
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanical improvement of calcium carbonate cements by in situ HEMA polymerization during hardening†
Barbara
Myszka
a,
Katrin
Hurle
b,
Kai
Zheng
a,
Stephan E.
Wolf
 *cd and
Aldo R.
Boccaccini
*ad
*cd and
Aldo R.
Boccaccini
*ad
aInstitute of Biomaterials, Friedrich-Alexander University Erlangen-Nürnberg (FAU), Cauerstrasse 6, 91058 Erlangen, Germany. E-mail: aldo.boccaccini@ww.uni-erlangen.de
bGeoZentrum Nordbayern – Mineralogy, Friedrich-Alexander University Erlangen-Nürnberg (FAU), Schlossgarten 5a, 91054 Erlangen, Germany
cInstitute of Glass and Ceramics, Friedrich-Alexander University Erlangen-Nürnberg (FAU), Martensstraße 5, 91058 Erlangen, Germany. E-mail: stephan.e.wolf@fau.de
dInterdisciplinary Center for Functional Particle Systems (FPS), Friedrich-Alexander University Erlangen-Nürnberg (FAU), 91058 Erlangen, Germany
First published on 25th April 2019
Abstract
Calcium carbonate cements are a promising alternative for calcium phosphate cements. Due to the higher solubility of calcium carbonate compared to apatite, it is expected to have an improved biodegradation rate; a clear advantage for prospective biomedical applications in a clinical context. As of yet, the poor mechanical performance of calcium carbonate cements precludes their exploitation as a bioceramic. Herein, we report a mechanically enhanced calcium carbonate cement, formulated of vaterite and magnesium-doped amorphous calcium carbonate, which was strengthened by in situ polymerization of incorporated 2-hydroxyethylmethacrylate (HEMA) during setting in simulated body fluid (SBF). This approach allowed us to generate a pronouncedly strengthened polymer-ceramic composite whose mechanical properties can be tuned by the monomer fraction. Varying cement compositions were characterized with respect to their mechanical performance, morphology, and quantitative phase composition. HEMA polymer reinforcement strengthened the calcium carbonate cement distinctly in four-point bending test while, in stark contrast, the brittleness of the polymer-free cement simply made such tests impossible. Our results demonstrate that the chosen polymer-enforcement strategy is an efficient and straightforward approach and allows for tuning the mechanical properties of an otherwise brittle and thus unusable calcium carbonate cement towards biomedical application.
Introduction
The development of biomaterials, which are suitable for filling bone defects and which feature tailored properties concerning biodegradability, osteoconductivity, and osteoinduction, is a prevailing challenge in the field of orthopaedic and dental surgery. Hetero- or autografts or implants of natural origin (e.g. calcareous corals) are often used,1,2 however, they come with major drawbacks, such as biological variability and risk of viral or bacterial contaminations. In a warming world, we even may have to face an increasing uncertainty in supply when it comes to corals.3–5 To address these problems which are associated with biogenic calcareous biomaterials, suitable synthetic materials are needed but their synthesis and design protocols have to meet the demands and requirements of clinical applications. In this context, fast-setting ionic cements are an attractive material class for bone filling. In the last several years, a range of calcium phosphate (CaP) cements have been developed which are suitable as a bone replacement material due to their biocompatibility and bioactivity.6 However, it is still a challenge to further augment the rates of bioresorption of such cements to improve bone formation and regeneration.7–9The biological resorption of bone replacement materials depends on the solubility of their constituting mineral phases. Since calcium carbonate shows higher solubility than apatite, turning from calcium phosphate-based to calcium carbonate-based cements is an auspicious approach to manufacture cements with improved and tuneable biodegradation rates.10,11 Cements composed of a blend of calcium carbonate and calcium phosphate have already been tested,12,13 but only a marginal number of studies focussed on cements which contain calcium carbonate as the only mineral component.14 Exploitation of such calcareous cements is, as of yet, impaired by their inferior mechanical properties. In this contribution, we explore a strategy to augment the mechanical performance of calcium carbonate cements by in situ polymerization during setting of the cement.
Calcium carbonate occurs in three anhydrous crystalline phases: calcite, aragonite and vaterite. It also exists in form of an extremely reactive and unstable amorphous calcium carbonate (ACC) which plays a fundamental role in biogenic and biomimetic calcification processes.10,15–18 In the biosphere, calcium carbonate is near-to omnipresent and is precipitated by a wide range of organisms in which it serves as load-bearing skeleton or damage-tolerant weapon. In these biogenic ceramics, the brittleness of pure calcium carbonate is overcome by a hybrid and composite material approach which grants strength and toughness at the same time to the biomineral. For more than two decades, nacre- and coral-based substitutes are already exploited as bioactive and biocompatible biomaterials, e.g. as powders and pseudomorphically transformed porous ceramics or in the form of mineral-enhanced gels.2,19,20In vivo studies have demonstrated that calcium carbonate-based bioceramics derived from corals promote bone ingrowth and resorption.21,22 However, the mechanical properties of such coral-based grafts are poor.3 Moreover, sintering of calcium carbonate bioceramics is challenging since calcium carbonate requires very low sintering temperatures due to its low thermal decomposition temperature.23 In this context, calcium carbonate cements are promising for designing calcareous bioceramics via low-temperature routes.24 Combes and co-workers have recently studied the feasibility of preparing an initially biphasic calcium carbonate cement.14 Employing one highly reactive amorphous phase and a metastable crystalline calcium carbonate phase, this first calcareous cement recrystallized – fully in agreement with Ostwald step rule – during setting.14 The calcium carbonate cement was shown to be non-toxic for human osteoprogenitor cells suggesting good cytocompatibility. However, the inferior mechanical properties of their cement effectively impeded clinical application; the compressive strength of the cement was below 13 MPa. To push the mechanical properties of calcium carbonate cements into the range at which a clinical application is feasible and reasonable, we take inspiration from biominerals and reinforce the brittle cement with a ductile polymeric phase which shall provide similar toughness like the intracrystalline biopolymers in biominerals. This method is already established strategy in the field of calcium phosphate cements.25,26 Here, we add a monomeric substance, 2-hydroxyethylmethacrylate (HEMA), to the unsettled cement powder and allow polymerization during cement hardening.25 HEMA is one of the most common polymers in biomedical applications which renders it suitable for this feasibility study.27
The work presented herein studies a new type of bone cement solely made of calcium carbonate serving as the inorganic/ceramic majority component. The quality and purity of the initial calcium carbonate powders was characterized by a series of techniques, i.e., X-ray diffraction (XRD), Fourier transform infrared (FTIR) spectroscopy, Raman spectroscopy, and scanning electron microscopy (SEM). The setting of calcium carbonate cement was triggered by the presence of water. Simulated body fluid was used to account for the distinct ion composition in human body fluids and to assure comparable conditions to hardening in vivo. The mechanical characteristics of the cement were enhanced by in situ polymerization of HEMA during setting of the cement paste, generating a mechanical stable polymer-ceramic composite. We compared the mechanical properties and quantitative phase composition of the HEMA-enhanced cements to a non-reinforced and polymer-free calcium carbonate cement with otherwise identical composition and hardening conditions.
Materials and methods
Synthesis of CaCO3 powders
Cement and powder characterization
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif)
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
The absolute quantities of the crystalline CaCO3 phases were then determined by the G-factor method, an external standard method. The G-factor, a device specific calibration constant, is obtained from measurement of an appropriate external standard according to eqn (1).33
 | (1) |
 | (2) |
Results and discussion
Powder synthesis and characterization
In order to prepare the calcium carbonate-based cements, phase-pure calcium carbonate polymorphs were freshly synthesized. Phase-pure vaterite was synthesized by a double-composition technique which yielded polycrystalline and ellipsoidal aggregates with particle sizes ranging from 1 μm to 6 μm and a diameter of about 2.5 μm.36–38 At higher magnification, micrographs show that these ellipsoidal particles are composed of aligned needle-like nanocrystallites with a mean length of 200 nm (Fig. 1A). Further analytical details, especially on the phase purity and composition of the synthesized starting material is given in the ESI† (Fig. S1). | ||
| Fig. 1 Scanning electron micrographs of synthesized (A) vaterite and (B) Mg-doped amorphous calcium carbonate (ACC). | ||
Pure amorphous calcium carbonate (ACC) is unstable and transforms to a crystalline phase when brought into contact with water so fast that it is not suitable for a reasonable cement formulation. Hence, we adjusted the rate of ACC phase transformation by using magnesium as phase stabilizer and crystallization inhibitor. Mg-ACC powders were prepared with varying magnesium content, i.e., with 10% of Mg (ACC_10Mg), with 20% of Mg (ACC_20Mg) and 40% of Mg (ACC_40Mg). The morphology of the Mg-ACC powder varies only marginally as a function of magnesium content (Fig. 1B). All variants of Mg-doped ACC consist of spheroidal and aggregated nanoparticles whose average size ranges from 120 to 200 nm and are X-ray amorphous (Fig. S2A, ESI†). After preparation, Mg-ACC slowly recrystallizes to calcite even when stored dry, as evidenced by a representative XRD pattern recorded 7 days after a sample of ACC_10Mg was prepared (Fig. S2B, ESI†). After 31 days of storage in a desiccator, this specific sample had completely transformed to crystalline calcite whereas samples of ACC_20Mg and ACC_40Mg were still amorphous (Fig. S2C, ESI†). ICP measurements showed that the rate of Mg2+ incorporation does not mirror the exact Mg/Ca ratio of the mother solution but increases with increasing Mg concentration (Fig. S2D, ESI†). A further DTA-TGA is given in the ESI† which further corroborates the purity of the synthesized phase (Fig. S3, ESI†).
Cement preparation and characterization
Cement pastes were prepared by mixing the powders of Mg-doped ACC and vaterite in a ratio of 1 to 2 with an appropriate amount of liquid phase (SBF) to give a liquid to powder ratio (LPR) of 1 mL g−1. To mechanically enhance the cements by in situ HEMA polymerization during cement setting, 2-hydroxyethylmethacrylate (HEMA) was added in varying amounts (10, 25, and 50 wt%) to the SBF solution. Polymerization was triggered by addition of APS and TEMED to the cement mixture. We followed the reaction in situ by isothermal calorimetry which demonstrated that the hardening reaction proceeded very rapidly for both the reference sample in absence of HEMA and a sample containing 50 wt% HEMA. Major heat release is only observed in the first hour of hardening (Fig. 2A) and, for both samples, a single and sharp heat flow maximum occurred already after some few minutes of reaction time. This pronounced heat maximum was significantly higher for the polymer-containing sample, for which the over-all reaction time also lasted distinctly longer. Thus, the total heat release during setting and hardening was significantly higher for the samples containing 50 wt% HEMA in comparison with those without polymer which we attribute to an additional heat release as a result of polymerization.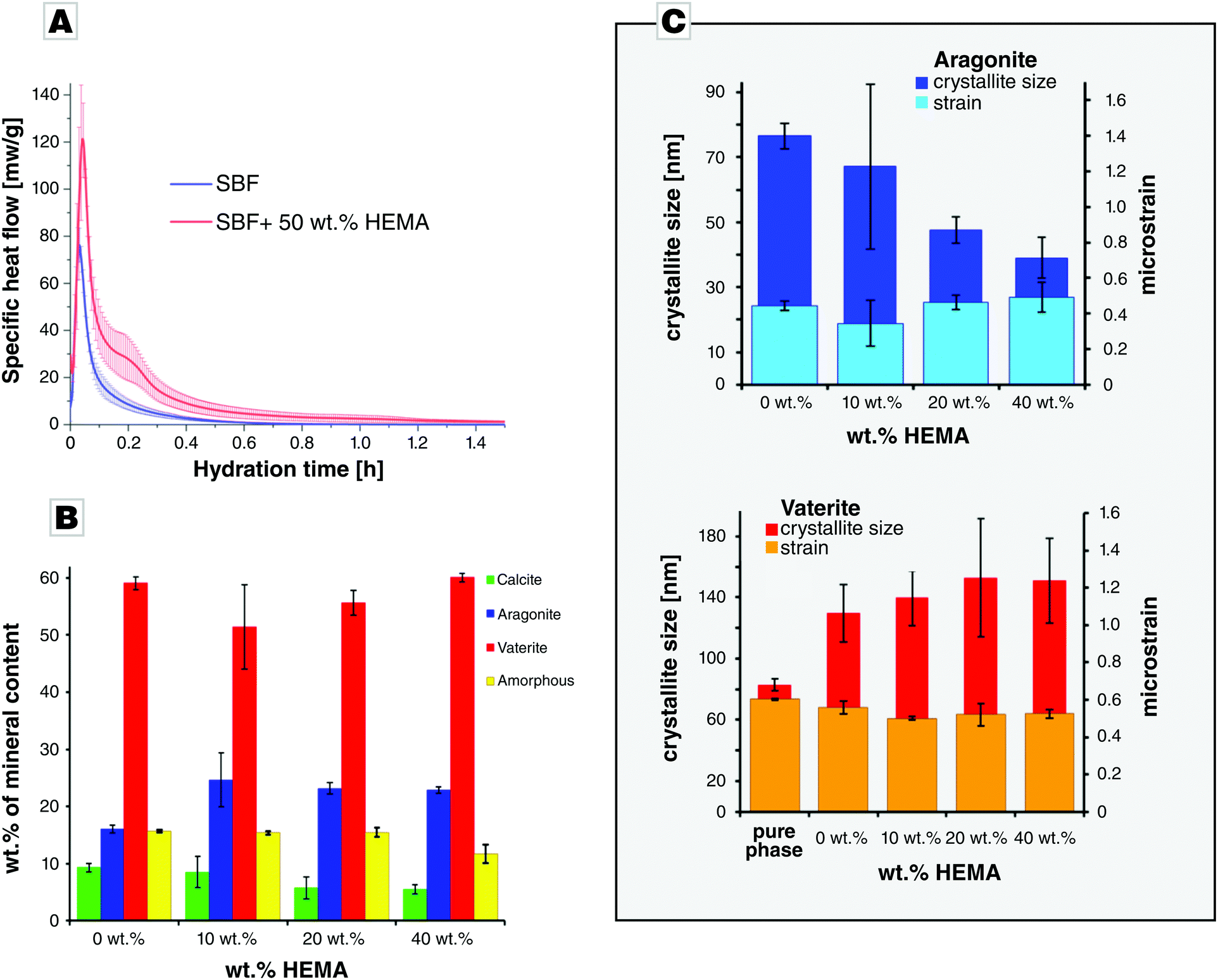 | ||
Fig. 2 Characterization of the cement setting. (A) Calorimetry of a cement prepared of ACC_Mg20/vaterite in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, prepared in absence and in presence of 50 wt% HEMA; the mean values of three independent measurements are presented and the error bars give the standard deviation. (B) Phase analysis of cements (ACC_Mg20/vaterite = 1/2) after 24 h, as a function of HEMA content; mean values of three independently prepared measurements are shown. (C) Crystallite sizes and microstrain analysis of aragonite and vaterite in cements (ACC_Mg20/V = 1/2, LPR = 1 mL g−1) prepared at different HEMA concentrations after 24 h of hardening at 37 °C; the mean values of three independently prepared measurements are shown. :2, prepared in absence and in presence of 50 wt% HEMA; the mean values of three independent measurements are presented and the error bars give the standard deviation. (B) Phase analysis of cements (ACC_Mg20/vaterite = 1/2) after 24 h, as a function of HEMA content; mean values of three independently prepared measurements are shown. (C) Crystallite sizes and microstrain analysis of aragonite and vaterite in cements (ACC_Mg20/V = 1/2, LPR = 1 mL g−1) prepared at different HEMA concentrations after 24 h of hardening at 37 °C; the mean values of three independently prepared measurements are shown. | ||
In all hardened cement samples investigated by XRD, vaterite was the main phase, while calcite and aragonite were present in smaller amounts along with traces of sodium chloride present in all samples. G-Factor quantification indicated the presence of a residual amorphous fraction. In order to compare the changes of CaCO3 phase composition as a function of HEMA content, the data was normalized to the sum of carbonate phases, including the amorphous content (Fig. 2B). While the vaterite content was practically identical for all samples within the error range, the amount of aragonite was significantly higher for the HEMA containing samples, compared to the reference. Accordingly, calcite content significantly decreased in case of 25% HEMA and 50% HEMA, compared to the reference. The normalized amorphous content was nearly identical to the reference except for 50% HEMA, which had a slightly smaller amorphous fraction. The crystallite size of aragonite decreased in presence of HEMA, compared to the reference samples hardened in absence of HEMA (Fig. 2C). In contrast to aragonite, the crystallite size of vaterite increased slightly in presence of HEMA but was unaffected by a change in HEMA content. The initial crystallite size of vaterite was lower than that of the vaterite present in the hardened cement samples, while its microstrain decreased during hardening, indicating crystal growth. The crystallite size of calcite exceeded the range in which reliable interpretation on basis of the Scherrer equation is possible.
For calcite, both the cell parameters for the a- and c-axes were significantly larger in case of the calcite standard, compared to the calcite that formed from reaction of the carbonate cements (Fig. S4A, ESI†). For aragonite, no variation in cell parameters was observed (Fig. S4B, ESI†). The cell parameters for the a- and b-axes of vaterite did not vary significantly, while the c parameter was significantly higher in case of pure vaterite (Fig. S4C, ESI†). The shorter cell parameters of calcite formed during hardening might indicate that Mg was incorporated into the calcite lattice. Mg2+ has a smaller ionic radius of 0.72 Å compared to Ca2+ with 1.00 Å (both values given for six fold coordination)39 and thus would lead to a reduction in cell parameters. Since aragonite did not show a change in cell parameters, it is plausible to assume that Mg2+ was incorporated into the calcite lattice or remained in the residual amorphous phase. ACC phases are well-known to be stabilized by Mg incorporation which also accounts for the unexpected long-time stability of a residual amorphous phase.40 Moreover, magnesium is known to impede calcite growth while favouring aragonite formation which explains the presence of aragonite in the hardened cements.41,42
From the phase analysis combined with the results of the crystal size determination, we can infer that calcite and aragonite formation was driven by vaterite transformation only to a minor extend. Instead, ACC redissolution mainly fed the formation and growth of aragonite and calcite. The slight increase in the average crystallite size of vaterite indicates that the vaterite fraction of lower crystallinity has transformed to either aragonite or calcite, leading to an overall increase of the average crystallite size. From the cement's phase composition, one may conclude that the polymer addition slightly influences the crystallization pathway. Higher polymer content seems to impair formation of calcite; higher amounts of HEMA lead to a preferred aragonite formation.
The morphological evolution in time of samples which contain 50 wt% of HEMA is shown in Fig. 3. It demonstrates that, in the first 6 hours of hardening, the cement appears to be composed of poorly crystalline phases. After 6 h of hardening, rhombohedral calcite and ellipsoidal vaterite crystals appeared; thus, the overall fraction of crystalline phases increased with time. EDS analysis showed that a calcareous precipitate had formed (Fig. S5, ESI†).
 | ||
| Fig. 3 SEM micrographs of fracture surfaces of cements (ACC_Mg20:V = 1:2, LPR = 1 mL g−1, 50 wt% HEMA) after different times of setting. The samples were gently crushed so that individual particles and crystallites can be easily distinguished. Since the polymerized HEMA produces only a low contract in comparison to the mineral matrix, the polymer matrix is barely visible. | ||
The compressive strength of the calcium carbonate cements (Fig. 4A) increased with HEMA content. Reference samples, which contained no polymer and were hardened for 6 hours were still too brittle to perform any measurements; only reference samples hardened for longer periods of time could be evaluated. Even after a hardening period of 14 days, the compressive strength of the reference samples was very weak (below 1 MPa). In contrast, the addition of 10 and 25 wt% of HEMA already improved the compressive strength of the calcium carbonate cements considerably. For hardening periods not longer than 6 hours, the determined compressive strength was below 1 MPa, and, after 1 day of setting, the samples with 10 wt% HEMA already reached a compressive strength of 2 MPa which further increased to 4 MPa after 14 days. By increasing the content of HEMA to 25 wt%, the compressive strength was essentially increased by a twofold; after 14 days a compressive strength of approx. 9 MPa was determined. The addition of 50 wt% HEMA strengthened the cement most pronouncedly. Already after 2 hours of setting, the compressive strength was already at 14 MPa, and it slowly increased reaching a value of about 25 MPa within a week's time. After two weeks of hardening, the compressive strength of the enhanced cement rose to 33 MPa. Overall, it seems clear that strengthening by addition of 50 wt% HEMA polymer to the cement remarkably improves the compressive strength of our calcium carbonate cement.
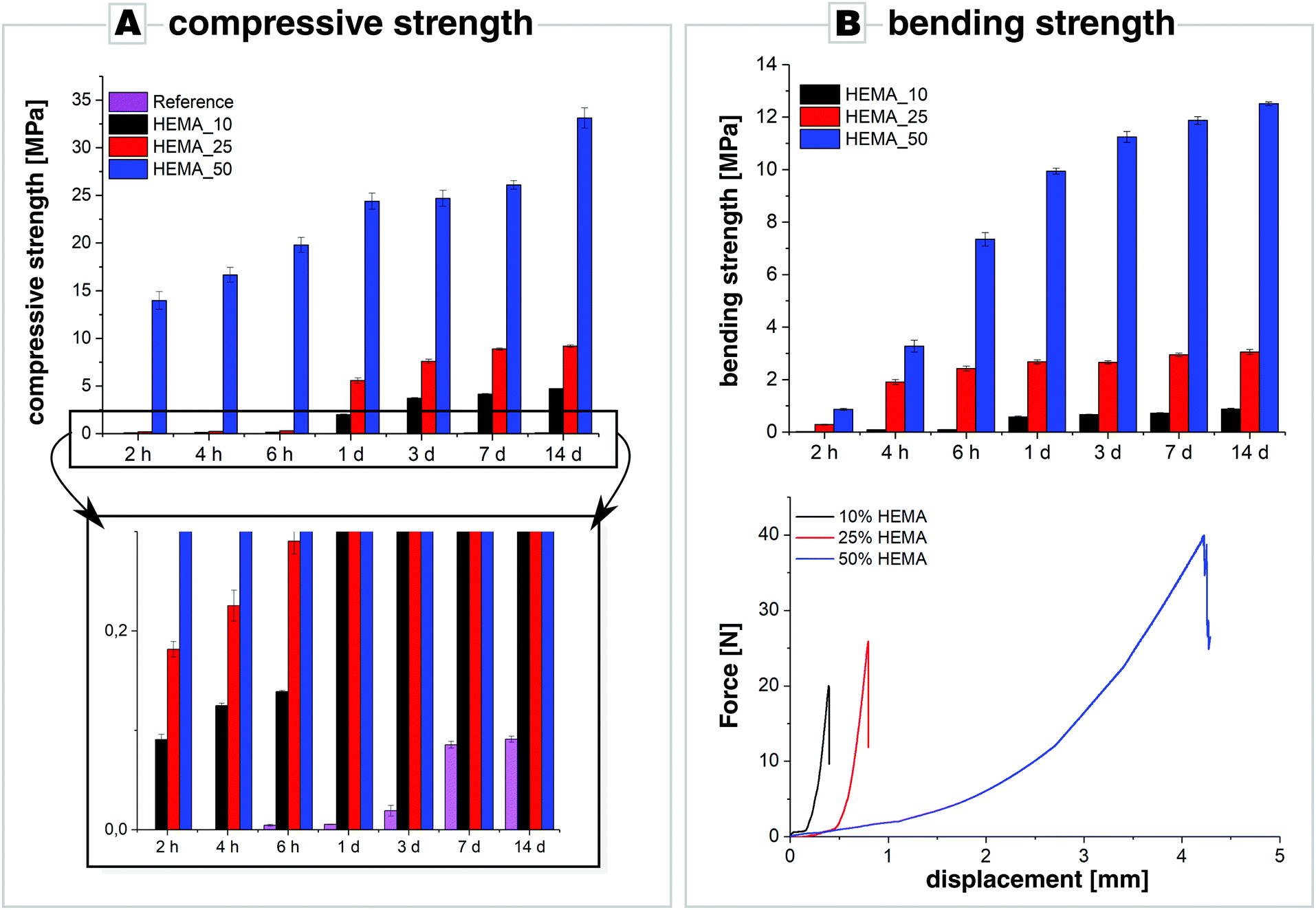 | ||
| Fig. 4 (A) Compressive strength and (B) four-point bending tests along with force displacements curves of cements (ACC_20Mg/vaterite = 1/2, LPR = 1 mL g−1) with various contents of HEMA (10, 25, 50 wt%) as a function of hardening time. For the most part, reference samples were too brittle to perform measurements. | ||
The mechanical performance was further tested by means of four-point bending tests and showed that incorporation of HEMA improved also the cement's bending strength at the same time (Fig. 4B). HEMA-free reference samples were too brittle to perform such measurements, whereas samples enhanced with 10 wt% HEMA already showed a bending strength of approx. 0.1 MPa after 2 hours of hardening which further increased to 0.9 MPa after 14 days of hardening. Addition of 25 wt% HEMA further enhanced the cement's bending strength: after 2 hours of setting, the bending strength was 0.3 MPa and increased by a six-fold within the next two hours, to reach a value of about 2.5 MPa after 6 hours of hardening. Increasing the HEMA content even further to 50 wt%, the four-point bending strength rose from about 1 MPa after 2 hours of hardening, to about 13 MPa after 14 days of hardening. While cements with a HEMA content of 10 and 25 wt% still showed the characteristic brittle fracture of ceramics and a maximum bending deformation of the samples of less than 0.8 mm (Fig. 4B), samples with 50 wt% of polymer exhibited pronounced flexibility as bending up to 4 mm was possible before fracture (see Fig. S6 in the ESI†). In fact, Christel and co-workers obtained similar bending strength values by adding 50 wt% HEMA to α-tricalcium phosphate cements. These cements showed a maximum bending displacement of 0.4–1.5 mm.26 In contrast, our calcium carbonate cement with 50 wt% polymer was much more flexible, as bending was possible for 2–4 mm before the samples underwent fracture. This might be caused by the difference in crystal morphology as elongated aragonite crystals may reinforce the cement by fibre toughening and fibre pull-out. The large particle size distribution, which can be seen in Fig. 3 in which agglomerations of relatively large particles can be spotted, might have a similar effect by causing tortuous crack paths further augmenting the mechanical response in four-point-bending tests.
The actual role and mechanisms, by which the polymer reinforces such cements, still have to be analysed in detail. Our calorimetric analysis already an insight on how the toughening and strengthening effect might take place although HEMA polymers do not strongly bind to mineral surfaces as they lack anionic side chains. The polymerization of HEMA quickly takes place at an early stage of the cement setting, and thus the morphology of the formed HEMA polymer matrix fills the intercrystalline volume and voids of the cement. After completion of the polymerization, the setting reaction still continues so that the formed polymer strands are incorporated into the crystals while Ostwald-ripening, phase transformation, and crystal growth further proceeds. Upon loading, the intracrystalline polymer matrix can then augment the mechanical response of the reinforced cement by a range of mechanisms, e.g., by fibre pull-out, ligament formation upon crack extension, or crack bridging mechanisms and other mechanisms which enhance stress distribution.43,44 In order to optimize the mechanical response of the polymer-enhanced cements, detailed and ultrastructural analyses have to be carried out in future. For instance, we expect that by crosslinking of the HEMA polymer matrix and by further introduction of optimized mineral binding sites into the polymer matrix, the mechanical properties of our cements can be further increased.
Conclusions
We developed a non-brittle Mg-doped polymer-reinforced cement with appealing mechanical properties which contains only calcium carbonate as the mineral component. By taking inspiration from biogenic ceramics, we addressed the issue of the poor mechanical performance of calcium carbonate-based cements which made their broad exploitation impossible although calcareous cements represent attractive and auspicious alternatives to CaP cements due to a higher expected biodegradation rate. This feasibility study involved also the optimization of the phase transformation rate of the highly reactive component amorphous calcium carbonate by stabilization via Mg-doping.Since the mechanical performance of the cement was significantly improved by adding 50 wt% of HEMA polymer, the cement–polymer composites may be suitable even for minimally invasive injection to cure bone defects. By optimizing the cement's particle size distribution, as well as the liquid to powder ratio, further improvements of the system may be achieved. For further evaluation of such polymer-enhanced calcium carbonate cements as new bioceramics, further studies to characterize cell compatibility are envisaged.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. B. and B. M. received support by the European ITN NanoHeal (Grant Agreement #642976). S. E. W. acknowledges financial support by an Emmy Noether starting grant issued by the German Research Foundation (DFG, no. WO1712/3-1)References
- C. Lorin, A. M. Melin, J. P. Chenu, A. Perromat and G. Déléris, Appl. Spectrosc., 2004, 58, 332–337 CrossRef CAS PubMed
.
- G. Guillemin, J. L. Patat, J. Fournie and M. Chetail, J. Biomed. Mater. Res., 1987, 21, 557–567 CrossRef CAS PubMed
- J. Vuola, T. Böhling, J. Kinnunen, E. Hirvensalo and S. Asko-Seljavaara, J. Biomed. Mater. Res., 2000, 51, 117–122 CrossRef CAS PubMed
- O. Hoegh-Guldberg, J. Geophys. Res.: Oceans, 2005, 110, 1–11 CrossRef
- F. Fujian, J. Fuzhou Teach. Coll., 2002, 5–8 Search PubMed
- W. Habraken, P. Habibovic, M. Epple and M. Bohner, Mater. Today, 2016, 19, 69–87 CrossRef CAS
- C. H. Tsai, R. M. Lin, C. P. Ju and J. H. Chern Lin, Biomaterials, 2008, 29, 984–993 CrossRef CAS PubMed
- R. O’Neill, H. O. McCarthy, E. B. Montufar, M. P. Ginebra, D. I. Wilson, A. Lennon and N. Dunne, Acta Biomater., 2017, 50, 1–19 CrossRef PubMed
-
Z. Sheikh, M. N. Abdallah, A. A. Hanafi, S. Misbahuddin, H. Rashid and M. Glogauer, Materials, 2015, 8, 7913–7925 Search PubMed
- L. Brecevic and A. E. Nielsen, J. Cryst. Growth, 1989, 98, 504–510 CrossRef CAS
- J. C. Heughebaert and G. H. Nancollas, J. Phys. Chem., 1984, 88, 2478–2481 CrossRef CAS
- I. Khairoun, M. G. Boltong, F. C. M. Driessens and J. A. Planell, J. Biomed. Mater. Res., 1997, 38, 356–360 CrossRef CAS PubMed
- I. Khairoun, M. G. G. Boltong, F. C. M. C. M. Driessens and J. A. A. Planell, Biomaterials, 1997, 18, 1535–1539 CrossRef CAS PubMed
- C. Combes, B. Miao, R. Bareille and C. Rey, Biomaterials, 2006, 27, 1945–1954 CrossRef CAS PubMed
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS PubMed
-
S. E. Wolf and L. B. Gower, in New Perspectives on Mineral Nucleation and Growth, ed. M. Kellermeier, A. van Driessche, L. Benning and D. Gebauer, Springer International, Cham, Switzerland, 2017, pp. 43–75 Search PubMed
- J. Ihli, W. C. Wong, E. H. Noel, Y.-Y. Kim, A. N. Kulak, H. K. Christenson, M. J. Duer and F. C. Meldrum, Nat. Commun., 2014, 5, 3169 CrossRef PubMed
- J. Harris, I. Mey, M. Hajir, M. Mondeshki and S. E. Wolf, CrystEngComm, 2015, 17, 6831–6837 RSC
- A. Piattelli, G. Podda and A. Scarano, Biomaterials, 1997, 18, 623–627 CrossRef CAS PubMed
- M. Roudier, C. Bouchon, J. L. Rouvillain, J. Amédée, R. Bareille, F. Rouais, J. Ch. Fricain, B. Dupuy, P. Kien, R. Jeandot and B. Basse-Cathalinat, J. Biomed. Mater. Res., 1995, 29, 909–915 CrossRef CAS PubMed
- F. Souyris, C. Pellequer and C. Payrot, J. Oral Maxillofac. Surg., 1985, 13, 64–69 CAS
- W. R. Walsh, P. J. Chapman-Sheath, S. Cain, J. Debes, W. J. M. Bruce, M. J. Svehla and R. M. Gillies, J. Orthop. Res., 2003, 21, 655–661 CrossRef CAS PubMed
- A. Lucas-Girot, P. Langlois, J. C. Sangleboeuf, A. Ouammou, T. Rouxel and J. Gaude, Biomaterials, 2002, 23, 503–510 CrossRef CAS
- K. Hosoi, T. Hashida, H. Takahashi, N. Yamasaki and T. Korenaga, J. Mater. Sci. Lett., 1996, 15, 812–814 CrossRef CAS
- K. Hurle, T. Christel, U. Gbureck, C. Moseke, J. Neubauer and F. Goetz-Neunhoeffer, J. Mater. Sci.: Mater. Med., 2016, 27, 1–13 CrossRef CAS PubMed
- T. Christel, M. Kuhlmann, E. Vorndran, J. Groll and U. Gbureck, J. Mater. Sci. Mater. Med., 2013, 24, 573–581 CrossRef CAS PubMed
- I. N. Savina, V. Cnudde, S. D’Hollander, L. Van Hoorebeke, B. Mattiasson, I. Y. Galaev and F. Du Prez, Soft Matter, 2007, 3, 1176–1184 RSC
- Y. Politi, D. R. Batchelor, P. Zaslansky, B. F. Chmelka, J. C. Weaver, I. Sagi, S. Weiner and L. Addadi, Chem. Mater., 2010, 22, 161–166 CrossRef CAS
- E. N. Maslen, V. A. Streltsov, N. R. Streltsova and N. Ishizawa, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 929–939 CrossRef
- A. D. Negro and L. Ungaretti, Am. Mineral., 1971, 56, 768–772 Search PubMed
-
E. Mugnaioli, I. Andrusenko, T. Schüler, N. Loges, R. E. Dinnebier, M. Panthöfer, W. Tremel and U. Kolb, Angew. Chem., Int. Ed., 2012, 51, 7041–7045 Search PubMed
- W. E. Nickels, J. E. Fineman and M. A. Wallace, J. Phys. Chem., 1949, 53, 625–628 CrossRef PubMed
- D. Jansen, C. Stabler, F. Goetz-Neunhoeffer, S. Dittrich and J. Neubauer, Powder Diffr., 2011, 26, 31–38 CrossRef CAS
-
S. Hall and B. Mcmahon, International Tables for Crytallography, Vol. C Mathematical, Physical and Chemical Tables, 3rd edn, 2004 Search PubMed
- S. T. Bergold, F. Goetz-Neunhoeffer and J. Neubauer, Cem. Concr. Res., 2015, 76, 202–211 CrossRef CAS
- G. T. Zhou, J. C. Yu, X. C. Wang and L. Z. Zhang, New J. Chem., 2004, 28, 1027–1031 RSC
- Y. Levi-Kalisman, S. Albeck, A. Brack, S. Weiner and L. Addadi, Chem. – Eur. J., 1998, 4, 389–396 CrossRef
- Y. Y. Wang, Q. Z. Yao, H. Li, G. T. Zhou and Y. M. Sheng, Cryst. Growth Des., 2015, 15, 1714–1725 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, J. Cryst. Growth, 2003, 254, 206–218 CrossRef CAS
- M. M. Reddy and G. H. Nancollas, J. Cryst. Growth, 1976, 35, 33–38 CrossRef CAS
- K. J. Davis, P. M. Dove and J. J. J. De Yoreo, Science, 2000, 290, 1134–1137 CrossRef CAS PubMed
- F. F. Barthelat, Philos. Trans. R. Soc., A, 2007, 365, 2907–2919 CrossRef CAS PubMed
- M. Mirkhalaf, A. K. Dastjerdi and F. Barthelat, Nat. Commun., 2014, 5, 3166 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tb00237e |
| This journal is © The Royal Society of Chemistry 2019 |