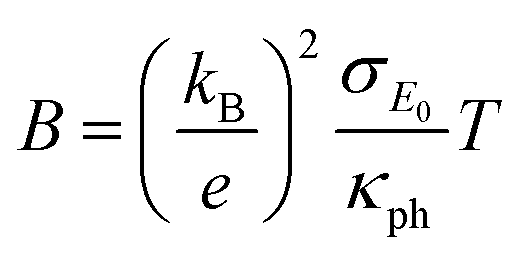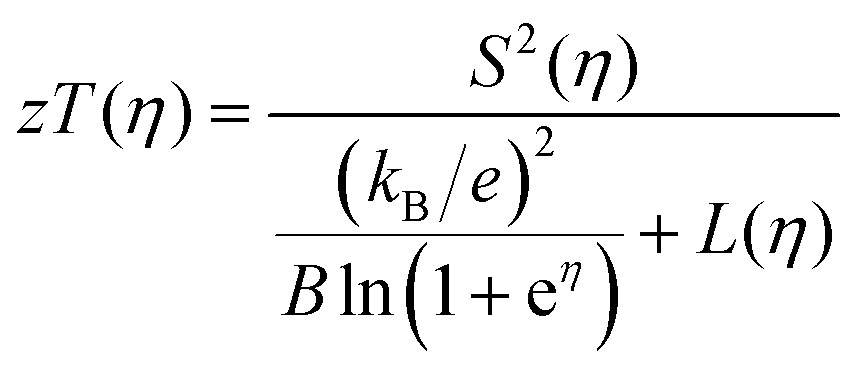Layered manganese bismuth tellurides with GeBi4Te7- and GeBi6Te10-type structures: towards multifunctional materials†
Received
19th February 2019
, Accepted 18th July 2019
First published on 18th July 2019
1 Introduction
Layered chalcogenides have received much attention due to their exceptional properties. These include topologically non-trivial electronic structures, charge-density waves, superconductivity or promising thermoelectric properties.1–7 For thermoelectric waste-heat recovery and Peltier cooling near room temperature (RT), the layered semiconductor Bi2Te3 and its substitution variants with Sb and Se are used in technical applications.8 The thermoelectric performance of Bi2Te3 has been optimized e.g. by Se substitution and Cu doping.9 In addition to its thermoelectric properties,10,11 angle-resolved photoemission spectroscopy (ARPES) has proved that Bi2Se3 and Bi2Te3 are topological insulators.3,12–14 Variants of Bi2Te3 or Bi2Se3 doped with transition metals such as Cr or Fe exhibit different magnetic interactions caused by the dopants; they may represent a new class of magnetic topological insulators in the field of spintronics.15,16 Probably motivated by its natural abundance, Mn, has widely been used as a dopant in Bi2Te3; and structural, electrical and magnetic properties have been studied.17–22 For example, thin-film topological insulators based on Mn- or Cr-doped Bi2Te3 are potential candidates for highly sensitive anomalous Hall effect sensors.17,23 In contrast to the ordinary Hall effect caused by the Lorentz force, the anomalous Hall effect arises from the spin–orbit interaction of spin-polarized charge carriers in conductive ferromagnetic materials and provides an alternative approach for magnetic sensing.
Layered Bi2Te3 exhibits a 15R stacking sequence of three slabs per unit cell separated by van der Waals gaps (cf. Section 3.3, Fig. 5), each slab composed of double layers of [BiTe6] octahedra often called “quintuple layer” as it consists of five atom layers. Many compounds can be derived from Bi2Te3 by formally inserting TtTe (Tt = Pb, Ge, Sn) units.24 In addition to its thermoelectric behavior, SnBi2Te4 shows a pressure-induced electronic topological transition between 3.5 and 5.0 GPa.25 ARPES yields evidence for topological surface states with Dirac dispersion in the indirect bandgap of bulk Pb(Bi2−xSbx)Te4.26 The GeBi2Te4-type structure of these compounds is described by a 21R stacking sequence, whose “septuple” layers comprise three triple layers of condensed octahedra as described by 2∞[Te3/3M(μ-Te3/6)M(μ-Te3/6)MTe3/3] (M = Ge, Bi; usually disordered), again separated by van der Waals gaps.27,28 Formally adding one Bi2Te3-type quintuple layer per GeBi2Te4-type septuple building block results in the GeBi4Te7 structure type with a 12P stacking sequence.27,29 Insertion of a second Bi2Te3-type layer results in the GeBi6Te10 structure type, described by a 51R stacking sequence. From another point of view, these structures can be understood as consisting of distorted rocksalt-type slabs similar to those in GeBi2Te4 and in Bi2Te3, separated by van der Waals gaps.24 Mn doping and its influence on the thermoelectric properties was also reported for the layered compound Ge4Sb2Te7 crystallizing with a Ge3As2Te6-type structure.30 MnBi2Te4 has been reported to be the first compound of the series MTe(Bi2Te3)n where M is a transition metal represented by manganese.31 Single-crystal data of MnBi2Te4 revealed Mn/Bi antisite disorder; the magnetic properties are strongly anisotropic and antiferromagnetic ordering is observed below 24 K.32,33 Investigations on the tentative phase diagram of the system Mn/Bi/Te indicated that there are further layered compounds in that system,34 and estimates concerning their structures were suggested based on the interpretation of multi-phase powder diffraction patterns. In heterostructured Bi2Se3/MnBi2Se4 thin films, which were grown by means of molecular beam epitaxy, MnBi2Se4 crystallizes in the same structure type as bulk MnBi2Te4.35 We have now synthesized further members of the homologous series (MnTe)(Bi2Te3)n with GeBi4Te7-type27,29 or GeBi6Te10-type24 structures. According to single-crystal X-ray data, single-phase syntheses and chemical analyses, their chemical compositions significantly deviate from the idealized formulae given in ref. 34. However, our substances might be the same or rather similar compounds as the ones discussed in this parallel work even though the authors claim that they are stable, whereas our samples are metastable as discussed below (in addition, DSC curves in ref. 34 are different from ours; and as the authors did not obtain single-phase samples and do not show chemical analyses, a detailed comparison is impossible). Measurements of transport properties with respect to thermoelectric behavior represent one step towards characterizing these potential multifunctional materials.
2 Experimental
2.1 Synthesis
Bulk samples were obtained from the elements Mn (chunks, Onyxmet, 99.9%), Bi (Alfa Aesar, 99.999%) and Te ingots (Haines & Maassen, 99.9%). Mn was heated under vacuum with small silica glass powder at 1000 °C for 1 day to completely remove brownish MnO2 from the surface. After this cleaning procedure, Mn was stored under argon to avoid oxidation. Stoichiometric mixtures (approximately 2 g) of the elements according to the various sum formulae mentioned in the following sections were sealed in graphitized flat-bottomed silica glass ampules (inner diameter: 12 mm, wall thickness: 1.5 mm) under argon atmosphere, placed in tube furnaces and heated from RT to 950 °C. The samples were kept at these conditions for 1 d and subsequently cooled to RT by quenching at air. The resulting ingots were then annealed at 575 °C for 1 week, again followed by quenching at air. Disc-shaped ingots used for thermal conductivity measurements were obtained by using flat-bottomed silica-glass ampules. The samples were polished to a plane-parallel thickness of 1.07 mm (Mn0.81Bi6.13Te10) or 1.29 mm (Mn0.85Bi4.10Te7), by using SiC grinding powder. Cuboid-shaped samples for measurements of the electrical conductivity and the Seebeck coefficient were fabricated using a diamond wire saw and polished with SiC grinding powder. The samples are dark gray with metallic luster. Upon exposure to air over months, no hydrolysis, oxidation or decomposition were observed, even for powder samples.
2.2 Powder diffraction
For powder X-ray diffraction (PXRD) at RT, the samples were crushed and spread on Mylar foil. Data were recorded on a Huber G670 diffractometer (Guinier geometry with imaging-plate detector and integrated read-out system) using Cu-Kα1 radiation [Ge(111) monochromator, λ = 1.54051 Å]. WINXPOW36 was used for data evaluation and TOPAS37 for Rietveld refinements. Reflection profiles were described with fundamental parameters using a direct convolution approach. Crystallite strain was described using a Voigt function, and preferred orientation was refined with spherical harmonics of the 4th order. The background of each diffraction pattern was modelled by a set of 18 parameters (shifted Chebychev polynomial). Structure models with their equivalent isotropic displacement parameters were taken from the single-crystal data. Temperature-dependent measurements in rotating silica glass capillaries under dry Ar atmosphere were obtained with synchrotron radiation (beamline ID11, ESRF, Grenoble) on a Huber diffractometer (λ = 0.18972 Å) with a FReLoN 2K CCD detector.44 A hot-gas blower (GSB 1300, FMB Oxford) with a maximal heating rate of 20 K min−1 was used. For temperature calibration, hexagonal BN was used since its c lattice parameter increases with temperature almost linearly.38 In order to adjust the measured temperatures of the heating and cooling curves according to the calibration, a second-order polynomial fit was used for interpolation.
2.3 Single-crystal X-ray diffraction
X-ray diffraction data of a Mn0.85Bi4.10Te7 single-crystal were obtained on a STOE STADIVARI diffractometer (Ag-Kα1 radiation, λ = 0.56086 Å) with a Dectris Pilatus 300k detector. Integration was done with X-Area39 and numerical absorption correction using X-RED40 and X-SHAPE.41,42 Single-crystal diffraction data of Mn0.73Bi6.18Te10 were obtained using microfocused synchrotron radiation (beamline ID11, ESRF, Grenoble)43 on a Huber diffractometer (λ = 0.30996 Å) with a FReLoN 2K CCD detector.44 Integration was done using CrysAlis Pro45 and semiempirical absorption correction was applied with SADABS.46 Incomplete absorption in the CCD phosphor as a function of beam incidence was taken into account.47 Solution and refinement of both structures was done with SHELX-2014.48 Further details on the crystal structure analyses can be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: +49-7247-808-666; e-mail: crysdata@fiz-karlsruhe.de) on quoting the depository numbers 1891486 (Mn0.85Bi4.10Te7) and 1891488 (Mn0.73Bi6.18Te10). Crystal structures were visualized with Diamond.49
2.4 Thermal analysis
Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10 powders were analyzed by means of differential scanning calorimetry (DSC) using a Setaram Labsys ATD-DSC device with k-probe (Ni–Cr/Ni–Al; Tmax = 800 °C) thermocouples and Al2O3 as a reference compound. Weighted amounts of each specimen (ca. 50 mg) were sealed in small evacuated silica ampules and exposed to two consecutive heating and cooling cycles from RT up to 800 °C at the rate of 2 K min−1.
2.5 Electron microscopy
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) were done using a Zeiss LEO 1530 instrument (acceleration voltage 20 kV) equipped with an EDX detector (INCA software,50 Oxford Instruments). Selected-area electron diffraction (SAED), high-resolution transmission electron microscopy (HRTEM) and further EDX were executed on a Philips CM-200 STEM (LaB6 cathode, 200 kV, super-twin lens, point resolution 0.23 nm) equipped with RTEM 136-5 EDX detector (EDAX, Genesis software51). A double-tilt low-background sample holder (Gatan) was used. For TEM investigations, the bulk material was manually cut with a diamond saw, glued on a copper ring, mechanically ground to a thickness of approximately 30 μm, polished using a dimple grinder (Gatan) and finally Ar-ion-thinned (Duo-Mill, Gatan). Evaluation of SAED data was done using the AnalySIS software.52 For HRTEM and SAED simulations, jEMS53 was used. Micrographs and diffraction patterns were evaluated using Digital Micrograph.54
2.6 Thermoelectric characterization
Thermal diffusivity (κ) was measured under He atmosphere using a Linseis LFA1000 apparatus equipped with an InSb detector (aperture diameter 3 mm). Simultaneous heat loss and finite pulse corrections were applied using Dusza's model.55 Values were averaged from five measurement points at each temperature. For κ calculation, they were multiplied with the Dulong-Petit heat capacity (Mn0.85Bi4.10Te7: Cp = 0.1659 J g−1 K−1; Mn0.81Bi6.13Te10: Cp = 0.1624 J g−1 K−1) and the density as derived by the weight and the volume determined by Archimedes’ principle with a precision of 0.03 g cm−3 (Mn0.85Bi4.10Te7: 7.53 g cm−3; Mn0.81Bi6.13Te10: 7.65 g cm−3). Densities were at least 98.5% of the corresponding X-ray densities. DSC measurements (cf. Fig. S1 – S denotes items in the ESI†) confirm that Cp corresponds to the Dulong–Petit value within experimental error although DSC measurements of Bi2Te356 indicate that Cp near RT is still below the Dulong–Petit value. However, other related compounds such as (Sn1−xPbx)Bi2Te457 or GeBi4Te758 reach this value near RT. The Seebeck coefficient S (in μV K−1) and the electrical conductivity σ (in S cm−1) were measured on cuboid samples (Mn0.81Bi6.13Te10: 8.13 mm × 1.25 mm × 1.76 mm, Mn0.85Bi4.10Te7: 6.92 mm × 1.09 mm × 1.91 mm) simultaneously under He atmosphere using a Linseis LSR-3 instrument with NiCr/Ni and Ni contacts and a continuous reversal of the polarity of the thermocouples (bipolar setup). Measurements comprised three heating/cooling cycles 50–400–50 °C with 25 °C steps at 10 K min−1 heating rate and three data points per temperature. The errors of S and σ are smaller than 10%; for κ, they are ca. 5%. The combined uncertainty may amount to ∼15% for the zT value.
2.7 Magnetic measurements
Temperature- and field-dependent magnetic measurements were performed on pressed powder samples (ca. 10 mg) using a Superconducting Quantum Interference Device (SQUID) magnetometer (MPMS®3) from Quantum Design. Temperature-dependent zero-field-cooled (ZFC) and field-cooled (FC) warming magnetization measurements were performed at magnetic fields of 0.02 T and 1 T in the temperature range from 2–320 K and field-dependent magnetization measurements were done at 2 K and at 300 K.
3 Results and discussion
3.1 Phase composition and homogeneity
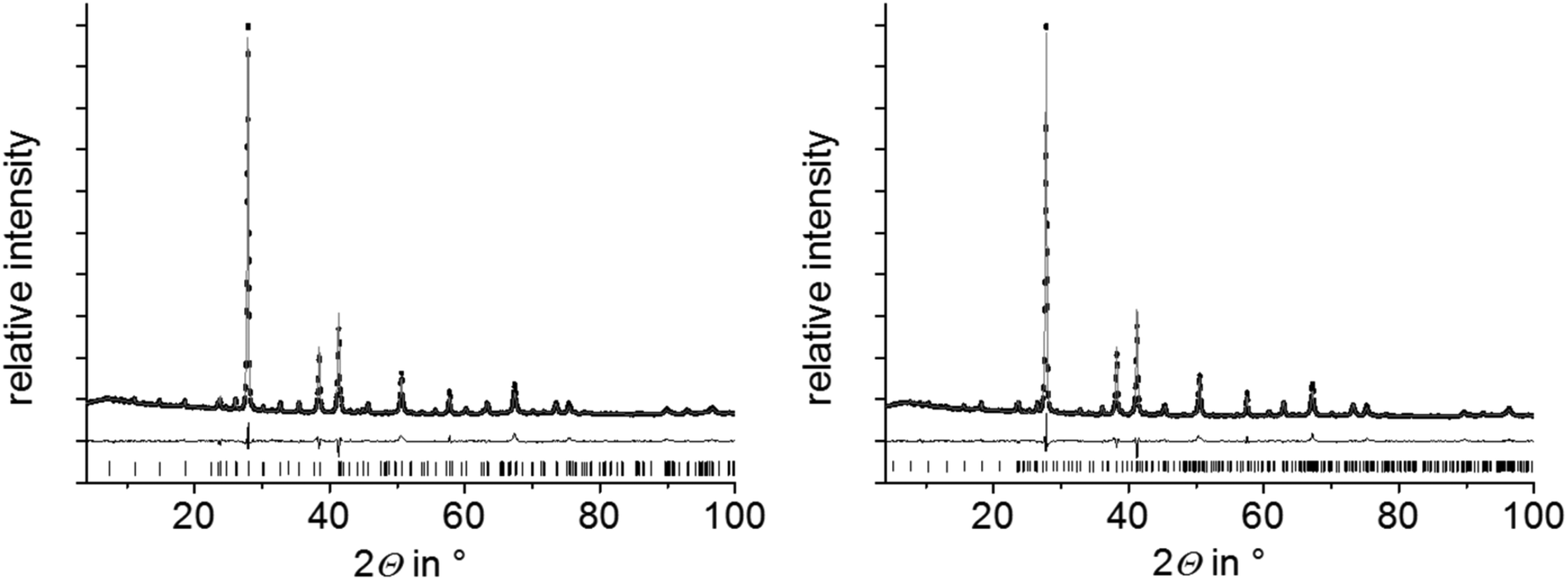
Compounds with nominal compositions MnTe(Bi2Te3)n with n = 2 or 3 may be expected to crystallize in the GeBi4Te727,29 or GeBi6Te1024 structure types. Corresponding reactions, however, did not lead to single-phase samples. Samples with the nominal composition MnBi4Te7 contain two phases, i.e. a mixture of GeSb2Te4-type and GeBi4Te7-type compounds (Fig. S2, ESI†). The nominal composition MnBi6Te10 yields a mixture GeBi4Te7-type and GeBi6Te10-type phases (Fig. S3, ESI†). In contrast, nominal compositions such as Mn0.85Bi4.10Te7 (derived from single-crystal data, cf. Section 3.3) and Mn0.81Bi6.13Te10 (which was suggested by preliminary single-crystal data) lead to samples that appear to be single-phase according to Rietveld refinements based on PXRD data (Fig. 1 and Tables S1–S3 in the ESI†). In both cases, the refined parameters of the single-crystal structure analyses were used for the Rietveld refinement and only lattice parameters were refined. For GeBi6Te10-type samples, they agreed with those of the single-crystal. For Mn0.85Bi4.10Te7, lattice parameters from PXRD differ slightly from those of the single crystal but correspond to those of the GeBi4Te7-type phase in heterogeneous samples with the nominal composition MnBi6Te10, from which the single crystal was isolated (Table S4, ESI†). The deviations from the stoichiometric compositions GeBi4Te7 and GeBi6Te10 are consistent with the EDX data obtained from Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10 bulk samples (Table S5, ESI†). The measured Mn concentrations match the nominal compositions significantly better than those expected for stoichiometric MnBi4Te7 and MnBi6Te10. Very small amounts of a manganese telluride side phase were detected in the bulk sample Mn0.85Bi4.10Te7 by SEM-EDX; in Mn0.81Bi6.13Te10, however, appeared to be phase-pure (Fig. S4, ESI†). Note that nominal compositions Mn0.92Bi4.08Te7 and Mn0.85Bi6.15Te10 derived from refinements not assuming charge neutrality (Section 3.3) but full occupation of all Wyckoff positions yield no single phases according to Rietveld refinements, at least not with the synthesis route described above (Fig. S5, ESI†).
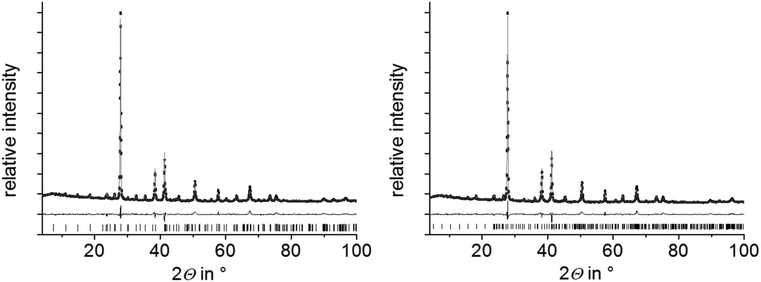 |
| | Fig. 1 PXRD patterns of Mn0.85Bi4.10Te7 (left) and Mn0.81Bi6.13Te10 (right); Rietveld refinements taking into account structure models based on SCXRD refinements (Section 3.3) for Mn0.85Bi4.10Te7 (GeBi4Te7-type structure), Mn0.73Bi6.18Te10 (GeBi6Te10-type structure, the slightly different Mn content has no influence on the Rietveld refinement); experimental data (black points), fitted curves (gray) and difference plots (black, below); reflection markers correspond to the GeBi4Te7 structure type (left) and the GeBi6Te10 structure type (right), respectively. | |
The composition has been varied in order to check whether the compounds form solid solutions. Charge neutrality assuming MnII, BiIII, Te−II and cation vacancies was taken into account as these compounds exhibit small bandgaps (Section 3.6). The composition can be varied in the range of x = 0.15–0.20 for Mn1−x□x/3Bi4+2x/3Te7 and in the range of x = 0.19–0.26 for Mn1−x□x/3Bi6+2x/3Te10. The homogeneity of the corresponding bulk samples has been confirmed by Rietveld refinements and EDX measurements (see Fig. S6 and Table S6, ESI†). Lattice parameters do not change significantly (Tables S4 and S7, ESI†). This might be due to vacancies compensating the effect of different atom radii.
3.2 Stability of manganese bismuth tellurides with GeBi4Te7 or GeBi6Te10 structure type as a function of particle size
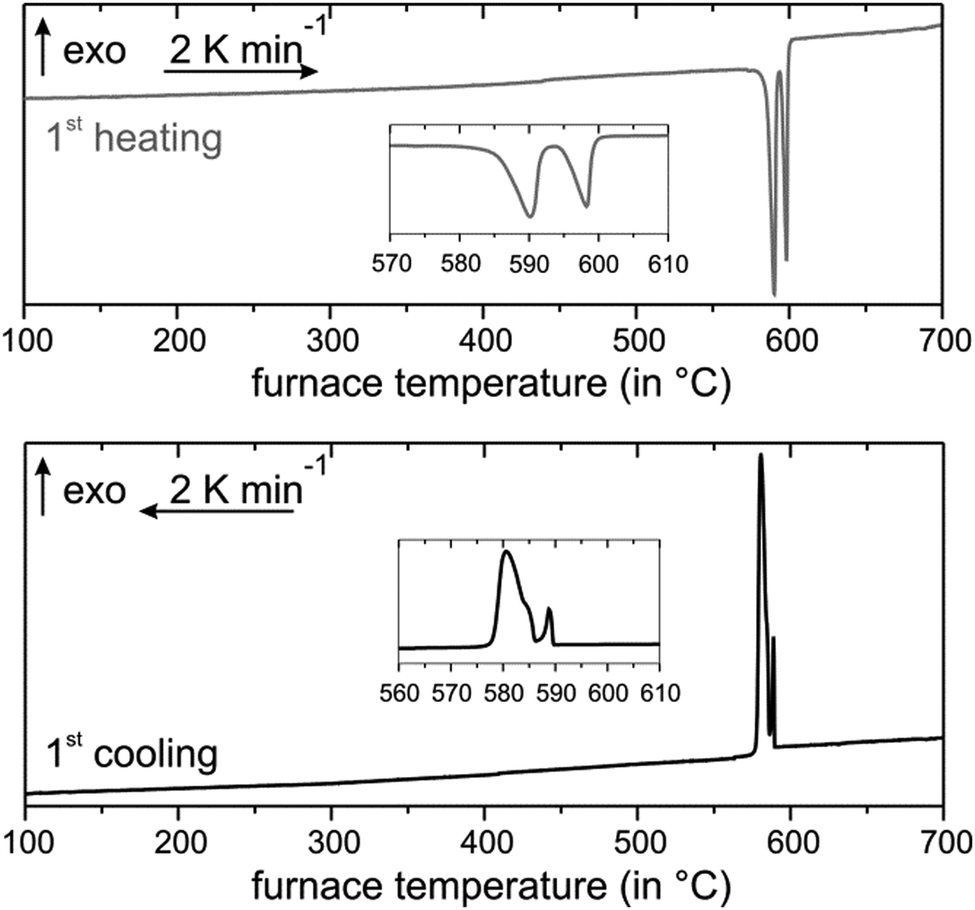
Annealing at 585 °C and subsequent quenching affords phase-pure samples of Mn0.85Bi4.10Te7 with GeBi4Te7-type structure and Mn0.81Bi6.13Te10 with GeBi6Te10-type structure according to PXRD patterns. Temperature-programmed PXRD data show no changes except thermal expansion up to ca. 210 °C (Fig. 2). At this temperature, both compounds start decomposing into phases with Bi2Te3 structure type and a Mn-rich phase as visible in samples after thermoelectric measurements (Section 3.6, Fig. S7, ESI†). The decomposition of fine powdered samples is complete at 270 °C for Mn0.85Bi4.10Te7 and at 295 °C for Mn0.81Bi6.13Te10. Fig. 2 shows the disappearance of characteristic low-angle reflections of the long-periodic structures and the emergence of typical reflections of the Bi2Te3 structure type upon decomposition (Fig S8, ESI†). Temperature-programmed PXRD data of MnBi2Te4 show similar decomposition.31,32Ref. 34 does not report this decomposition as temperature-dependent PXRD was not used but is crucial to detect the metastability. In addition, the decomposition of bulk samples is much slower as discussed later in this chapter. As this decomposition extends over a wide temperature range, it cannot be detected in DSC measurements (Fig. 3 and 4), which has been discussed in detail for MnBi2Te4.32 However, there are characteristic DSC signals for other effects, whose tentative assignment is listed for Mn0.85Bi4.10Te7 in Table S8 and Fig. S9 (ESI†) and for Mn0.81Bi6.13Te10 in Table S9 and Fig. S10 (ESI†). Upon heating of Mn0.85Bi4.10Te7, two endothermic signals are observed (Fig. 3). We attribute the first signal to the melting of Bi2Te3 (a shoulder on the low-temperature side of the first signal) and of Mn0.85Bi4.10Te7, since the starting material was partially decomposed (Fig. S11, ESI†). It is followed by the melting of another compound, possibly MnBi2Te4, whose slowexothermic formation is unlikely to be detected.32,33 The first cooling curve shows three exothermic signals, which can be assigned to the solidification of Bi2Te3, Mn0.85Bi4.10Te7 and MnBi2Te4, respectively, as tentatively confirmed by PXRD (Fig. S12, ESI†). The second heating cycle is similar.
 |
| | Fig. 2 Temperature-programmed PXRD starting from phase-pure samples with the compositions (a) Mn0.85Bi4.10Te7 and (b) Mn0.81Bi6.13Te10, synchrotron radiation (λ = 0.18972 Å), heating rate 20 K min−1. | |
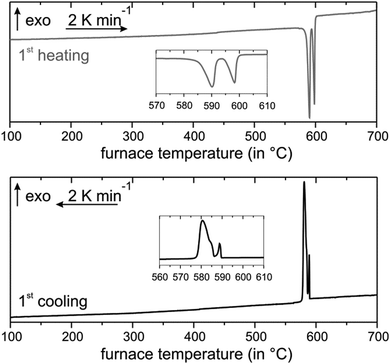 |
| | Fig. 3 DSC of Mn0.85Bi4.10Te7: first heating (top) and cooling (bottom) curves. | |
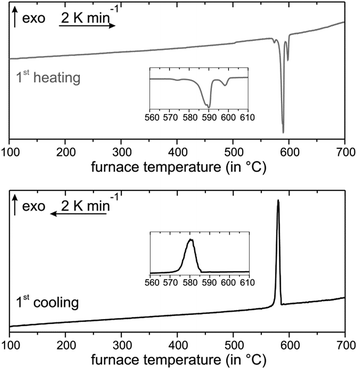 |
| | Fig. 4 DSC of Mn0.81Bi6.13Te10: first heating (top) and cooling (bottom) curves. | |
The DSC curve upon heating of Mn0.81Bi6.13Te10 shows four endothermic signals (Fig. 4). The first one might indicate small amounts of a Bi-rich bismuth telluride, which could form next to MnTe2 by redox decomposition (compare Fig. S13, ESI†). In analogy with the above interpretation for the Mn0.85Bi4.10Te7 sample, the following signals probably correspond to the almost simultaneous melting of Bi2Te3 and Mn0.81Bi6.13Te10, then of Mn0.85Bi4.10Te7, and of MnBi2Te4. The cooling curve hosts a broad unstructured signal that may indicate glassy behavior or partial crystallization of disordered compounds (Fig. 4). We speculate that just above the melting point of Bi2Te3, the Mn-containing ternaries undergo a series of stepwise structure transformations. The DSC experiment was probably too fast to resolve these transitions. PXRD after the DSC measurement indicates a Bi2Te3-type structure mixed with GeBi4Te7- and GeBi6Te10- and possibly GeBi2Te4-type structures (Fig. S14, ESI†). The second DSC heating cycle is similar but shows fewer signals during heating (Table S9 and Fig. S10, ESI†).
Due to these complex phase relationships, manganese bismuth tellurides with GeBi4Te7-type or GeBi6Te10-type structure cannot be obtained by quenching or slowly cooling the melt. Annealing in the existence range of the compounds is essential. As they constitute high-temperature phases that decompose at lower temperatures, quenching after the annealing step is beneficial.
However, ex situ SEM images and EDX measurements after three heating cycles up to 400 °C, which were performed for thermoelectric characterization (Section 3.6), show that the decomposition is much slower in compact ingots; the samples still consist mainly of the initial phase (Fig. S15 and Table S10, ESI†). Corresponding Rietveld refinements reveal ca. 80% of the main phase along with possibly Mn-doped Bi2Te359 (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and MnTe260 with pyrite-type structure (space group Pa) (Fig. S7, ESI†). Thus, the metastability with respect to decomposition is best shown using very fine powders.
m) and MnTe260 with pyrite-type structure (space group Pa) (Fig. S7, ESI†). Thus, the metastability with respect to decomposition is best shown using very fine powders.
As mentioned above, this metastability was also observed for MnBi2Te4, where the decomposition products should be non-stoichiometric Bi-rich bismuth tellurides61–63 “Bi2Te3−x” and MnTe2−y.31,32 According to temperature-programmed PXRD, GeBi4Te7- and GeBi6Te10-type compounds are not formed from decomposed samples during cooling (Fig. S16 and S17, ESI†). This could mean that the reaction is too slow at 500 °C and below, or that the decomposition products are spatially segregated, resulting in strongly inhibited solid-state reactivity. A possible reason for the increased stability of the bulk material compared to the finely ground samples could be that the metastable compounds decompose faster after many defects were brought into the crystal structure by crushing and that stresses in compact samples may hinder decomposition.
In order to exclude alternative explanations such as reactions with the silica-glass capillary or impurities, crushed samples of Mn0.81Bi6.13Te10 and Mn0.85Bi4.15Te7 were annealed in silica-glass ampules at 300 °C for 1 day, so that most of the single phases decomposed to a Bi-rich Bi2Te3-like phase and MnTe2 (pyrite type, Pa) (Fig. S18, ESI†). After this treatment, the samples were used as starting material for our synthesis route, i.e. they were melted again, followed by annealing and quenching. This again leads to phase-pure samples according to PXRD (Fig. S19, ESI†).
3.3 Crystal structures of Mn0.85(3)Bi4.10(2)Te7 and Mn0.73(4)Bi6.18(2)Te10
The crystal structures of representative crystals of manganese bismuth tellurides with GeBi4Te7-type and GeBi6Te10-type structures were elucidated from X-ray diffraction data. In the structure refinements, charge neutrality was used as a constraint assuming oxidation states of II for Mn and III for Bi. The presence of MnII is also in accordance with the magnetization measurements, i.e., the effective moment μeff is in agreement with the value expected for MnII for both compounds (cf. Section 3.5). Furthermore, other oxidation states seem unlikely as the oxidation potential of Te is not expected to enable higher oxidation states of the cations. Lower oxidation states of Bi are typically characterized by homoatomic bonds.64 In the related compound MnBi2Te4, the exclusive presence of Mn2+ was shown by X-ray absorption spectroscopy and confirmed by magnetic susceptibility measurements.32,33 Mixed occupancies of Mn and Bi on all cation sites were refined as indicated by the scattering density present and the resulting R-values. Cation vacancies were assumed to be located only on the Wyckoff positions 1b for Mn0.85Bi4.10Te7 and 3a for Mn0.73Bi6.18Te10, which represent the center of the GeBi2Te4-type building blocks, since vacancies close to the van der Waals gaps are very unfavorable due to a resulting very low coordination number 2 for Te atoms at the van der Waals gap.
Mn0.85(3)Bi4.10(2)Te7.
Crystallographic data and details of the structure refinement for a crystal with the refined composition Mn0.85(3)Bi4.10(2)Te7 isolated from a heterogeneous sample with the nominal composition MnBi6Te10 are listed in Table 1, atomic positions and displacement parameters are given in Table 2. In contrast to Mn/Bi intermixing on the cation sites, the vacancy concentration of 5.1(17)% on the 1b site seems not very significant; the assumption of an idealized composition MnBi4Te7 results in almost the same residuals (R1 = 0.0332 for I > 2σ(I)). However, such an idealized composition yields no single-phase samples (cf. Section 3.1). The EDX analysis of the investigated crystal (Fig. S20, ESI†) yields the composition Mn5.8(5)Bi35.9(5)Te58.3(3), which confirms a deficiency of Mn in good agreement with the nominal composition of Mn0.85Bi4.10Te7 = Mn7.1Bi34.3Te58.6 (Table S11, ESI†).
Table 1 Crystallographic data for the single-crystal structure determination of Mn0.85(3)Bi4.10(2)Te7 with GeBi4Te7-type structure and Mn0.73(4)Bi6.18(2)Te10 with GeBi6Te10-type structure
| Chemical formula |
Mn0.85(3)□0.05(2)Bi4.10(2)Te7 |
Mn0.73(4)□0.09(2)Bi6.18(2)Te10 |
|
M (in g mol−1) |
1796.72 |
2608.1 |
|
F(000) |
726 |
3154 |
| Crystal system/space group |
Trigonal/Pm1 (no. 164) |
Trigonal/Rm (no. 166) |
| Lattice parameters (in Å) |
a = 4.3591(3), c = 23.769(2) |
a = 4.3698(3), c = 101.829(8) |
|
V (in Å3) |
391.41(6) |
1683.9(3) |
|
Z
|
1 |
3 |
|
ρ (in g cm−3) |
7.63 |
7.72 |
|
μ (in mm−1) |
32.26 |
13.45 |
| Absorption correction |
Numerical |
Semiempirical46 |
| Radiation |
Ag-Kα, λ = 0.56086 Å |
Synchrotron, λ = 0.30996 Å |
| Independent reflections |
594 |
723 |
|
d
min (in Å) |
0.68 |
0.72 |
| Refined parameter/restraints |
27/3 |
36/4 |
|
θ
min/θmax (in °) |
2.03/24.30 |
1.83/12.43 |
| Extinction coefficient |
— |
0.0045(3) |
|
R
σ
/Rint |
0.0322/0.0649 |
0.0549/0.0706 |
|
R indices [I > 2σ(I)] |
R
1 = 0.0331, wR2 = 0.0874 |
R
1 = 0.0303, wR2 = 0.0703 |
|
R indices (all data) |
R
1 = 0.0357, wR2 = 0.0895 |
R
1 = 0.0312, wR2 = 0.0712 |
| GoF (all) |
1.08 |
1.20 |
| Δρmin/Δρmax (in e Å−3) |
−1.52/1.53 |
−3.19/2.10 |
Table 2 Wyckoff positions, atomic coordinates, site occupancies and coefficients of the anisotropic as well as equivalent isotropic displacement parameters (in Å2) of Mn0.85Bi4.10Te7 (Ueq = 1/3[(U11 + U22 + U33)])
| Atom |
W. p. |
x
|
y
|
z
|
Occ. |
| Mn1/Bi1/□ |
1b |
0 |
0 |
1/2 |
0.694(11)/0.255(6)/0.051(17) |
| Mn2/Bi2 |
2d |
1/3 |
2/3 |
0.3423 |
0.0414(4)/0.959(3) |
| Mn3/Bi3 |
2d |
1/3 |
2/3 |
0.0860 |
0.035(3)/0.965(3) |
| Te1 |
2d |
1/3 |
2/3 |
0.5680 |
1 |
| Te2 |
2c |
0 |
0 |
0.2703 |
1 |
| Te3 |
2d |
1/3 |
2/3 |
0.8413 |
1 |
| Te4 |
1a |
0 |
0 |
0 |
1 |
| Atom |
U
11 = U22 = 2U12 |
U
33
|
U
23 = U13 |
U
eq
|
| Mn1/Bi1/□ |
0.0334(7) |
0.0314(10) |
0 |
0.0328(6) |
| Mn2/Bi2 |
0.0315(2) |
0.0368(4) |
0 |
0.0333(2) |
| Mn3/Bi3 |
0.0300(3) |
0.0352(3) |
0 |
0.0318(2) |
| Te1 |
0.0336(3) |
0.0291(5) |
0 |
0.0321(3) |
| Te2 |
0.0295(3) |
0.0315(5) |
0 |
0.0302(3) |
| Te3 |
0.0290(3) |
0.0308(5) |
0 |
0.0296(3) |
| Te4 |
0.0269(4) |
0.0269(6) |
0 |
0.0269(3) |
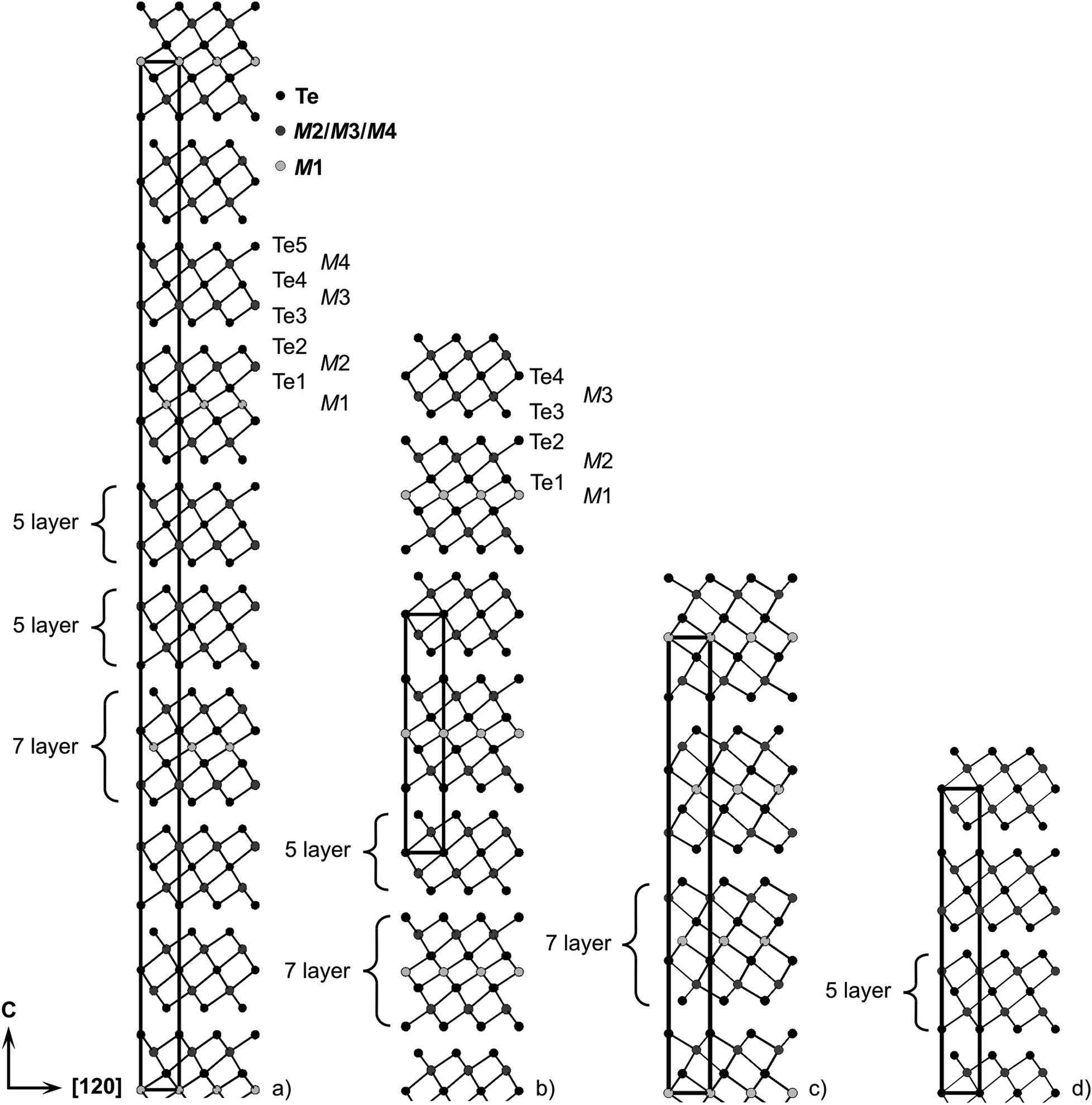
A refinement without assuming charge neutrality but full site occupancies (i.e., no vacancies) also results in similar R-values and a composition Mn0.92Bi4.08Te7; however, corresponding reactions also yield no single-phase products (cf. Section 3.1). Mn0.85Bi4.10Te7 crystallizes in the space group Pm1 and can be described by a 12P stacking sequence with two different building blocks (Fig. 5b): double layers of edge-sharing [BiTe3/3Te3/6] octahedra similar to those in Bi2Te324 (Fig. 5d) and GeBi2Te4-type (Fig. 5c) septuple layers of condensed octahedra described by 2∞[Te3/3M(μ-Te3/6)M(μ-Te3/6)MTe3/3]. The atomic interaction within each layer is much stronger than the interlayer van-der-Waals-like bonding. Similar lattice parameters were reported for Mn-doped BiTe (space group Pm1; a = 4.3783(1) Å, c = 23.8107(8) Å),65 whose BiSe-type structure consists of Bi2Te3-like layers separated by double layers of Bi atoms resembling those in elemental Bi. However, such a structure model cannot reproduce our diffraction data.
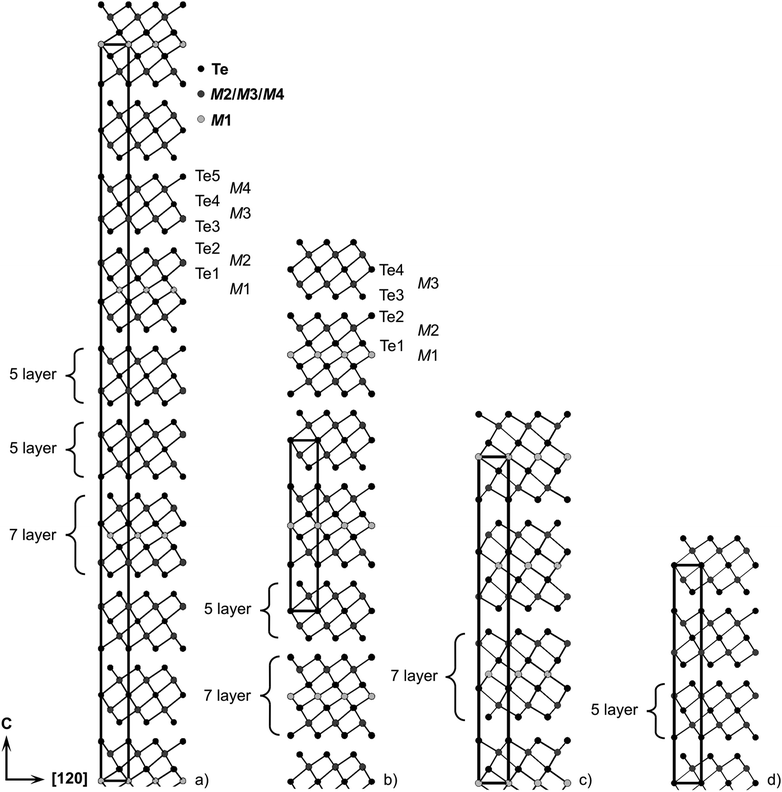 |
| | Fig. 5 Crystal structures of (a) Mn0.81Bi6.13Te10 with GeBi6Te10-type structure; (b) Mn0.85Bi4.10Te7 with GeBi4Te7-type structure; (c) Mn0.85Bi2.1Te432 with GeBi2Te4-type structure; (d) Bi2Te324 – for cation distribution, including mixed sites, cf.Tables 2 and 4 (M1 is the Mn-rich site). | |
Mn0.73(4)Bi6.18(2)Te10.
Crystallographic data and details of the structure refinement for a crystal with the refined composition Mn0.73(4)Bi6.18(2)Te10 isolated from a heterogeneous sample with the nominal composition MnBi6Te10 are given in Table 1; atomic positions and displacement parameters are listed in Table 3. Based on the above mentioned constraints, the refinement yields a significant vacancy concentration of 9(2)% on the site 1b. Constraining to the stoichiometric composition MnBi6Te10 leads to higher residuals (R1 = 0.0315 for I > 2σ(I)) and such an idealized composition does not lead to single-phase samples (cf. Section 3.1). Assuming full occupancies without constraining to charge neutrality yields a composition of Mn0.85Bi6.15Te10, again not suitable for the synthesis of single-phase samples. The EDX analysis of the crystal used for data collection (Fig. S21 and Table S12, ESI†) confirms a Mn deficiency and is in good agreement with the composition Mn0.73Bi6.18Te10 obtained in the structure refinement. In space group Rm, the asymmetric unit consists of distorted NaCl-type septuple building blocks similar to those in GeBi4Te7, which are separated by two Bi2Te3-like layers, resulting in a deficiency and is in good agreement with the composition 51R stacking sequence (Fig. 5a). Again, Mn/Bi intermixing, especially on the 3a, is a characteristic feature of this structure.
Table 3 Wyckoff positions, atomic coordinates, site occupancies and coefficients of the anisotropic as well as equivalent isotropic displacement parameters (in Å2) for Mn0.73Bi6.18Te10 (Ueq = 1/3[U11 + U22 + U33])
| Atom |
W. p. |
x
|
y
|
z
|
Occ. |
| Mn1/Bi1/□ |
3a |
0 |
0 |
0 |
0.634(16)/0.275(6)/0.09(2) |
| Mn2/Bi2 |
6c |
0 |
0 |
0.29647(2) |
0.017(4)/0.983(3) |
| Mn3/Bi3 |
6c |
0 |
0 |
0.23671(2) |
0.015(4)/0.985(3) |
| Mn4/Bi4 |
6c |
0 |
0 |
0.47010(2) |
0.014(4)/0.986(3) |
| Te1 |
6c |
0 |
0 |
0.34922(2) |
1 |
| Te2 |
6c |
0 |
0 |
0.05365(2) |
1 |
| Te3 |
6c |
0 |
0 |
0.41290(2) |
1 |
| Te4 |
6c |
0 |
0 |
0.11658(2) |
1 |
| Te5 |
6c |
0 |
0 |
0.17957(2) |
1 |
| Atom |
U
11 = U22 = 2U12 |
U
33
|
U
23 = U13 |
U
eq
|
| Mn1/Bi1/□ |
0.0221(5) |
0.0238(8) |
0 |
0.0227(4) |
| Mn2/Bi2 |
0.02252(17) |
0.0278(3) |
0 |
0.02428(16) |
| Mn3/Bi3 |
0.0208(2) |
0.0269(3) |
0 |
0.02283(19) |
| Mn4/Bi4 |
0.0208(2) |
0.0265(3) |
0 |
0.02269(19) |
| Te1 |
0.0233(2) |
0.0207(4) |
0 |
0.0224(2) |
| Te2 |
0.0191(2) |
0.0229(4) |
0 |
0.0204(2) |
| Te3 |
0.0192(2) |
0.0204(4) |
0 |
0.0196(2) |
| Te4 |
0.0174(2) |
0.0176(4) |
0 |
0.0175(2) |
| Te5 |
0.0190(3) |
0.0205(4) |
0 |
0.0195(2) |
Comparison of selected interatomic distances with Bi2Te3 and Mn0.85Bi2.1Te432.
Selected interatomic distances are listed in Table S13 (ESI†). Bond lengths in the Bi2Te3-like quintuple layers in Mn0.85Bi4.10Te7, i.e. d(M3–Te) = 3.0543(7) Å and d(M3–Te4) = 3.2410(4) Å (labelling cf.Fig. 5), deviate by only 0.4% from interatomic distances in Bi2Te366 itself [d(Bi–Te2) = 3.067 Å, d(Bi–Te1) = 3.254 Å]; they are slightly shorter by ∼0.01 Å as the cation position contains 3–4% of Mn2+, whose radius (0.81 Å for CN 6), which is smaller than that of Bi3+ (1.17 Å for CN 6).67 The quintuple layers in Mn0.73(4)Bi6.18(2)Te10 [d(M3–Bi3) = 3.0628(5) Å, d(M3–Te4) = 3.2395(5) Å, d(M4–Te4) = 3.2536(5) Å, d(M4–Te5) = 3.0594(5) Å] exhibit the same tendency and deviate only up to 0.4% from the interatomic distances in Bi2Te3. All cation–anion bond lengths in the septuple slab of Mn0.85Bi4.10Te7 [d(M2–Te2) = 3.0436(7) Å, d(M2–Te1) = 3.2987(9) Å, d(M1–Te1) = 2.9909(7) Å] and Mn0.73Bi6.18Te10 [d(M2–Te2) = 3.0473(5) Å, d(M2–Te1) = 3.3055(6) Å, d(M1–Te1) = 2.9969(4) Å] host more bismuth are thus slightly longer than the corresponding bond lengths in Mn0.85Bi2.1Te4 [d(M1–Te2) = 3.027(1) Å, d(M1–Te1) = 3.291(1) Å, d(M2–Te1) = 2.977(1) Å].32 On the other hand, the distances across the van der Waals gaps between the quintuple and the septuple slab in Mn0.85Bi4.10Te7 [d(Te2–Te3) = 3.6559(7) Å] and Mn0.73Bi6.18Te10 [d(Te2–Te3) = 3.6516(5) Å] decrease slightly with higher bismuth concentration.
3.4 TEM investigations
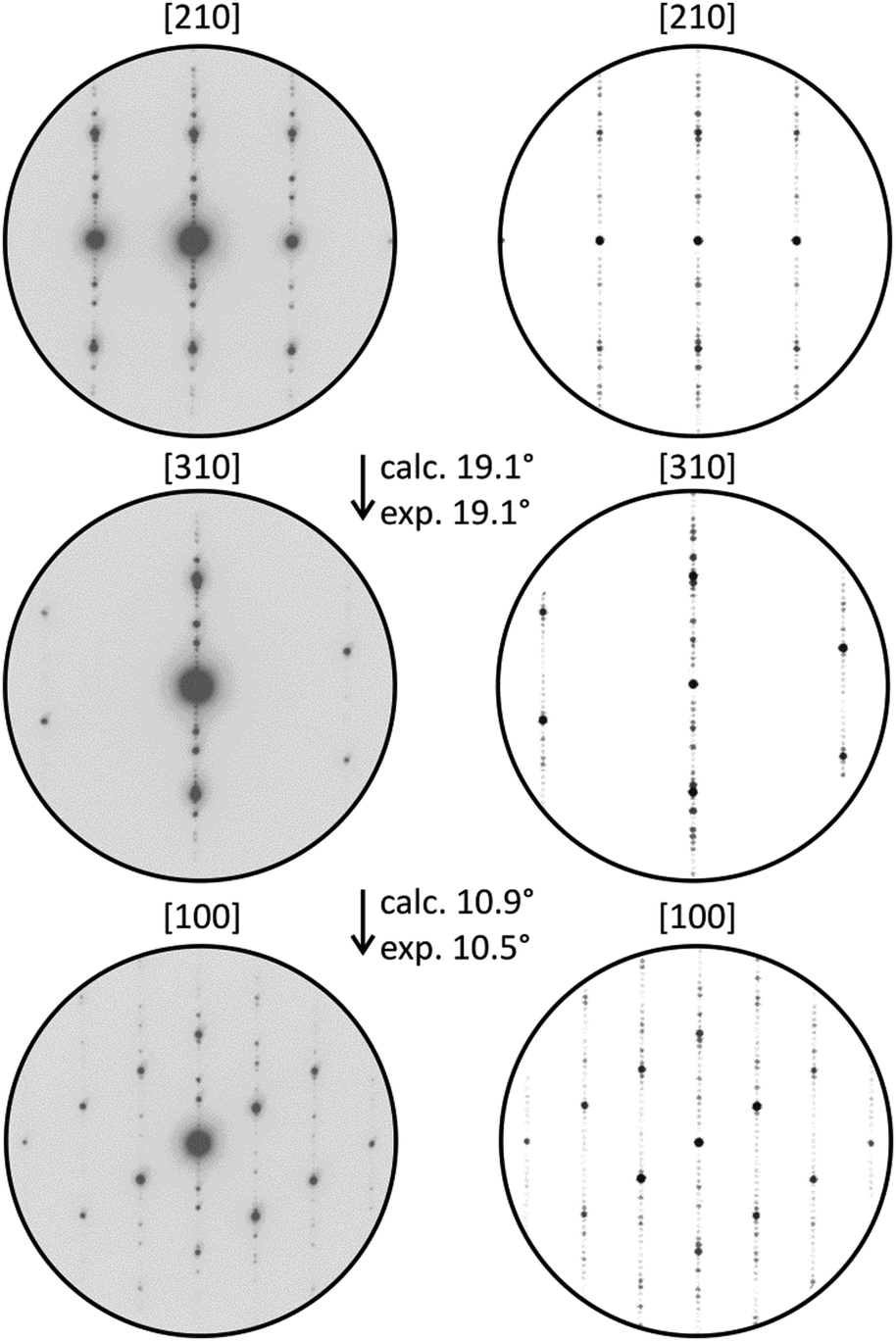
Structured diffuse scattering was not detected in single-crystal X-ray diffraction patterns of the manganese bismuth tellurides with GeBi4Te7 or GeBi6Te10 structure type (Fig. S22, ESI†), thus stacking disorder is not pronounced in these compounds. In order to further analyze possible stacking faults or superstructures along [001], selected area electron diffraction patterns (SAED) were recorded. Fig. 6 and 7 show experimental diffraction patterns and simulated ones based on the results of the crystal structure analyses (Section 3.3). The absence of diffuse streaks along [001]* indicates that there is no significant amount of stacking faults. Indexing of the patterns corresponds to the lattice symmetry and unit cell parameters found in the X-ray diffraction experiments. In order to visualize the long-range ordered stacking of the different slabs, HRTEM images were acquired (Fig. 8 and Fig. S23 and S24, ESI†). The structural entities of quintuple slabs as in Bi2Te3 and septuple slabs as in MnBi2Te4 can be clearly identified. Simulated HRTEM images based on the results of the crystal structure analyses fit the experimental ones quite well and thus confirm the structure models (Fig. 8 and 9). Note that single vacancies have very little influence on the contrasts of atom columns that consist of several atoms and can thus not be detected in such images.
 |
| | Fig. 6 SAED patterns of a manganese bismuth telluride (from a sample with the nominal composition MnBi6Te10) with the GeBi4Te7-type structure and corresponding simulated patterns (kinematical approximation based on the model for Mn0.85Bi4.10Te7 from the single-crystal structure analysis) with experimental and calculated tilt angles. | |
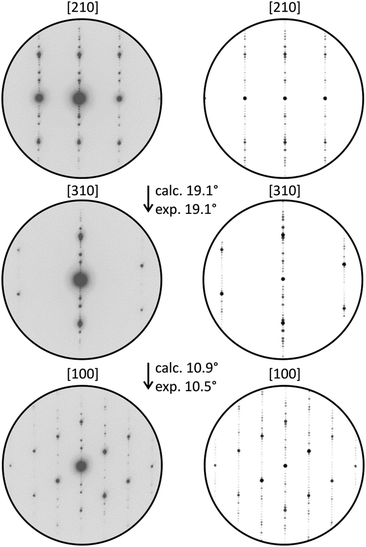 |
| | Fig. 7 SAED patterns of a manganese bismuth telluride (from a sample with the nominal composition Mn0.77Bi6.15Te10) with the GeBi6Te10-type structure and corresponding simulated diffraction patterns (kinematical approximation, based on the model for Mn0.73Bi6.18Te10 from the single crystal structure analysis) with experimental and calculated tilt angles. | |
 |
| | Fig. 8 HRTEM images (zone axis [100], Fourier filtered) of manganese bismuth tellurides with the GeBi4Te7 (bottom) and GeBi6Te10 (top) structure types, each with a corresponding SAED pattern; inset: simulation based on the single-crystal data of Mn0.85Bi4.10Te7 (Δf = −85 nm, t = 8.36 nm; beam semiconvergence 1.2 mrad, defocus spread 15 nm) and Mn0.73Bi6.18Te10 (Δf = −67 nm, t = 0.88 nm; beam semiconvergence 1.2 mrad, defocus spread 15 nm); white bars symbolize quintuple Bi2Te3-like slab and black lines symbolize septuple layers characteristic of MnBi2Te4. | |
 |
| | Fig. 9 HRTEM defocus series (zone axis [100], Fourier filtered) from a manganese bismuth telluride with the GeBi6Te10-type structure, insets: GeBi6Te10-type structure (green = Te; yellow = Bi-rich; red = Mn-rich: for the exact cation distribution, including mixed sites cf.Tables 2 and 4) and simulated images based on the single-crystal data of Mn0.73Bi6.18Te10, t = 0.88 nm; beam semiconvergence 1.2 mrad, defocus spread 15 nm, defocus values Δf are given in the images. | |
3.5 Magnetic properties
Temperature- and field-dependent magnetization was measured on pellets from single-phase powders with the compositions Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10. In low magnetic fields of μ0H = 0.02 T, both compounds demonstrate a temperature-driven phase transition from the paramagnetic into magnetically ordered phases. For Mn0.85Bi4.10Te7, an antiferromagnetic transition can clearly be discerned at TN = 13 K in low fields (see peak in the inset of Fig. 10a at 13 K), which is followed by a subsequent rearrangement of the microscopic spin alignment at lower temperatures, as indicated by an upturn of the magnetization curve. Given the ferromagnetic hysteresis at T = 2 K in Fig. 11a, a predominantly ferromagnetic alignment with a large ferromagnetic component of the spins can be envisaged at lowest temperatures. The microscopic spin arrangement and the origin of these two transitions for Mn0.85Bi4.10Te0 should, however, be elucidated in more detail in the future using single crystals. In comparison, for Mn0.81Bi6.13Te10 a very strong upturn of the magnetization below TC = 12 K together with the splitting of the zero-field-cooled and field-cooled magnetization data indicate a transition into a ferromagnetic phase in this compound.
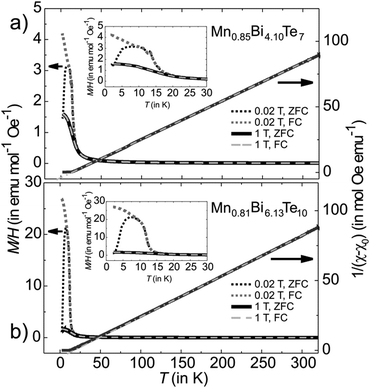 |
| | Fig. 10 Magnetization M/H and reciprocal susceptibility of (a) Mn0.85Bi4.10Te7 and (b) Mn0.81Bi6.13Te10 as a function of temperature measured at various applied magnetic fields. The extrapolated line on the right axis is a Curie–Weiss fit to the high-temperature data indicating a value θCW of 14(2) and 17(2) K for Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10, respectively. A small offset χ0 ∼ −0.0007(3) emu mol−1 Oe−1 was added to the data to account for the contribution of closed diamagnetic electronic shells. | |
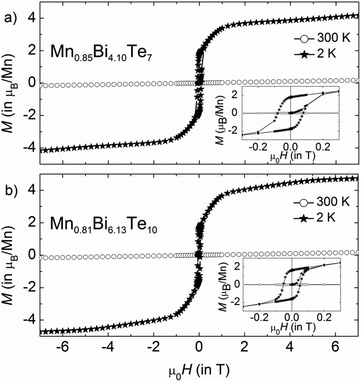 |
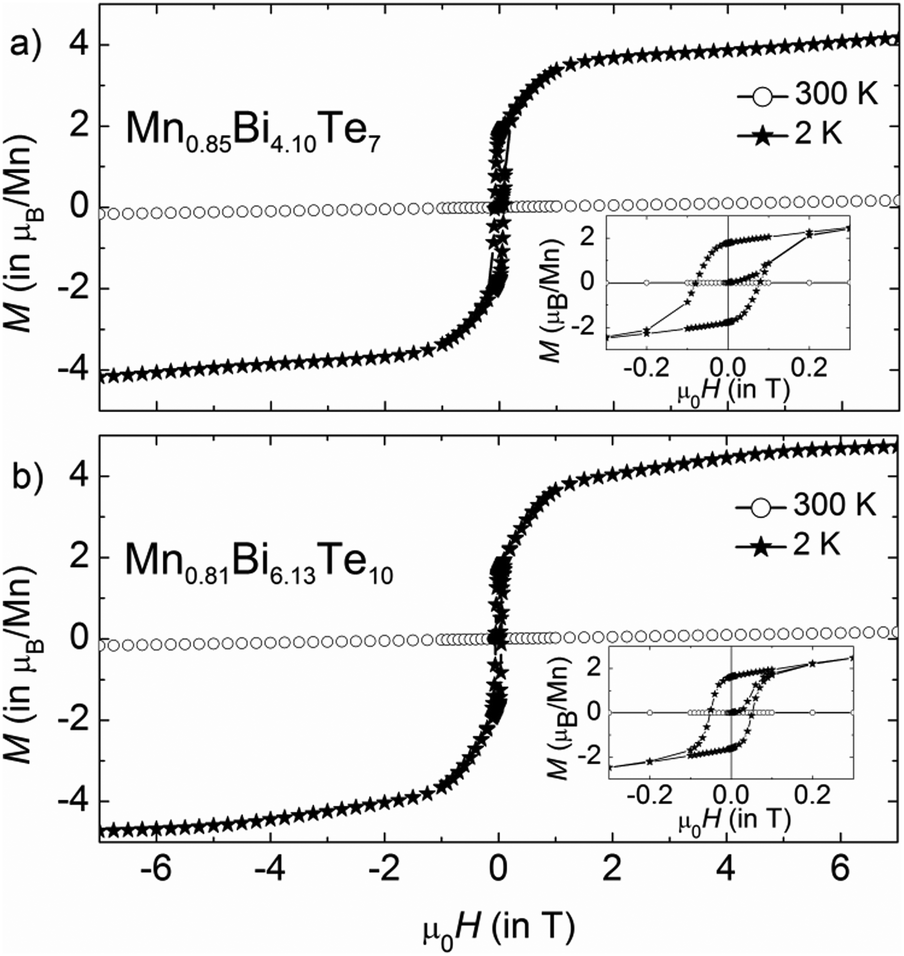
| | Fig. 11 Magnetic hysteresis curves of (a) Mn0.85Bi4.10Te7 and (b) Mn0.81Bi6.13Te10 at 300 and 2 K. The samples were cooled in zero field from 320 K. | |
The effective paramagnetic moment per formula unit can be estimated from the Curie–Weiss fit of the high-temperature data (Fig. 10) and constitutes 5.7(1) μB for Mn0.85Bi4.10Te7 and 5.8(1) μB for Mn0.81Bi6.13Te10, consistent with the high-spin d5 configuration of Mn2+ (S = 5/2). Positive values of θCW = 14(2) and 17(2) K for Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10, respectively, indicate predominantly ferromagnetic coupling. Magnetization curves as function of the magnetic field (Fig. 11) confirm dominating ferromagnetic interactions in both compounds with pronounced magnetic hysteresis at T = 2 K up to about μ0H = 0.2 T and an almost saturated magnetization close to the expected value for Mn(II) for fields above ∼2 T. The remanent magnetization and the coercive field are slightly larger for Mn0.85Bi4.10Te7 (μ0Hc = 0.08 T).
3.6 Thermoelectric properties
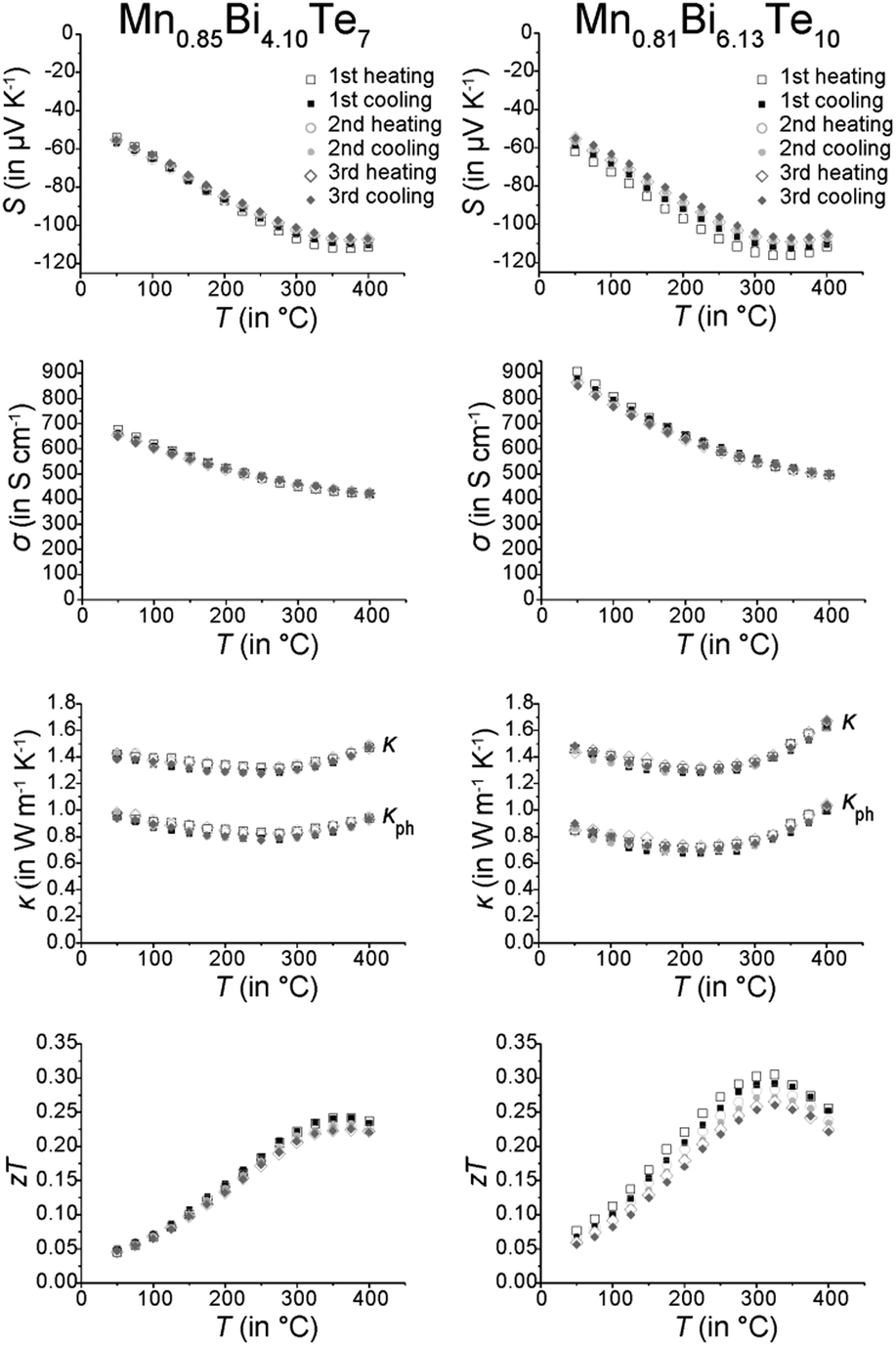
Thermoelectric measurements (Fig. 12) were conducted on parts of compact ingots between RT and 400 °C in three consecutive cycles. The temperature evolution of the electrical conductivity σ indicates metallic behavior above RT. Whereas MnBi2Te4 has been reported to be a p-type conductor with rather low σ (39 S cm−1 at RT),31 other investigations on comparable materials indicate n-type conduction, concluding from a negative Seebeck coefficient S and a much higher σ (450 S cm−1 at RT).32 Similarly, Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10 exhibit negative S and thus are n-type conductors. Despite the shallow minimum in the thermal evolution of S, the bandgap was roughly estimated using the Goldsmid–Sharp relationship,68Eg = 2eSmaxTmax, with the maximum absolute Seebeck coefficient Smax and the corresponding temperature Tmax. This yields narrow bandgaps of Eg ≈ 0.14 eV for both Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10. This bandgap corresponds with the high electrical conductivity and with the Eg of other compounds with layered GeBi4Te7- or GeBi6Te10-type structures (Table 4).
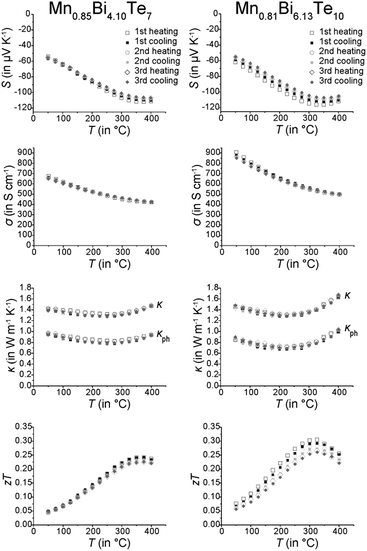 |
| | Fig. 12 Thermoelectric properties of Mn0.85Bi4.10Te7 (left column) and Mn0.81Bi6.13Te10 (right column); Seebeck coefficient (first row), electrical conductivity (second row), thermal conductivity and its phononic contribution (third row, calculated according to the Wiedemann–Franz relationship; the decrease of the Lorenz number L has been derived from the increase of the absolute value of S according to L = 1.5 + e(−|S|/116) where L is in 10−8 W Ω K−2 and S in μV K−1); this equation is accurate within 5% for single parabolic band/acoustic phonon scattering assumption.69 and zT values (fourth row); three subsequent heating and cooling cycles. | |
Table 4 Electronic properties of MBi4Te7, MBi6Te10 (M = Sn, Pb, Ge), Mn0.85Bi2.1Te4, Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10
| Compound |
S at RT (in μV K−1) |
σ at RT (in S cm−1) |
E
g (in eV) |
Ref. |
| MnBi2Te4 |
168 |
39 |
∼0.4 |
31
|
| Mn0.85Bi2.1Te4 |
−5 |
475 |
0.15 |
32
|
| Mn0.85Bi4.10Te7 |
−55 |
700 |
0.14 |
This work |
| GeBi4Te7 |
−127 |
435 |
0.21 |
76
|
| −160 |
230 |
24
|
| SnBi4Te7 |
−92 |
250 |
0.22 |
76
|
| PbBi4Te7 |
−68 |
1666 |
— |
76
|
| −66 |
1070 |
24
|
| Mn0.81Bi6.13Te10 |
−60 |
900 |
0.14 |
This work |
| GeBi6Te10 |
−65 |
920 |
— |
28
|
| PbBi6Te10 |
−47 |
870 |
— |
24
|
Thermal conductivities are rather low and almost constant up to 400 °C with 1.47 W m−1 K−1 for Mn0.85Bi4.10Te7 and 1.63 W m−1 K−1 for Mn0.81Bi6.13Te10. However, the small value of S limits the maximum zT values up to 0.25 at 375 °C and 0.28 at 325 °C for Mn0.85Bi4.10Te7 and Mn0.81Bi6.13Te10, respectively. The zT values decrease slightly from cycle to cycle, which could be attributed to the metastability of the manganese bismuth tellurides. The ex situ PXRD measurements performed after the thermoelectric measurements (Fig. S7, ESI†) show that still up to 80% of the main phase is present in both compounds. SEM investigations (Fig. S15 and Table S10, ESI†) confirm that the samples slowly decompose over several measurement cycles (cf. Sections 3.1 and 3.2).
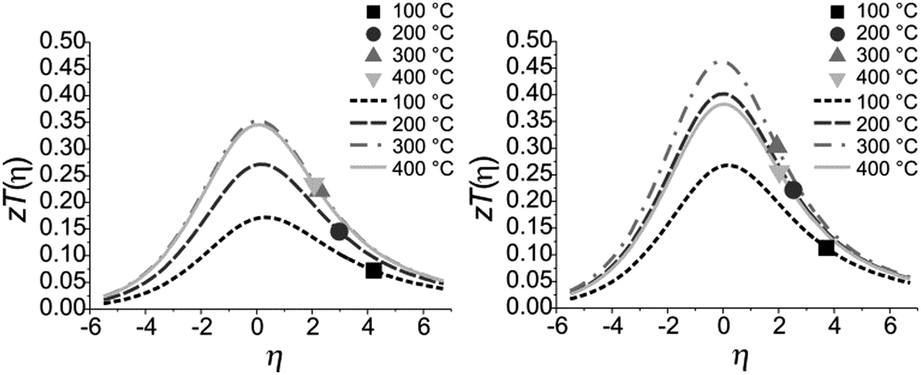
The quality factor B (eqn (1)), the figure of merit zT = S2σT/κ (eqn (2)) and the phononic part of the thermal conductivity κph were calculated as a function of the electrons’ chemical potential η from the measured transport properties σ, S and κ of the first cycle of the thermoelectric measurement (Fig. 12), which is only very little affected by the decomposition. An effective mass model was used to calculate the optimal carrier concentration zTopt. Details of the calculations are described at the end of the ESI† and can also be found in several references.70–75 This approach allows one to calculate the intrinsic electrical conductivity σE0 and κph and therefore also the material quality factor B (eqn (1)). These intrinsic properties are independent of doping and thus can be separated from doping and its optimization. The optimal zT values for different temperatures and therefore different B factors are shown in Fig. 13.
| | 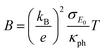 | (1) |
| | 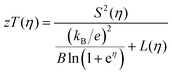 | (2) |
The measured
zT values of Mn
0.85Bi
4.10Te
7 and Mn
0.81Bi
6.13Te
10 indicate that in both compounds the carrier concentrations are higher than optimal as the measured
zT values are on the high-
η side of the maximal
zT(
η) (note that the carrier concentration is a monotonically increasing function of
η). With ideal carrier concentration (
i.e. optimal
η) the maximum of
zT for manganese bismuth tellurides with GeBi
4Te
7-type structure could increase up to 0.35 at 300 °C and up to 0.47 at 300 °C for the GeBi
6Te
10-type structure.
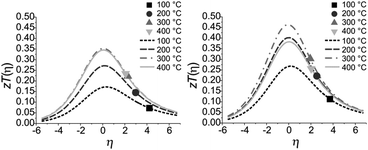 |
| | Fig. 13 Calculated and measured zT values for Mn0.85Bi4.10Te7 (left) and Mn0.81Bi6.13Te10 (right) as a function of η at 100 °C, 200 °C, 300 °C and 400 °C with B = 0.047, 0.078, 0.105 and 0.102 for Mn0.85Bi4.10Te7 and B = 0.076, 0.122, 0.144 and 0.115 for Mn0.81Bi6.13Te10. | |
4 Conclusions
The crystal-structure determinations of two new compounds in the manganese–bismuth–telluride system reveal unexpected combinations of chemical composition and structure type. Diffraction data of Mn0.85Bi4.10Te7 and Mn0.73Bi6.18Te10 indicate that the compounds are isostructural to GeBi4Te727,29 and GeBi6Te10,24 respectively. This is confirmed by a combination of X-ray diffraction, including the application of microfocused synchrotron radiation (for Mn0.73Bi6.18Te10), of TEM imaging and of EDX spectroscopy.43 According to the magnetization measurements, the oxidation state of MnII is established. This corresponds to the situation in MnBi2Te4.32,33 The structures contain significant amounts of cation vacancies. Intermixing of Mn and Bi on the cation sites is very pronounced and highly significant. Other layered chalcogenides of the GeBi2Te4, GeBi4Te7 and GeBi6Te10 structure types also exhibit very characteristic mixed cation occupancies.24,27,28,76–79 This phenomenon is also typical and, in principle, to be expected for manganese bismuth tellurides in these structure types. The non-stoichiometry of Mn1−x□x/3Bi4+2x/3Te7 (x = 0.15 – 0.2) (space group Pm1 with GeBi4Te7-type structure) and Mn1−x□x/3Bi6+2x/3Te10 (x = 0.19–0.26) (space group Rm with GeBi6Te10-type structure) is comparable to the situation in the related compound 33R-Ge4xSb2yTe7 (x, y ≈ 0.1), which is isostructural to Ge3Sb2Te6 despite a different stoichiometry.80 The manganese bismuth tellurides with GeBi4Te7- and GeBi6Te10-type structures are metastable and start decomposing at approximately 250 °C into a Mn-doped Bi2Te3-like structure, Bi-rich bismuth tellurides and a manganese-rich phase. This result is crucial concerning physical measurements at elevated temperatures. This decomposition was not observed in our DSC measurements, as it is very slow and extends over a wide temperature range (∼70 K). Yet, the DSC measurements show that these phases are not accessible by slow cooling of melts. An annealing step in the solid state followed by quenching to room temperature is essential to obtain phase pure samples, as kinetic effects impede the immediate decomposition at elevated temperatures. Small variations in the annealing temperature may lead to different structures. The rather broad DSC signal for the GeBi6Te10-type structure might indicate other structure variants. The zT values of these layered manganese bismuth tellurides indicate decent thermoelectric properties and may be affected by the metastability, but have plenty of room for optimization. The first step towards optimization would be to increase the stability, e.g. by partial substitution of manganese with group 14 elements as preliminary experiments indicate that such substitution is possible. For an increased thermoelectric performance, doping should decrease the concentration of the charge carriers. Much beyond thermoelectrics, the access to single-phase compounds in this ternary system is crucial for various more specialized analyses such as ARPES and detailed investigations of magnetic effects. MnBi2Te4 has attracted much attraction as the first antiferromagnetic topological insulator, with special focus on theoretical calculations.33,81 As these may be questionable if they are based on simple ordered models, the crystal-structure data presented in this study should be the basis for realistic calculations that take into account vancancies and Mn/Bi disorder. The new layered manganese bismuth tellurides may be promising candidates for topologically non-trivial materials and thus for studies of mutual influence between long-range magnetic ordering at low temperatures and non-trivial band-structure topology. The single-phase materials, which were only accessible by taking the non-stoichiometry into account, may also be a perspective for magnetic sensing if they should exhibit the anomalous Hall effect like, e.g., Mn-doped Bi2Te3.17,82
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the ESRF, Grenoble, for granting beamtime (experiments CH-5140 and CH-5142) as well as Dr Jonathan Wright, Lucien Eisenburger, Tobias Stollenwerk, Christina Fraunhofer and Dr Christopher Benndorf for help with the synchrotron measurements. Kristina Leger and Przemyslaw Blaschkewitz are acknowledged for support with the magnetization studies and the DSC measurements, respectively. We further thank Dr Ziya S. Aliev (Baku, Azerbaijan) for discussions. This work was supported by the German Research Foundation (DFG) by project OE 530/5-1 and in the framework of the Special Priority Program (SPP 1666, IS 250/2-1) “Topological Insulators”, by the ERA-Chemistry Program (RU 766/15-1), and by CRC “Correlated Magnetism: From Frustration to Topology” (SFB 1143).
Notes and references
- J. E. Moore, Nature, 2010, 464, 194–198 CrossRef CAS
 .
.
- M. Z. Hasan and C. L. Kane, Rev. Mod. Phys., 2010, 82, 3045–3067 CrossRef CAS .
- Y. L. Chen, J. G. Analytis, J.-H. Chu, Z. K. Liu, S.-K. Mo, X. L. Qi, H. J. Zhang, D. H. Lu, X. Dai, Z. Fang, S. C. Zhang, R. Z. Fisher, Z. Hussain and Z.-X. Shen, Science, 2009, 325, 178–181 CrossRef CAS PubMed .
- P. Jood and M. Ohta, Materials, 2015, 8, 1124–1149 CrossRef CAS PubMed .
- Y. Mizuguchi, Chem. Rec., 2016, 16, 633–651 CrossRef CAS PubMed .
- S. Sasaki, M. Kriener, K. Segawa, K. Yada, Y. Tanaka, M. Sato and Y. Ando, Phys. Rev. Lett., 2011, 107, 217001 CrossRef PubMed .
- T. V. Bay, T. Naka, Y. K. Huang, H. Luigjes, M. S. Golden and A. de Visser, Phys. Rev. Lett., 2012, 108, 57001 CrossRef CAS PubMed .
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS PubMed .
- W.-S. Liu, Q. Zhang, Y. Lan, S. Chen, X. Yan, Q. Zhang, H. Wang, D. Wang, G. Chen and Z. Ren, Adv. Energy Mater., 2011, 1, 577–587 CrossRef CAS .
- G. Sun, X. Qin, D. Li, J. Zhang, B. Ren, T. Zou, H. Xin, S. B. Paschen and X. Yan, J. Alloys Compd., 2015, 639, 9–14 CrossRef CAS .
- A. K. Bohra, R. Bhatt, A. Sigh, R. Basu, S. Bhattacharya, K. N. Meshram, S. Ahmad, A. K. Debnath, A. K. Chauhan, P. Bhatt, K. Shah, K. Bhotkar, S. Sharma, D. K. Aswal, K. P. Muthe and S. C. Gadkari, Energy Convers. Manage., 2017, 145, 415–424 CrossRef CAS .
- H. Zhang, C.-X. Liu, X.-L. Qi, X. Dai, Z. Fang and S.-C. Thang, Nat. Phys., 2009, 5, 438–442 Search PubMed .
- D. Hsieh, Y. Xia, D. Qian, L. Wray, F. Meier, J. H. Dil, J. Osterwalder, L. Patthey, A. V. Fedorov, H. Lin, A. Bansil, D. Grauer, Y. S. Hor, R. J. Cava and M. Z. Hansan, Phys. Rev. Lett., 2009, 103, 146401 CrossRef CAS PubMed .
- Y. Xia, D. Qian, D. Hsieh, L. Wray, A. Pal, H. Lin, A. Bansil, D. Grauer, Y. S. Hor, R. J. Cava and M. Z. Hasan, Nat. Phys., 2009, 5, 398–402 Search PubMed .
- C.-Z. Chang, J. Zhang, X. Feng, J. Shen, Z. Zhang, M. Guo, K. Li, Y. Ou, P. Wei, L.-L. Wang, Z.-Q. Ji, Y. Feng, S. Ji, X. Chen, J.-F. Jia, X. Dai, Z. Fang, S.-C. Zhang, K. He, Y. Wang, L. Lu, X.-C. Ma and Q.-K. Xue, Science, 2013, 340, 161–170 CrossRef .
- Y. H. Choi, N. H. Jo, K. J. Lee, J. B. Yoon, C. Y. You and M. H. Jung, J. Appl. Phys., 2011, 109, 07E312 CrossRef .
- Y. S. Hor, P. Roushan, H. Beidenkopf, J. Seo, D. Qu, J. G. Checkelsky, L. A. Wray, D. Hsieh, Y. Xia, S.-Y. Xu, D. Qian, M. Z. Hasan, N. P. Ong, A. Yazdani and R. J. Cava, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 195203 CrossRef .
- I. Vorobnik, U. Manju, J. Fujii, F. Borgatti, P. Torelli, D. Krizmancic, Y. S. Hor, R. J. Cava and G. Panaccione, Nano Lett., 2011, 11, 4079–4082 CrossRef PubMed .
- Y. Ni, Z. Zhang, I. C. Nlebedim, R. L. Hadimani and D. C. Jiles, IEEE Trans. Magn., 2015, 51, 1–4 CAS .
- J. Choi, S. Choi, J. Choi, Y. Park, H.-M. Park, H.-W. Lee, B.-C. Woo and S. Cho, Phys. Status Solidi B, 2004, 241, 1541–1544 CrossRef CAS .
- J. Růžička, O. Caha, V. Holý, H. Steiner, V. Volobuiev, A. Ney, G. Bauer, T. Duchoň, K. Veltruská, I. Khalakhan, V. Matolın, E. F. Schwier, H. Iwasawa, K. Shimada and G. Springholz, New J. Phys., 2015, 17, 013028 CrossRef .
- M. D. Watson, L. J. Collins-McIntyre, L. R. Shelford, A. I. Coldea, D. Prabhakaran, S. C. Speller, T. Mousavi, C. R. M. Grovenor, Z. Salman, S. R. Giblin and G. van der Laan, New J. Phys., 2013, 15, 103016 CrossRef .
- Y. Ni, Z. Zhang, I. C. Nlebedim and D. C. Jiles, IEEE Trans. Magn., 2016, 52, 1–4 Search PubMed .
- L. E. Shelimova, O. G. Karpinskii, P. P. Konstantinov, E. S. Avilov, M. A. Kretova and V. S. Zemskov, Inorg. Mater., 2004, 40, 451–460 CrossRef CAS .
- R. Vilaplana, J. A. Sans, F. J. Manjón, A. Andrada-Chacón, J. Sánchez-Benítez, C. Popescu, O. Gomis, A. L. J. Pereira, B. García-Domene, P. Rodríguez-Hernández, A. Munoz, D. Daisenberger and O. Oeckler, J. Alloys Compd., 2016, 685, 962–970 CrossRef CAS .
- S. Souma, K. Eto, M. Nomura, K. Nakayama, T. Sato, T. Takahashi, K. Segawa and Y. Ando, Phys. Rev. Lett., 2012, 108, 116801 CrossRef CAS PubMed .
- L. E. Shelimova, O. G. Karpinskii, M. A. Kretova and G. U. A. Lubman, Inorg. Mater., 1993, 29, 56–60 Search PubMed .
- L. E. Shelimova, P. P. Konstantinov, O. G. Karpinsky, E. S. Avilov, M. A. Kretova and V. S. Zemskov, J. Alloys Compd., 2001, 329, 50–62 CrossRef CAS .
- O. G. Karpinsky, L. E. Shelimova, M. A. Kretova and J.-P. Fleurial, J. Alloys Compd., 1998, 265, 170–175 CrossRef CAS .
- S. Welzmiller, F. Heinke, P. Huth, G. Bothmann, E.-W. Scheidt, G. Wagner, W. Scherer, A. Pöppl and O. Oeckler, J. Alloys Compd., 2015, 652, 74–82 CrossRef CAS .
- D. S. Lee, T.-H. Kim, C.-H. Park, C.-Y. Chung, Y. S. Lim, W.-S. Seo and H.-H. Park, CrystEngComm, 2013, 15, 5532–5538 RSC .
- A. Zeugner, F. Nietschke, A. U. B. Wolter, S. Gaß, R. C. Vidal, T. R. F. Peixoto, D. Pohl, C. Damm, A. Lubk, R. Hentrich, S. K. Moser, C. Fornari, C. H. Min, S. Schatz, K. Kißner, M. Ünzelmann, M. Kaiser, F. Scaravaggi, B. Rellinghaus, K. Nielsch, C. Heß, B. Büchner, F. Reinert, H. Bentmann, O. Oeckler, T. Doert, M. Ruck and A. Isaeva, Chem. Mater., 2018, 31, 2795–2806 CrossRef .
-
M. M. Otrokov, I. I. Klimovskikh, H. Bentmann, A. Zeugner, Z. S. Aliev, S. Gaß, A. U. B. Wolter, A. V. Koroleva, D. Estyunin, A. M. Shikin, M. BlancoRey, M. Hoffmann, A. Y. Vyazovskaya, S. V. Eremeev, Y. M. Koroteev, I. R. Amiraslanov, M. B. Babanly, N. T. Mamedov, N. A. Abdullayev, V. N. Zverev, B. Büchner, E. F. Schwier, S. Kumar, A. Kimura, L. Petaccia, G. Di Santo, R. C. Vidal, S. Schatz, K. Kiner, C.-H. Min, S. K. Moser, T. R. F. Peixoto, F. Reinert, A. Ernst, P. M. Echenique, A. Isaeva and E. V. Chulkov, 2018, arxiv.org/abs/1809.07389.
- Z. S. Aliev, I. R. Amiraslanov, D. I. Nasonova, A. V. Shevelkov, N. A. Abdullayev, Z. A. Jahangirli, E. N. Orujlu, M. M. Otrokov, N. T. Mamedov, M. B. Babanly and E. V. Chulkov, J. Alloys Compd., 2019, 789, 443–450 CrossRef CAS .
- J. A. Hagmann, X. Li, S. Chowdhury, S.-N. Dong, S. Rouvimov, S. J. Pookpanratana, K. Man Yu, T. A. Orlova, T. B. Bolin, C. U. Segre, D. G. Seiler, C. A. Richter, X. Liu, M. Dobrowolska and J. K. Furdyna, New J. Phys., 2017, 19, 085002 CrossRef .
-
WINXPOW, v. 2.2.1, Stoe & Cie. GmbH, Darmstadt, Germany, 2007 Search PubMed .
-
A. Coelho, TOPAS Academic, v. 5, Coelho Software, Brisbane, 2015 Search PubMed .
- R. S. Pease, Acta Crystallogr., 1952, 5, 356–361 CrossRef CAS .
-
X-Area, v. 1.77.1, STOE & Cie GmbH, Darmstadt, Germany, 2016 Search PubMed .
-
X-RED32, v. 1.63.1, STOE & Cie GmbH, Darmstadt, Germany, 2016 Search PubMed .
-
X-Shape, v. 2.18, STOE & Cie, Darmstadt, Germany, 2015 Search PubMed .
-
W. Herrendorf, PhD dissertation, Universität Karlsruhe, 1993 .
- F. Fahrnbauer, T. Rosenthal, T. Schmutzler, G. Wagner, G. B. M. Vaughan, J. P. Wright and O. Oeckler, Angew. Chem., Int. Ed., 2015, 54, 10020–10023 CrossRef CAS PubMed .
- J.-C. Labiche, O. Mathon, S. Pascarelli, M. A. Newton, G. G. Ferre, C. Curfs, G. Vaughan, A. Homs and D. F. Carreiras, Rev. Sci. Instrum., 2007, 78, 91301 CrossRef PubMed .
-
CrysAlis Pro 171.38.41, Agilent Technologies, Yarnton, Oxfordshire, England, 2015 Search PubMed .
-
SADABS, v. 2.05, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed .
- G. Wu, B. L. Rodrigues and P. Coppens, J. Appl. Crystallogr., 2002, 35, 356–359 CrossRef CAS .
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
-
K. Brandenburg, Diamond 3, Crystal Impact GbR, Bonn, Germany, 2014 Search PubMed .
-
INCA Suite, v. 4.09, Oxford Instruments Analytical Limited, Scotts Valley, USA, 1998–2007 Search PubMed .
-
Genesis, v. 6.1, EDAX, Mahwah, USA, 2010 Search PubMed .
-
analySIS, v. 2. 1, Olympus Soft Imaging Solutions, Münster, Germany, 1996 Search PubMed .
-
(a)
;
P. A. Stadelmann, jEMS, v. 4.4631 U2016, CIME-EPFL, Lausanne, Switzerland, 2016 Search PubMed;
(b) P. A. Stadelmann, Ultramicroscopy, 1987, 21, 131–145 CrossRef CAS .
-
Digital Micrograph, v. 3.6.5, Gatan, 1999 Search PubMed .
- L. Dusza, High Temp. – High Pressures, 1995/1996, 27–28, 467–473 Search PubMed .
- L. Yang, Z.-G. Chen, M. Hong, G. Han and J. Zou, ACS Appl. Mater. Interfaces, 2015, 7, 23694–23699 CrossRef CAS PubMed .
- L. Pan, J. Li, D. Berardan and N. Dragoe, J. Solid State Chem., 2015, 168, 168–173 CrossRef .
- A. S. Skoropanov, B. L. Valevxky, V. F. Skums, G. I. Samal and A. A. Vecher, Thermochim. Acta, 1985, 90, 331–334 CrossRef CAS .
- V. V. Atuchin, T. A. Gavrilova, K. A. Kokh, N. V. Kuratieva, N. V. Pervukhina and N. V. Surovtsev, Solid State Commun., 2012, 152, 1119–1122 CrossRef CAS .
- H. Fjellvåg, A. Kjekshus, T. Chattopadhyay, H. D. Hochheimer, W. Hönle and H. G. von Schnering, Phys. Lett. A, 1985, 112, 411–413 CrossRef .
- R. F. Brebrick, J. Appl. Crystallogr., 1968, 1, 241–246 CrossRef CAS .
- K. Yamana, K. Kihara and T. Matsumoto, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 147–149 CrossRef .
- Y. Feutelais, B. Legendre, N. Rodier and V. Agafonov, Mater. Res. Bull., 1993, 28, 591–596 CrossRef CAS .
-
M. Ruck, Between Covalent and Metallic Bonding: From Clusters to Intermetallics of Bismuth, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, ed. J. Reedijk, Elsevier, Waltham, MA, 2015, pp. 11445–11443 DOI:10.1016/B978-0-12-409547-2 .
- J. W. G. Bos, M. Lee, E. Morosan, H. W. Zandbergen, W. L. Lee, N. P. Ong and R. J. Cava, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 184429 CrossRef .
- A. N. Mansour, W. Wong-Ng, W. Huang, W. Tang, A. Tompson and J. Sharp, J. Appl. Phys., 2014, 116, 083513 CrossRef .
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef .
- H. J. Goldsmid and J. W. Sharp, J. Electron. Mater., 1999, 28, 869–872 CrossRef CAS .
- H.-S. Kim, Z. M. Gibbs, Y. Tang, H. Wang and G. J. Snyder, Appl. Mater., 2015, 3, 041506 CrossRef .
- R. P. Chasmar and R. Stratton, J. Electron. Control, 1959, 7, 52–72 CrossRef CAS .
-
H. J. Goldsmid, Introduction to Thermoelectricity, Springer- Verlag, Berlin, Heidelberg, 2016, ch. 4, pp. 45–66 Search PubMed .
-
H. Wang, Y. Pei, A. D. LaLonde and G. J. Snyder, in Thermoelectric Nanomaterials: Materials Design and Applications, ed. K. Koumoto, T. Mori, Springer, Berlin, Heidelberg, 2013, ch. 1, pp. 3–32 Search PubMed .
- S. D. Kang and G. J. Snyder, Nat. Mater., 2017, 16, 252–257 CrossRef PubMed .
-
A. F. May and G. J. Snyder, in Thermoelectrics and its Energy Harvesting, ed. D. M. Rowe, CRC Press, Boca Raton, FL, 2012, ch. 11, pp. 1–18 Search PubMed .
-
S. D. Kang and G. J. Snyder, 2018, arXiv:1710.06896v2.
- L. A. Kuznetsova, V. L. Kuznetsov and D. M. Rowe, J. Phys. Chem. Solids, 2000, 61, 1269–1274 CrossRef CAS .
- T. B. Zhukova and A. I. Zaslavskii, Sov. Phys. Crystallogr., 1972, 16, 796–800 Search PubMed .
- O. G. Karpinsky, L. E. Shelimova, M. A. Kretova and J.-P. Fleurial, J. Alloys Compd., 1998, 265, 170–175 CrossRef CAS .
- T. Matsunaga, R. Kojima, N. Yamada, T. Fujita, K. Kifune, Y. Kubota and M. Takata, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 407–411 CrossRef CAS PubMed .
- M. N. Schneider and O. Oeckler, Z. Anorg. Allg. Chem., 2008, 634, 2557–2561 CrossRef CAS .
- M. M. Otrokov, T. V. Menshchikova, M. G. Vergniory, I. P. Rusinov, A. Yu. Vyazovskaya, Yu. M. Koroteev, G. Bihlmayer, A. Ernst, P. M. Echenique, A. Arnau and E. V. Chulkov, 2D Mater., 2017, 4, 025082 CrossRef .
-
S. H. Lee, Y. Zhu, Y. Wang, L. Miao, T. Pillsbury, S. Kempinger, D. Graf, N. Alem, C.-Z. Chang, N. Samarth and Z. Mao, 2018, arXiv:1812.00339.
Footnote |
| † Electronic supplementary information (ESI) available: Elemental analyses (SEM-EDX) and SEM images, tables for Rietveld refinements of Mn0.85Bi4.10Te7 and Mn0.73Bi6.18Te10, PXRD of heterogeneous samples, PXRD of Mn1−x□x/3Bi4+2x/3Te7 with x = 0.15, 0.167 and 0.2 and Mn1−x□x/3Bi6+2x/3Te10 with x = 0.19, 0.23 and their lattice parameters, PXRD of Mn0.89Bi4.11Te7 and Mn0.85Bi6.15Te10 (refinement results without assuming charge neutrality), PXRD after thermoelectric measurements, temperature-dependent PXRD, PXRD after DSC measurements, DSC with possible assignment of their signals, HRTEM images of manganese bismuth tellurides with GeBi4Te7 and GeBi6Te10 structure type, information concerning the modeling of thermoelectric data. CCDC 1891486 and 1891488. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9tc00979e |
|
| This journal is © The Royal Society of Chemistry 2019 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Alexander
Zeugner
c,
Anna
Isaeva
b,
Alexander
Zeugner
c,
Anna
Isaeva
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: