
High-efficiency non-fullerene polymer solar cell fabricated by a simple process using new conjugated terpolymers†
Mai Ha
Hoang‡
a,
Gi Eun
Park‡
b,
Suna
Choi
b,
Chang Geun
Park
b,
Su Hong
Park
b,
Tuyen Van
Nguyen
a,
Sangjun
Kim
b,
Kyungwon
Kwak
b,
Min Ju
Cho
*b and
Dong Hoon
Choi
 *b
*b
aInstitute of Chemistry, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
bDepartment of Chemistry, Research Institute for Natural Sciences, Korea University, 145 Anam-Ro, Sungbuk-gu, Seoul 136-701, Korea. E-mail: chominju@korea.ac.kr; dhchoi8803@korea.ac.kr
First published on 29th November 2018
Abstract
A series of conjugated terpolymers, P2–P4, based on 4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene as the electron-donating unit, methyl-3-thiophenecarboxylate (3MT) as the weak electron-accepting unit, and 1,3-bis(2-ethylhexyl)benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD) as the strong electron-accepting unit were synthesized to investigate the effect of the contents of 3MT and BDD on their optical and electrochemical properties and the performance of the derived fullerene-free polymer solar cell (PSC) devices. By varying the contents of 3MT and BDD in the terpolymer structure, the solubility, absorption range, and energy levels could be efficiently tuned. Among the three terpolymers, the as-cast blend film of P3:acceptor with complementary absorption and favorable internal morphology exhibited the highest power conversion efficiency of 10.26% with a high open circuit voltage of 0.94 V and short circuit current density of 17.18 mA cm−2 in the PSC. Although the three repeating units were randomly arranged along the polymer backbone, their PSCs exhibited an excellent shelf-life of over 1000 h under ambient conditions, which is as good as those of binary D–A copolymers.
Introduction
A variety of non-fullerene acceptors are currently in use for bulk heterojunction (BHJ) polymer solar cells (PSCs), and their practical application in organic solar cells is very promising.1–8 Owing to their high power conversion efficiencies (PCEs) of over 10%, non-fullerene-type BHJ PSCs have been considered to be ideal for application in large-area flexible devices fabricated via low-cost practical solution processes such as spin-coating, ink-jet, and roll-to-roll printing techniques.1–3 Furthermore, as compared to the BHJ PSCs that are based on fullerene derivatives, non-fullerene-type PSCs have been reported to show not only effective complementary absorption behavior with high molar extinction coefficients but also relatively high PSC performance and long-term stability.4,5Among the various types of non-fullerene acceptors reported in the literature, acceptor–donor–acceptor (A–D–A) type small molecules have generated much interest as alternatives to fullerene derivatives.6–10 However, the new polymer donors are relatively less developed than the acceptors, which underscores the necessity of further research attempts to match the energy levels and absorption properties of the A–D–A type molecules. The light harvesting property of materials in the range of visible to near-infrared (NIR) wavelengths is important for PSCs and is dependent on a suitable selection of both accepting and donating materials, in contrast to the cases involving traditional fullerene-based PSCs.
A well-defined morphology of the active layer in a BHJ PSC is known to be an important factor for achieving high PCEs. Several researchers have attempted to produce blend films for optimized devices using either one or a combination of two or more processing techniques and strategies such as the treatment of the blend active-layer films with thermal or solvent vapor annealing,11,12 use of co-solvents and solvent additives for a well-defined nanophase segregated morphology in blend films,13,14 the introduction of interlayers for efficient charge transport between the active layer and electrodes,15–17 and control of the blend composition and film thickness for efficient exciton generation and diffusion.18,19 In order to maximize the efficiency of PSCs as described above, considerable effort has been devoted to optimizing the internal morphology of the donor and acceptor blend films and to designing a device with a new multi-layered structure.
However, these methods can sometimes have a negative effect on the stability and reproducibility of the device performance and comprise time-consuming complicated device fabrication.20,21 Therefore, an ideal strategy to overcome these disadvantages is to develop new donor and acceptor materials with good intrinsic semiconducting properties. Nevertheless, in the high-performance PSCs published in many previous literature reports, various treatments such as the aforementioned ones were applied, and various solvent additives were added to the active layer. The corresponding blend films have to produce a well-defined nanophase-separated morphology without the use of specific processing techniques and solvent additives.22 If high-performance PSCs can be implemented using a simple process, i.e., without using the aforementioned complicated process, they will have many advantages in the practical application of the PSC.
A promising design strategy for further improving the advantageous properties of copolymers involves the use of terpolymers comprising three types of monomers in the conjugated polymer backbone.23,24 Terpolymers exhibit several desirable attributes-such as high solubility, broad absorption range, finely tuned energy levels, and improved nanophase internal morphology and orientation of polymer chains in the blends over binary copolymers bearing one each of the donor and acceptor monomeric units.25,26
Recently, from among a few promising polymer donors, it was found that the medium-bandgap PBDB-T copolymer consisting of 4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene (BDT) and 1,3-bis(2-ethylhexyl)benzo[1,2-c:4,5-c′]dithiophene-4,8-dione (BDD) units, which was prepared by Hou and coworkers, demonstrated a high PCE (9.15%) in a non-fullerene PSC fabricated using a simple process. Moreover, an enhanced PCE (>11%) was achieved using a thermal annealed blend film containing a 1,8-diiodooctane (DIO) solvent additive.27
In order to fabricate a high-performance PSC using a simple process, it is necessary to slightly modify the structure of the PBDB-T such that the inherent semiconductor characteristics do not deteriorate greatly. A terpolymer system with appropriate third monomeric units could serve as an effective design strategy. In addition, these polymers are expected to provide favorable physical/optical properties and device performance.
In this study, we designed and synthesized a series of random terpolymers, P2–P4, containing BDT as the electron-donating unit, methyl-3-thiophenecarboxylate (3MT) as the weak electron-accepting unit, and BDD as a stronger electron-accepting unit than 3MT. The absorption spectra and energy levels of the terpolymers could be easily tuned by changing the molar ratio of the 3MT and BDD monomers. As compared to the copolymers P1 and P5, the terpolymer-based blend films without solvent additives and thermal treatment showed a relatively improved internal morphology in which the face-on polymer chain orientation profiles were maintained. After optimizing the contents of BDT, 3MT, and BDD in the various terpolymers, it was found that the non-fullerene PSC made of the as-cast P3 blend film exhibited the highest PCE of 10.26% with a high Voc of 0.95 V and Jsc of 17.18 mA cm−2. The above device performance is quite similar to that of a device in which a solvent additive is added to an active layer, which is then subjected to thermal treatment. Therefore, the performance of the PSC prepared using the simple process comprising the use of new conjugated terpolymers was superior to that of the PSC prepared using the binary conjugated copolymer.
Results and discussion
Design strategy, synthesis, and characterization
The design strategy of the new conjugated terpolymers involved employing donor and acceptor monomers used for two polymers, namely, 3MT-Th, which was composed of BDT and 3MT. PBDB-T was composed of BDT and BDD. As shown in Scheme 1, three random terpolymers P2 (y = 0.25), P3 (y = 0.50), and P4 (y = 0.75) with one donor and two different acceptor units were readily synthesized via Stille coupling polymerization using different feeding molar ratios of 3MT and BDD. Two D–A binary copolymers, P1 and P5, were synthesized as control polymers according to a previously used procedure.28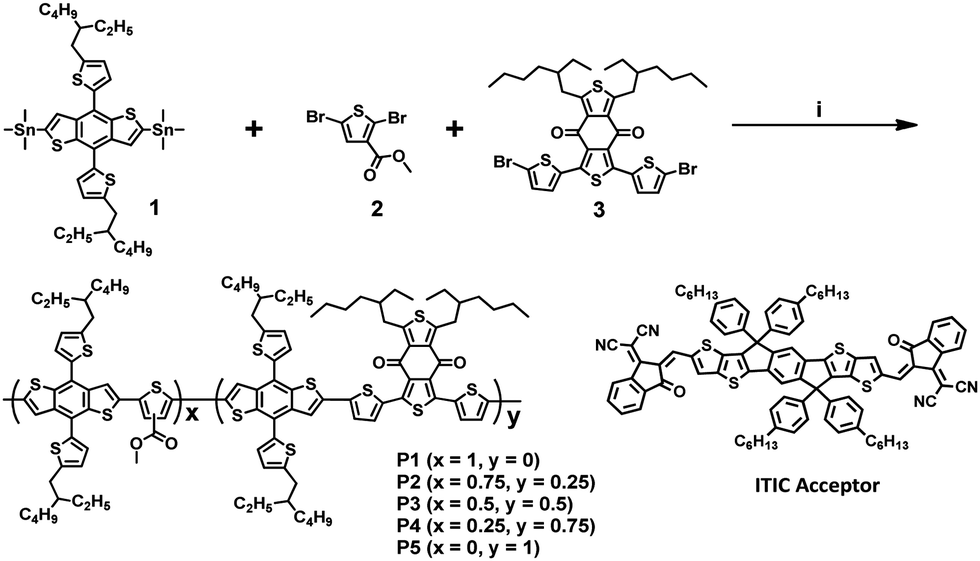 | ||
Scheme 1 Synthetic procedures for binary copolymers and random terpolymers. (i) Pd2(dba)3, P(o-tolyl)3, toluene/DMF (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 100 °C. ITIC is an A–D–A-type non-fullerene acceptor. :1), 100 °C. ITIC is an A–D–A-type non-fullerene acceptor. | ||
In contrast to the polymerization process for P1–P3, insoluble solids were precipitated in the toluene solvent when P4 and P5 were polymerized. When these insoluble solids were visually observed in the solution, the polymerization reaction was terminated. The observed precipitation indicated that the solubility of the terpolymer was highly dependent on the concentration of the BDD monomer units owing to its relatively rigid and flat structure, which was induced by strong interactions between the sulfur atom of the thiophene unit and the oxygen atom of the ketone group (Fig. S1, ESI†).29,30
The synthesized crude copolymers and terpolymers were purified via Soxhlet extraction using acetone, hexane, and chloroform in succession. All the synthesized polymers were adequately soluble in chloroform, chlorobenzene, and o-dichlorobenzene at 25 °C. The molecular weights and polydispersity indices (PDIs) of the polymers were determined using gel permeation chromatography with o-dichlorobenzene as the eluent and polystyrene as the standard at 80 °C (Table 1). The molecular weights (Mn) of P1–P5 were determined to be 26.7, 24.9, 24.4, 22.9, and 20.5 kDa, respectively.
| Polymer | M n (kDa) | PDI | Absorption (nm) | λ cut-off (nm) | E g , (eV) | E ox (V) | Energy levels (eV) | ||
|---|---|---|---|---|---|---|---|---|---|
| Solution | Film | HOMOd | LUMOe | ||||||
| a Thin film. b Energy bandgap evaluated using UV-vis absorption spectra. c Onset oxidation potentials obtained from cyclic voltammograms obtained in the film state. d HOMO = −(e(Eox − 0.37 V (ferrocene)) + 4.8 eV)). e LUMO energies were calculated from the difference between the HOMO energy and optical bandgap. | |||||||||
| P1 | 26.7 | 2.52 | 504 | 539 | 625 | 1.98 | 0.99 | −5.42 | −3.44 |
| P2 | 24.9 | 2.69 | 523 | 569 | 664 | 1.87 | 0.95 | −5.38 | −3.51 |
| P3 | 24.4 | 3.52 | 559 | 574 | 677 | 1.83 | 0.94 | −5.37 | −3.54 |
| P4 | 22.9 | 2.84 | 575 | 616 | 681 | 1.82 | 0.94 | −5.37 | −3.55 |
| P5 | 20.5 | 4.13 | 613 | 623 | 687 | 1.80 | 0.91 | −5.34 | −3.54 |
Optical and electrochemical properties
In order to investigate the optical properties of the synthesized polymers, their UV-vis absorption spectra were recorded in a solution and in the thin-film state (Fig. 1a and b).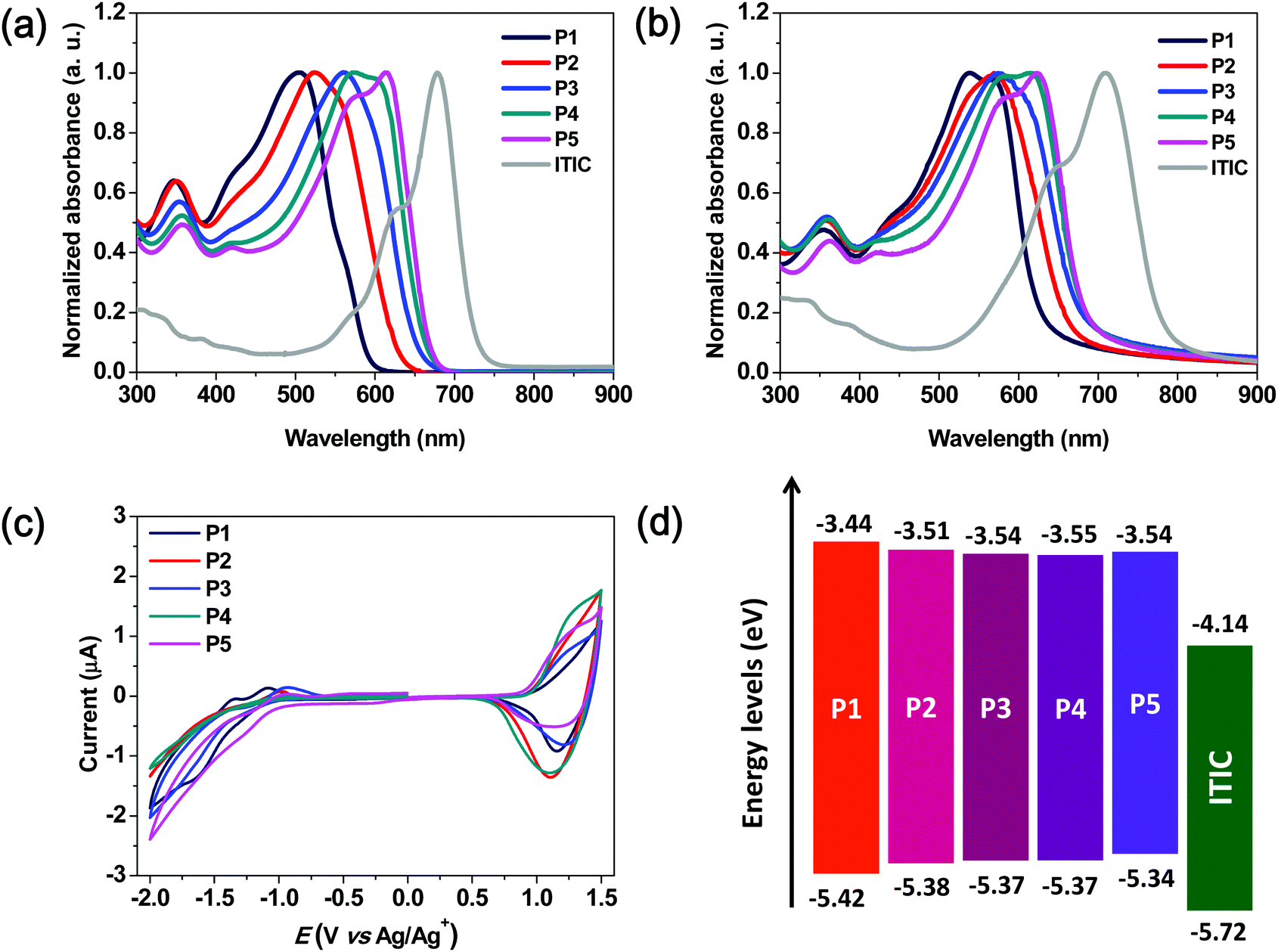 | ||
| Fig. 1 UV-vis absorption spectra of P1–P5 in (a) chloroform solutions and (b) the thin-film state. (c) Cyclic voltammograms of the film samples and (d) energy levels of all the polymers serving as donors and that of ITIC serving as the acceptor. | ||
The corresponding parameters are summarized in Table 1. In the solution, the strong intramolecular charge transfer absorption bands of terpolymers P2–P4 were observed at 523, 559, and 575 nm, respectively. These bands became increasingly red-shifted as the content of the BDD unit was increased and were located in between those of P1 and P5. This indicated that the absorption spectral ranges of the terpolymers could be easily tuned by controlling the molar ratio of the 3MT and BDD units, which were used as the weak and relatively strong accepting units, respectively. As compared to the absorption spectra of the polymer solutions, those of the polymer thin films exhibited a red-shift phenomenon owing to intermolecular interactions between the polymer backbones. Although random terpolymers lacked the regio-regularity of repeating units, their films also exhibited red-shifted absorption spectra. These phenomena were similar to those of binary D–A copolymers.
For investigating the aggregation behavior of the polymer chains in solution, the temperature-dependent absorption spectra of P1–P5 in chlorobenzene were acquired.31,32 As shown in Fig. S2 (ESI†), the P5 solution showed a distinct 0–0 vibronic peak at 613 nm at 20 °C. The absorption spectra changed significantly as the temperature was increased to 100 °C, thus indicating that P5 showed strong aggregation even in the solution. In contrast, the spectrum of P1 did not show any significant change as compared to that of P5. Thus, the three terpolymers simultaneously exhibited the properties of the two copolymers comprising the two repeating units, 3MT and BDD, respectively. This intrinsic polymer properties depend on the contents of the repeating units of 3MT and BDD.
The optical bandgaps of the P1–P5 films were 1.98, 1.87, 1.83, 1.82, and 1.80 eV, respectively, which were estimated from the absorption edges in the absorption spectra shown in Fig. 1b. As expected, an increase in the number of BDD units in the polymer backbone resulted in a gradual decrease in the optical bandgap.
The highest occupied molecular orbital (HOMO) energy levels of P1–P5 were determined using cyclic voltammetry, and their lowest unoccupied molecular orbital (LUMO) values were calculated using the optical bandgaps and HOMO energy values. The energy diagrams and corresponding parameters for the synthesized polymers are displayed in Fig. 1c and Table 1, respectively. The onset oxidation potentials (Eox) of P1–P5 were 0.99, 0.95, 0.94, 0.94, and 0.91 V, respectively, and the corresponding energies of the HOMO levels were calculated to be −5.42, −5.38, −5.37, −5.37, and −5.34 eV, respectively. The energies of the LUMO levels calculated from the HOMO levels and optical bandgaps are −3.44, −3.51, −3.54, −3.55, and −3.54 eV for P1–P5, respectively. As the content of the BDD-bearing repeating unit in the polymer backbone was increased, the HOMO and LUMO gradually moved to higher-lying and lower-lying positions, respectively, with a corresponding lowering of the bandgap.
This suggests that the energy levels of the terpolymers can be easily tuned by changing the molar ratio of the 3MT to BDD in the polymer backbone structure. As shown in the inset of Fig. S2f (ESI†), the five polymer solutions have very different colors with varying contents of the BDD-based repeating units, which indicates their different absorption properties. The observed energy level offsets, as compared to those of ITIC serving as the non-fullerene acceptor, guarantee a strong photo-induced charge transfer between the terpolymer and ITIC.
Non-fullerene PSCs based on terpolymers and ITIC
Non-fullerene BHJ PSCs were fabricated using the synthesized terpolymers P2–P4 and ITIC. In addition, P1- and P5-based PSC devices were also fabricated using the same device configuration to serve as control devices. We chose an inverted PSC device structure with the configuration of indium tin oxide (ITO)/ZnO/polymer:ITIC/MoO3/Ag as presented in literature reports.33,34 To explore the possibility of the use of simple devices based on the synthesized terpolymers, two types of active layers were fabricated; an as-cast blend film was fabricated with the terpolymer:ITIC without DIO and the other layer was composed of a ternary blend of the terpolymer:ITIC:DIO additive and was thermally annealed according to a previously reported procedure.34 Thus, PSCs containing active layers that were prepared under different conditions were fabricated, and their performance and stability (i.e., shelf-life and operational stability) were investigated.28 The various device fabrication conditions used for optimizing the performance and the corresponding PSC parameters are presented in Tables S1 and S2 (ESI†).As shown in Fig. 2a, all the terpolymer-based simple PSCs exhibited Voc and Jsc values of over 0.9 V and 15 mA cm−2, respectively. Furthermore, as the number of BDD units in the terpolymer backbone was increased, the value of Voc gradually decreased from 0.95 to 0.90 V, which is in good agreement with the differences between the HOMO energy levels of the polymers and the LUMO energy level of ITIC, as shown in Fig. 1d. Among the terpolymer-based PSCs fabricated using simple processed-blend films, the P3:ITIC (1:1 wt%) device exhibited the highest PCE of 10.26% with a high Voc of 0.94 V and Jsc of 17.18 mA cm−2. As compared to the binary copolymer-based PSCs (PCE = 8.22% for P1:ITIC and PCE = 8.82% for P5:ITIC), the terpolymer-based PSCs showed higher PCE values ranging from 8.95 to 10.26% without the use of specific treatments (Table 2).
 | ||
| Fig. 2 (a) Current density–voltage (J–V) curves and (b) external quantum efficiency (EQE) spectra of PSCs comprising the as-cast blend films of the five polymers and ITIC (1:1 wt%). The integrated Jsc curves were included. PCE parameters vs.P1–P5-based PSCs (c and d). | ||
:ITIC (1:1 wt%)
| Polymer | V oc (V) | J sc (mA cm−2) | FF (%) | PCEa (%) | J sc (mA cm−2) |
|---|---|---|---|---|---|
| a Average PCEs in parentheses for ten devices. b J sc values are calculated by integrating the EQE spectra. | |||||
| P1 | 0.97 | 15.59 | 54.38 | 8.22 (8.07 ± 0.14) | 14.81 |
| P2 | 0.95 | 15.76 | 59.76 | 8.95 (8.84 ± 0.11) | 14.73 |
| P3 | 0.94 | 17.18 | 63.52 | 10.26 (10.14 ± 0.12) | 16.52 |
| P4 | 0.90 | 17.19 | 61.77 | 9.56 (9.28 ± 0.28) | 16.01 |
| P5 | 0.88 | 16.30 | 61.48 | 8.82 (8.60 ± 0.23) | 14.97 |
The change in the performance of the P3-based PSC using a thermally annealed active layer and solvent additive of DIO with respect to that of the simple PSC mentioned above was also assessed. A slightly enhanced PCE value of 11.02% (Voc = 0.92 V, Jsc = 17.22 mA cm−2, and FF = 69.56%) was obtained for P3:ITIC with a 0.25 wt% DIO that was thermally annealed at 140 °C for 10 min, as shown in Fig. S3 and Table S1 (ESI†).
It is interesting to note that even though various processing conditions such as different contents of P3 and ITIC, annealing temperatures, and types/amounts of additive were used in the fabrication of the active layer for P3:ITIC-based PSCs as shown in Table S1 (ESI†), the performances of these devices did not differ significantly. Thus, the high-efficiency terpolymer-based PSCs introduced in this study have several advantages such as a simple fabrication process and reduced device fabrication time.
The external quantum efficiency (EQE) spectra of the terpolymer:ITIC-based PSCs are shown in Fig. 2b. All the devices exhibited high EQEs from the visible to NIR wavelengths, which indicates that the active layers showed efficient exciton generation. Furthermore, both the donor polymer and acceptor ITIC contribute to these high-EQE values in the spectral range of 400–700 nm, as the desired complementary absorption behavior is achieved by combining the large- and medium-bandgap monomeric segments as donors.5,35
The hole and electron mobilities of the blend films were measured using the space charge limited current method. The configuration of the hole-only devices was ITO/PEDOT:PSS/polymers:ITIC (1:1)/Au and that of the electron-only devices was ITO/ZnO/polymers:ITIC (1:1)/LiF/Al.
As shown in Fig. S4 and Table S3 (ESI†), the hole and electron mobilities of the blend films increased gradually from P1 to P5. Among them, P3:ITIC showed a similar hole and electron mobility of 5.25 × 10−5 cm2 V−1 s−1 and 1.56 × 10−5 cm2 V−1 s−1, respectively. Thus, the P3:ITIC blend film exhibited a more balanced charge transport (μh/μe = 3.37), which likely contributed to the higher FF values of the corresponding devices.
Morphological studies of the blend films of the polymer and ITIC
The surface and internal morphologies of the active layers with the as-cast blend films were investigated using atomic force microscopy (AFM) and transmission electron microscopy (TEM), respectively. As shown in Fig. 3, the active layers comprising terpolymer:ITIC (1:1) blends showed very fine domains with root-mean-square roughness values of less than 0.4 nm, which are smaller than those observed for the P5:ITIC blend. It is likely that the random terpolymers favorably hindered the regular ordering of the polymer backbones and alleviated the degree of crystallization of the polymer backbones.36 Thus, it can be expected that the ITIC molecules and their fine aggregates could be easily intercalated into the polymer chain network.
 | ||
| Fig. 3 (a–e) AFM height images (5 μm × 5 μm) and (f–j) TEM images of the as-cast polymer:ITIC (1:1) films. P1:ITIC (a and f), P2:ITIC (b and g), P3:ITIC (c and h), P4:ITIC (d and i), and P5:ITIC (e and j). Scale bar = 200 nm. No solvent additive was used in the active layer. | ||
The TEM images also showed the internal morphology of the optimized polymer:acceptor blend films. As shown in Fig. 3f–j, as compared to the blend films containing P1 and P5, the internal morphology of the P3:ITIC blend film was more homogeneous with fine domains. This structure is known to be favorable for effective exciton generation, diffusion, charge separation, and dissociated charge transport.37,38
Thus, these results validated the relatively high Jsc and FF values achieved for the PSCs based on terpolymers as compared to the other two copolymers, P1 and P5.
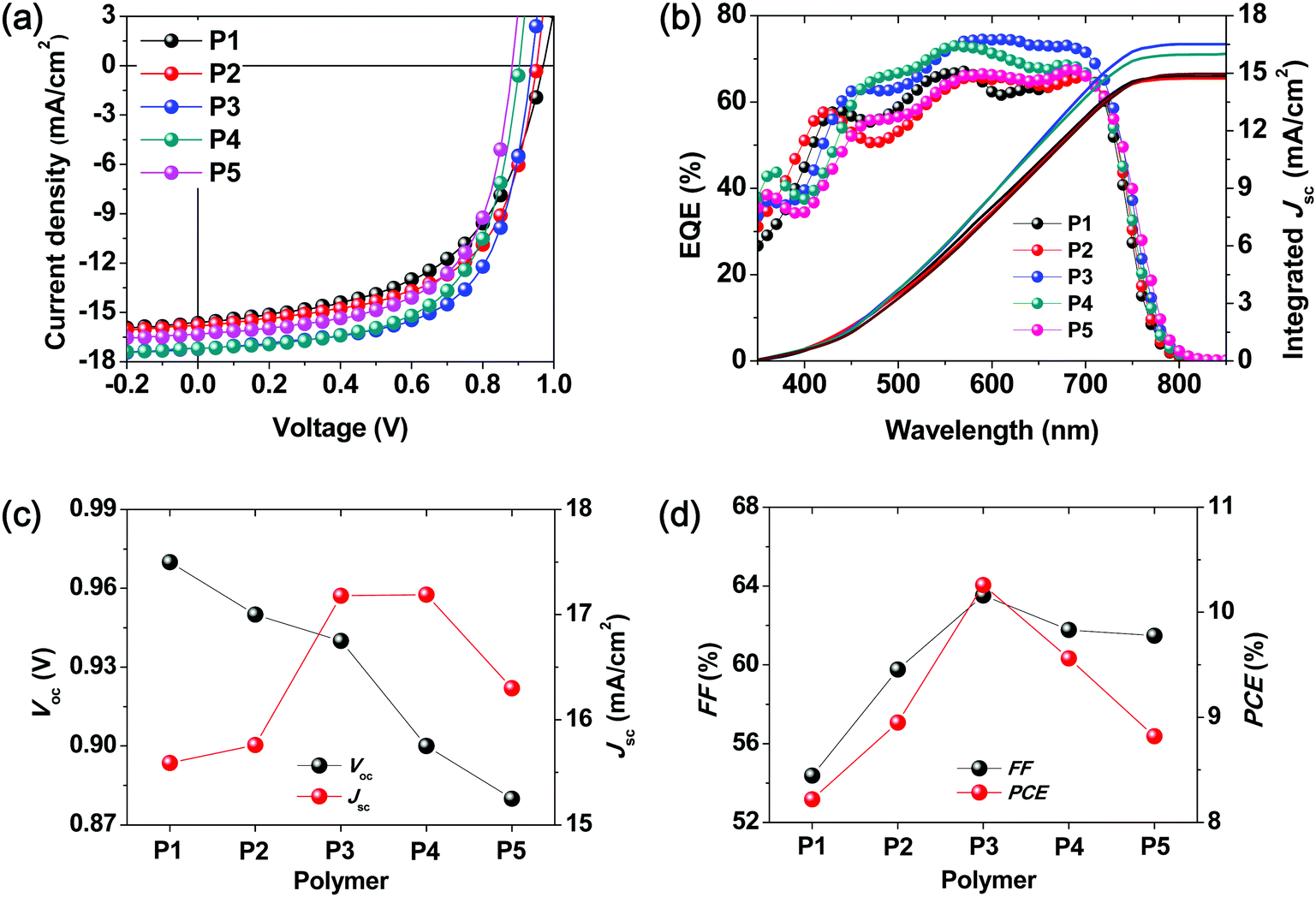
To investigate the change in crystallinity and chain orientation in the terpolymer system, grazing incidence wide-angle X-ray diffraction (GIWAXD) measurements were performed on both the neat films of individual polymers and the as-cast polymer:ITIC blend films (1:1 wt%). The GIWAXD patterns for the neat polymer films and blend films are shown in Fig. S5 and S6 (ESI†), and Fig. 4, respectively. In the case of the neat films, all five polymers displayed a face-on orientation, with the (010) π–π-stacking peak in the out-of-plane direction and a distinct (100) diffraction peak in the in-plane direction. Based on these results, it was concluded that the chain orientation of the terpolymers bearing randomly ordered repeating units exhibited a tendency similar to that of the binary D–A copolymers.
 | ||
| Fig. 4 2D grazing incidence wide-angle X-ray diffraction (GIWAXD) patterns of the blend films of P1–P5 with ITIC; (a) out-of-plane and (b) in-plane profile. | ||
As shown in Fig. S5 and Table S5 (ESI†), the full width at half maximum of the (100) diffraction peaks of the neat polymer films in the in-plane direction decreased progressively with the decreasing content of the 3MT unit. Furthermore, the (200) diffraction peak of the P5 film also decreased progressively with the increasing content of the 3MT unit and disappeared completely from P1 and P2 as shown in Fig. S5c (ESI†). Thus, introducing 3MT moieties tethered randomly into donors perturbed the chain regularity and reduced the crystallinity in the film. From these results, it was found that the crystallinity of the terpolymer can be easily tuned by controlling the ratio of the 3MT and BDD units.
The P2–P4 blend films with ITIC exhibited strong (010) diffraction peaks of π–π stacking in the out-of-plane profiles, as shown in Fig. 4 and Fig. S6 (ESI†). The terpolymer chains in the blend films were found to predominantly form a face-on orientation on the substrate, which maintained the morphological characteristics of the neat films of terpolymers, as mentioned above. This is similar to the diffraction results obtained for the blend films of copolymer:ITIC, and consequently, the face-on orientation is likely the most favorable orientation to induce facile exciton dissociation and efficient hole/electron generation, and transport the charge carriers to the vertical direction in sandwich-type PSCs.39
According to the performance of the PSC measured previously, the degree of crystallization of the P3:ITIC blend film was conjectured to constitute an internal morphology in which exciton diffusion is most favorable. Based on the results of the GIWAXD, it was found that the predominant face-on orientation behavior and proper crystallinity of the blend films can be used to obtain high-performance PSCs.40
Stability of the PSCs
For the purpose of comparison with binary D–A copolymers having a high shelf-life, as reported in previous studies, random terpolymer P3-based PSC devices were stored under ambient conditions for more than 1000 h without encapsulation, and their shelf-life was assessed using the ISOS-D-1 (Shelf) protocol.41,42Fig. S7 and S8 (ESI†) show the changes in the photovoltaic parameters according to the J–V characteristics of the polymer:ITIC-based PSCs. The stability of the PSCs was evaluated as a function of storage time in terms of their photovoltaic parameters (e.g., PCE, Jsc, Voc, and FF).
Fig. 5a clearly shows the effect of aging on the PCEs during long-term storage. The PCEs of the PSCs with the P3:ITIC as-cast film were similar to the initial values for 1056 h. Although the repeating units of P3 were randomly arranged, no significant changes were observed. However, it was observed that P3-based PSCs with DIO had a larger decrement of 13%. To confirm this phenomenon in terms of morphology change, AFM was used to analyze the topography of the films during the measurement, as shown in Fig. S9 (ESI†).43–45 As compared to the P3-based active layer with the DIO additive, the DIO-free blend films retained their less-changed morphology even after 1056 h under ambient conditions. In brief, the shelf-life of the PSC was confirmed to be influenced by the DIO additive added to the active layer instead of being dependent on the donor polymer.
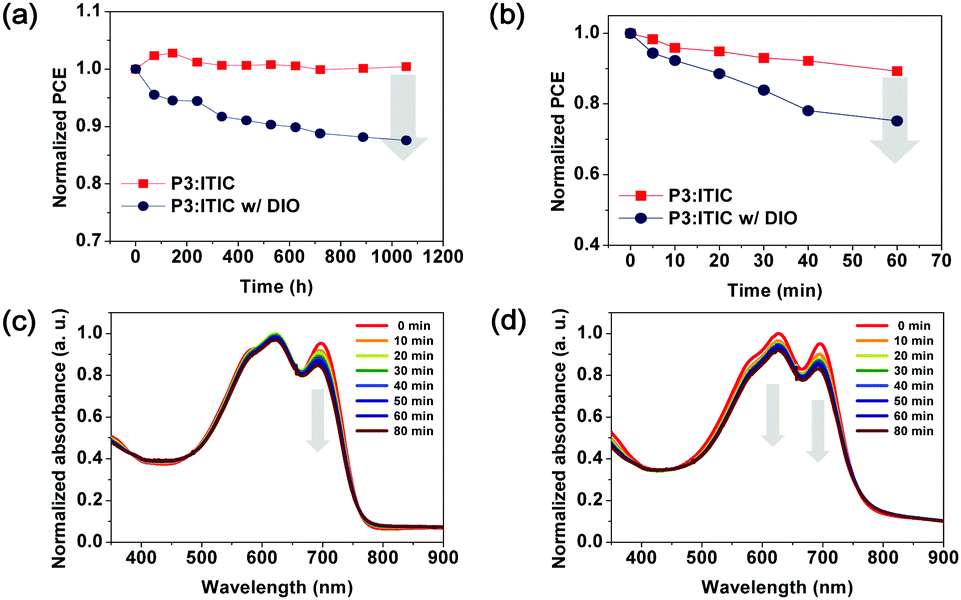 | ||
| Fig. 5 Shelf-life (a) and operational stability (b) of the P3-based PSCs without DIO and with DIO. UV-vis absorption spectra of P3-based PSCs without DIO (c) and with DIO (d) during continuous illumination (AM 1.5 G illumination at 100 mW cm−2). | ||
The PSC devices were subjected to continuous illumination (AM 1.5 G illumination at 100 mW cm−2) to investigate the operational stability of the polymer:ITIC blend films. The device performance was evaluated periodically to observe the illumination-dependent degradation effect. As shown in Fig. 5c, the absorption intensity of ITIC at λmax (709 nm) decreased more significantly (13%), whereas that of P3 maintained up to 97% of the initial absorption intensities even after 1 h of illumination. This indicates that the operational stability of the P3-based PCE is mainly dependent on the photostability of the ITIC acceptor. In contrast, when the P3:ITIC blend film contains a DIO additive, the absorption intensities of the P3 donor (λ = 574–610 nm) and ITIC acceptor (λ = 709 nm) are simultaneously decreased. This, along with the previously observed ITIC photodegradation phenomenon, indicates that the DIO can affect the photostability of all the components of the active layer.46
To better understand the effect of the degradation of PSCs, we conducted acceleration experiments to monitor the photodegradation of the pristine films of P1, P3, P5, and ITIC in situ by cumulative exposure to a 532 nm excitation laser. Under ambient conditions, there was a relatively small change in the Raman spectra (see Fig. 6) of the P1, P3, and P5 pristine films even after 35 min of light exposure. However, the intensity of the Raman peak of the ITIC film changed significantly after exposure, as shown in Fig. 6d. From these results, it was concluded that the decomposition of the ITIC is more facile and contributes predominantly to the deteriorating performance of the PSCs.
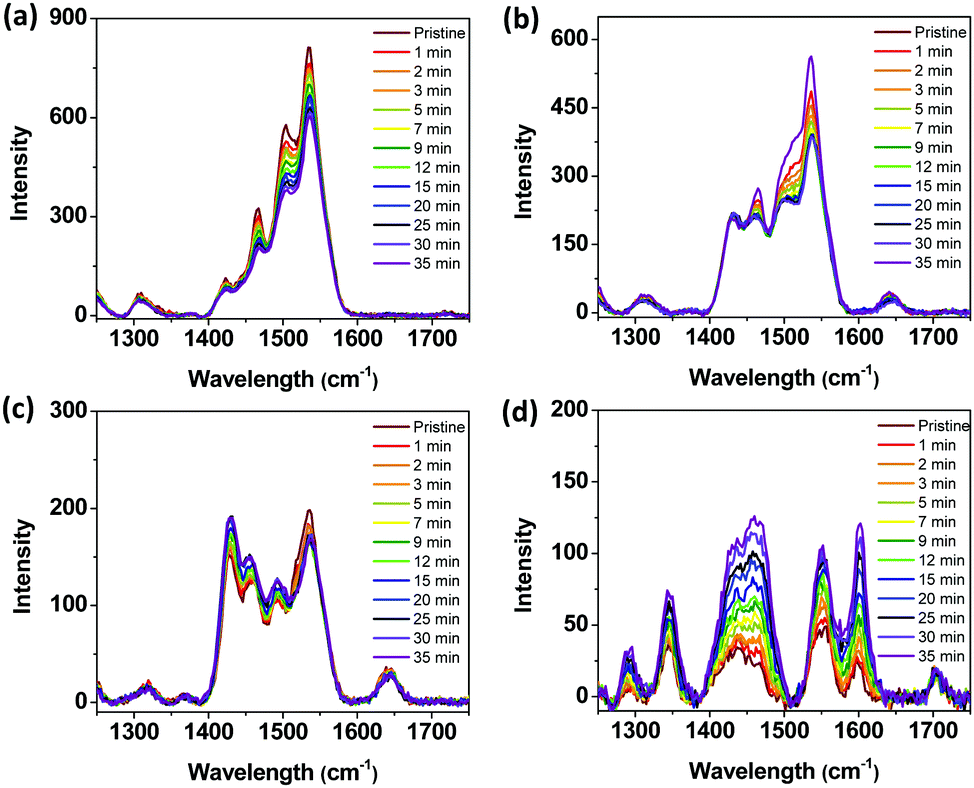 | ||
| Fig. 6 Raman spectra of the as-cast films of (a) P1, (b) P3, (c) P5, and (d) ITIC during in situ degradation in air as a function of the total in situ laser exposure time. | ||
Low-temperature processable flexible PSCs
Finally, we fabricated a simple and low-temperature-processable PSC on a flexible polymer substrate (poly(ethylene 2,6-naphthalate (PEN)/ITO/ZnO/P3:ITIC/MoO3/Ag). As compared with the PSC fabrication method involving the use of the ITO glass, the as-cast blend film should be used in this case because the polymer substrate is expected to be deformed during the thermal treatment.Fig. 7 shows the J–V curves for the PSCs using two different substrate materials. The ITO-coated PEN-based device exhibited a PCE (9.59%) similar to that of the PSC fabricated using conventional ITO-coated glass substrates (PCE = 10.26%). Thus, the P3 terpolymer developed in this study is a promising material for future application in flexible PSCs fabricated using typically simple and low-temperature-process technology.
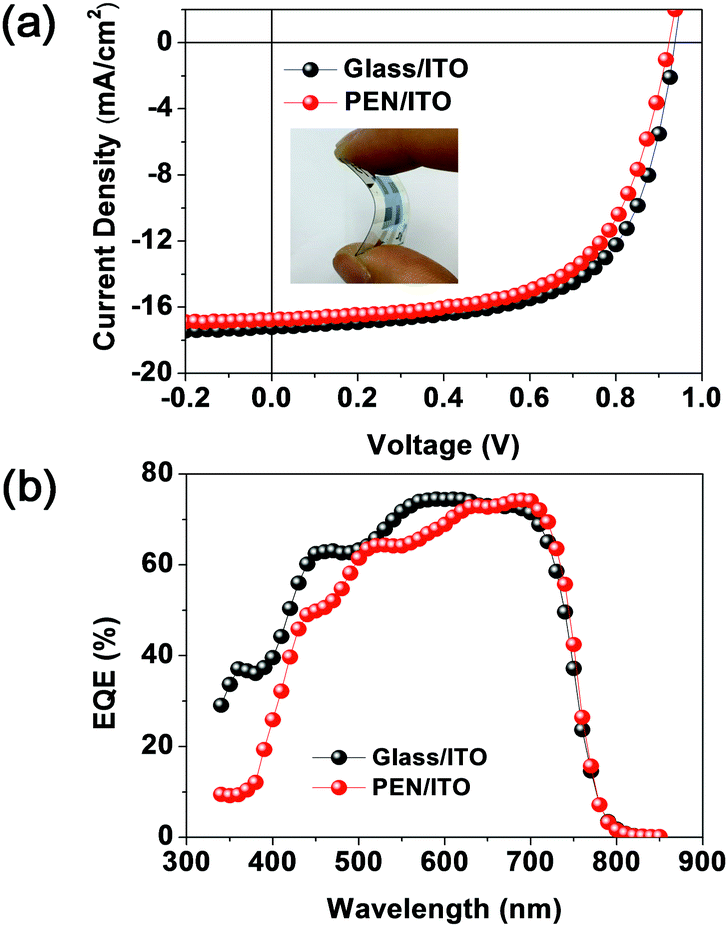 | ||
| Fig. 7 (a) J–V curves and (b) EQE spectra of P3:ITIC-based PSCs using a glass substrate and flexible PEN substrate. | ||
Conclusions
In this study, a series of terpolymers based on BDT, 3MT, and BDD were synthesized, and the effects of the contents of 3MT and BDD on the performance of the corresponding PSCs with an inverted-type device configuration were studied. Furthermore, the optical and electrochemical properties could be tuned effectively by adding a third BDD monomer into the polymer structure. Among the three terpolymers studied, the as-cast blend film of P3:ITIC (1:1 wt%) without an additive under the optimal conditions exhibited the highest PCE of 10.26% with a high Voc of 0.95 V and Jsc of 17.18 mA cm−2 in the PSC. In addition, the stability of the terpolymer-based PSCs was less affected by the regularity of the arrangement of the repeating units in the terpolymer backbone. Conclusively, more complex random terpolymers with 3MT and BDD as acceptors make it possible to produce PSCs with a high efficiency and high stability using inexpensive, time saving, and simple processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF20100020209 and NRF2018R1D1A1B07042533). This research was funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.02-2015.89. We are grateful to Pohang Accelerator Laboratory (Pohang, Korea) for allowing us to conduct the grazing incidence wide angle X-ray diffraction (GIWAXD) measurements.Notes and references
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef CAS PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423 CrossRef CAS PubMed.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- G. Zhang, K. Zhang, Q. Yin, X.-F. Jiang, Z. Wang, J. Xin, W. Ma, H. Yan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2017, 139, 2387 CrossRef CAS PubMed.
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298 CrossRef CAS PubMed.
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef CAS PubMed.
- H. Yao, L. Ye, J. Hou, B. Jang, G. Han, Y. Cui, G. M. Su, C. Wang, B. Gao, R. Yu, H. Zhang, Y. Yi, H. Y. Woo, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1700254 CrossRef PubMed.
- J. Zhua, S. Li, X. Liu, H. Yao, F. Wang, S. Zhang, M. Sun and J. Hou, J. Mater. Chem. A, 2017, 5, 15175 RSC.
- Q. Fan, Y. Wang, M. Zhang, B. Wu, X. Guo, Y. Jiang, W. Li, B. Guo, C. Ye, W. Su, J. Fang, X. Ou, F. Liu, Z. Wei, T. C. Sum, T. P. Russell and Y. Li, Adv. Mater., 2018, 30, 1704546 CrossRef PubMed.
- B. Guo, W. Li, X. Guo, X. Meng, W. Ma, M. Zhang and Y. Li, Adv. Mater., 2017, 29, 1702291 CrossRef PubMed.
- Y. Li, D. H. Lee, J. Lee, T. L. Nguyen, S. Hwang, M. J. Park, D. H. Choi and H. Y. Woo, Adv. Funct. Mater., 2017, 27, 1701942 CrossRef.
- J. Wang, W. Wang, X. Wang, Y. Wu, Q. Zhang, C. Yan, W. Ma, W. You and X. Zhan, Adv. Mater., 2017, 29, 1702125 CrossRef PubMed.
- T. Yu, X. Xu, G. Zhang, J. Wan, Y. Li and Q. Peng, Adv. Funct. Mater., 2017, 27, 1701491 CrossRef.
- W. Gao, Q. An, R. Ming, D. Xie, K. Wu, Z. Luo, Y. Zou, F. Zhang and C. Yang, Adv. Funct. Mater., 2017, 27, 1702194 CrossRef.
- C. Sun, Z. Wu, Z. Hu, J. Xiao, W. Zhao, H.-W. Li, Q.-Y. Li, S.-W. Tsang, Y.-X. Xu, K. Zhang, H.-L. Yip, J. Hou, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1784 RSC.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591 CrossRef.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170 CrossRef CAS PubMed.
- K. Kawashima, T. Fukuhara, Y. Suda, Y. Suzuki, T. Koganezawa, H. Yoshida, H. Ohkita, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 10265 CrossRef CAS PubMed.
- R. Yu, S. Zhang, H. Yao, B. Guo, S. Li, H. Zhang, M. Zhang and J. Hou, Adv. Mater., 2017, 29, 1700437 CrossRef PubMed.
- P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544 RSC.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- E. Zhou, M. Nakano, S. Izawa, J. Cong, I. Osaka, K. Takimiya and K. Tajima, ACS Macro Lett., 2014, 3, 872 CrossRef CAS.
- T. E. Kang, K.-H. Kim and B. J. Kim, J. Mater. Chem. A, 2014, 2, 15252 RSC.
- X. Guo, Q. Fan, C. Cui, Z. Zhang and M. Zhang, Acta Phys.-Chim. Sin., 2018, 34, 1279 Search PubMed.
- J. Zhang, K. Jiang, G. Yang, T. Ma, J. Liu, Z. Li, J. Y. L. Lai, W. Ma and H. Yan, Adv. Energy Mater., 2017, 7, 1602119 CrossRef.
- S. Chen, H. J. Cho, J. Lee, Y. Yang, Z.-G. Zhang, Y. Li and C. Yang, Adv. Energy Mater., 2017, 7, 1701125 CrossRef.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734 CrossRef CAS PubMed.
- G. E. Park, S. Choi, S. Y. Park, D. H. Lee, M. J. Cho and D. H. Choi, Adv. Energy Mater., 2017, 7, 1700566 CrossRef.
- D. Qian, W. Ma, Z. Li, X. Guo, S. Zhang, L. Ye, H. Ade, Z. Tan and J. Hou, J. Am. Chem. Soc., 2013, 135, 8464 CrossRef CAS PubMed.
- S. Zhang, Y. Qin, M. A. Uddin, B. Jang, W. Zhao, D. Liu, H. Y. Woo and J. Hou, Macromolecules, 2016, 49, 2993 CrossRef CAS.
- Y. Liu, S. Chen, G. Zhang, P. C. Y. Chow and H. Yan, J. Mater. Chem. A, 2017, 5, 15017 RSC.
- H. Yao, R. Yu, T. J. Shin, H. Zhang, S. Zhang, B. Jang, M. A. Uddin, H. Y. Woo and J. Hou, Adv. Energy Mater., 2016, 6, 1600742 CrossRef.
- G. E. Park, H. J. Kim, S. Choi, D. H. Lee, M. A. Uddin, H. Y. Woo, M. J. Cho and D. H. Choi, Chem. Commun., 2016, 52, 8873 RSC.
- G. E. Park, S. Choi, D. H. Lee, M. Godumala, M. A. Uddin, H. Y. Woo, M. J. Cho and D. H. Choi, J. Mater. Chem. A, 2017, 5, 663 RSC.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973 CrossRef CAS PubMed.
- T. E. Kang, H.-H. Cho, H. J. Kim, W. Lee, H. Kang and B. J. Kim, Macromolecules, 2013, 46, 6806 CrossRef CAS.
- G. E. Park, H. J. Kim, D. H. Lee, M. J. Cho and D. H. Choi, Polym. Chem., 2016, 7, 5069 RSC.
- K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk and R. A. J. Janssen, Angew. Chem., 2013, 125, 8499 CrossRef.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403 CrossRef CAS.
- Z. Li, K. Jiang, G. Yang, J. Y. L. Lai, T. Ma, J. Zhao, W. Ma and H. Yan, Nat. Commun., 2016, 7, 13094 CrossRef CAS PubMed.
- N. K. Elumalai, A. Saha, C. Vijila, R. Jose, Z. Jie and S. Ramakrishna, Phys. Chem. Chem. Phys., 2013, 15, 6831 RSC.
- F. J. Lim, A. Krishnamoorthy and G. W. Ho, ACS Appl. Mater. Interfaces, 2015, 7, 12119 CrossRef CAS PubMed.
- E. Y. Ko, G. E. Park, J. H. Lee, H. J. Kim, D. H. Lee, H. Ahn, M. A. Uddin, H. Y. Woo, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 8838 CrossRef CAS PubMed.
- S. Savagatrup, A. D. Printz, T. F. O'Connor, A. V. Zaretski, D. Rodriquez, E. J. Sawyer, K. M. Rajan, R. I. Acosta, S. E. Root and D. J. Lipomi, Energy Environ. Sci., 2015, 8, 55 RSC.
- T. Kim, J. Choi, H. J. Kim, W. Lee and B. J. Kim, Macromolecules, 2017, 50, 6861 CrossRef CAS.
- B. J. Tremolet de Villers, K. A. O’Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28, 876 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc05035j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |