
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and helical supramolecular organization of discotic liquid crystalline dibenzo[hi,st]ovalene†
Qiang
Chen‡
a,
Wojciech
Zajaczkowski‡
a,
Johannes
Seibel‡
b,
Steven
De Feyter
 b,
Wojciech
Pisula
*ac,
Klaus
Müllen
*ad and
Akimitsu
Narita
*ae
b,
Wojciech
Pisula
*ac,
Klaus
Müllen
*ad and
Akimitsu
Narita
*ae
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: pisula@mpip-mainz.mpg.de; muellen@mpip-mainz.mpg.de; narita@mpip-mainz.mpg.de
bKU Leuven, Department of Chemistry, Division of Molecular Imaging and Photonics, Celestijnenlaan 200F, 3001 Leuven, Belgium
cLodz University of Technology, Department of Molecular Physics, Faculty of Chemistry, Zeromskiego 116, 90-924, Lodz, Poland
dInstitute of Physical Chemistry, Johannes Gutenberg-University, Duesbergweg 10-14, 55128, Mainz, Germany
eOrganic and Carbon Nanomaterials Unit, Okinawa Institute of Science and Technology Graduate University, Okinawa 904-0495, Japan
First published on 27th August 2019
Abstract
Dibenzo[hi,st]ovalene (DBOV) has emerged as a new polycyclic aromatic hydrocarbon (PAH) with intriguing optical properties with strong red emission. Nevertheless, DBOV derivatives thus far synthesized either had mesityl groups that hinder the π–π stacking of the aromatic cores or showed low solubility, and the self-assembly of DBOVs has not been investigated. In this work, two 3,4,5-tris(dodecyloxy)phenyl (TDOP) groups are introduced at the meso-positions of DBOV in order to enhance its solubility without compromising the intermolecular interactions. The obtained DBOV-TDOP forms at elevated temperatures a discotic liquid crystalline phase. Due to π–π-stacking interactions as well as local phase separation between the aromatic cores and the flexible alkoxy side chains, the DBOV-TDOP molecules self-assemble into columnar stacks. In the low temperature phase, the space demanding TDOP substituents induce a rotation of the discs towards each other leading to an exceptionally long helical pitch within the columnar structures. Moreover, scanning tunnelling microscopy (STM) elucidates intriguing self-assembly of DBOV-TDOP at the interface of highly oriented pyrolytic graphite (HOPG) and 1-phenyloctane (PO). Individual molecules are visualized by STM, revealing that the alkyl chains protrude from the surface into the solution and suggesting that a self-assembled bilayer film structure is predominantly formed.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are attractive molecules not only because of their intriguing (opto)electronic and photophysical properties, but also for their unique self-assembly behaviour induced by the large aromatic core structures. A wide range of applications has thus been proposed for PAHs, not only as semiconductor materials in organic field-effect transistors and light-emitting devices, but also as fluorescent dyes in bioimaging.1–14 The supramolecular organization of the disc-shaped PAH molecules is of particular interest, leading to various self-assembled structures such as columns, nanotubes, and micelles.15–23 The combination of the rigid PAH cores with flexible aliphatic chains at the peripheral positions can induce a phase separation between these two components at the molecular level, resulting in the formation of columnar structures due to the strong intermolecular π–π interactions, and discotic liquid crystalline (LC) properties.24–35 Triphenylene, perylene and hexa-peri-hexabenzocoronene (HBC) derivatives are representative examples, which have been extensively studied with a variety of peripheral substituents, demonstrating controllable self-assembly behaviour and high charge-carrier mobility along the column direction.21,36–38 Their intermolecular packing can be tuned by attaching soft chains to provide supramolecular structures with specific organization, such as columnar self-assemblies with controlled rotation angle as well as intra- and intercolumnar distances.21,32 PAHs larger than HBC, as well as non-planar bowl-shaped PAHs, such as corannulene and sumanene were demonstrated to be also applicable as discotic LC materials.23,39–42 Nevertheless, the LC behaviours of PAHs other than triphenylenes, HBCs and perylenes are still underexplored and there is so far only a limited number of PAHs that are known to show LC properties. In particular, LC PAHs with smaller HOMO–LUMO gaps are seldom reported, despite the interest in their longer-wavelength absorption and emission as well as potential to achieve higher charge-carrier mobilities.43Further, two-dimensional (2D) self-assembly of PAHs has been studied at the liquid–solid interface between an organic solvent and highly-oriented pyrolytic graphite (HOPG). For studies at the liquid–solid interface PAHs are often substituted with long or bulky aliphatic groups at the periphery to increase their solubility. For example, alkoxy-substituted extended triphenylenes were found to form ordered monolayers at the 1,2,4-trichlorobenzene (TCB)/HOPG interface with a hexagonal structure.44 A representative result with a larger PAH was reported in 1995, demonstrating that HBC substituted with six alkyl chains can form 2D self-assembled structures at the liquid–solid interface.45 The HBC core extended with four additional K-regions has been shown to form a highly ordered monolayer at the TCB/HOPG interface, extending defect-free area over several hundred square nanometers, but with occasional molecular stacking on top of the first layer.46 In contrast thereto, unsubstituted HBC has been shown to form complete bilayers at the TCB/HOPG interface.47 Under ultra-high vacuum (UHV) conditions and at low temperatures, perchlorinated HBC has been studied on the Au(111) surface, revealing formation of islands of highly ordered supramolecular networks even at coverages lower than a full monolayer.48 More recently, we have observed preferential bilayer formation of triply-fused nanographene–porphyrin–nanographene conjugates at the TCB/HOPG interface.49 However, reported self-assemblies of PAHs at liquid–solid interfaces are predominantly monolayers, and formation of such self-assembled bilayers, where intermolecular interactions are stronger than the molecule–surface interactions, still remains rare.50
We recently reported the synthesis of a new type of nanographene, namely dibenzo[hi,st]ovalene (DBOV) with both zigzag and armchair edge structures, showing intensive absorption and emission in the visible light region.51,52 The molecules also revealed stimulated emission in a polystyrene matrix, which is crucial for potential applications in organic lasers or optical amplifiers.53 Moreover, this molecule has high chemical stability under ambient conditions, allowing easy processing and potentially long-term device application. However, derivatives of DBOV that have so far been synthesized were substituted with mesityl groups, which prevent the π–π stacking of the aromatic cores, or had low solubility.51,52,54,55 Packing mode in the solid state and self-assembly behaviour at solid/liquid interface of substituted DBOV derivatives with a new type of aromatic core remained so far unknown, although such information will be essential for further implementation of them in (opto)electronic devices.
Herein, we report a synthesis of a DBOV derivative functionalized with two 3,4,5-tris(dodecyloxy)phenyl substituents (DBOV-TDOP, Scheme 1). TDOP groups were selected in order to enhance the solubility without significantly affecting the π–π interactions between the DBOV cores, in comparison to the mesityl group with two ortho-methyl groups. The structure of DBOV-TDOP was unambiguously characterized by a combination of nuclear magnetic resonance (NMR) spectroscopy, high resolution matrix-assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF MS), and the optoelectronic properties were studied by UV-vis absorption and fluorescence spectroscopy. The large rigid aromatic core has strong tendency to aggregate with the long n-dodecyl chains stretched to the periphery, enabling the formation of columnar self-assembly structures. The phase formation and self-assembly in the solid state was investigated by differential scanning calorimetry (DSC), polarized optical microscopy (POM), and two-dimensional wide- and small-angle X-ray scattering (2D-WAXS and -SAXS), which clearly indicated its packing into helical stacks with an exceptionally long pitch. It has been proven that the helical packing can be well controlled by attaching side alkyl chains with a suitable steric demand, while a tight packing and good intracolumnar order were maintained. In addition, this molecule tends to form bilayer structures at the interface of HOPG and 1-phenyloctane (PO) due to strong intermolecular interactions.
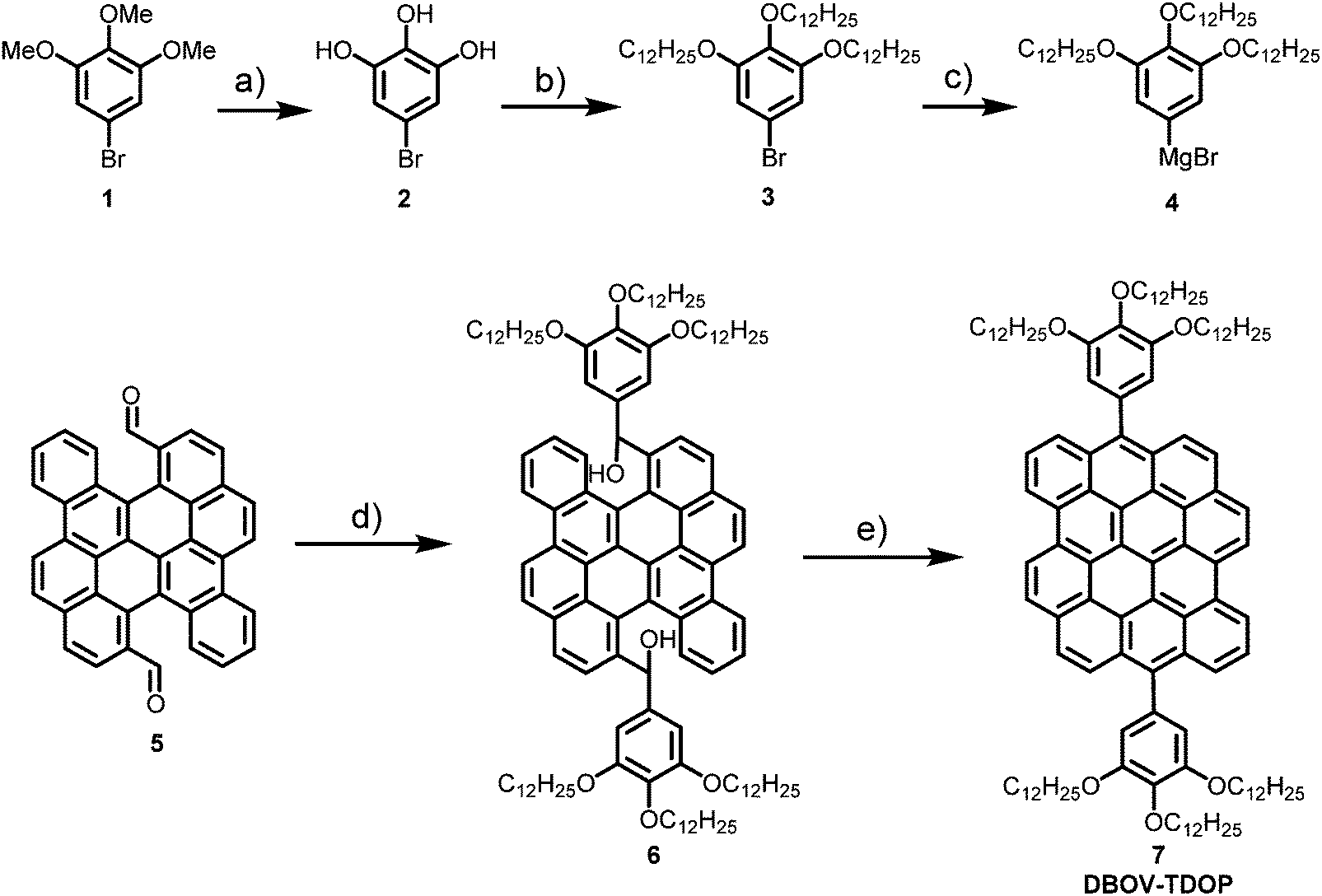 | ||
| Scheme 1 Synthetic route towards 6,14-bis{3,4,5-tris(dodecyloxy)phenyl}dibenzo-[hi,st]ovalene (DBOV-TDOP). Reaction conditions: (a) BBr3, −78 °C to r.t., overnight, 98% yield; (b) C12H25Br, K2CO3, N,N-dimethylformamide, 80 °C, 20 h, 63% yield; (c) Mg, I2, tetrahydrofuran, reflux, overnight; (d) 4, tetrahydrofuran, 4 h, room temperature; (e) BF3·OEt2, dichloromethane, room temperature, overnight, then p-chloranil, 2 h, 85% yield in three steps. | ||
Results and discussion
Synthesis and characterization of DBOV-TDOP
DBOV-TDOP was synthesized as shown in Scheme 1. First, 5-bromo-1,2,3-tris(dodecyloxy)benzene (3) was synthesized from 5-bromo-1,2,3-trimethoxybenzene (1) through a sequence of demethylation with BBr3 and three-fold nucleophilic substitution with 1-bromododecane. Then Grignard reagent 4 was prepared by refluxing 3 and magnesium turning in tetrahydrofuran with I2 as initiator. 5,14-Diformylbenzo[a]dinaphtho[2,1,8-cde:1′,2′,3′,4′-ghi]perylene (5)52,56 was subsequently treated with Grignard reagent 4 at room temperature followed by quenching with NH4Cl to give diol intermediate 6, which was subjected to BF3·OEt2 promoted Friedel–Crafts reaction and oxidation by p-chloranil to afford DBOV-TDOP with a high yield of 85% in three steps. This compound showed high solubility in dichloromethane, tetrahydrofuran or toluene, which allowed unambiguous characterization of its structure in solution.As shown in Fig. 1a, well-resolved sharp peaks are observed in its 1H NMR spectrum, which could be assigned with two-dimensional NMR techniques (see Fig. S1 and S2, ESI†). High-resolution (HR) MALDI-TOF MS spectra (Fig. 1b) exhibited one intense peak with m/z of 1729.2822, in very good agreement with the calculated value of 1729.2835. The UV-vis absorption was measured in toluene at a concentration of 10−6 M (Fig. 1c). Similar to other DBOV derivatives,51,52,54,55 the maximum absorption wavelength (λmax) is located at 609 nm with very high extinction coefficient of 3.85 × 105 M−1 cm−1. Three characteristic shoulder peaks are observed at shorter wavelength, arising from the vibronic coupling involving C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch. The solution showed very strong red fluorescence with maximum emission peak wavelength at 615 nm. Small Stokes shift of only 6 nm indicates its rigid core structure and minor energy loss in the excited state. Moreover, high relative fluorescence quantum yield (Φ) of 0.89 was measured with Nile blue A perchlorate as standard,57 indicating the potential of DBOV-TDOP for optoelectronic applications.
C stretch. The solution showed very strong red fluorescence with maximum emission peak wavelength at 615 nm. Small Stokes shift of only 6 nm indicates its rigid core structure and minor energy loss in the excited state. Moreover, high relative fluorescence quantum yield (Φ) of 0.89 was measured with Nile blue A perchlorate as standard,57 indicating the potential of DBOV-TDOP for optoelectronic applications.
 | ||
Fig. 1 (a) 1H NMR spectra of DBOV-TDOP in [D8]THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CS2 = 2:1; (b) HR MALDI-TOF MS spectrum of DBOV-TDOP, inset: magnified spectrum showing isotopic distribution pattern in comparison with theoretical calculation; (c) UV-vis absorption and fluorescence spectra of DBOV-TDOP measured in toluene solution with concentration of 10−6 M, inset shows pictures of toluene solution under room light (left) and UV light (right). :CS2 = 2:1; (b) HR MALDI-TOF MS spectrum of DBOV-TDOP, inset: magnified spectrum showing isotopic distribution pattern in comparison with theoretical calculation; (c) UV-vis absorption and fluorescence spectra of DBOV-TDOP measured in toluene solution with concentration of 10−6 M, inset shows pictures of toluene solution under room light (left) and UV light (right). | ||
Solid-state self-assembly into helical columns
The thermal stability of DBOV-TDOP was investigated by thermogravimetric analysis (TGA), which showed a high decomposition temperature of up to 365 °C (5% weight loss, Fig. S3, ESI†). The attachment of the six dodecyloxy chains induced distinct thermotropic properties which were studied by DSC and POM. The 2nd DSC heating scan showed two prominent peaks (Fig. 2a). The first peak at 145 °C was attributed to the reorganization of the side chains, while the second at 158 °C was related to the melting into the liquid crystalline phase. The clearing temperature for the compound was found at 252 °C and was confirmed by POM. Cooling the compound between two glass slides under cross-polarized microscopy from its isotropic into the liquid crystalline phase resulted in a characteristic birefringent fan-shaped focal conic texture (Fig. 2b). A typical supramolecular liquid crystalline organization was also found in the liquid crystalline phase by 2D-WAXS and -SAXS on extruded fibers. The hexagonal liquid crystalline phase typically leads to π-stacking reflections in the wide-angle range on the meridional plane of the 2DWAXS pattern and to equatorial reflections in the small angle region that are related to the hexagonal intercolumnar arrangement.58 As derived from the positions at the ratio of 1: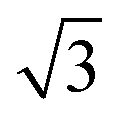 :2 of the equatorial scattering intensities (Fig. 3a and c), the DBOV-TDOP self-assembled into columnar stacks arranged in a hexagonal lattice. The equatorial reflections were indexed according to a hexagonal lattice parameter of ahex = 3.07 nm (Fig. 3e). The meridional reflections were assigned to the intracolumnar π-stacking distance of 0.36 nm. In the liquid crystalline phase, the molecular dynamics are high and the aromatic discs can perform an axial motion around the column axes. As the thermal energy decreases at lower temperature, this molecular motion is reduced leading to higher intracolumnar order and the formation of a more complex organization.59 Cooling the sample to the crystalline phase at 30 °C changed the supramolecular organization of DBOV-TDOP as indicated by the complex pattern in Fig. 3b. The large number of new scattering intensities confirmed the higher long-range order in the crystalline phase. The π-stacking distance slightly increased to 0.37 nm, while the intercolumnar arrangement turned into a square lattice with asqr = 5.35 nm. The reflections with mixed indices out of the equator and of the meridian located on scattering lines was characteristic for helical packing within the stacks.60–62 Additional off-meridional reflections in the small-angle region implied a helical intracolumnar packing of the molecules. The helical pitch of 5.55 nm was determined from the first scattering line (Fig. 3d) and corresponded to 15 molecules per helical pitch including a 180° rotation (Fig. 4). This exceptionally long helical pitch, in comparison to the typical pitch of around 1 nm for other PAHs, was related to good intracolumnar packing of the molecules.63 A rotation of 12° between adjacent molecules in the stack was induced to overcome the steric hindrance of the out-of-plane arranged TDOP group in the periphery of the aromatic DBOV core. The strategy to control the helical organization in supramolecular columns of disc-shaped molecules by employing sterically demanding substituents has been also realized for other nanographenes,35,38,64 but for the first time for a dibenzo[hi,st]ovalene of this aromatic shape. Interestingly, this concept could be applied for a DBOV core by attaching only two bulky substituents as proven in this work. Comparison to other core extended discotic PAHs, we confirmed the exceptionally long helical pitch of the DBOV molecules.63 Since the corresponding reflections of mixed indices are distinct and well defined, the molecular ordering in the stacks is pronounced and the helical twist between neighboring discs is well defined. Fluctuations in the twist angle would lead to poor or none reflections related to the helical packing. The high intracolumnar order of the helical structure with such long pitch is attributed to good intermolecular π-interactions.
:2 of the equatorial scattering intensities (Fig. 3a and c), the DBOV-TDOP self-assembled into columnar stacks arranged in a hexagonal lattice. The equatorial reflections were indexed according to a hexagonal lattice parameter of ahex = 3.07 nm (Fig. 3e). The meridional reflections were assigned to the intracolumnar π-stacking distance of 0.36 nm. In the liquid crystalline phase, the molecular dynamics are high and the aromatic discs can perform an axial motion around the column axes. As the thermal energy decreases at lower temperature, this molecular motion is reduced leading to higher intracolumnar order and the formation of a more complex organization.59 Cooling the sample to the crystalline phase at 30 °C changed the supramolecular organization of DBOV-TDOP as indicated by the complex pattern in Fig. 3b. The large number of new scattering intensities confirmed the higher long-range order in the crystalline phase. The π-stacking distance slightly increased to 0.37 nm, while the intercolumnar arrangement turned into a square lattice with asqr = 5.35 nm. The reflections with mixed indices out of the equator and of the meridian located on scattering lines was characteristic for helical packing within the stacks.60–62 Additional off-meridional reflections in the small-angle region implied a helical intracolumnar packing of the molecules. The helical pitch of 5.55 nm was determined from the first scattering line (Fig. 3d) and corresponded to 15 molecules per helical pitch including a 180° rotation (Fig. 4). This exceptionally long helical pitch, in comparison to the typical pitch of around 1 nm for other PAHs, was related to good intracolumnar packing of the molecules.63 A rotation of 12° between adjacent molecules in the stack was induced to overcome the steric hindrance of the out-of-plane arranged TDOP group in the periphery of the aromatic DBOV core. The strategy to control the helical organization in supramolecular columns of disc-shaped molecules by employing sterically demanding substituents has been also realized for other nanographenes,35,38,64 but for the first time for a dibenzo[hi,st]ovalene of this aromatic shape. Interestingly, this concept could be applied for a DBOV core by attaching only two bulky substituents as proven in this work. Comparison to other core extended discotic PAHs, we confirmed the exceptionally long helical pitch of the DBOV molecules.63 Since the corresponding reflections of mixed indices are distinct and well defined, the molecular ordering in the stacks is pronounced and the helical twist between neighboring discs is well defined. Fluctuations in the twist angle would lead to poor or none reflections related to the helical packing. The high intracolumnar order of the helical structure with such long pitch is attributed to good intermolecular π-interactions.
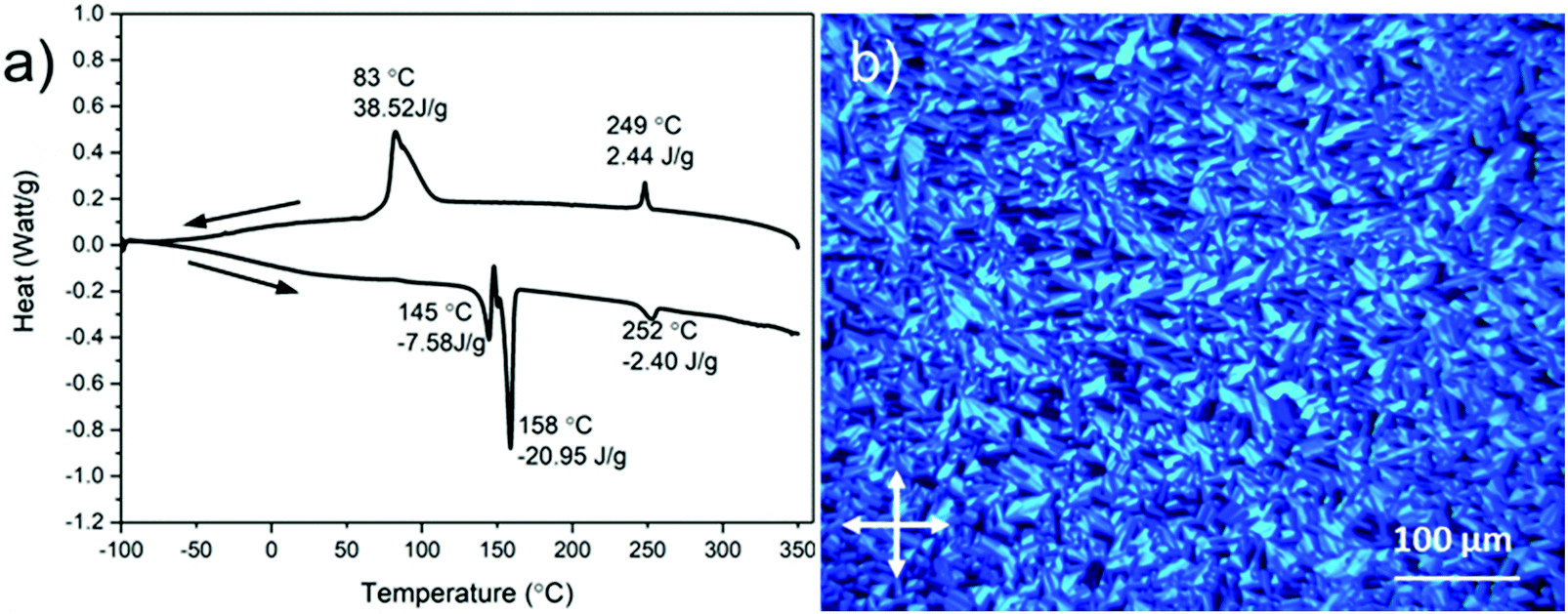 | ||
| Fig. 2 (a) Differential scanning calorimetry (DSC) scans (1st cooling and 2nd heating at 10 °C min−1) and (b) polarized optical microscopy (POM) photograph at 150 °C of DBOV-TDOP. | ||
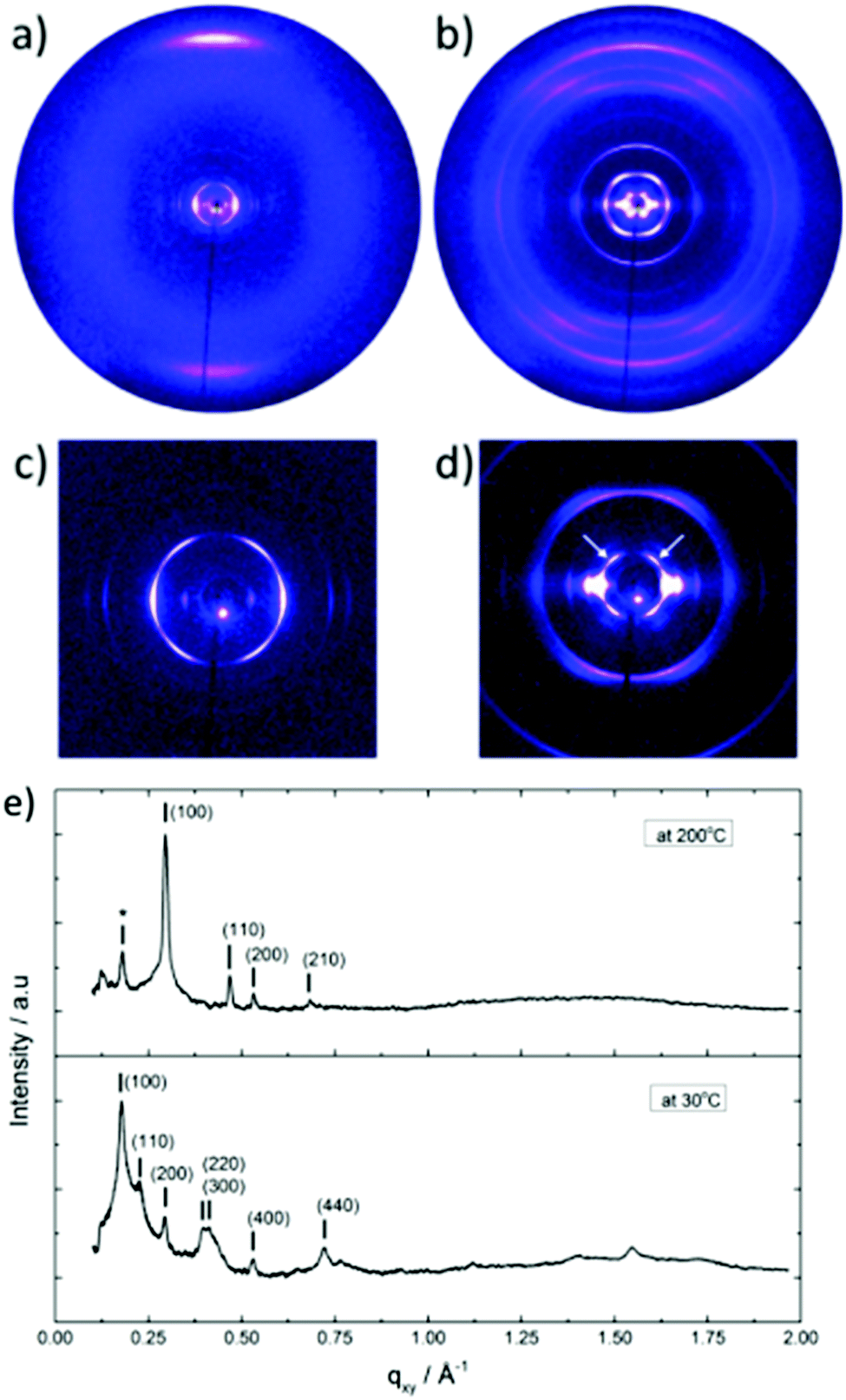 | ||
| Fig. 3 2D-WAXS and -SAXS patterns of DBOV-TDOP obtained at (a), (c) 200 °C and (b), (d) 30 °C (* indicates remaining reflection from the crystalline phase; white arrows assign reflections of the first scattering line) and (e) equatorial integration for both measurements (reflections are indexed by Miller indices). | ||
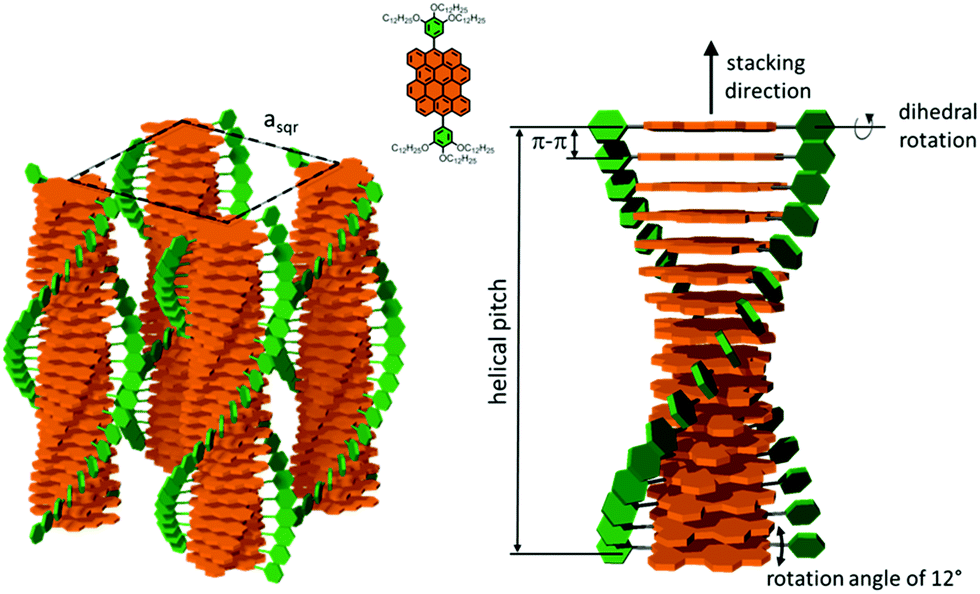 | ||
| Fig. 4 Schematic illustration of the helical organization of DBOV-TDOP at 30 °C. Due to π-stacking interactions, the molecules self-assemble in columnar structures that are arranged in a hexagonal fashion. In the stacks, the molecules are rotated by 12° with respect to each other and form a helix. Alkoxy side chains are omitted for clarity. | ||
STM characterization of DBOV-TDOP
Scanning tunnelling microscopy (STM) was used to study the 2D self-assembly behaviour of DBOV-TDOP at the interface between PO and HOPG. A solution of DBOV-TDOP in PO was drop-casted on freshly-cleaved HOPG and subsequently imaged by STM at room temperature, at the liquid–solid interface. Already at a concentration of c = 6 × 10−5 mol L−1 the HOPG surface was found to be covered by a molecular layer, appearing as a row-like structure, with only a few single-molecule defects and small non-covered areas (Fig. 5a). The apparent height of the layer was found to be approximately 0.7 nm (see inset in Fig. 5a). The unit cell (a = 2.32 ± 0.05 nm, b = 1.49 ± 0.04 nm, α = 74.8 ± 2.0°) was determined with several drift-corrected high-resolution STM images, i.e. by imaging first the molecular layer and subsequently the HOPG surface underneath by changing the tunnelling parameters (Fig. S4, ESI†). A tentative model of the monolayer structure, based on high-resolution STM images and unit cell dimensions, is shown in Fig. 5d. The molecules assemble with the DBOV cores aligned in a row-like fashion with the long molecular axes oriented perpendicular to the row direction. Adjacent rows are shifted half a molecular width, so that interdigitation of the peripheral phenyl groups allows close-packing. Single-molecule defects thereby help to identify the position in the assembly (Fig. 5d and Fig. S5, ESI†). The unit cell parameters indicate that the space between adjacent aromatic cores is not sufficient for the adsorption of the alkyl-chains. Thus, they are most likely desorbed from the surface and back-folded in the supernatant solution. More dilute solutions (c = 6 × 10−6 mol L−1) lead to the formation of similar molecular assemblies, albeit with much lower surface coverage, while maintaining the same apparent height (∼0.7 nm) (Fig. 5b and Fig. S6, ESI†).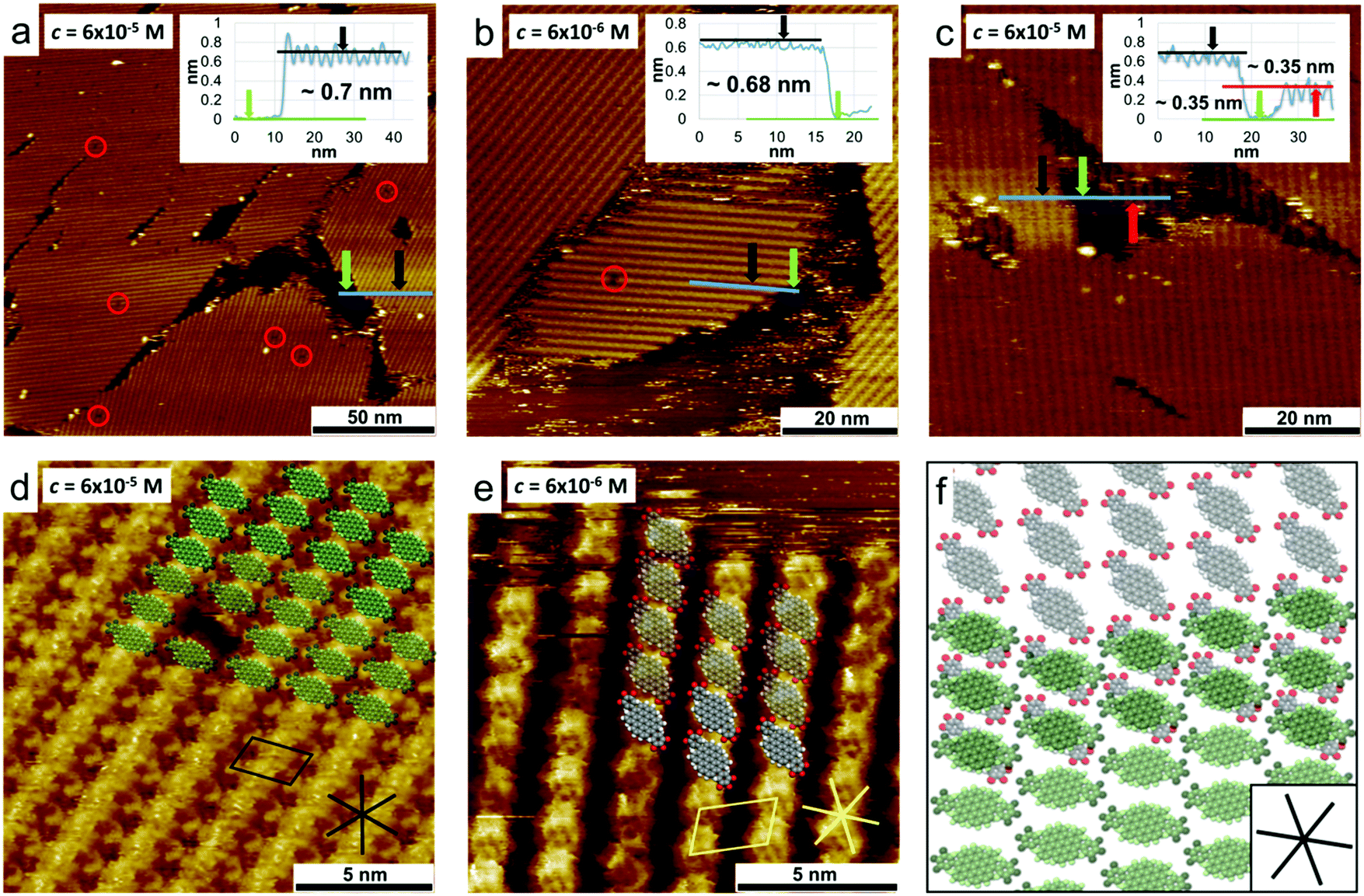 | ||
| Fig. 5 STM characterization of DBOV-TDOP at the PO/HOPG interface. (a) Large-scale STM image showing the long-range order with few single-molecule defects (red circles) and non-covered areas (c = 6 × 10−5 mol L−1). The line profile across a non-covered area reveals an apparent layer height of approximately 0.7 nm (see inset). (b) Large-scale STM image at lower concentration (c = 6 × 10−6 mol L−1) showing an increase of disordered areas and the same layer height (see inset). (c) STM image revealing double layer formation with an apparent height of about 0.35 nm for each layer. (d) High-resolution STM images superimposed with a tentative model of the molecular assembly. (e) High-resolution STM image and tentative model of the molecular assembly in the lower layer. The alkyl chains are presumably desorbed from the surface and omitted in the model for clarity. (f) Superposition of the tentative molecular models for the first and second layer. Imaging parameters: (a) Vbias = −1.1 V, Iset = 80 pA, (b) Vbias = −1.2 V, Iset = 60 pA, (c) Vbias = −1.1 V, Iset = 60 pA, (d) Vbias = −1.1 V, Iset = 230 pA, (e) Vbias = −0.9 V, Iset = 50 pA. | ||
Closer inspection of the molecular layers adjacent to the uncovered areas revealed that the molecules assemble in a bilayer instead of a monolayer as evidenced by the STM image provided in Fig. 5c, which clearly shows a lower molecular layer that extends beneath the second layer. Furthermore, high-resolution images of this lower layer show a different molecular orientation, i.e. the long axes of the DBOV cores are not perpendicular, but instead rotated by approximately 40° with respect to the row propagation (Fig. 5e). A superposition of the tentative molecular assembly of the first and second layer is shown in Fig. 5f. The DBOV cores of the molecules in the first and second layer are proposed to be at the same positions, while the long molecular axes are rotated in a way that the peripheral aryl moieties, which are assumed to be out-of-plane compared to the DBOV core, are not on top of each other, allowing a closer distance between the cores and thus increased π–π interactions. The observed preferential double layer formation even at low concentrations indicates that the π–π interactions between the DBOV cores are stronger than the DBOV–HOPG interactions. Note that additional layers above the double layer may form, which, however, could be removed by the STM tip during the imaging process.65 Additional layers would decrease the tunnelling current and thus the STM tip would have to move closer to the surface to maintain the current setpoint, thereby removing the additional layers again while scanning. In the liquid crystalline phase, these strong π–π interactions lead to the observed columnar structures. The apparent layer height of about 0.35 nm is also comparable with the π-stacking distance of 0.36 nm found in the columnar stacks as is the relative orientation of the molecules in the first and second layer.
Conclusion
In conclusion, we reported the synthesis of the discotic dibenzo[hi,st]ovalene molecule functionalized with two 3,4,5-tris(dodecyloxy)phenyl groups at the meso-positions. The self-assembly behavior of this molecule was extensively investigated. The attachment of 1,2,3-tris(dodecyloxy)phenyl substituents induced thermotropic properties and self-assembly into columnar structures of DBOV-TDOP. Due to the distinct steric demand of the side groups, the molecules arranged in a helical fashion in the stacks. Interestingly, an especially long helical pitch is found indicating a tight molecular packing and high intracolumnar order despite the pronounced steric hindrance of the substituents. A well-defined rotation of the aromatic cores together with long-range ordering in the columnar stacks are prerequisites for an unhindered intrinsic charge carrier transport in discotic liquid crystals. Additionally, at the liquid–solid interface, large-area bilayer film formation was observed even at low concentrations as a result of the strong intermolecular π–π interactions. The tendency to form self-organized and well-defined supramolecular structures, both columns and film, makes DBOV-TDOP a promising candidate for fabricating electronic devices.Experimental methods
General
All reactions working with air- or moisture-sensitive compounds were carried out under argon atmosphere using standard Schlenk line techniques. Thin layer chromatography (TLC) was done on silica gel coated aluminum sheets with F254 indicator and column chromatography separation was performed with silica gel (particle size 0.063–0.200 mm). Nuclear Magnetic Resonance (NMR) spectra were recorded using Bruker DPX 300 MHz NMR spectrometers. Chemical shifts (δ) were expressed in ppm relative to the residual of solvents (CD2Cl2, 1H: 5.32 ppm, 13C: 53.84 ppm; THF-d8, 1H: 3.58 ppm, 13C: 67.57 ppm). Coupling constants (J) were recorded in Hertz. High resolution mass spectra (HR MS) were recorded on a Bruker Reflex II-TOF spectrometer by matrix-assisted laser decomposition/ionization (MALDI) using 7,7,8,8-tetracyanoquinodimethane (TCNQ) as matrix and calibrated against poly(ethylene glycol). Thermogravimetric analysis (TGA) was measured on a Mettler Toledo TGA-851 system with a heating speed of 10 K min−1 under nitrogen atmosphere. Differential scanning calorimetry (DSC) was recorded on a Mettler DSC 30 instrument with a heating speed of 10 K min−1 under nitrogen atmosphere. UV-vis absorption spectra were recorded on a PerkinElmer Lambda 900 spectrometer at room temperature using a 10 mm quartz cell. Photoluminescence spectra were recorded on a J&MTIDAS spectrofluorometer. The fluorescence quantum yield (Φ) was measured using nile blue A perchlorate (in ethanol under air, Φ = 0.27) as a reference.57Materials
Unless otherwise noted, all starting materials and reagents were purchased from commercial sources and used without further purification. The synthesis of 5,14-diformylbenzo[a]dinaphtho[2,1,8-cde:1′,2′,3′,4′-ghi]perylene (5) is described in our previously report,52 and we also used an improved synthetic route, which we have more recently developed.56Synthesis of 5-bromo-1,2,3-tris(dodecyloxy)benzene (3)
5-Bromo-1,2,3-trimethoxybenzene (1) (2.5 g, 10 mmol) was dissolved in dry dichloromethane (30 mL), and the solution was cooled down to −78 °C. BBr3 (8.26 g, 33.0 mmol) was added dropwise to the solution then the reaction mixture was gradually warmed up to room temperature and stirred overnight. After completion of reaction, the mixture was poured into 100 mL of ice water and extracted with ethyl acetate (100 mL) for three times. The organic layers were combined, washed with brine (100 mL) and dried over Na2SO4. After evaporation of the solvent and drying under reduced pressure, a crude product of 5-bromobenzene-1,2,3-triol (2) (2.0 g, 98%) was obtained as white solid, which was used directly for the next step without further purification. To a 100 mL Schlenk flask was added the crude product of 5-bromobenzene-1,2,3-triol (2) (2.0 g, 9.8 mmol), 1-bromododecyl (9.92 g, 40.0 mmol) and K2CO3 (5.52 g, 40.0 mmol). The flask was evacuated and backfilled with argon for three times before N,N-dimethylformamide (50 mL) was added. After stirring at 80 °C for 20 h, the mixture was cooled down to room temperature and diluted with ethyl acetate (200 mL), washed with water (50 mL) and brine (50 mL), dried over Na2SO4 and evaporated. The obtained residue was purified by silica gel column chromatography (eluent: n-hexane) and recrystallized with ethanol to give 5-bromo-1,2,3-tris(dodecyloxy)benzene (3) (4.5 g, 63% yield over two steps) as white solid. 1H NMR (300 MHz, methylene chloride-d2) δ 6.68 (s, 2H), 3.97–3.83 (m, 6H), 1.85–1.72 (m, 4H), 1.72–1.62 (m, 2H), 1.51–1.39 (m, 6H), 1.39–1.22 (m, 48H), 0.94–0.82 (m, 9H); 13C NMR (75 MHz, methylene chloride-d2) δ 154.3, 137.8, 115.8, 110.3, 73.8, 69.7, 32.4, 30.7, 30.2, 30.2, 30.1, 30.1, 30.1, 30.0, 29.8, 29.8, 29.7, 26.5, 26.5, 23.1, 14.3; HRMS (MALDI-TOF): m/z calcd for C42H77BrO3: 708.5056 [M]+, found: 708.5005.Synthesis of DBOV-TDOP
To a 25 mL Schlenk tube was added magnesium turnings (27 mg, 1.1 mmol), I2 (3 mg, 0.01 mmol) and tetrahydrofuran (1 mL). The mixture was gently heated while approximately 1 mL of 5-bromo-1,2,3-tris(dodecyloxy)benzene (3) (568 mg, 0.800 mmol) dissolved in tetrahydrofuran (THF) (3 mL) was added. As soon as the solution became colorless, the remaining solution was added dropwise under mild reflux and stirring was continued overnight. The obtained solution of Grignard reagent (4 mL) was transferred to a solution of 5,14-diformylbenzo[a]dinaphtho[2,1,8-cde:1′,2′,3′,4′-ghi]perylene (5) (25 mg, 0.049 mmol) in dry THF (30 mL). After stirring at room temperature for 4 h, the reaction was quenched by addition of saturated NH4Cl solution (15 mL). After stirring for 15 min, the solution was extracted with ethyl acetate (50 mL) for three times. The combined organic layers were washed with brine, dried with Na2SO4 and evaporated. The residue was dried under vacuum for 2 h and dissolved in anhydrous dichloromethane (30 mL). After bubbling with argon saturated with dichloromethane vapour for 15 min, BF3·OEt2 (0.1 mL) was added and the stirring was continued overnight. After quenching with methanol (2 mL), p-chloranil (12 mg, 0.049 mmol) was added and the mixture was stirred for 2 h at room temperature. The solvent was evaporated and the residue was purified by silica gel column chromatography (eluent: n-hexane:dichloromethane = 3:1 to 0:1) to give the title compound (72 mg, 85% yield) as blue solid. 1H NMR (300 MHz, THF-d8) δ 9.27 (d, J = 8.5 Hz, 2H), 9.02 (d, J = 7.7 Hz, 2H), 8.36 (d, J = 8.4 Hz, 2H), 8.10 (d, J = 8.3 Hz, 2H), 7.97 (t, J = 9.1 Hz, 4H), 7.89 (d, J = 9.3 Hz, 2H), 6.87 (s, 4H), 4.16 (t, J = 6.2 Hz, 4H), 4.05 (t, J = 6.2 Hz, 8H), 1.97–1.79 (m, 15H), 1.69–1.61 (m, 4H), 1.61–1.15 (m, 119H), 0.97–0.80 (m, 18H); 13C NMR (75 MHz, THF-d8) δ 139.1, 137.5, 134.9, 132.7, 132.5, 131.3, 130.3, 130.1, 128.9, 127.9, 127.5, 127.3, 126.5, 125.5, 125.1, 124.7, 124.3, 123.6, 123.4, 122.1, 121.4, 111.0, 74.0, 70.0, 33.2, 33.1, 31.9, 31.1, 31.1, 31.1, 31.0, 30.9, 30.9, 30.8, 30.7, 30.7, 30.6, 27.6, 27.4, 23.9, 23.9, 14.9, 14.9; HRMS (MALDI-TOF): m/z calcd for C122H168O6: 1729.2841 [M]+, found: 1729.2822.
POM
The optical textures of the compound were investigated using a Zeiss microscope with polarizing filters equipped with a Hitachi KP-D50 colour digital CCD camera. The samples were sandwiched between two glass slides and thermally treated on a Linkam hot stage regulated with a Linkam TMS 91 temperature controller.2DWAXS and -SAXS
2DWAXS and -SAXS measurements were performed using a setup consisting of copper solid-anode X-ray tube (Bruker AXS Krystalloflex 760, operated at 35 kV and 30 mA), Osmic confocal MaxFlux optics and a three pin-hole collimation system. The fiber samples for the measurements were prepared by extrusion at 25 °C and were positioned perpendicular to the incident X-ray beam and vertical to the 2D detector. The scattering intensity was detected on a 2-D image plate (MAR-345) with a pixel size of 100 μm (2345 × 2345 pixels). Scattering data are expressed as a function of the scattering vector: q = 4π/λ sin(Θ), where Θ is a half the scattering angle and λ = 0.154 Å is the wavelength of the incident radiation. All X-ray scattering measurements were performed under vacuum (∼20 mbar) to reduce air scattering and beam damage to the sample. All 2DWAXS data processing and analysis was performed by using the software package Datasqueeze (http://www.datasqueezesoftware.com).STM
STM measurements were performed with a Molecular Imaging or Agilent PicoLE system in constant-current mode (PicoSPM) at the 1-phenyloctane/highly-oriented pyrolytic graphite (PO/HOPG) interface at room temperature (20–22 °C). The imaging parameters Iset (tunneling current) and Vbias (substrate bias) are indicated in the figure captions. STM tips were obtained by mechanical cutting of a Pt/Ir wire (80%/20%, diameter 0.25 mm). Immediately before use, the HOPG substrate (grade ZYB, Advanced Ceramics) was freshly cleaved using adhesive tape. The solvent, 1-phenyloctane (Sigma-Aldrich, 98%), was used without further purification. Scanning Probe Imaging Processor (SPIP 6.5.1) software from Image Metrology ApS was used for STM image processing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the Max Planck Society and the Fund of Scientific Research Flanders (FWO) under EOS 30489208. J. S. acknowledges financial support through a Marie Skłodowska-Curie Individual Fellowship (EU project 789865 – EnSurf). Open Access funding provided by the Max Planck Society.References
- M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267–1300 CrossRef CAS.
- A. Narita, X. Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- W. Pisula, X. Feng and K. Müllen, Chem. Mater., 2011, 23, 554–567 CrossRef CAS.
- Q. Ye and C. Chi, Chem. Mater., 2014, 26, 4046–4056 CrossRef CAS.
- J. Li and Q. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28049–28062 CrossRef CAS PubMed.
- L. Zhang, Y. Cao, N. S. Colella, Y. Liang, J. L. Bredas, K. N. Houk and A. L. Briseno, Acc. Chem. Res., 2015, 48, 500–509 CrossRef CAS PubMed.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed.
- R. K. Gupta and A. A. Sudhakar, Langmuir, 2019, 35, 2455–2479 CrossRef CAS PubMed.
- D. Meng, G. Liu, C. Xiao, Y. Shi, L. Zhang, L. Jiang, K. K. Baldridge, Y. Li, J. S. Siegel and Z. Wang, J. Am. Chem. Soc., 2019, 141, 5402–5408 CrossRef CAS.
- X. Liu, M. Chen, C. Xiao, N. Xue and L. Zhang, Org. Lett., 2018, 20, 4512–4515 CrossRef CAS PubMed.
- X. Cui, C. Xiao, T. Winands, T. Koch, Y. Li, L. Zhang, N. L. Doltsinis and Z. Wang, J. Am. Chem. Soc., 2018, 140, 12175–12180 CrossRef CAS PubMed.
- J. Li, K. Zhou, J. Liu, Y. Zhen, L. Liu, J. Zhang, H. Dong, X. Zhang, L. Jiang and W. Hu, J. Am. Chem. Soc., 2017, 139, 17261–17264 CrossRef CAS PubMed.
- C. Ruiz, U. K. Pandey, R. Termine, E. M. Garcia-Frutos, G. Lopez-Espejo, R. P. Ortiz, W. Huang, T. J. Marks, A. Facchetti, M. C. Ruiz Delgado, A. Golemme and B. Gomez-Lor, ACS Appl. Mater. Interfaces, 2016, 8, 26964–26971 CrossRef CAS PubMed.
- H. A. Lin, Y. Sato, Y. Segawa, T. Nishihara, N. Sugimoto, L. T. Scott, T. Higashiyama and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 2874–2878 CrossRef CAS PubMed.
- B. R. Kaafarani, Chem. Mater., 2011, 23, 378–396 CrossRef CAS.
- S. Xiao, M. Myers, Q. Miao, S. Sanaur, K. Pang, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390–7394 CrossRef CAS.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef CAS.
- S. Xiao, J. Tang, T. Beetz, X. Guo, N. Tremblay, T. Siegrist, Y. Zhu, M. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2006, 128, 10700–10701 CrossRef CAS PubMed.
- Z. Chen, A. Lohr, C. R. Saha-Moller and F. Wurthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- E. Moulin, J. J. Cid and N. Giuseppone, Adv. Mater., 2013, 25, 477–487 CrossRef CAS.
- W. Jin, Y. Yamamoto, T. Fukushima, N. Ishii, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 9434–9440 CrossRef CAS PubMed.
- L. Chen, S. R. Puniredd, Y. Z. Tan, M. Baumgarten, U. Zschieschang, V. Enkelmann, W. Pisula, X. Feng, H. Klauk and K. Müllen, J. Am. Chem. Soc., 2012, 134, 17869–17872 CrossRef CAS PubMed.
- W. Yuan, X. K. Ren, M. Li, H. Guo, Y. Han, M. Wu, Q. Wang, M. Li and Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 6161–6165 CrossRef CAS PubMed.
- S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hagele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS PubMed.
- S. K. Pal, S. Setia, B. S. Avinash and S. Kumar, Liq. Cryst., 2013, 40, 1769–1816 CrossRef CAS.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- N. Boden, R. J. Bushby, J. Clements and B. Movaghar, J. Mater. Chem., 1999, 9, 2081–2086 RSC.
- S. Kumar, Chem. Soc. Rev., 2006, 35, 83–109 RSC.
- T. Wohrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- W. Pisula, M. Zorn, J. Y. Chang, K. Müllen and R. Zentel, Macromol. Rapid Commun., 2009, 30, 1179–1202 CrossRef CAS.
- J. M. Warman, J. Piris, W. Pisula, M. Kastler, D. Wasserfallen and K. Müllen, J. Am. Chem. Soc., 2005, 127, 14257–14262 CrossRef CAS PubMed.
- D. Adam, P. Schuhmacher, J. Simmerer, L. Häussling, K. Siemensmeyer, K. H. Etzbachi, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141–143 CrossRef CAS.
- R. J. Bushby and O. R. Lozman, Curr. Opin. Colloid Interface Sci., 2002, 7, 343–354 CrossRef CAS.
- M. Kastler, W. Pisula, F. Laquai, A. Kumar, R. J. Davies, S. Baluschev, M. C. Garcia–Gutiérrez, D. Wasserfallen, H. J. Butt, C. Riekel, G. Wegner and K. Müllen, Adv. Mater., 2006, 18, 2255–2259 CrossRef CAS.
- L. Chen, K. S. Mali, S. R. Puniredd, M. Baumgarten, K. Parvez, W. Pisula, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2013, 135, 13531–13537 CrossRef CAS PubMed.
- A. M. van de Craats, J. M. Warman, A. Fechtenkötter, J. D. Brand, M. A. Harbison and K. Müllen, Adv. Mater., 1999, 11, 1469–1472 CrossRef CAS.
- X. Feng, W. Pisula, T. Kudernac, D. Wu, L. Zhi, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2009, 131, 4439–4448 CrossRef CAS.
- W. Pisula, Z. Tomovic, M. D. Watson, K. Müllen, J. Kussmann, C. Ochsenfeld, T. Metzroth and J. Gauss, J. Phys. Chem. B, 2007, 111, 7481–7487 CrossRef CAS PubMed.
- T. Nagano, K. Nakamura, Y. Tokimaru, S. Ito, D. Miyajima, T. Aida and K. Nozaki, Chem. – Eur. J., 2018, 24, 14075–14078 CrossRef CAS PubMed.
- Y. Shoji, T. Kajitani, F. Ishiwari, Q. Ding, H. Sato, H. Anetai, T. Akutagawa, H. Sakurai and T. Fukushima, Chem. Sci., 2017, 8, 8405–8410 RSC.
- D. Miyajima, K. Tashiro, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2009, 131, 44–45 CrossRef CAS PubMed.
- A. Idelson, C. Sterzenbach, S. S. Jester, C. Tschierske, U. Baumeister and S. Hoger, J. Am. Chem. Soc., 2017, 139, 4429–4434 CrossRef CAS PubMed.
- C. Aumaitre and J. F. Morin, Chem. Rec., 2019, 19, 1142–1154 CrossRef CAS PubMed.
- T. Yatabe, M. A. Harbison, J. D. Brand, M. Wagner, K. Müllen, P. Samorí and J. P. Rabe, J. Mater. Chem., 2000, 10, 1519–1525 RSC.
- A. Stabel, P. Herwig, K. Müllen and J. P. Rabe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1609–1611 CrossRef CAS.
- T. Dumslaff, B. Yang, A. Maghsoumi, G. Velpula, K. S. Mali, C. Castiglioni, S. De Feyter, M. Tommasini, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 4726–4729 CrossRef CAS PubMed.
- P. Samorí, N. Severin, C. D. Simpson, K. Müllen and J. P. Rabe, J. Am. Chem. Soc., 2002, 124, 9454–9457 CrossRef.
- Y. Zhang, Y. Zhang, G. Li, J. Lu, X. Lin, Y. Tan, X. Feng, S. Du, K. Müllen and H. J. Gao, J. Chem. Phys., 2015, 142, 101911 CrossRef PubMed.
- Q. Chen, L. Brambilla, L. Daukiya, K. S. Mali, S. De Feyter, M. Tommasini, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2018, 57, 11233–11237 CrossRef CAS PubMed.
- P. Samorí, A. Fechtenkötter, F. Jäckel, T. Böhme, K. Müllen and J. P. Rabe, J. Am. Chem. Soc., 2001, 123, 11462–11467 CrossRef PubMed.
- G. M. Paterno, Q. Chen, X. Y. Wang, J. Liu, S. G. Motti, A. Petrozza, X. Feng, G. Lanzani, K. Müllen, A. Narita and F. Scotognella, Angew. Chem., Int. Ed., 2017, 56, 6753–6757 CrossRef CAS PubMed.
- D. M. Coles, Q. Chen, L. C. Flatten, J. M. Smith, K. Müllen, A. Narita and D. G. Lidzey, Nano Lett., 2017, 17, 5521–5525 CrossRef CAS PubMed.
- A. J. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- Q. Chen, D. Wang, M. Baumgarten, D. Schollmeyer, K. Müllen and A. Narita, Chem. – Asian J., 2019, 14, 1703–1707 CrossRef CAS PubMed.
- G. M. Paternò, L. Nicoli, Q. Chen, K. Müllen, A. Narita, G. Lanzani and F. Scotognella, J. Phys. Chem. C, 2018, 122, 25007–25013 CrossRef.
- Q. Chen, S. Thoms, S. Stöttinger, D. Schollmeyer, K. Müllen, A. Narita and T. Basché, 2019, under review.
- R. Sens and K. H. Drexhage, J. Lumin., 1981, 24–25, 709–712 CrossRef CAS.
- K. Ban, K. Nishizawa, K. Ohta, A. M. van de Craats, J. M. Warman, I. Yamamoto and H. Shirai, J. Mater. Chem., 2001, 11, 321–331 RSC.
- I. Fischbach, T. Pakula, P. Minkin, A. Fechtenkötter, K. Müllen, H. W. Spiess and K. Saalwächter, J. Phys. Chem. B, 2002, 106, 6408–6418 CrossRef CAS.
- R. Martín-Rapún, D. Byelov, A. R. A. Palmans, W. H. de Jeu and E. W. Meijer, Langmuir, 2009, 25, 8794–8801 CrossRef.
- E. Beltran, E. Cavero, J. Barbera, J. L. Serrano, A. Elduque and R. Gimenez, Chem. – Eur. J., 2009, 15, 9017–9023 CrossRef CAS.
- M. Peterca, M. R. Imam, C. H. Ahn, V. S. Balagurusamy, D. A. Wilson, B. M. Rosen and V. Percec, J. Am. Chem. Soc., 2011, 133, 2311–2328 CrossRef CAS PubMed.
- W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634–3649 CrossRef CAS PubMed.
- X. Feng, W. Pisula and K. Müllen, J. Am. Chem. Soc., 2007, 129, 14116–14117 CrossRef CAS PubMed.
- W. Azzam, Appl. Surf. Sci., 2016, 371, 562–570 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tc03350e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |