FOtbpa)3-based devices than those of Ir(Ftbpa)3 (0.80 V). To understand the above result, the morphologies of the two blend films are studied, which excludes the influence of morphology. Furthermore, radiative and non-radiative recombination in two devices is quantitatively investigated, which suggests that a higher Voc can be attributed to reduced radiative and non-radiative recombination loss for the Ir(FOtbpa)3-based devices.
Introduction
Solar energy is considered to be a promising renewable energy source to address the increasing worldwide energy demands. In particular, solution processed bulk-heterojunction (BHJ) organic photovoltaic devices (OPVs) have been identified as promising candidates because of their potential in low-cost, large-area, light-weight and flexible productions. To date, power conversion efficiencies (PCE) over 15% have been achieved for single junction OPVs with the emergence of non-fullerene acceptors,1,2 which makes OPVs feasible for industrialization. The voltage losses in OPVs have been regarded as the major challenge remaining to further improve the PCE comparable with inorganic or hybrid perovskite PVs.The open-circuit voltage (Voc) in OPVs is proportional to the energy of the charge transfer (CT) state (ECT) between the donor and acceptor.3 It has been found that the energetic difference between the highest occupied molecular orbital (HOMO) of the donor and the lowest unoccupied molecular orbital (LUMO) of the acceptor is roughly equal to ECT.4–6 Therefore, many reports are focused on increasing the Voc through increasing ECT by minimizing the energetic offset between donors and acceptors.7–9 Increasing ECT will however lead to a small driving force (defined as the energy difference between optical gap of the neat donor or acceptor and ECT) for exciton dissociating to free charges. Generally, fullerene based OPVs tend to show low PCEs with small driving forces (<0.3 eV), whereas, a reasonably high IQE (>85%) was obtained for P3TI:PC71BM blends with a small driving force of 0.1 eV.10 Recently, non-fullerene based OPVs have exhibited efficient exciton dissociation despite a negligible driving force.11–14 Furthermore, the voltage loss between ECT/q to Voc is due to radiative and non-radiative recombination. An empirical relation of 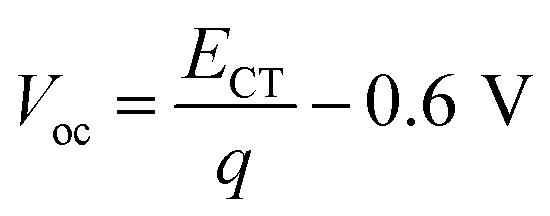 , has been found for fullerene based OPVs, of which radiative recombination at donor/acceptor interfaces via the CT state causes ∼0.25 V loss and non-radiative recombination causes ∼0.35 V loss.3,15 Thus, reducing recombination losses is another important strategy to obtain a high Voc.16 It was reported that decreasing the donor/acceptor interfacial area is an effective way to reduce voltage losses.17 Therefore, high Voc can be achieved for organic materials with long exciton diffusion lengths, which will enable a reduced optimum interfacial area. Furthermore, reducing non-radiative recombination losses (<0.3 V) enabled high Voc for materials with high photoluminescence (PL) yields, which have also been reported.18,19
, has been found for fullerene based OPVs, of which radiative recombination at donor/acceptor interfaces via the CT state causes ∼0.25 V loss and non-radiative recombination causes ∼0.35 V loss.3,15 Thus, reducing recombination losses is another important strategy to obtain a high Voc.16 It was reported that decreasing the donor/acceptor interfacial area is an effective way to reduce voltage losses.17 Therefore, high Voc can be achieved for organic materials with long exciton diffusion lengths, which will enable a reduced optimum interfacial area. Furthermore, reducing non-radiative recombination losses (<0.3 V) enabled high Voc for materials with high photoluminescence (PL) yields, which have also been reported.18,19
At present, the photo-induced charges mainly originate from singlet exciton dissociation in high performance OPVs. Triplet excitons, which have longer lifetimes or diffusion lengths than singlets, may provide a favorable approach to increase the photocurrent of OPVs due to the forbidden nature of recombination from the triplet state.20,21 In addition, the long diffusion lengths are beneficial to have large domains with decreased interfaces, which will further improve Voc.17 In general, the excitons generated by absorbing photons in organic materials are singlet due to the selection rule in the electronic dipole transition processes.22 The triplet excitons can be obtained by flipping the spin orientation of singlet excitons through the effective intersystem crossing (ISC) or by bimolecular singlet fission.23,24 Enlarging spin–orbit coupling (SOC) by chemically or physically introducing heavy atoms into the conjugated materials has been proposed to enhance ISC rate.25–27 So far, some research studies have been done on triplet material-based OPVs (T-OPVs)28–31 and the highest PCE for small-molecule Ir complexes is 3.81%.32 However, the voltage losses in T-OPVs were rarely investigated.33 In terms of recombination losses, the long exciton diffusion lengths and high emissive properties of triplet materials are beneficial for large Voc.
Here, we therefore investigate the voltage losses in T-OPVs via radiative and non-radiative recombination losses by employing highly sensitive external quantum efficiency and electroluminescence (EL) measurements. Two homoleptic iridium (Ir) complexes, tris(1-(2,4-bis(trifluoromethyl)phenyl)-4-(thiophen-2-yl)benzo[g]phthalazine) Ir(III) ((Ir(Ftbpa)3) and tris(1-(2,5-bis(trifluoromethyl)phenoxy)-4-(thiophen-2-yl)benzo[g]phthalazine) Ir(III) (Ir(FOtbpa)3), are designed as electron donors and phenyl-C71-butyric acid methyl ester (PC71BM) is used as the electron acceptor. OPVs based on Ir(Ftbpa)3 and Ir(FOtbpa)3 donors exhibit PCEs of 3.17% and 3.56%, which are decent performances regarding the studies on T-OPVs to date, and also showed great enhancement compared to poor photovoltaic performance of the 1-chloro-4-(thiophen-2-yl)benzo[g]phthalazine (Ftbpa) (0.001%) and 1-(2,5-bis(trifluoromethyl)phenoxy)-4-(thiophen-2-yl)benzo[g]phthalazine (FOtbpa) (0.007%) ligands as donors. More importantly, a higher Voc is achieved for Ir(FOtbpa)3-based devices despite a lower ECT, which is attributed to the reduced radiative and non-radiative recombination loss.
Experimental section
Synthesis and characterization
All commercially available reagents and chemicals were used without further purification. All reactions involving air-sensitive reagents were carried out under an atmosphere of nitrogen.1-Chloro-4-(thiophen-2-yl)benzo[g]phthalazine, Ftbpa and Ir(Ftbpa)3 was synthesized according to the literature reports.34
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Then, the crude product was recrystallized from dichloromethane/hexane to give FOtbpa as a yellow solid. Yield: 82%. 1H NMR (600 MHz, CDCl3): δ 9.10 (s, 1H), 9.07 (s, 1H), 8.27 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 7.95 (s, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.86 (d, J = 3.3 Hz, 1H), 7.82–7.75 (m, 2H), 7.67 (d, J = 8.3 Hz, 1H), 7.64 (d, J = 4.9 Hz, 1H), 7.32 (t, J = 4.3 Hz, 1H). HRMS (ESI+) m/z: calcd for C24H13F6N2OS+ [M + H]+: 491.0653, found: 491.0692.
:1, v/v) as the eluent to give Ir(FOtbpa)3 as a black solid. Yield: 25%. 1H NMR (600 MHz, CDCl3): δ 9.16 (s, 3H), 8.69 (s, 3H), 8.28 (d, J = 8.3 Hz, 3H), 8.17 (d, J = 8.2 Hz, 3H), 7.82–7.78 (m, 3H), 7.78–7.73 (m, 3H), 7.34 (d, J = 4.2 Hz, 3H), 7.12 (s, 3H), 6.64 (d, J = 4.7 Hz, 3H), 6.54 (d, J = 8.2 Hz, 3H), 5.71 (d, J = 8.1 Hz, 3H). HRMS (MALDI-TOF) m/z: calcd for C72H33F18IrN6O3S3+ [M]+: 1660.1118, found: 1660.2736. Elemental analysis calcd for C72H33F18IrN6O3S3: C, 52.08; H, 2.00; N, 5.06; found: C, 52.08; H, 2.28; N, 5.19.
:1, v/v). Then, the crude product was recrystallized from dichloromethane/hexane to give FOtbpa as a yellow solid. Yield: 82%. 1H NMR (600 MHz, CDCl3): δ 9.10 (s, 1H), 9.07 (s, 1H), 8.27 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 7.95 (s, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.86 (d, J = 3.3 Hz, 1H), 7.82–7.75 (m, 2H), 7.67 (d, J = 8.3 Hz, 1H), 7.64 (d, J = 4.9 Hz, 1H), 7.32 (t, J = 4.3 Hz, 1H). HRMS (ESI+) m/z: calcd for C24H13F6N2OS+ [M + H]+: 491.0653, found: 491.0692.
:1, v/v) as the eluent to give Ir(FOtbpa)3 as a black solid. Yield: 25%. 1H NMR (600 MHz, CDCl3): δ 9.16 (s, 3H), 8.69 (s, 3H), 8.28 (d, J = 8.3 Hz, 3H), 8.17 (d, J = 8.2 Hz, 3H), 7.82–7.78 (m, 3H), 7.78–7.73 (m, 3H), 7.34 (d, J = 4.2 Hz, 3H), 7.12 (s, 3H), 6.64 (d, J = 4.7 Hz, 3H), 6.54 (d, J = 8.2 Hz, 3H), 5.71 (d, J = 8.1 Hz, 3H). HRMS (MALDI-TOF) m/z: calcd for C72H33F18IrN6O3S3+ [M]+: 1660.1118, found: 1660.2736. Elemental analysis calcd for C72H33F18IrN6O3S3: C, 52.08; H, 2.00; N, 5.06; found: C, 52.08; H, 2.28; N, 5.19.
Characterization
1H NMR spectra were measured using a JEOLAL-600 MHz spectrometer at ambient temperature. High resolution mass spectra were recorded using a Thermo-Electron Corporation Finnigan LTQ mass spectrometer (ESI-MS) and LCMS-IT/TOF (HRMS). The laser desorption ionization time-of flight mass spectrometry (LDI-TOF-MS) data were obtained using a Shimadzu AXIMA Performance MALDI-TOF instrument in both positive and negative detection modes with an applied voltage of 25 kV between the target and the aperture of the time-of-flight analyzer. Elemental analysis was performed using a flash EA 1112 spectrometer. The single crystal of Ir(FObpa)3 was obtained from the diffusion of a chloroform/hexane mixture. The low temperature (104.6 K) single-crystals X-ray experiments were performed using a Rigaku RAXIS-SPIDER IP diffractometer with graphite-monochromatized MoKα radiation (λ = 0.71073 Å). Data collection and reduction, cell refinement, and experiential absorption correction were performed with the Rigaku RAPID AUTO software package (Rigaku, 1998, Version 2.30). CCDC 1916919.† Electrochemical measurement was performed with a Potentiostat/Galvanostat Model 283 (Princeton Applied Research) electrochemical workstation, using Pt as the working electrode, platinum wire as the auxiliary electrode, and a Ag wire as the reference electrode standardized against ferrocene/ferrocenium. The reduction/oxidation potentials were measured in anhydrous DMF solution containing 0.1 M n-Bu4NPF6 as the supporting electrolyte at a scan rate of 150 mV s−1.Device fabrication and characterization
The OPVs were fabricated with the structure of ITO/poly(3,4-ethylenedioxythiophene) doped with poly(styrene-sulfonate) (PEDOT:PSS)/active layer/LiF/Al. The ITO substrates were cleaned with detergent and TL-1 (NH3:H2O2:H2O = 1:1:5) for 30 min. PEDOT:PSS was spin-coated on the cleaned ITO substrates followed by annealing at 150 °C for 15 min. The active layers (total 20 mg mL−1) were spin-coated from chloroform (CF) solutions on top of the PEDOT:PSS at 2000 rpm for 40 s in a glovebox filled with N2. The substrates were moved into a vacuum chamber where 0.6 nm LiF and 90 nm Al were thermally evaporated at a pressure less than 2.0 × 10−4 Pa with a shadow mask to define the active area to be 0.047 cm2. Hole only devices were fabricated with the structure of ITO/PEDOT:PSS/active layer/MoO3/Al. Electron only devices were fabricated with the structure of ITO/ZnO/active layer/LiF/Al. The hole or electron mobilities of the BHJ blends were measured using the space-charge-limited current (SCLC) method according to the Murgatroyd law and using eqn (1) to fit the trap-free regions of the dark J–V curves from the hole or electron only devices.35,36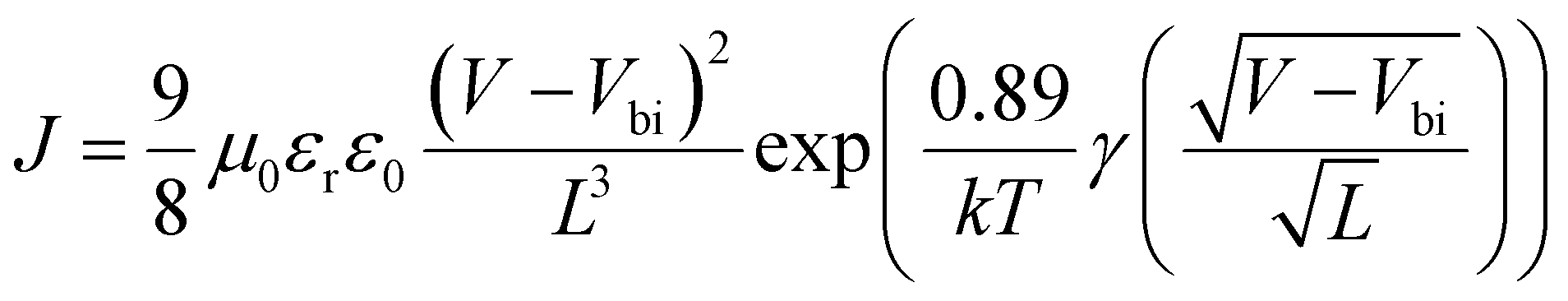 | (1) |
Current density–voltage (J–V) curves are measured by using a Keithley 2400 Source Meter under an illumination of AM 1.5 simulated by a solar simulator (LSH-7320 LED Solar Simulator, Newport). External quantum efficiency (EQE) spectra were obtained using a QE-R system (Enli Technology Co. Ltd, Taiwan). UV-vis absorption spectra were recorded using a PerkinElmer Lambda 900 spectrometer. Photoluminescence (PL) and EL spectra were recorded using an Andor spectrometer (Shamrock sr-303i-B, coupled to a Newton EMCCD silicon detector cooled to −60 °C). For the EL measurements, a Keithley 2400 Source Meter was utilized for applying an external electric field. EQEEL was measured using a homebuilt system using a calibrated large area Si photodiode 1010B from Oriel, a Keithley 2400 Source Meter to provide voltage and record injected current, and a Keithley 485 Picoammeter to measure the emitted light intensity. Fourier-transform photocurrent spectroscopy (FTPS)-EQE was carried out using a Vertex 70 from Bruker optics, equipped with a QTH lamp, quartz beamsplitter and external detector option. A low noise current amplifier (SR570) was used to amplify the photocurrent produced upon illumination of the devices with light modulated by the FTIR. The output voltage of the current amplifier was fed back into the external detector port of the FTIR, Atomic force microscopy (AFM) was performed using a Dimension 3100 system (Digital Instruments/Veeco) with antimony (n) doped silicon cantilevers (SCM-PIT, Veeco) in tapping mode. The active layer thickness was determined using a Veeco Dektak 6M Stylus profilometer.
Results and discussions
The incorporation of the heavy-atom Ir in the organic framework could largely enhance the SOC and lead to an effective ISC rate. As a near-infrared (NIR) phosphorescent material, Ir(Ftbpa)3 possess UV-vis-NIR absorption with edge over 750 nm, long phosphorescent lifetime and good solubility, which makes it a promising donor material for T-OPVs. Although a long excited-state lifetime could be obtained in these noble-metal based dyes, the notorious bimolecular triplet–triplet annihilation between dyes, along with aggregation caused quenching (ACQ) in solid films would enhance the non-radiative rate and thus reduce the excited-state lifetime, which would shorten the exciton diffusion length. The usage of inert substituents could protect and isolate the excitons in the aggregation state and alleviate ACQ. Based on Ir(Ftbpa)3, insertion of oxygen-bridges between the benzo[g]phthalazine moiety and bis(trifluoromethyl)phenyl group generate bis(trifluoromethyl)phenoxy groups as flexible inert δ-spacers to protect the exciton, and thus alleviate the ACQ and maintain long phosphorescent lifetimes for the aggregation states. As a result, Ir(FOtbpa)3 (Fig. 1a) was designed and synthesized with the structure fully characterized by 1H NMR, high-resolution mass spectrometry, elemental analysis and single-crystal X-ray diffraction measurements.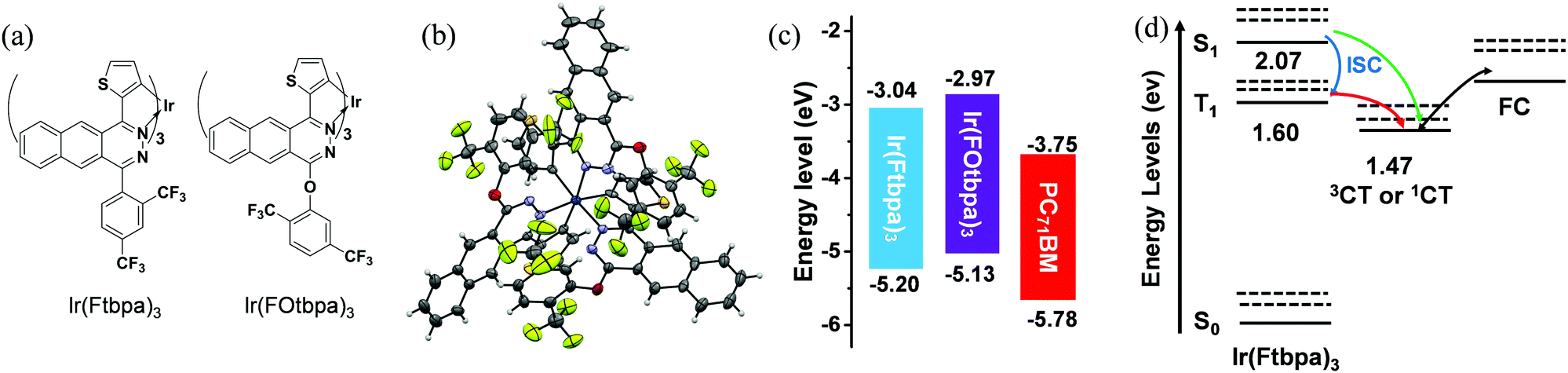 | ||
| Fig. 1 (a) Chemical structures of Ir(Ftbpa)3 and Ir(FOtbpa)3; (b) single-crystal structure of Ir(FOtbpa)3 with thermal ellipsoids plotted at 50% probability level; (c) energy levels of Ir(Ftbpa)3, Ir(FOtbpa)3 and PC71BM; (d) a schematic Jablonski diagram for the charge generation process of Ir(Ftbpa)3:PC71BM blend under photoexcitation. ISC: intersystem crossing; ground state (S0), lowest singlet state (S1), lowest triplet state (T1), singlet charge transfer state (1CT), triplet charge transfer state (3CT), and free charges (FC). | ||
The single crystals of Ir(FOtbpa)3 were readily grown from a chloroform/methanol mixture. As show in Fig. 1b, the single-crystal X-ray diffraction measurement verified that Ir(FOtbpa)3 possesses a facial configuration around the Ir center. The average C–O–C angles and the dihedral angles between the bis(trifluoromethyl)phenyl groups and the benzo[g]phthalazine cores are 117° and 86°. Consequently, the bis(trifluoromethyl)phenoxy groups could protect the benzo[g]phthalazine moieties and Ir center at one side.
The energy levels of Ir(Ftbpa)334 and Ir(FOtbpa)3 were estimated by cyclic voltammogram (CV) measurements (Fig. S1, ESI†). The LUMO/HOMO energy levels of Ir(Ftbpa)3, Ir(FOtbpa)3, and PC71BM are calculated to be −3.04/−5.20, −2.97/−5.13, and −3.75/−5.78 eV (Fig. 1c), respectively. It indicates that insertion of an oxygen-bridge has no obvious effect on the electrochemical LUMO–HOMO gap while both LUMO and HOMO levels are elevated slightly.
To give readers an intuitive understanding of the charge generation process in T-OPVs, the energetic states of the Ir(Ftbpa)3:PC71BM blend is presented in Fig. 1d where the singlet and triplet states of Ir(Ftbpa)3 were calculated in a previous report,34 and the energies of the CT states is obtained from the FTPS-EQE measurement. In the charge generation process of the singlet system, the CT states are formed directly from the S1 before being separated into free charges. While in the Ir(Ftbpa)3:PC71BM system, excitons go through a fast ISC from S1 to T1 (blue arrow in Fig. 1d). The energy offset between T1 and 3CT may be beneficial for triplet excitons to form 3CT and then dissociate into free charges (red arrow). However, this is also a possibility even in the triplet system, CT excitons might generate from S1 without going through T1 (green line).
The UV-vis absorption spectra of Ftbpa and FOtbpa ligands showed absorption bands below 450 nm (Fig. S2a, ESI†), which could be ascribed to the π–π* transition. Ir complexes, Ir(Ftbpa)3 and Ir(FOtbpa)3, exhibited significantly enhanced and broadened absorption compared to Ftbpa and FOtbpa ligands shown in Fig. 2a. The bands below 450 nm are attributed to the ligands’ absorption, while the absorption bands at 450–700 nm correspond to the mixed transitions of 1MLCT (metal-to-ligand charge transfer) and 3MLCT. The weak absorption band extending over 700 nm could be the excitation from the ground states to the lowest triplet state (S0 → T1). After blending with PC71BM, the blend films with a weight ratio of 1:1.5 showed similar absorption spectra due to the overlapped absorptions between Ir complexes and PC71BM. Compared with Ir(Ftbpa)3, Ir(FOtbpa)3 displayed similar NIR phosphorescence with an emission peak at 767 nm, but a lower PL quantum yield (ΦPL) of 10.8% and a shorter phosphorescent lifetime (τp) of 489 ns in degassed CH2Cl2 (Table S1 and Fig. S2b, ESI†), which are attributed to its slightly enlarged radiative transition rate constant (kr = 2.2 × 105 s−1) and significantly increased non-radiative transition rate constant (knr = 1.8 × 106 s−1). The significantly increased knr of Ir(FOtbpa)3 could be ascribed to the rotation of pendent bis(trifluoromethyl)phenoxy groups in the solution.
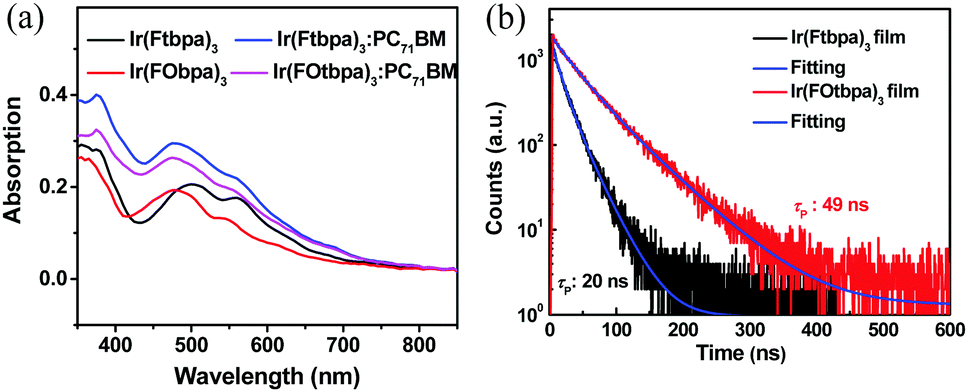 | ||
| Fig. 2 (a) Absorption spectra of Ir(Ftbpa)3, Ir(FOtbpa)3 and corresponding blend films with PC71BM in a weight ratio of 1:1.5; (b) transient PL decay curves of Ir(Ftbpa)3 and Ir(FOtbpa)3 neat films. | ||
In neat films, the Ir(Ftbpa)3 complex showed slightly red-shifted emissions with peaks at 784 nm compared to that of Ir(FOtbpa)3 with peaks at 780 nm (Fig. S2c, ESI†), which should correspond to phosphorescence characteristics of the triplet excited states. Accordingly, the energies of T1 were estimated, by the highest energy vibronic band of the phosphorescence spectra, to be 1.58 eV and 1.59 eV for Ir(Ftbpa)3 and Ir(FOtbpa)3, respectively. The complete elimination of the ligand fluorescence emissions indicated the strong SOC and efficient ISC rate from S1 to T1. The ΦPL of Ir(FOtbpa)3 and Ir(Ftbpa)3 reduced to 2.4% and 2.6% (Table S1, ESI†), respectively, which could be ascribed to the ACQ with enlarged knr caused by the interactions of triplet excitons such as triplet–triplet annihilation. Also, the τp of Ir(FOtbpa)3 and Ir(Ftbpa)3 reduced to 49 ns and 19 ns, respectively (Fig. 2b). The knr of Ir(FOtbpa)3 and Ir(Ftbpa)3 were calculated to be 2.0 × 107 s−1 and 5.1× 107 s−1 in neat films, respectively, which are about 11 times and 43 times larger than their knr in degassed CH2Cl2. The values of kr were calculated to be 4.9 × 105 s−1 and 1.4 × 106 s−1 for Ir(FOtbpa)3 and Ir(Ftbpa)3 neat films, respectively. Since the only difference of Ir(FOtbpa)3 and Ir(Ftbpa)3 molecules is the pendent group, the much smaller enhancement of knr for Ir(FOtbpa)3 is ascribed to the usage of the bis(trifluoromethyl)phenoxy groups as δ-spacers, which hamper the interactions of triplet excitons in aggregated state and alleviate the reductions of ΦPL and τp. Thus, Ir(FOtbpa)3 displays longer τp in the pristine film, which is beneficial for the exciton diffusion.
To study the voltage losses in T-OPVs, the Ir complexes were evaluated using PC71BM as the electron acceptor with weight ratios of 2:1, 1:1.5 and 1:3. Photovoltaic parameters of the T-OPVs based on Ir(Ftbpa)3 and Ir(FOtbpa)3 are summarized in Table 1. For Ir(Ftbpa)3:PC71BM devices, a PCE of 3.17% with a short-circuit current density (Jsc) of 8.70 mA cm−2, Voc of 0.80 V, and fill factor (FF) of 0.46 is obtained at a weight ratio of 1:1.5. For Ir(FOtbpa)3:PC71BM devices, the best PCE increases to 3.56% with a Voc of 0.88 V, Jsc of 8.58 mA cm−2, and FF of 0.47 at the same weight ratio (1:1.5). On the other hand, the Ftbpa and FOtbpa ligands showed very poor performance with low PCEs of 0.001% and 0.007% in similar device structures (Table S2, ESI†), which confirms the significant contribution of Ir to the performance of corresponding T-OPVs. The typical J–V and EQE curves for Ir complex-based devices with a weight ratio of 1:1.5 are shown in Fig. 3a and b. The EQE curves of these Ir complex-based devices showed a spectral response from both donor and acceptor absorption regions (300 to 700 nm). The integrated Jsc values from the EQE curves are 8.26 and 8.11 mA cm−2 for Ir(Ftbpa)3:PC71BM and Ir(FOtbpa)3:PC71BM devices, respectively, which are consistent with the values from J–V measurement. The J–V characteristics of the hole-only and electron-only devices are shown in Fig. S3a and b (ESI†). The hole and electron mobilities are 6.6 × 10−7 and 1.76 × 10−4 cm2 V−1 s−1 for Ir(Ftbpa)3 blends (ratio 1:1.5) and 1.5 × 10−6 and 1.5 × 10−4 cm2 V−1 s−1 for Ir(FOtbpa)3 blends (ratio 1:1.5), as found through the SCLC measurements. The lower hole mobilities than the singlet materials resulted in unbalanced mobilities and the smaller FFs here.
| Donor | Ratio | V oc (V) | J sc (mA cm−2) | FF | PCE (%) |
|---|---|---|---|---|---|
| Ir(Ftbpa)3 | 2:1 |
0.85 (0.85 ± 0.01) | 6.43 (6.47 ± 0.1) | 0.39 (0.38 ± 0.01) | 2.13 (2.07 ± 0.19) |
| 1:1.5 |
0.80 (0.80 ± 0.01) | 8.70 (8.72 ± 0.19) | 0.46 (0.43 ± 0.02) | 3.17 (3.01 ± 0.19) | |
| 1:3 |
0.78 (0.78 ± 0.01) | 8.62 (8.58 ± 0.07) | 0.42 (0.41 ± 0.01) | 2.97 (2.71 ± 0.05) | |
| Ir(FOtbpa)3 | 2:1 |
0.93 (0.93 ± 0.01) | 5.07 (4.67 ± 0.23) | 0.32 (0.31 ± 0.01) | 1.51 (1.34 ± 0.09) |
| 1:1.5 |
0.88 (0.88 ± 0.01) | 8.58 (8.41 ± 0.51) | 0.47 (0.45 ± 0.02) | 3.56 (3.30 ± 0.26) | |
| 1:3 |
0.85 (0.85 ± 0.02) | 8.11 (8.14 ± 0.44) | 0.46 (0.41 ± 0.03) | 3.15 (2.80 ± 0.23) | |
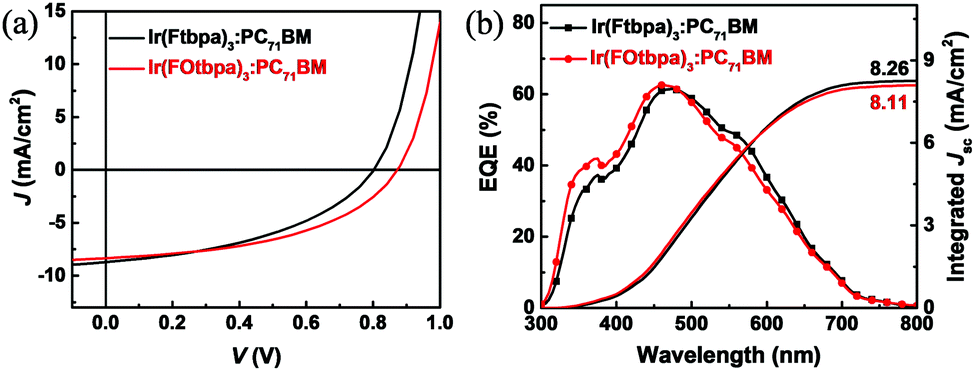 | ||
| Fig. 3 (a) J–V characteristics of the T-OPVs based on Ir(Ftbpa)3:PC71BM and Ir(FOtbpa)3:PC71BM blends with a weight ratio of 1:1.5; (b) EQE and integrated Jsc of Ir(Ftbpa)3:PC71BM and Ir(FOtbpa)3:PC71BM blends with a weight ratio of 1:1.5. | ||
Comparing the devices based on these two Ir complexes with different weight ratios, we find that the Voc increases with increasing content of the Ir complexes. Similar phenomena have been reported and attributed to the changes in the interfacial area of the donor/acceptor.17,37 Atomic force microscopy (AFM) was used to investigate the morphologies of the blend films with different weight ratios. As shown in the images (Fig. S4, ESI†), there seem to be minor morphological differences between the different blend ratios for both Ir(Ftbpa)3:PC71BM and Ir(FOtbpa)3:PC71BM blend films. While AFM only examines the surface morphology, the phase separation of the whole active layer could be investigated by PL measurement. Steady state PL spectra of the pristine Ir(Ftbpa)3 and Ir(FOtbpa)3 films are compared with their corresponding blends with different weight ratios (Fig. S5, ESI†). The PL intensities from Ir(Ftbpa)3 and Ir(FOtbpa)3 triplet excitons are strongly quenched by PC71BM in all blends, indicating efficient excitons dissociation and charge transfer between the two Ir complex donors and PC71BM acceptor with highly mixed donors and acceptors. The CT state PL from 2:1, 1:1.5, and 1:3 Ir(Ftbpa)3:PC71BM blend films are presented in Fig. 4a. The interfacial CT state emission is observed at ∼950 nm, which is clearly red-shifted compared to Ir(Ftbpa)3 exciton emission at 784 nm. Furthermore, it shows a clear trend of suppressed CT PL from the films with a higher PC71BM content. Similar results have also been found in Ir(FOtbpa)3:PC71BM blends (Fig. 4b). Since the CT PL intensities are generally very low, EL measurement is a much more sensitive method to determine the ECT. Therefore, the EL emission from devices based on pristine Ir complexes and their blends are also recorded. As shown in Fig. 4c and d, these electrically generated CT state EL emissions are consistent with the CT state PL emissions generated by photoexcitation. The Ir(Ftbpa)3:PC71BM blend films showed red-shift EL emissions at around 950 nm compared to 780 nm for the pristine Ir(Ftbpa)3 devices (Fig. 4c). Similar red-shift EL emissions are observed in the Ir(FOtbpa)3:PC71BM blends (Fig. 4d) at around 973 nm. These indicate that the triplet energy of Ir(Ftbpa)3 and Ir(FOtbpa)3 are much higher than the ECT in the blends, which confirms the effective utilization of triplet excitons in the charge generation process.
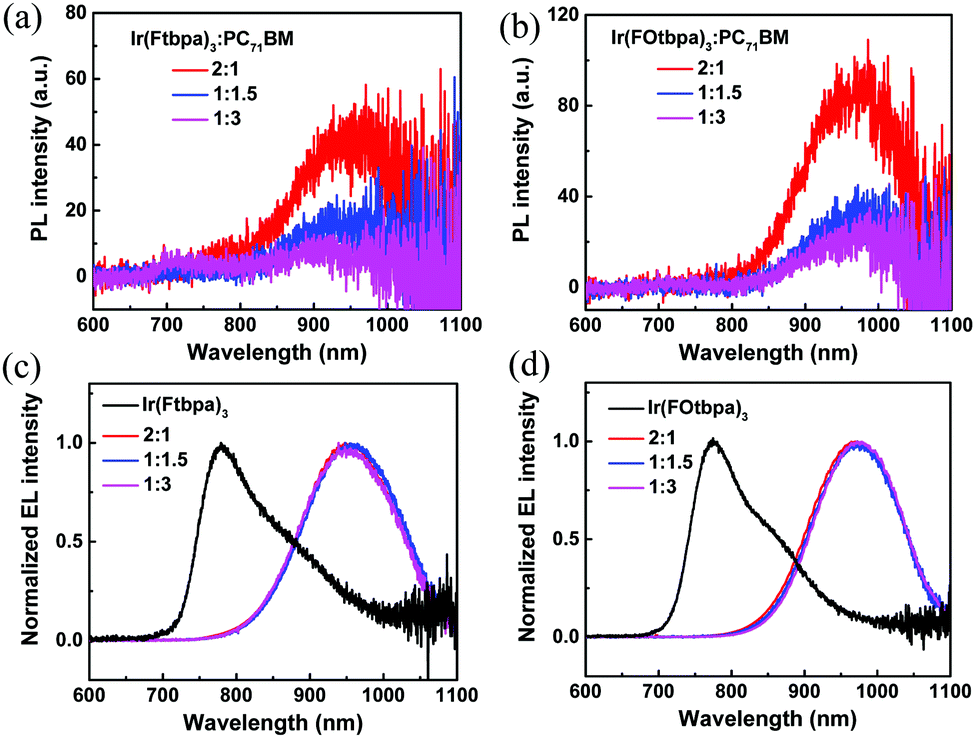 | ||
| Fig. 4 Sub-band-gap PL spectra from CT transitions of (a) Ir(Ftbpa)3:PC71BM and (b) Ir(FOtbpa)3:PC71BM blends with different weight ratios. The films were excited by a 532 nm laser; (c) EL spectra for pristine Ir(Ftbpa)3 and Ir(Ftbpa)3:PC71BM blends with different weight ratios; (d) the EL spectra of pristine Ir(FOtbpa)3 and Ir(FOtbpa)3:PC71BM blends with different weight ratios. | ||
More specifically, the ECT can be determined through fitting the FTPS-EQE spectra according to the model developed by Vandewal based on Marcus theory.
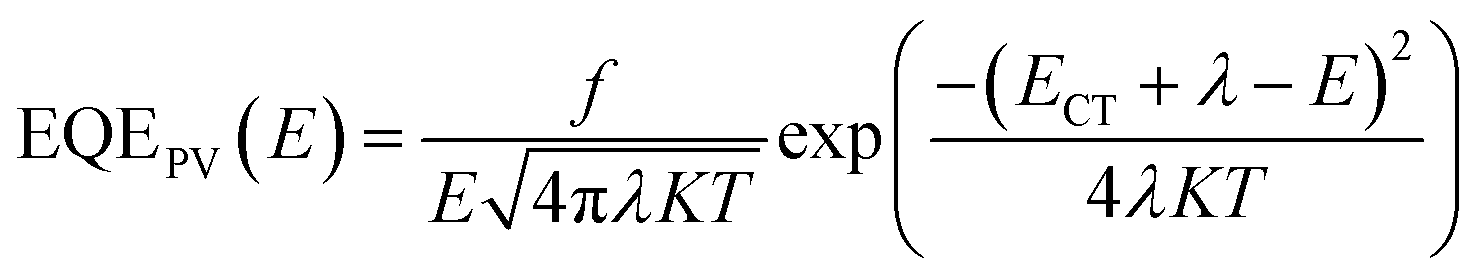 | (2) |
:1, 1:1.5, and 1:3 blends. For the Ir(FOtbpa)3-based devices, ECT values of 1.41 eV, 1.38 eV, and 1.38 eV are obtained for the 2:1, 1:1.5, and 1:3 blends.
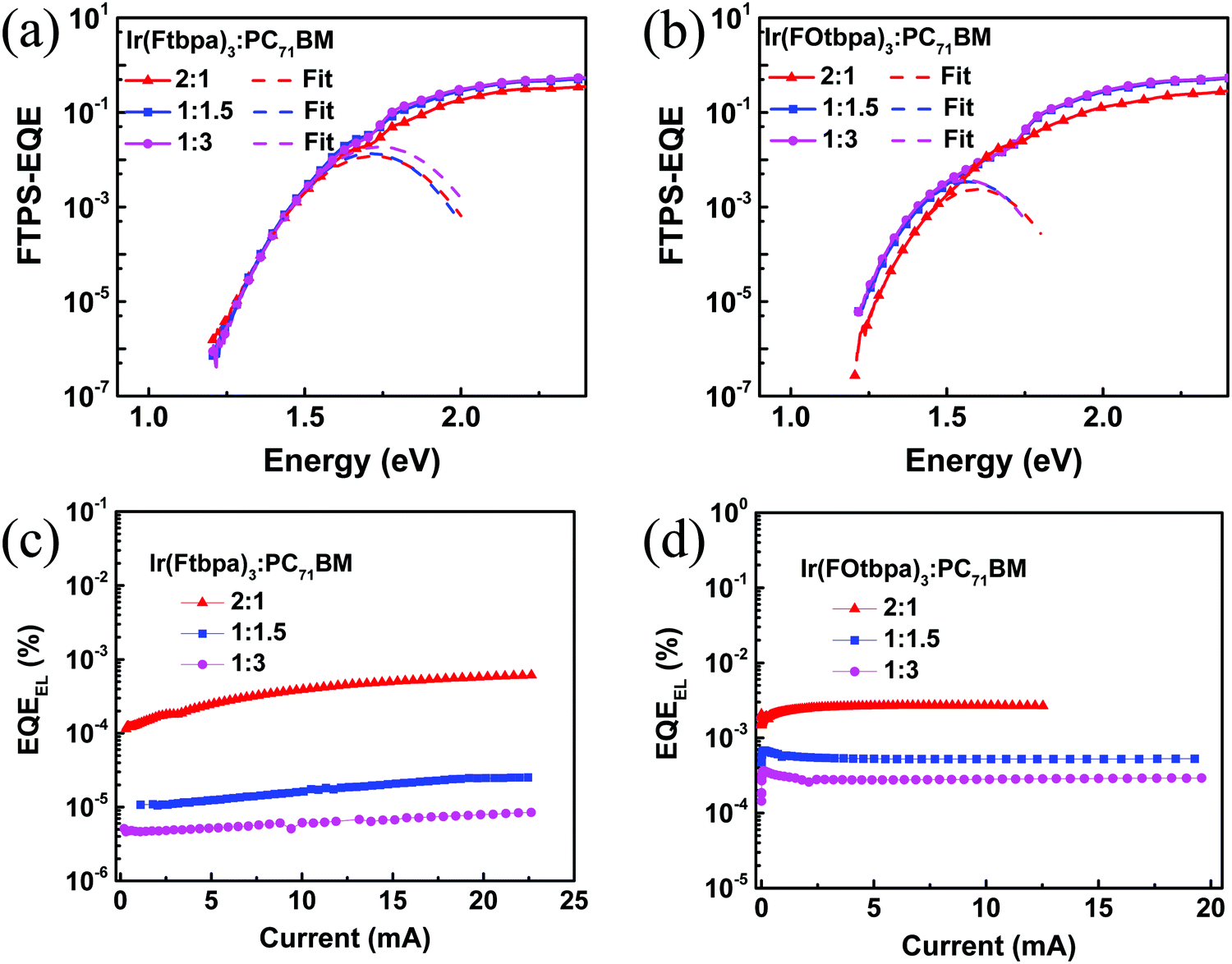 | ||
| Fig. 5 FTPS-EQE spectra of (a) Ir(Ftbpa)3:PC71BM and (b) Ir(FOtbpa)3:PC71BM. The dash curves are fits of the FTPS-EQE spectra using eqn (2); (c) EQEEL of the Ir(Ftbpa)3:PC71BM and (d) Ir(FOtbpa)3:PC71BM. | ||
As shown in Table 1, the Voc of the OPVs based on Ir(Ftbpa)3 are in the range of 0.85–0.78 V and the Voc of the OPVs based on Ir(FOtbpa)3 are in the range of 0.93–0.85 V. The contradiction between ECT and Voc for different blend ratios motivates us to further understand the voltage losses. Considering the detailed balance theory, the Voc of OPVs is then determined by eqn (3), where radiative (qΔVrad) and non-radiative (qΔVnon-rad) recombination losses can be experimentally determined by the fitting parameters and measured EQEEL.
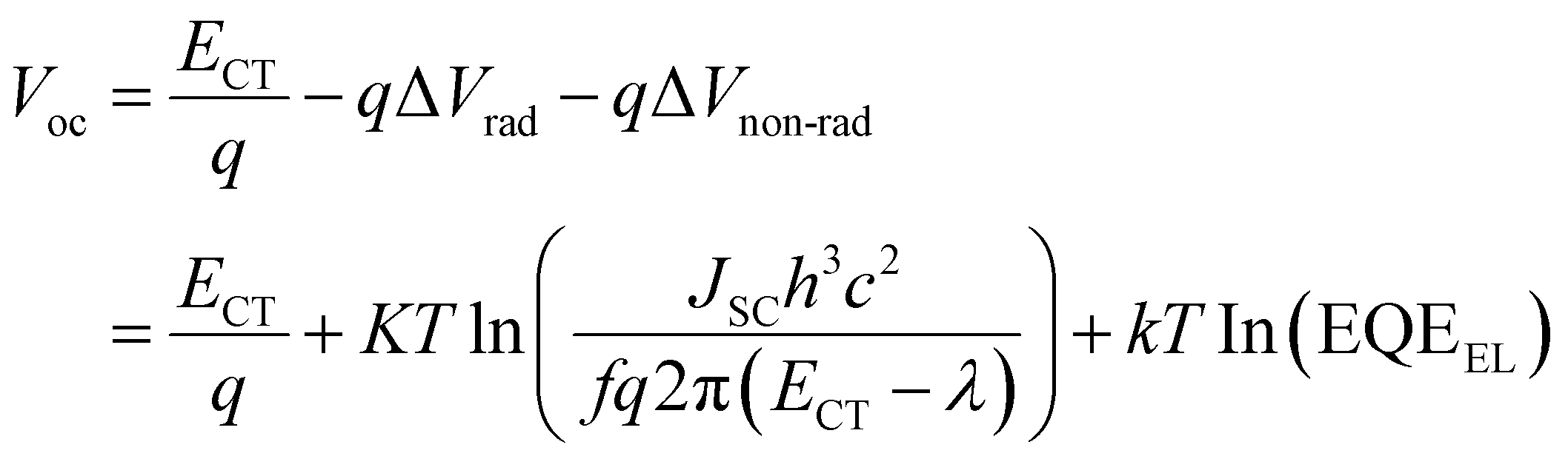 | (3) |
The qΔVrad and qΔVnon-rad for blends with different ratios were calculated (Table 2). The qΔVrad for both Ir(Ftbpa)3 and Ir(FOtbpa)3-based devices is independent with blend ratios. From the EQEEL measurements (Fig. 5c, d and Table 2), the EQEEL of the Ir(Ftbpa)3 and Ir(FOtbpa)3-based devices decreased with increasing content of PC71BM. These lead to low qΔVnon-rad for both Ir(Ftbpa)3 and Ir(FOtbpa)3-based devices resulting in a higher Voc with a low PC71BM content.
| Donor | Ratio | qV oc (eV) | f 1 (eV2) | E CT (eV) | λ (eV) | qΔVrad (eV) | EQEEL (%) | qΔVnon-rad (eV) |
|---|---|---|---|---|---|---|---|---|
| Ir(Ftbpa)3 | 2:1 |
0.85 | 6 × 10−3 | 1.46 | 0.27 | 0.25 | 1 × 10−4 | 0.36 |
| 1:1.5 |
0.80 | 6 × 10−3 | 1.47 | 0.25 | 0.25 | 1 × 10−5 | 0.42 | |
| 1:3 |
0.78 | 9 × 10−3 | 1.48 | 0.27 | 0.26 | 5 × 10−6 | 0.44 | |
| Ir(FOtbpa)3 | 2:1 |
0.93 | 9 × 10−4 | 1.41 | 0.19 | 0.21 | 2 × 10−3 | 0.27 |
| 1:1.5 |
0.88 | 6 × 10−4 | 1.38 | 0.12 | 0.19 | 7 × 10−4 | 0.31 | |
| 1:3 |
0.85 | 1 × 10−3 | 1.38 | 0.18 | 0.20 | 3 × 10−4 | 0.33 |
For the best device performances based on Ir(Ftbpa)3 and Ir(FOtbpa)3 blends (1:1.5), as shown in Table 1, the difference in the PCEs is mainly due to the difference in Vocs. When we compare the energy levels of these two donors, the HOMO level of Ir(Ftbpa)3 is lower than that of Ir(FOtbpa)3 (Fig. 1b), which indicates that the Ir(Ftbpa)3 blend may have a higher Voc. However, the Voc of Ir(Ftbpa)3-based devices is 0.08 V lower than that of the Ir(FOtbpa)3-based devices. The Ir(Ftbpa)3-based devices have a higher ECT of 1.47 eV compared with the value of 1.38 eV for the Ir(FOtbpa)3-based devices, which is consistent with the HOMO level difference. The qΔVrad for Ir(Ftbpa)3-based devices is 0.25 eV, which is higher than the value of 0.19 eV for the Ir(FOtbpa)3-based devices. The EQEEL of the device based on Ir(FOtbpa)3 is more than one order of magnitude higher than that of the Ir(Ftbpa)3. This leads to a calculated qΔVnon-rad of 0.31 eV for the Ir(FOtbpa)3-based devices, about 0.11 eV lower than that of the Ir(Ftbpa)3-based devices. Both radiative and non-radiative recombinations for the Ir(FOtbpa)3-based devices are lower than those of the Ir(Ftbpa)3-based devices, which results in a higher Voc for the Ir(FOtbpa)3-based devices. The calculated data fit well with Voc in these two blends.
Contradictory to the energy gap law (the non-radiative decay rate is exponentially increasing with decreasing energy difference between the excited and ground states), the Ir(FOtbpa)3-based device has a lower ECT, but a higher EQEEL. Considering the photophysical properties of the two Ir complexes, the larger kr (1.4 × 106 s−1) of Ir(Ftbpa)3 than that of Ir(FOtbpa)3 (kr = 4.9 × 105 s−1) in solid state may correlate with the larger radiative recombination loss in Ir(Ftbpa)3-based devices. The longer exciton lifetime (τ = 49 ns) and much smaller knr (2.0 × 107 s−1) compared with those of Ir(Ftbpa)3 (τ = 19 ns and knr = 5.1 × 107 s−1) in pristine films due to the flexible inert δ-spacer may decrease the non-radiative recombination loss in Ir(FOtbpa)3-based devices. In addition to the above reasons, some other charge carrier loss mechanisms may coexist in the Ir(Ftbpa)3-based devices.
The recombination mechanism was further studied by measuring the light intensity dependencies of Jsc and Voc (Fig. S6, ESI†). The Ir(Ftbpa)3 and Ir(FOtbpa)3-based devices (1:1.5) show figure-of-merit (α) values of 0.93 and 0.92, respectively, indicating that bimolecular recombination occurs in both systems at short circuit conditions. At open circuit conditions, a slope of 2 kBT/q for monomolecular (trap-assisted) recombination and a slope of 1 kBT/q for bimolecular recombination exist. In some cases, surface recombination would make the slope less than 1 kBT/q. The Ir(FOtbpa)3-based devices (1:1.5) show a slope of 1.03 kBT/q, while the Ir(Ftbpa)3-based devices (1:1.5) show a slope less than 1 kBT/q (0.95 kBT/q). Thus, the Ir(Ftbpa)3-based devices (1:1.5) is more dominated by surface recombination than the Ir(FOtbpa)3-based devices (1:1.5), which is consistent with the non-radiative recombination losses from EQEEL calculations.
Conclusions
In summary, the voltage losses in T-OPVs based on two Ir complexes and PC71BM are studied from the aspects of radiative and non-radiative recombination. Firstly, significantly increased PCE from 0.007% (devices based on ligands) to 3.56% (the Ir(FOtbpa)3-based devices) was observed, which confirms the major contribution by introducing Ir. Secondly, a trend of increasing Voc with increasing donor contents was found in two Ir complex systems by varying the weight ratios between the donors and acceptors. Thirdly, T-OPVs based on Ir(FOtbpa)3 exhibited a higher Voc compared to Ir(Ftbpa)3, which could be attributed to supressed non-radiative recombination losses due to the relatively small knr for Ir(FOtbpa)3. Furthermore, the additional surface recombination in the Ir(Ftbpa)3-based devices also has an impact on the non-radiative recombination losses, which results in a lower Voc.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. Zhang and Y. Jin acknowledge funding from the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) for the Joint China-Sweden Mobility programme, the Knut and Alice Wallenberg foundation under contract 2016.0059, the Swedish Government Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 200900971) and the China Scholarship Council (CSC). J. Qiao would like to thank the financial support from the NSFC of China (51711530040 and 51473086). J. Xue thanks the National Postdoctoral Program for Innovative Talents (BX20180159) for the financial support.References
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. V. Manca, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 125204 CrossRef.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- J. Widmer, M. Tietze, K. Leo and M. Riede, Adv. Funct. Mater., 2013, 23, 5814–5821 CrossRef CAS.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef CAS.
- S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932–14944 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 2231–2234 CrossRef CAS PubMed.
- K. Vandewal, Z. Ma, J. Bergqvist, Z. Tang, E. Wang, P. Henriksson, K. Tvingstedt, M. R. Andersson, F. Zhang and O. Inganäs, Adv. Funct. Mater., 2012, 22, 3480–3490 CrossRef CAS.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 16089 CrossRef CAS.
- S. Li, L. Zhan, C. Sun, H. Zhu, G. Zhou, W. Yang, M. Shi, C.-Z. Li, J. Hou, Y. Li and H. Chen, J. Am. Chem. Soc., 2019, 141, 3073–3082 CrossRef CAS PubMed.
- S. Chen, Y. Wang, L. Zhang, J. Zhao, Y. Chen, D. Zhu, H. Yao, G. Zhang, W. Ma, R. H. Friend, P. C. Y. Chow, F. Gao and H. Yan, Adv. Mater., 2018, 30, 1804215 CrossRef PubMed.
- H. Fu, Y. Wang, D. Meng, Z. Ma, Y. Li, F. Gao, Z. Wang and Y. Sun, ACS Energy Lett., 2018, 3, 2729–2735 CrossRef CAS.
- Z. Tang, J. Wang, A. Melianas, Y. Wu, R. Kroon, W. Li, W. Ma, M. R. Andersson, Z. Ma, W. Cai, W. Tress and O. Inganäs, J. Mater. Chem. A, 2018, 6, 12574–12581 RSC.
- J. Benduhn, K. Tvingstedt, F. Piersimoni, S. Ullbrich, Y. Fan, M. Tropiano, K. A. McGarry, O. Zeika, M. K. Riede, C. J. Douglas, S. Barlow, S. R. Marder, D. Neher, D. Spoltore and K. Vandewal, Nat. Energy, 2017, 2, 17053 CrossRef CAS.
- K. Vandewal, J. Widmer, T. Heumueller, C. J. Brabec, M. D. McGehee, K. Leo, M. Riede and A. Salleo, Adv. Mater., 2014, 26, 3839–3843 CrossRef CAS PubMed.
- D. Qian, Z. Zheng, H. Yao, W. Tress, T. R. Hopper, S. Chen, S. Li, J. Liu, S. Chen, J. Zhang, X.-K. Liu, B. Gao, L. Ouyang, Y. Jin, G. Pozina, I. A. Buyanova, W. M. Chen, O. Inganäs, V. Coropceanu, J.-L. Bredas, H. Yan, J. Hou, F. Zhang, A. A. Bakulin and F. Gao, Nat. Mater., 2018, 17, 703–709 CrossRef CAS PubMed.
- X. Liu, X. Du, J. Wang, C. Duan, X. Tang, T. Heumueller, G. Liu, Y. Li, Z. Wang, J. Wang, F. Liu, N. Li, C. J. Brabec, F. Huang and Y. Cao, Adv. Energy Mater., 2018, 8, 1801699 CrossRef.
- P. Heremans, D. Cheyns and B. P. Rand, Acc. Chem. Res., 2009, 42, 1740–1747 CrossRef CAS PubMed.
- O. V. Mikhnenko, R. Ruiter, P. W. M. Blom and M. A. Loi, Phys. Rev. Lett., 2012, 108, 137401 CrossRef PubMed.
- Z. Xu, B. Hu and J. Howe, J. Appl. Phys., 2008, 103, 043909 CrossRef.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- H. Zhen, Q. Hou, K. Li, Z. Ma, S. Fabiano, F. Gao and F. Zhang, J. Mater. Chem. A, 2014, 2, 12390–12396 RSC.
- M. H. Yun, E. Lee, W. Lee, H. Choi, B. R. Lee, M. H. Song, J.-I. Hong, T.-H. Kwon and J. Y. Kim, J. Mater. Chem. C, 2014, 2, 10195–10200 RSC.
- M. Qian, R. Zhang, J. Hao, W. Zhang, Q. Zhang, J. Wang, Y. Tao, S. Chen, J. Fang and W. Huang, Adv. Mater., 2015, 27, 3546–3552 CrossRef CAS PubMed.
- Y.-N. Liu, S.-F. Wang, Y.-T. Tao and W. Huang, Chin. Chem. Lett., 2016, 27, 1250–1258 CrossRef CAS.
- I. A. Wright, Polyhedron, 2018, 140, 84–98 CrossRef CAS.
- L. Xu, C.-L. Ho, L. Liu and W.-Y. Wong, Coord. Chem. Rev., 2018, 373, 233–257 CrossRef CAS.
- Y. Jin, Y. Zhang, Y. Liu, J. Xue, W. Li, J. Qiao and F. Zhang, Adv. Mater., 2019, 31, 1900690 CrossRef PubMed.
- Q. Wu, Y. Cheng, Z. Xue, X. Gao, M. Wang, W. Yuan, S. Huettner, S. Wan, X. Cao, Y. Tao and W. Huang, Chem. Commun., 2019, 55, 2640–2643 RSC.
- J. Benduhn, F. Piersimoni, G. Londi, A. Kirch, J. Widmer, C. Koerner, D. Beljonne, D. Neher, D. Spoltore and K. Vandewal, Adv. Energy Mater., 2018, 8, 1800451 CrossRef.
- J. Xue, L. Xin, J. Hou, L. Duan, R. Wang, Y. Wei and J. Qiao, Chem. Mater., 2017, 29, 4775–4782 CrossRef CAS.
- P. N. Murgatroyd, J. Phys. D: Appl. Phys., 1970, 3, 151–156 CrossRef.
- N. Felekidis, A. Melianas and M. Kemerink, Org. Electron., 2018, 61, 318–328 CrossRef CAS.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1916919. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9tc04914b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |