
A phosphorus and carbon composite containing nanocrystalline Sb as a stable and high-capacity anode for sodium ion batteries†
Miao
Zhang
ab,
Liuzhang
Ouyang
 *ab,
Min
Zhu
a,
Fang
Fang
*c,
Jiangwen
Liu
a and
Zongwen
Liu
*d
*ab,
Min
Zhu
a,
Fang
Fang
*c,
Jiangwen
Liu
a and
Zongwen
Liu
*d
aSchool of Materials Science and Engineering, Guangdong Provincial Key Laboratory of Advanced Energy Storage Materials, South China University of Technology, Guangzhou, 510641, PR China. E-mail: meouyang@scut.edu.cn
bChina-Australia Joint Laboratory for Energy & Environmental Materials, Key Laboratory of Fuel Cell Technology of Guangdong Province, Guangzhou, 510641, PR China
cDepartment of Materials Science, Fudan University, Shanghai, 200433, PR China
dSchool of Chemical and Biomolecular Engineering, The University of Sydney, NSW 2006, Australia
First published on 2nd December 2019
Abstract
Sodium ion batteries are a potential alternative to lithium ion batteries due to the low cost and natural abundance of sodium. In this study, we demonstrate the synthesis of a ternary Sb/P–C composite by using facile discharge plasma-assisted milling (P-milling) to combine Sb and high theoretical specific capacity red phosphorus (RP). The high hardness Sb particles facilitated the refinement of the P particles and exfoliated expanded graphite during the P-milling process, while the rapid heating by plasma promoted the refinement of Sb and P particles. Ultimately, the crystalline Sb nanograins were well-dispersed in the amorphous P and C matrix, and the P and C components were connected by the P–C bonds. This unique structure ensures a strong electrode structural integrity and ultra-fast electron transport during cycling. As a result, the Sb/P–C composite delivered a high average initial coulombic efficiency of 73.5%, and a reversible capacity of 596 mA h g−1 after 300 cycles at 1.0 A g−1, a very impressive performance among the reported P-based materials. The present approach provides a new strategy for the preparation of anode materials of high capacity and long cycle life for sodium-ion batteries and other energy storage systems.
1. Introduction
As a prospective component for large scale energy storage devices, sodium-ion batteries (SIBs) have recently received extensive attention. However, compared with Li+, larger and heavier Na+ (with an ion radius of 0.98 Å) makes it difficult for smooth insertion/extraction in anode materials of SIBs.1–3 Thus, it is imperative to design new Na-based anode materials with high capacity and long cycle life. To date, a series of Na-storage anode materials, including alloy-type materials (Ge, Sn, P, Sb etc.),4–8 conversion reaction materials (Fe2O3, CuO, MoS2, etc.),9–12 and carbonaceous materials (hard carbon, doped carbon, etc.), have been reported.13–15 Among all these anode materials, red phosphorus (P) is a very promising candidate because it has the highest ever known theoretical specific capacity (2596 mA h g−1 with Na3P),16 but it suffers from exceptional capacity fading because of its intrinsic low electrical conductivity (∼10−14 S cm−1) and huge volume expansion (∼490%) during the sodiation process.17 Thus, many studies have devoted to alloying red P with conductive metals to stabilize P and improve the electrical conductivity of the electrode.18–20 However, most of the conductive elements that have been used so far (Fe, Co, Ni, etc.) are inactive for Na-storage, resulting in low specific capacities of the electrode materials.19–22 On this account, Sn–P compounds have attracted great interest as both Sn and P are active elements and the intermediate products can synergetically facilitate the sodiation/desodiation processes. As reported, Sn4P3 showed a reversible capacity of 718 mA h g−1 at 0. 1 A g−1, and SnP3@C exhibited a high reversible capacity of 810 mA h g−1 at 0.15 A g−1.18,23 However, they are limited by poor cycling stability because of server volume variation (520%) and coarsening of Sn during cycling. Thus, it is necessary to explore new P-based materials with high specific capacity and good structural stability during cycling.The element Sb is an attractive anode material for SIBs due to its high theoretical specific capacity (660 mA h g−1 with Na3Sb) and a suitable reaction potential (∼0.5–0.8 V vs.Na+/Na).24 In addition, compared with Sn, its volume expansion (390%) during the sodiation process is much smaller.25 Recently, Darwiche et al. reported that pure micrometric Sb could be directly used as the anode material in SIBs, maintaining a capacity close to 600 mA h g−1 at C/2 over 160 cycles.7 Thus, combining Sb and P to form a Sb/P composite is believed to be a good approach, but it still faces the problem of volume expansion as both components accompany huge volume change during the sodiation process. In view of this, carbon was introduced into the composite as the matrix to accommodate the volume changes of Sb and P and also to enhance the electrical conductivity of the composite.16,17 To achieve a better dispersion of Sb and P in the carbon matrix, it is very crucial to choose a suitable carbon source and an efficient preparation method for making the composite. Our previous work demonstrated that expanded graphite (EG) was an excellent carbon source due to its high specific surface area and that it can be easily exfoliated by discharge plasma-assisted milling (P-milling).26,27 Compared with the conventional mechanical milling, P-milling is a very novel and high efficient ball-milling method. By introducing plasma in the milling process, both the stress effect and the heat effect could be realized by the impact stress of the milling and the rapid heating of the plasma. Through this approach, a series of active materials of carbon composites have been synthesized as electrode materials.26–28
Here we report the successful synthesis of a Sb and amorphous P–C composite by one-step P-milling using Sb, P and EG as raw materials. The crystalline Sb grains were well-dispersed in the amorphous P–C matrix. When sodium was inserted, the well-dispersed intermediate discharge/charge products of antimony nanoparticles acted as conducting pathways for activating the reversible Na storage reaction of the P component. Meanwhile the P–C bonds formed during the P-milling process would strengthen the structural resistance against the volume stress during Na+ insertion/retraction. As a result, the Sb/amorphous P–C composite delivered a high sodium overall storage capacity and superior rate capability with excellent long-term cycling performance.
2. Results and discussion
Fig. 1a shows the XRD patterns of red P (RP), EG and the composites prepared by P-milling. After 20 h of P-milling, the diffraction peaks of RP and EG were dramatically weakened in the P70EG30-P-milling 20 h sample, accompanied by the appearance of black P (BP) in the pattern.29 For the Sb70EG30-P-milling 20 h sample, the peaks associated with EG disappeared and a broad peak from 20° to 35° was formed in the pattern, suggesting that the Sb particles could facilitate the exfoliation and fracture of the EG during the P-milling process. Besides this broad peak, all other distinguishable diffraction peaks in the Sb70EG30-P-milling 20 h composite correspond to the Sb phase. For the Sb30P40EG30-P-milling 20 h sample, both the EG and P peaks disappeared, reflecting the generation of a disordered structure in the composite prepared by P-milling of P, EG and Sb together. The structural information on the samples was further reflected by the Raman spectra as shown in Fig. 1b. The raw Sb powder shows two characteristic vibrational Raman peaks at 111 and 147 cm−1. Additional peaks at 190 and 254 cm−1 are attributed to Sb2O3.30 The EG exhibits a weak D-band at 1340 cm−1 and a strong G-band at 1580 cm−1, demonstrating a highly graphitic structure. The pristine RP displays three peaks between 300 and 500 cm−1, consistent with the results reported in the literature.17 After 20 h of P-milling, the P bands could not be seen in both the P70EG30-P-milling 20 h and the Sb30P40EG30-P-milling 20 h samples. This was due to the wrapping of C on P in the composites, leading to the decrease in the intensity of the P peaks.31 In addition, the Sb peaks were weakened and broadened in the Sb70EG30-P-milling 20 h sample and disappeared in the Sb30P40EG30-P-milling 20 h sample. These changes were believed to be due to the wrapping of the nanostructures by other components. In contrast to the EG, the integral area ratio of the D and G bands (ID/IG) was apparently increased for the samples after P-milling, indicating a significant reduction in size of the EG along the c-direction and an accumulation of defects. By calculation we found that the value of ID/IG was different for various samples. It was 1.44 for P70EG30-P-milling 20 h, 2.12 for Sb70EG30-P-milling 20 h and 2.51 for Sb30P40EG30-P-milling 20 h. This demonstrates that the milling of a combination of P and Sb could more effectively destruct the layered structure of EG, with the P acting as a grinding aid while the Sb served as a milling medium. | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra for different samples. (c) P 2p XPS spectra (bottom) of the Sb30P40EG30-P-milling 20 h and P70EG30-P-milling 20 h samples and Sb 3d and O 1s XPS spectra (top) of the Sb30P40EG30-P-milling 20 h and Sb70EG30-P-milling 20 h samples. SEM images of the (d) P70EG30-P-milling 20 h, (e) Sb70EG30-P-milling 20 h and (f) Sb30P40EG30-P-milling 20 h composite powders. | ||
In order to further probe the electronic states and electronic interactions of the surface elements in different samples, X-ray photoelectron spectroscopy (XPS) studies were performed. As shown in Fig. 1c, the broad peak at 134.7 eV in the P 2p spectra of the P70EG30-P-milling 20 h sample could be assigned to the oxidized phosphorus, resulting from the slight oxidation on the top of the powders in air.32 The rest of the peaks in the P70EG30-P-milling 20 h sample could be fitted to the 2p1/2 and 2p3/2 doublets, which were split by 0.9 eV with an integrated intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.33 The peak located at 129.9 eV was the P 2p3/2 doublet from the P–P bond, while the peak located at 130.3 eV was the P 2p3/2 doublet from the P–C bond. These results were consistent with the previous findings in the composite of BP and carbon.29 Similar to the P70EG30-P-milling 20 h sample, two features related to Sb could also been found in the Sb 3d spectra of the Sb70EG30-P-milling 20 h sample. The two peaks at 528.6 (3d5/2) and 538.0 eV (3d3/2) correspond to that of metallic Sb, and the peaks at 530.7 (3d5/2) and 540.1 eV (3d3/2) correspond to Sb oxide due to the slight oxidation of the Sb particles.34 Because of the overlapping of the Sb 3d and O 1s core peaks, the component at 532.0 eV was attributed to the O 1s orbital.35 Notably, for the Sb30P40EG30-P-milling 20 h sample, these peaks exhibited a shift to lower binding energies (BE) by 0.2 eV for the P 2p orbital, 0.1 eV for the P–C bond and 1.0 eV for phosphide, as well as a shift to higher BE by 0.6 eV for the Sb 3d orbital and 1.0 eV for Sb oxide. The decrease of BE indicates the enhanced electron screening effect because of the increase in electron density, whereas the increase of BE suggests a decrease in electron density.36 Hence, in our case, it was believed that the shifts of BE in the Sb30P40EG30-P-milling 20 h sample were attributed to the increased electron density of P and decreased electron density of Sb. These characteristics could be ascribed to electron transfer from Sb to P through the Sb/P contact interface.32 Moreover, no extra peaks could be found in the XPS image of the Sb30P40EG30-P-milling 20 h sample, and this indicates that the van der Waals force was the driving force to enable the formation of the Sb/P interfaces as confirmed by FTIR spectroscopy (Fig. S1, ESI†).
:2.33 The peak located at 129.9 eV was the P 2p3/2 doublet from the P–P bond, while the peak located at 130.3 eV was the P 2p3/2 doublet from the P–C bond. These results were consistent with the previous findings in the composite of BP and carbon.29 Similar to the P70EG30-P-milling 20 h sample, two features related to Sb could also been found in the Sb 3d spectra of the Sb70EG30-P-milling 20 h sample. The two peaks at 528.6 (3d5/2) and 538.0 eV (3d3/2) correspond to that of metallic Sb, and the peaks at 530.7 (3d5/2) and 540.1 eV (3d3/2) correspond to Sb oxide due to the slight oxidation of the Sb particles.34 Because of the overlapping of the Sb 3d and O 1s core peaks, the component at 532.0 eV was attributed to the O 1s orbital.35 Notably, for the Sb30P40EG30-P-milling 20 h sample, these peaks exhibited a shift to lower binding energies (BE) by 0.2 eV for the P 2p orbital, 0.1 eV for the P–C bond and 1.0 eV for phosphide, as well as a shift to higher BE by 0.6 eV for the Sb 3d orbital and 1.0 eV for Sb oxide. The decrease of BE indicates the enhanced electron screening effect because of the increase in electron density, whereas the increase of BE suggests a decrease in electron density.36 Hence, in our case, it was believed that the shifts of BE in the Sb30P40EG30-P-milling 20 h sample were attributed to the increased electron density of P and decreased electron density of Sb. These characteristics could be ascribed to electron transfer from Sb to P through the Sb/P contact interface.32 Moreover, no extra peaks could be found in the XPS image of the Sb30P40EG30-P-milling 20 h sample, and this indicates that the van der Waals force was the driving force to enable the formation of the Sb/P interfaces as confirmed by FTIR spectroscopy (Fig. S1, ESI†).
Fig. 1d–f show the typical SEM images of the P70EG30-P-milling 20 h, Sb70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h samples. The morphologies of the raw materials of the irregular shaped Sb particles, bulky P particles and layer-structured EG powders changed dramatically after the P-milling processes (Fig. S2, ESI†). The particles in the P70EG30-P-milling 20 h composite were mainly on the micrometer scale, while most particles in the Sb70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h composites have sizes of a few hundred nanometers. The size change suggests that the high hardness of the Sb particles helped the refining of P particles during the P-milling process, which would further enhance the sodium storage performances.
Transmission electron microscopy (TEM) observations were carried out to further investigate the dispersion state of different components in the as-prepared samples. Fig. 2a shows the low-magnification of the P70EG30-P-milling 20 h sample. The milled EG and P particles aggregated together to form a dense structure. The high resolution TEM (HRTEM) image in Fig. 2b reveals that the graphite nanosheets have a thickness ranging from a few to a dozen nanometers. Meanwhile, in the HRTEM image shown in Fig. 2c, the spacing of the lattice fringes was determined to be 0.257 nm, consistent with the (111) lattice spacing of BP. Fig. 2d shows the low-magnification TEM image of the Sb70EG30-P-milling 20 h sample, and the Sb nanoparticles and carbon matrix were packed together to form a cotton-like structure. And instead of the long-range order of the graphite nanosheets, only basic structural units (BSUs) with sizes of about several unit cells thick and 2–3 nm of lateral extent of graphite sheets could be found in the Sb70EG30-P-milling 20 h sample, as shown in Fig. 2e.37 This further demonstrates that the Sb particles could facilitate the pulverization of the well-graphitized EG powder during the P-milling processes. Besides, in the HRTEM image shown in Fig. 2f, the lattice fringes with a spacing of 0.311 nm could be observed which corresponds to the (012) lattice spacing of Sb in the Sb70EG30-P-milling 20 h sample. As for the Sb30P40EG30-P-milling 20 h sample, the low-magnification TEM image in Fig. 2g shows a similar structure to that of the Sb40EG30-P-milling 20 h sample. The HRTEM images in Fig. 2h and i reveal the Sb crystalline grains with sizes of several to dozens of nanometers distributed uniformly in the amorphous matrix. The existence of nanocrystalline Sb particles was further confirmed by selected-area electron diffraction (SAED). In the SAED pattern as shown in Fig. 2j, the diffraction rings could be indexed to the (012), (104), (110), (202), (116) and (122) planes of Sb. In addition, the results of elemental mapping by energy-dispersive X-ray spectroscopy (EDX) as shown in Fig. 2k–n display the homogeneous distribution of Sb, P and C in the Sb30P40EG30-P-milling 20 h sample.
 | ||
| Fig. 2 TEM observations of the P-milling samples with different compositions: (a) low-magnification TEM image of the P70EG30-P-milling 20 h sample. (b and c) High-resolution TEM (HRTEM) images of the arrowed areas in (a). (d) Low-magnification TEM image of the Sb70EG30-P-milling 20 h sample. (e and f) HRTEM images of the arrowed areas in (d). (g) Low-magnification TEM image of the Sb30P40EG30-P-milling 20 h sample. (h and i) HRTEM images of the zone arrowed in (g). (j) SAED pattern. (k) Scanning TEM (STEM) image. (l–n) Elemental mapping images of the Sb30P40EG30-P-milling 20 h sample. | ||
The electrochemical performance of the synthesized materials was evaluated with half cells, using Na as the counter electrode. To explore the effects of the ball milling time on the sodiation/desodiation behavior of the Sb30P40EG30 composite, the mixed powders of Sb, RP and EG with a weight ratio of 30:40:30 were milled for different times of 10 h, 20 h and 30 h (Fig. S3, ESI†). Although the Sb30P40Eg30-P-milling 10 h electrode showed higher initial discharge capacity (1062 mA h g−1) and initial coulombic efficiency (ICE, 76.0%) than the Sb30P40Eg30-P-milling 20 h electrode (996 mA h g−1, 74.0%), the reversible capacity of the Sb30P40Eg30-P-milling 10 h electrode (661 mA h g−1) was lower than that of the Sb30P40Eg30-P-milling 20 h electrode (722 mA h g−1) after 100 cycles. This was because the Sb and P particles could be more homogenously dispersed and embedded in the carbon matrix with prolonged milling time, leading to the improvement of cycling stability. However, prolonged milling would deteriorate the initial discharge capacity and initial coulombic efficiency due to the increased oxidation of the air-sensitive nanoparticles. Therefore, the reversible capacity of the Sb30P40Eg30-P-milling 30 h electrode (499 mA h g−1) was lower than that of the Sb30P40Eg30-P-milling 20 h electrode.
To explore the effects of discharge plasma in the P-milling process, we synthesized an Sb30P40Eg30-C-milling 20 h composite for comparison. The XRD patterns, SEM images and cycling performance of the Sb30P40Eg30-C-milling 20 h and Sb30P40Eg30-P-milling 20 h composites are shown in the ESI (Fig. S4†). As shown in Fig. S4a,† the EG peaks disappeared and a broad peak from 20° to 35° formed in both samples. Besides this broad peak, all other distinguishable diffraction peaks in the two samples correspond to the Sb phase. Even so, the particle sizes of the two composites were entirely different. The particle size of the P-milled composite was much smaller than that of the C-milled composite (Fig. S7b and c†). This was because the very rapid heating caused by the charged particles of plasma led to an explosion of Sb and P to release the thermal stress, which was beneficial to refine the Sb and P particles.28 The smaller particle size of the Sb30P40EG30-P-milling 20 h composite was beneficial to improve the Na+ diffusion kinetics and maintain the structural integrity of the electrode during the cycling. Thus, the cycling performance of the Sb30P40EG30-P-milling 20 h electrode was obviously better than that of the Sb30P40EG30-C-milling 20 h electrode. After 100 cycles, the reversible capacity of the Sb30P40EG30-P-milling 20 h electrode was 722 mA h g−1, whereas the value was only 339 mA h g−1 for the Sb30P40EG30-C-milling 20 h electrode (Fig. S7d†).
To verify the differences of initial discharge capacity and ICE from the P70EG30-P-milling 20 h, Sb70EG30-P-milling 20 h and the Sb30P40EG30-P-milling 20 h electrodes, a total of six half-cells were tested at a current density of 0.2 A g−1 for each electrode. The initial discharge/charge curves are given in the ESI (Fig. S5†), and the corresponding initial discharge capacity and ICE for each cell are shown in Fig. 3a. Compared with the Sb30P40EG30-P-milling 20 h electrode, the P70EG30-P-milling 20 h electrode has a much higher weight percent of P, so it was expected that the P70EG30-P-milling 20 h electrode would show a much higher initial discharge capacity as P has a much higher theoretical specific capacity than Sb. The measured data, however, show the opposite. The P70EG30-P-milling 20 h electrode delivered an average initial discharge capacity of 847 mA h g−1, while the Sb30P40EG30-P-milling 20 h electrode achieved an average initial discharge capacity of 964 mA h g−1. This phenomenon might be related to both the enhanced Na+ diffusion kinetics and the improved electronic conductivity of the sample as a result of the good refinement of the P particles, such that more P atoms could react with Na-ions at the same current density. For the Sb70EG30-P-milling 20 h composite, due to the relatively low theoretical specific capacity of Sb, it exhibited the lowest initial discharge capacity of 633 mA h g−1. In addition, the average ICE of the P70EG30-P-milling 20 h electrode was only 51.3%. The low capacity might be related to the formation of a layer of solid-electrolyte interphase (SEI) on the surface of the active material. This large capacity fading was also due to the large volume expansion of the P particles and the subsequent loss of contact with the conductive matrix. However, for the Sb70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h electrodes, the ICE was 75.6% and 73.5%, respectively. These values suggest that the reversibility of the P was improved in the Sb30P40EG30-P-milling 20 h electrode. This improvement could be due to the unique structure of the Sb30P40EG30-P-milling 20 h composite. In the sample, the uniformly distributed Sb nanoparticles in the amorphous P–C matrix provided sufficient electronic conductivity that promoted good connections between the RP particles which reduced the influence of volume expansion on the connections between the RP and the current collector.
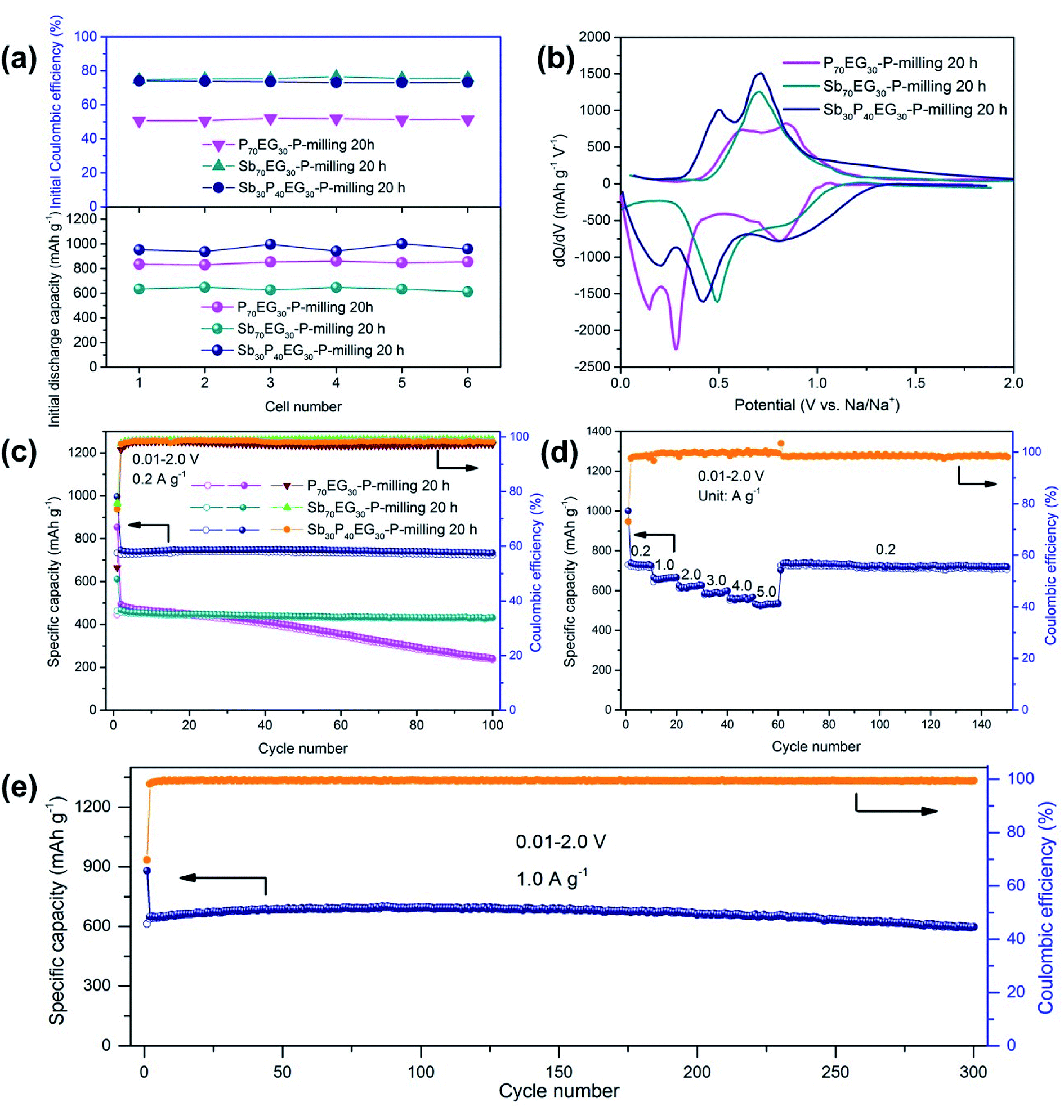 | ||
| Fig. 3 (a) Summary of the initial discharge capacities and ICEs of the P70EG30-P-milling 20 h, Sb70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h electrodes. (b) Differential capacity versus voltage curves at the 1st cycle. (c) Cycling performance of the P70EG30-P-milling 20 h, Sb70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h electrodes. (d) Rate capability and (e) long cycling performance at 1.0 A g−1 of the Sb30P40EG30-P-milling 20 h electrode. | ||
To clarify the reactions of the P70EG30-P-milling 20 h, Sb70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h electrodes in SIBs, the dQ/dV curves of the three electrodes are displayed in Fig. 3b. One large cathodic peak (∼0.8 V) could be seen in all three electrodes that could be attributed to the decomposition of electrolyte in the formation of the SEI layer on the surface of the active materials. The occurrence of another two cathodic peaks below 0.6 V in the P70EG30-P-milling 20 h electrode was a result of the Na+ insertion reaction that formed the NaxP compounds.38 For the Sb70EG30-P-milling 20 h electrode, the cathodic peak located at about 0.5 V was attributed to the Na3Sb alloy formation.7 In the reverse process, two small peaks at around 0.6 and 0.9 V could be observed in the P70EG30-P-milling 20 h electrode, demonstrating the transformation of the NaxP phase into the P phase through a two-step reaction.38 As for the Sb70EG30-P-milling 20 h electrode, the large anodic peak at about 0.7 V was ascribed to the desodiation process of the Na3Sb.7 Compared with the P70EG30-P-milling 20 h electrode, the final cathodic peak and the initial anodic peak of the Sb30P40EG30-P-milling 20 h electrode shifted to higher and lower potentials, respectively. This phenomenon was due to the enhancement of reaction kinetics and the electronic conductivity after Sb addition.
Fig. 3c presents the cycling performance of the P70EG30-P-milling 20 h, Sb70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h electrodes. The capacity of the P70EG30-P-milling 20 h electrode declined dramatically to 235 mA h g−1 after 100 cycles. This capacity fading was mainly due to the structure failure caused by the large volume changes of the P particles and the subsequent lost contact with the current collector during long-term cycling. The Sb70EG30-P-milling 20 h electrode exhibited excellent cycling stability, but a relatively low reversible capacity of 429 mA h g−1 after 100 cycles. In contrast, the Sb30P40EG30-P-milling 20 h electrode showed superior cycle performance, retaining a reversible capacity of 722 mA h g−1 with a stable CE above 98% throughout 100 cycles. This demonstrated that the Sb30P40EG30-P-milling 20 h composite was able to maintain its structural integrity of the particles during the cycling processes. To further examine the structural evolution of the three electrodes, the surface morphologies of the three electrodes before and after the 100 cycles were observed by SEM (Fig. S6, ESI†). It was found that the P70EG30-P-milling 20 h electrode had been seriously destructed after continuous lithiation/delithiation, while the Sb70EG30-P-milling 20 h and the Sb30P40EG30-P-milling 20 h electrodes could maintain their integrity after 100 cycles.
The electrochemistry impedance measurements were conducted to further investigate the conductivities of the electrodes. Fig. S7a and b in the ESI† show the Nyquist plots of the Sb30P40EG30-P-milling 20 h electrode compared with the P70EG30-P-milling 20 h electrode at the fully charged state after the first cycle and 100 cycles. All impedance curves show a semicircle in the medium-to-low frequency region, which could be assigned to the charge-transfer resistance (Rct).26 The Rct value was calculated using the equivalent circuit shown in the inset of Fig. S7a.† The Rct values of the P70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h electrodes were 118 and 48 Ω, respectively, so a higher electron transfer rate was achieved in the Sb30P40EG30-P-milling 20 h electrode. After 100 cycles, the Rct value of the Sb30P40EG30-P-milling 20 h was 55 Ω. The tiny change of the Rct value after cycling indicated the excellent structural stability of the Sb30P40EG30-P-milling 20 h electrode. In comparison, the Rct value was increased to 253 Ω after 100 cycles for the P70EG30-P-milling 20 h electrode. This great change after cycling revealed the destruction of electrical connections during sodiation/desodiation processes.
To ensure reasonable comparisons, we prepared Sb10P60EG30-P-milling 20 h, Sb50P20EG30-P-milling 20 h, Sb40P40EG20-P-milling 20 h and Sb20P40EG40-P-milling 20 h composites under the same experimental conditions as those of the Sb30P40EG30-P-milling 20 h composite. The galvanostatic charge/discharge cycling tests demonstrated that the weight ratio of Sb, P and EG in the Sb30P40EG30-P-milling 20 h composite was the optimum ratio for achieving the best electrochemical performance (Fig. S8, ESI†). The low content of the Sb or EG component was not beneficial to obtain a composite with good structural stability, while the high content of the Sb or EG component would cause low reversible capacity. Besides the superior cycling stability, the Sb30P40EG30-P-milling 20 h electrode also exhibited excellent rate capability. As shown in Fig. 3d, the electrode achieved reversible capacities of 718, 664, 626, 598, 568 and 533 mA h g−1 at current densities of 0.2, 1, 2, 3, 4 and 5 A g−1, respectively. When the current rate returned to 0.2 A g−1, the initial capacity was completely recovered, demonstrating the excellent tolerance for the rapid sodium ion insertion/extraction processes. To further evaluate the superior capacity output and cycling stability of the Sb30P40EG30-P-milling 20 h electrode, we performed galvanostatic charge/discharge cycling tests at a high current density of 1 A g−1. As shown in Fig. 3e, the Sb30P40EG30-P-milling 20 h electrode reached a high reversible capacity of 596 mA h g−1 after 300 cycles. The charge/discharge curves of the Sb30P40EG30-P-milling 20 h electrode at a high current density of 1 A g−1 are given in the ESI (Fig. S9†). The electrochemical performances of the Sb/P–C composite in terms of initial discharge capacity, initial Coulombic efficiency and cycling stability were further compared with previously reported P-based materials as listed in Table S1.†
To gain more information about the phase evolution of the Sb30P40EG30-P-milling 20 h electrode during the electrochemical reaction processes, ex situ XRD measurements were performed on the discharged/charged electrodes. As shown in Fig. 4a and b, the characteristic peaks of the Sb phase disappeared after discharging to 0.3 V, and a small peak appeared at 38.6°, which could be indexed to the (112) plane of the Na3Sb phase (PDF No. 74-1162). As the potential was further lowered to 0.01 V, no new peaks were observed in the XRD pattern, indicating that an amorphous NaxP phase was formed during the sodiation process.18 In the desodiation process, the diffraction peak of the Na3Sb phase disappeared after charging to 2.0 V, and no peaks corresponding to the Sb phase were observed because of the change of Sb from the crystalline to amorphous state after the initial discharge/charge process. In the second cycle, the XRD patterns at fully discharged and charged states were similar to those in the first cycle. Furthermore, ex situ TEM images confirmed the structural evolution of the Sb30P40EG30-P-milling 20 h electrode during the electrochemical reaction processes. As shown in Fig. 4c, the electrode material at an initial discharge state of 0.01 V had a loose structure with agglomerated particles from dozens to hundreds of nanometers. The HRTEM image in Fig. 4d shows the well-dispersed nanocrystallites with the lattice fringes of 0.233 nm dotted in the amorphous matrix. This lattice spacing agrees well with the (112) plane of the Na3Sb phase. Compared with the original morphology of the Sb30P40EG30-P-milling 20 h composite (Fig. 2d), the electrode material (Fig. 4e) shows obvious structural evolution (slight pulverization) after the initial discharge/charge process. Despite this, the nanoparticles were still tightly jointed together and this enabled the electron transport from the current collector to the active material. Besides, no nanocrystals could be found in the HRTEM image shown in Fig. 4f and this is consistent with the ex situ XRD results.
 | ||
| Fig. 4 (a) Initial potential vs. the specific capacity curve of the Sb30P40EG30-P-milling 20 h electrode with five electrochemical reaction states for ex-XRD measurements, namely open cell potential (OCP), discharge to 0.3 V and 0.01 V. (b) Ex situ XRD patterns of the Sb30P40EG30-P-milling 20 h electrode at different states for the initial cycle. (c) Low-magnification TEM image and (d) HRTEM image of the Sb30P40EG30-P-milling 20 h electrode at an initial discharge state of 0.01 V. (e) Low-magnification TEM image and (f) HRTEM image of the Sb30P40EG30-P-milling 20 h electrode at an initial charge state of 2.0 V. (g) A schematic illustration of the electrochemical reaction processes of the P70EG30-P-milling 20 h and the Sb30P40EG30-P-milling 20 h electrodes. | ||
On the basis of the above analysis, a schematic illustration is presented to summarise the electrochemical reaction processes of the P70EG30-P-milling 20 h and Sb30P40EG30-P-milling 20 h electrodes. As illustrated in Fig. 4g, without the involvement of an effective buffer matrix, severe pulverization occurred in the P70EG30-P-milling 20 h electrode during the electrochemical reaction process due to the huge volume change during the phase transition between P and NaxP. The pulverization of the electrode would cause some P particles to lose contact with the current collector, resulting in capacity decay. Besides, because of the poor sodiation kinetics of the bulky P particles, the alloying reaction of the P70EG30-P-milling 20 h electrode was incomplete. However for the Sb30P40EG30-P-milling 20 h electrode, the well-dispersed intermediate discharge/charge products of Sb nanoparticles acted as electronic channels to enable electrochemical activation of the P component, and the P–C bonds formed during the P-milling process would strengthen the structural resistance against the volume stress during Na+ insertion/retraction, therefore achieving excellent cycling stability and rate capability.
3. Conclusions
In summary, we have synthesized a variety of Sb/C, P/C and Sb/P–C composites through regulating the P-milling time and weight ratio of the raw materials. The galvanostatic charge/discharge cycling tests demonstrated that the Sb30P40EG30-P-milling 20 h composite achieved the best electrochemical performance. The structural information of the Sb30P40EG30-P-milling 20 h composite was obtained by a series of characterization techniques. The results of XRD and Raman spectroscopy indicate that both P and EG were amorphous in structure and the P–C bonds were detected by XPS. The TEM results show that the amorphous P and disordered C network were uniformly mixed together, and the Sb nanoparticles were well dispersed in the P–C mixture. When reacted with sodium, the well-dispersed intermediate discharge/charge products of the Sb nanoparticles acted as electronic channels, enabling the electrochemical activation of the phosphorus component. The P–C bonds formed during the P-milling process can strengthen the structural resistance against the volume stress during Na+ insertion/retraction. As a result, the Sb30P40EG30-P-milling 20 h composite achieved a high reversible capacity of 722 mA h g−1 after 100 cycles at 0.2 A g−1, a superior rate capability with a reversible capacity of 533 mA h g−1 at 5 A g−1 and stable long-term cycling performance with a reversible capacity of 596 mA h g−1 after 300 cycles at 1 A g−1.4. Experimental
4.1. Materials preparation
Expandable graphite (99.9% purity, 100 mesh, Qingdao Xinghua Graphite Products Co., Ltd) was calcined at 950 °C for 2 min in a muffle furnace to obtain the worm-like expanded graphite (EG). Sb (99.5%, Aladdin), RP (98.5%, Aladdin) and EG powders were mixed at different weight ratios in a cemented carbide vial. The mass ratio of the ball to the mixed powder was 100:1. The P-milling process was conducted under an argon atmosphere with a vibration type mill. More details of the P-milling were described in our previous papers. The samples synthesized by P-milling were denoted as SbaPbEGc-P-milling x, where a, b and c are the mass percents of different components, and x is the P-milling time. For comparison, the powder mixture was also treated by C-milling using the same milling equipment as the P-milling except for lack of discharge plasma assistance.
4.2. Materials characterization
The crystal structures of the samples were identified using an X-ray diffractometer (XRD, PANalytical Empyrean) with Cu Kα radiation. Raman spectra were obtained using a HORIBA LabRAM Aramis spectromicroscopy system with a 633 nm wavelength. The electronic states of elemental and surface composition of the samples were determined using an X-ray photoelectron spectrometer (XPS, Thermo Scientific ESCALAB 250) using an Al Kα source. The chemical bonds of the samples were identified using a Fourier transform infrared spectroscope (FTIR, Nicolet IS50) in the transmission mode. The sample morphologies were characterized by field-emission SEM (FESEM Carl Zeiss Supra 40) and TEM (JEOL JEM-2100). For the ex situ XRD and TEM analyses, we collected the electrodes by separating the test cells in an argon-filled glove box. The electrodes were washed with DEC several times and then vacuum dried in an antechamber of the glove box. The reacted electrodes were sealed inside a Kapton membrane to minimize air contact during the XRD measurements.4.3. Electrochemical measurements
The sodium-storage performances of the samples were measured with CR2025 coin cells assembled in an argon-filled glove box. The working electrodes consisted of 70 wt% of active materials, 15 wt% of Super P (TIMCAL Graphite & Carbon) and 15 wt% of carboxymethyl cellulose (CMC, Mw = 700000)/styrene butadiene rubber (SBR, BM-451B, Zeon) (1:1 m/m). The load density of the active materials was about 1 mg cm−2. Sodium foil was used as both the counter and reference electrodes. The electrolyte was 1 M NaClO4 in a mixture of ethylene carbonate (EC)/diethyl carbonate (DEC) (1:1 v/v) with 5 wt% FEC. Galvanostatic charge/discharge experiments were performed at different current rates in a potential range between 0.01 and 2 V on a battery testing system (LAND CT2001A). Cyclic voltammetry (CV) over the potential range of 0–2 V at 0.1 mV s−1 and electrochemical impedance spectroscopy (EIS) at a 5 mV amplitude signal in a frequency range of 1 MHz to 0.1 Hz were taken on an electrochemical workstation (Gamry Interface 1000).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51431001, 51271078 and U120124). ZL acknowledges the funding support from the Australian Research Council (ARC DP180102976).References
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 RSC.
- Q. Li, Z. Zhang, S. Dong, C. Li, X. Ge, Z. Li, J. Ma and L. Yin, Part. Part. Syst. Charact., 2017, 34, 1600115 CrossRef.
- Y. Liu, N. Zhang, L. Jiao, Z. Tao and J. Chen, Adv. Funct. Mater., 2015, 25, 214–220 CrossRef CAS.
- W. Li, S. Hu, X. Luo, Z. Li, X. Sun, M. Li, F. Liu and Y. Yu, Adv. Mater., 2017, 29, 1605820 CrossRef PubMed.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805–20811 CrossRef CAS PubMed.
- L. Wang, C. Wang, F. Li, F. Cheng and J. Chen, Chem. Commun., 2017, 54, 38–41 RSC.
- Z. Jian, B. Zhao, P. Liu, F. Li, M. Zheng, M. Chen, Y. Shi and H. Zhou, Chem. Commun., 2014, 50, 1215–1217 RSC.
- X. Wang, Y. Liu, Y. Wang and L. Jiao, Small, 2016, 12, 4865–4872 CrossRef CAS PubMed.
- C. Zhu, X. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., 2014, 53, 2152–2156 CrossRef CAS PubMed.
- C. Wu, Y. Jiang, P. Kopold, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2016, 28, 7276–7283 CrossRef CAS PubMed.
- H. G. Wang, Z. Wu, F. L. Meng, D. L. Ma, X. L. Huang, L. M. Wang and X. B. Zhang, ChemSusChem, 2013, 6, 56–60 CrossRef CAS PubMed.
- K. Tang, L. Fu, R. J. White, L. Yu, M.-M. Titirici, M. Antonietti and J. Maier, Adv. Energy Mater., 2012, 2, 873–877 CrossRef CAS.
- R. Alcantara, J. M. Jimenez-Mateos, P. Lavela and J. L. Tirado, Electrochem. Commun., 2001, 3, 639–642 CrossRef CAS.
- W. Li, Z. Yang, M. Li, Y. Jiang, X. Wei, X. Zhong, L. Gu and Y. Yu, Nano Lett., 2016, 16, 1546–1553 CrossRef CAS PubMed.
- Y. J. Zhu, Y. Wen, X. L. Fan, T. Gao, F. D. Han, C. Luo, S. C. Liou and C. S. Wang, ACS Nano, 2015, 9, 3254–3264 CrossRef CAS PubMed.
- Y. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N. S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139–4144 CrossRef CAS PubMed.
- W. Zhang, M. Dahbi, S. Amagasa, Y. Yamada and S. Komaba, Electrochem. Commun., 2016, 69, 11–14 CrossRef CAS.
- W.-J. Li, Q.-R. Yang, S.-L. Chou, J.-Z. Wang and H.-K. Liu, J. Power Sources, 2015, 294, 627–632 CrossRef CAS.
- J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050–2059 RSC.
- S. O. Kim and A. Manthiram, Chem. Commun., 2016, 52, 4337–4340 RSC.
- X. Fan, J. Mao, Y. Zhu, C. Luo, L. Suo, T. Gao, F. Han, S.-C. Liou and C. Wang, Adv. Energy Mater., 2015, 5, 1500174 CrossRef.
- F. Wan, J. Z. Guo, X. H. Zhang, J. P. Zhang, H. Z. Sun, Q. Yan, D. X. Han, L. Niu and X. L. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 7790–7799 CrossRef CAS PubMed.
- J. Liu, L. Yu, C. Wu, Y. Wen, K. Yin, F. K. Chiang, R. Hu, J. Liu, L. Sun, L. Gu, J. Maier, Y. Yu and M. Zhu, Nano Lett., 2017, 17, 2034–2042 CrossRef CAS PubMed.
- D. Cheng, J. Liu, X. Li, R. Hu, M. Zeng, L. Yang and M. Zhu, J. Power Sources, 2017, 350, 1–8 CrossRef CAS.
- W. Sun, R. Hu, H. Zhang, Y. Wang, L. Yang, J. Liu and M. Zhu, Electrochim. Acta, 2016, 187, 1–10 CrossRef CAS.
- H. Liu, R. Hu, M. Zeng, J. Liu and M. Zhu, J. Mater. Chem., 2012, 22, 8022–8028 RSC.
- J. Sun, G. Zheng, H. W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS PubMed.
- X. Sun, Z. Lu, Y. Xiang, Y. Wang, J. Shi, G. C. Wang, M. A. Washington and T. M. Lu, ACS Nano, 2018, 12, 6100–6108 CrossRef CAS PubMed.
- Y. Kim, Y. Park, A. Choi, N. S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- F. Shi, Z. Geng, K. Huang, Q. Liang, Y. Zhang, Y. Sun, J. Cao and S. Feng, Adv. Sci., 2018, 5, 1800575 CrossRef PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- A. Darwiche, L. Bodenes, L. Madec, L. Monconduit and H. Martinez, Electrochim. Acta, 2016, 207, 284–292 CrossRef CAS.
- V. Sudarsan, K. P. Muthe, J. C. Vyas and S. K. Kulshreshtha, J. Alloys Compd., 2002, 336, 119–123 CrossRef CAS.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- C. S. Wang, G. T. Wu and W. Z. Li, J. Power Sources, 1998, 76, 1–10 CrossRef CAS.
- W. J. Li, S. L. Chou, J. Z. Wang, H. K. Liu and S. X. Dou, Nano Lett., 2013, 13, 5480–5484 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: One step synthesis of an Sb/amorphous P–C composite as a stable and high-capacity anode for sodium ion batteries by plasma-assisted milling. Supplementary figures (Fig. S1–S9). See DOI: 10.1039/c9ta07508a |
| This journal is © The Royal Society of Chemistry 2020 |