
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNiobium disulphide (NbS2)-based (heterogeneous) electrocatalysts for an efficient hydrogen evolution reaction†
Leyla
Najafi‡
a,
Sebastiano
Bellani‡
a,
Reinier
Oropesa-Nuñez
 b,
Beatriz
Martín-García
a,
Mirko
Prato
c,
Vlastimil
Mazánek
d,
Doriana
Debellis
e,
Simone
Lauciello
e,
Rosaria
Brescia
e,
Zdeněk
Sofer
d and
Francesco
Bonaccorso
*ab
b,
Beatriz
Martín-García
a,
Mirko
Prato
c,
Vlastimil
Mazánek
d,
Doriana
Debellis
e,
Simone
Lauciello
e,
Rosaria
Brescia
e,
Zdeněk
Sofer
d and
Francesco
Bonaccorso
*ab
aGraphene Labs, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy. E-mail: francesco.bonaccorso@iit.it
bBeDimensional Spa., Via Albisola 121, 16163 Genova, Italy
cMaterials Characterization Facility, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy
dDepartment of Inorganic Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague 6, Czech Republic
eElectron Microscopy Facility, Istituto Italiano di Tecnologia, Via Morego 30, 16163 Genova, Italy
First published on 31st October 2019
Abstract
The design of efficient and cost-effective catalysts for the hydrogen evolution reaction (HER) is the key for molecular hydrogen (H2) production from electrochemical water splitting. Transition metal dichalcogenides (MX2), most notably group-6 MX2 (e.g., MoS2 and WS2), are appealing catalysts for the HER alternative to the best, but highly expensive, Pt-group elements. However, their HER activity is typically restricted to their edge sites rather than their basal plane. Furthermore, their semiconducting properties hinder an efficient electron transfer to the catalytic sites, which impedes a high rate of H2 production. Herein, we exploit liquid-phase exfoliation-produced metallic (1H, 2H and 3R) NbS2 nanoflakes, belonging to the class of metallic layered group-5 MX2, to overcome the abovementioned limitations. Both chemical treatment with hygroscopic Li salt and electrochemical in operando self-nanostructuring are exploited to improve the NbS2 nanoflake HER activity. The combination of NbS2 with other MX2, in our case MoSe2, also provides heterogeneous catalysts accelerating the HER kinetics of the individual counterparts. The designed NbS2-based catalysts exhibit an overpotential at a cathodic current of 10 mA cm−2 (η10) as low as 0.10 and 0.22 V vs. RHE in 0.5 M H2SO4 and 1 M KOH, respectively. In 0.5 M H2SO4, the HER activity of the NbS2-based catalysts is also superior to those of the Pt/C benchmark at current densities higher than 80 mA cm−2. Our work provides general guidelines for a scalable and cost-effective exploitation of NbS2, as well as the entire MX2 portfolio, for attaining a viable H2 production through electrochemical routes.
Introduction
Molecular hydrogen (H2) represents an ideal high-energy density source (between 120 and 140 MJ kg−1),1,2 since it can be produced via electrochemical water splitting from renewable sources3,4 and its consumption is sustainable and environmentally friendly.5,6 To speed up the rate of the hydrogen evolution reaction (HER) (4H+ + 4e− → 2H2 in acidic media; 4H2O + 4e− → 2H2 + 4OH− in alkaline media), commercial electrolyzers demand effective catalysts based on Pt-group elements.7–9 However, the cost10 and the scarcity11 of these noble metal-based electrocatalysts hinder their massive use in a move toward a hydrogen economy.12,13 Therefore, great efforts have been made towards the development of cost effective noble-metal-free alternatives, including transition metal alloys,14,15 nitrides,16,17 chalcogenides,18–23 phosphides,24,25 carbides,26–28 and carbonaceous nanomaterials.29–31 In this context, transition metal dichalcogenides –MX2– (M = transition metal; X = S, Se, Te), composed of covalently bonded X–M–X blocks,32,33 are appealing alternative catalysts for the HER.34–38 Theoretical39–41 and experimental studies41–43 for the most investigated group-6 MX2 (e.g., H-MoS2 and H-WS2) have shown that the scarce unsaturated X-edges are the HER-active sites, since they have a near zero Gibbs free energy of adsorbed atomic H (ΔG0H), while the more abundant basal plane is catalytically inert. Furthermore, the semiconducting properties of group-6 MX2 limit the electron transfer to the catalytic edge sites,44,45 thus hindering the high H2 production rate required in electrolyzer systems. The design of nanostructured MX2 (ref. 42 and 46–49) has been pursued to exploit their high per site activity, achieving overpotential versus the reversible hydrogen electrode (RHE) at 10 mA cm−2 cathodic current density (η10), approaching those of noble metal-based electrocatalysts (i.e., <0.1 V).28,50,51 However, MX2 nanostructuring inevitably raises scalability and cost issues, impeding their facile implementation. Recently, metallic MX2 based on group-5 metals (i.e., vanadium (V), niobium (Nb), tantalum (Ta)) attracted utmost interest for the HER process due to the predicted HER activity of their basal planes (especially for sulphides),52–58 beyond that of the chalcogen and metal edges.55,56 Additionally, group-5 MX2 have been theoretically proposed as ideal supports for anchoring single metal atom catalysts (e.g., Pt, Ni and Pd), boosting the HER activity of the latter.59 Recent experimental studies have validated the theoretical predictions, producing chemical vapour deposition (CVD)-synthetized 2H-TaS2 and 2H-NbS2 nanoplatelets displaying an η10 of ∼50–60 mV with a low catalyst loading of only 10–55 μg cm−2.54 Similar results have also been achieved by another NbS2 polytype, i.e., chemically exfoliated 3R-NbS2 flakes.57 However, the aforementioned performances were always reached after an electrochemical pre-conditioning of the electrodes consisting of thousands of cyclic voltammetry (CV) scans.54,57 The latter cause a progressive self-optimizing evolution of the NbS2 morphology, i.e., a reduction in the thickness of the nanoplatelets.54,57 This morphology change speeds up the electron transport towards the HER-active sites by shortening the interlayer electron-transfer pathways and facilitating the access of the aqueous protons (H3O+) to the catalytic films.54,57 Meanwhile, it increases the electrochemically accessible surface area of the electrode films.54,55,57 Parallel investigations also claimed that during the cycling process, the oxides naturally formed on the ambient-exposed H-TaS2 surface are peeled off by the H2 bubbles and the real HER activity of H-TaS2 is therefore exhibited subsequently.55 Despite these breakthrough performances, such self-optimizing fragmentation of the catalyst films could negatively affect their adhesion to the electrode substrates, raising severe doubts about their reliability for high-rate H2 production.60 Therefore, novel insight into the processing of metallic group-5 MX2 is urgently required for their practical validation for the HER. Prospectively, optimized electrode morphologies obtained during the deposition of the electrode films could be “freezed” by using catalyst binders, such as sulfonated tetrafluoroethylene-based fluoropolymer copolymers (e.g., Nafion).In this work, we report the HER activity of single/few-layer NbS2 flakes produced by an environmentally friendly liquid phase exfoliation (LPE) of synthetized material crystals (mixture of 3R- and 2H-polytypes). In contrast to CVD growth61 and mechanical exfoliation,62 LPE is promptly scalable63–68 and does not require expensive nanostructuring of NbS2 materials. Moreover, LPE does not use hazardous chemicals,69 which could affect the physico-chemical properties of the chemically exfoliated materials.70–72 Finally, LPE does not face the issues related to electrochemical exfoliation methods, e.g., the material oxidation during anodic exfoliation in aqueous electrolytes and the ion intercalation-induced morphology degradation effects.73,74 To improve the catalytic activity of the NbS2 flakes, a solution-based chemical treatment with hygroscopic bis(trifluoromethane)sulfonimide lithium salt (Li-TFSI) was used to enhance the electrolyte accessibility to the HER-active surface of NbS2 flakes, overcoming the electrically insulating oxidation of the flake surfaces.75,76In operando electrochemical cycling treatment, as recently reported in the literature54,55 and licensed document,77 was also applied to the LPE-produced NbS2 flakes. Furthermore, the chemical and the electrochemical treatments were combined to positively benefit from possible synergistic effects on the HER activity of the electrodes. Finally, on the basis of published density functional theory (DFT) calculations and ab initio molecular dynamics (AIMD) simulations,56 NbS2 flakes were hybridized with MoSe2 flakes to tune the ΔG0H of the basal planes of NbS2 flakes to optimal thermo-neutral values (i.e., 0 eV) in the resulting heterogeneous configurations for both edge and basal sites. In acidic solution (0.5 M H2SO4), the designed heterogeneous group-5 TMD catalysts (named NbS2:MoSe2) indeed outperform their individual components (η10 of 0.10 V for NbS2:MoSe2, 0.42 V for NbS2 and 0.28 V for MoSe2). For the first time, the HER activity was also investigated in alkaline (1 M KOH) solutions, showing a promising η10 of 0.22 V. The HER activity of our best electrocatalysts was tested over 12 h of continuous operation at a fixed potential corresponding to an initial cathodic current density of 80 mA cm−2, validating the electrochemical durability of NbS2-based electrocatalysts. Our results aim to provide key guidelines to efficiently exploit two-dimensional (2D) NbS2 and, more generally, 2D metallic group-5 MX2 for the HER via scalable material and electrode processing.
Results and discussion
Synthesis and exfoliation of NbS2 crystals
NbS2 crystals were synthesized by direct reaction from Nb and S elements.78 In more detail, Nb powder and S granules at a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 elemental stoichiometry (1 wt% excess of S to avoid Nb-intercalated structures,79) were loaded in a quartz glass ampoule were heated at 450 °C for 12 h and subsequently at 600 °C for 48 h. The products were then treated at 900 °C for 48 h and cooled down to room temperature over a period of 24 h. The as-produced NbS2 crystals were characterized by scanning electron microscopy (SEM) coupled with energy-dispersive X-ray spectroscopy (EDS) (Fig. 1a–c), revealing a near-ideal stoichiometric composition of the NbS2 crystals (S:Nb atomic ratio ∼1.8, see ESI, Table S1†), which is in agreement with previous studies.78 The layered structure of the NbS2 crystals is clearly visible on the edges, as shown by the SEM image in Fig. 1d. The NbS2 flakes were produced by LPE63,80 of the synthetized crystals in 2-propanol (IPA) followed by sedimentation-based separation (SBS)81,82 to remove un-exfoliated crystals by ultracentrifugation (see the Experimental section). The use of IPA as a solvent is effective to remove possible S impurities from the sample during the LPE, SBS and filtration processes in the supernatant, since S is slightly soluble in alcohols (solubility > 0.05 mg mL−1).83 Moreover, starting from cost-effective artificial crystals, the LPE method does not require complex material processing or time-consuming bottom-up nanomaterial synthesis (e.g., CVD).84 The morphology of the as-produced NbS2 flakes was characterized by bright-field transmission electron microscopy (BF-TEM) and atomic force microscopy (AFM) in order to evaluate their lateral dimension and thickness, respectively. Fig. 1e shows the BF-TEM image of representative NbS2 flakes, displaying wrinkled surfaces with irregular shapes, but sharp edges. Fig. 1f shows an AFM image of a representative NbS2 flake, together with its height profile showing a step edge of ∼1.5 nm.
:2 elemental stoichiometry (1 wt% excess of S to avoid Nb-intercalated structures,79) were loaded in a quartz glass ampoule were heated at 450 °C for 12 h and subsequently at 600 °C for 48 h. The products were then treated at 900 °C for 48 h and cooled down to room temperature over a period of 24 h. The as-produced NbS2 crystals were characterized by scanning electron microscopy (SEM) coupled with energy-dispersive X-ray spectroscopy (EDS) (Fig. 1a–c), revealing a near-ideal stoichiometric composition of the NbS2 crystals (S:Nb atomic ratio ∼1.8, see ESI, Table S1†), which is in agreement with previous studies.78 The layered structure of the NbS2 crystals is clearly visible on the edges, as shown by the SEM image in Fig. 1d. The NbS2 flakes were produced by LPE63,80 of the synthetized crystals in 2-propanol (IPA) followed by sedimentation-based separation (SBS)81,82 to remove un-exfoliated crystals by ultracentrifugation (see the Experimental section). The use of IPA as a solvent is effective to remove possible S impurities from the sample during the LPE, SBS and filtration processes in the supernatant, since S is slightly soluble in alcohols (solubility > 0.05 mg mL−1).83 Moreover, starting from cost-effective artificial crystals, the LPE method does not require complex material processing or time-consuming bottom-up nanomaterial synthesis (e.g., CVD).84 The morphology of the as-produced NbS2 flakes was characterized by bright-field transmission electron microscopy (BF-TEM) and atomic force microscopy (AFM) in order to evaluate their lateral dimension and thickness, respectively. Fig. 1e shows the BF-TEM image of representative NbS2 flakes, displaying wrinkled surfaces with irregular shapes, but sharp edges. Fig. 1f shows an AFM image of a representative NbS2 flake, together with its height profile showing a step edge of ∼1.5 nm.
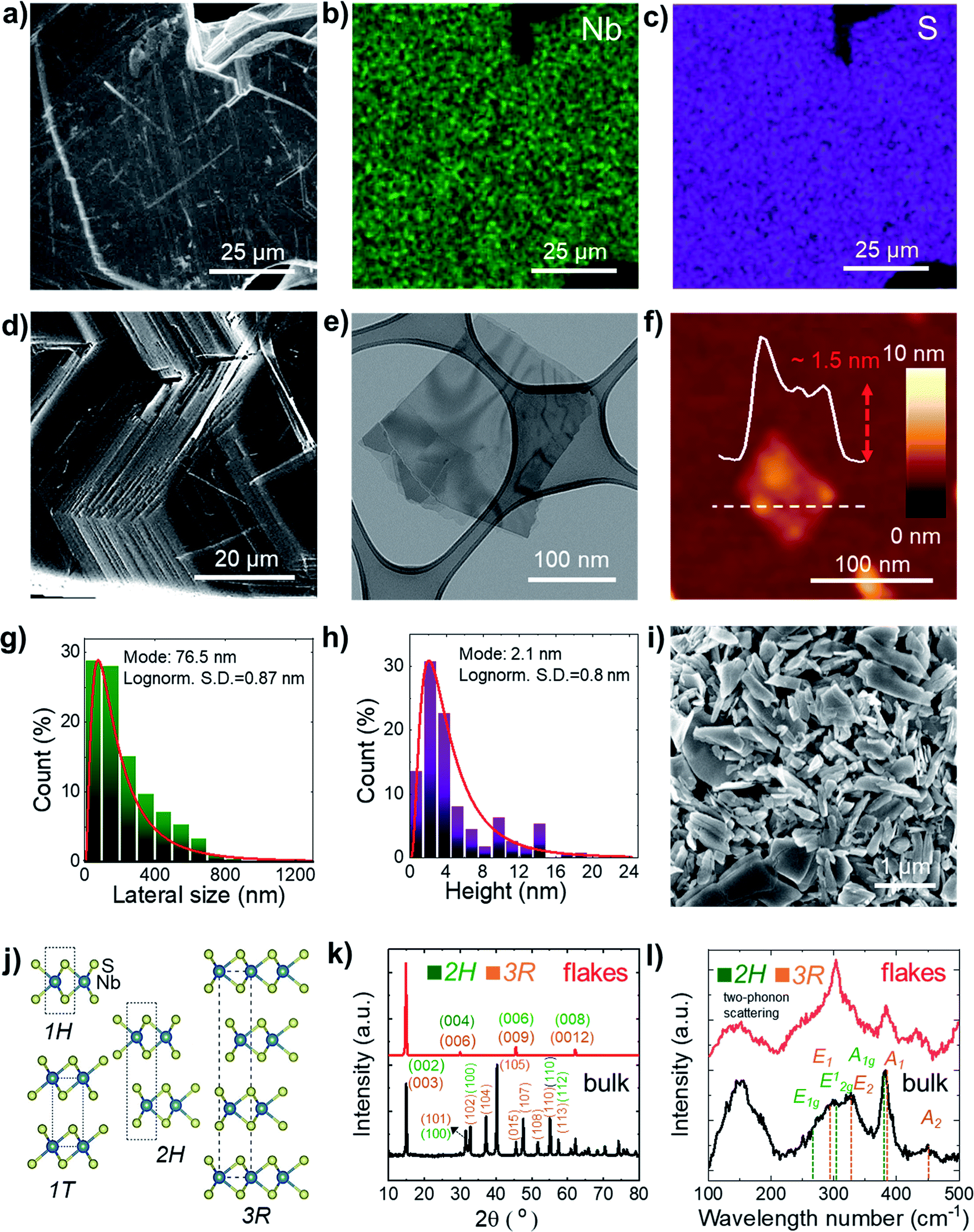 | ||
| Fig. 1 (a) SEM image of the as-synthetized NbS2 crystals and the corresponding quantitative EDS maps for (b) Nb (Lα peak at 2.17 keV, in green) and (c) S (Kα peak at 2.31 keV, in violet). (d) SEM image of a NbS2 crystal edge showing its layered structure. (e) BF-TEM image of LPE-produced NbS2 flakes. (f) AFM image of LPE-produced NbS2 flakes. (g) BF-TEM-based statistical analysis of the lateral dimension of the NbS2 flakes. (h) AFM statistical analysis of the NbS2 flakes. (i) SEM image of a film of NbS2 flakes filtered through a microporous nylon membrane, showing the presence of NbS2 flakes with a lateral size >1 μm. (j) Structure of the NbS2 polytypes experimentally observed in the literature.79 (k) XRD and (l) Raman spectra of the synthetized NbS2 crystals and the LPE-produced NbS2 flakes. The XRD and Raman peaks assigned to 2H- and 3R-NbS2 are also shown. | ||
Statistical BF-TEM-based analysis (Fig. 1g) indicates that the lateral size of the flakes follows a log-normal distribution peaked at ∼76 nm, with maximum values above 1 μm. The statistical AFM analysis (Fig. 1h) shows that the sample is mainly composed of few-layer NbS2 flakes (the AFM thickness of a NbS2 monolayer lies generally between 0.6 nm and 0.8 nm, depending on the substrate and the AFM instrumentation;85–87 the NbS2 interlayer distance is ∼0.6 nm (ref. 88–91)), with a log-normal distribution peaked at ∼2.1 nm.
Additional SEM measurements were carried out on NbS2 flakes filtered through a microporous nylon membrane (Fig. 1i). Such a material deposition method produce films exhibiting NbS2 flakes with a lateral size larger than 1 μm. The crystal structure of the NbS2 crystals and flakes was evaluated by X-ray diffraction measurements (XRD). Fig. 1j shows the structure of the NbS2 polytypes experimentally observed in the literature.79 Typically, NbS2 crystals are found in two possible hexagonal polytypes with a distinctive stacking order of the NbS6 prisms.92,93 The first one, 2H-NbS2, is composed of two NbS2 layer per unit cell (space group: P63/mmc), whereas the second one, 3R-NbS2, consists of three layers per unit cell (space group: R3m).92 Other phases of NbS2 are thermodynamically unfavoured (trigonal 1T-NbS2; space group: P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1, derived under special deposition conditions in a monolayer or thin film form),94,95 or only theoretically predicted (Haeckelite 1S-NbS2; space group: P4/mbm;96 at pressure >20 GPa, and tetragonal NbS2; space group: I4/mmm (ref. 93)). The XRD data (Fig. 1k) show that the as-synthetized NbS2 crystals consist of a mixture of both 2H and 3R polytypes (indexed to PDF cards no. 04-005-8447 (ref. 97 and 98) and no. 04-004-7343,91,98 respectively). After exfoliation, the sample shows XRD peaks corresponding to the diffractions on NbS2 crystals aligned along the (001) plane99,100 (or to 1H-NbS2, i.e., the monolayer form of both 2H and 3R-NbS2). This indicates that the NbS2 flakes are arranged parallel to the c-axis perpendicular to the substrate and retain their native crystal structure.48,101 Raman spectroscopy analysis (Fig. 1l) further confirms the crystallinity of the exfoliated samples, well displaying the nondegenerate Raman active modes of the native crystals predicted by group theory for 2H-NbS2 (space group: D46h)102,103 and 3R-NbS2 (space group: C53v).102,104 In more detail, 2H-NbS2 exhibits the peaks attributed to E1g, E12g and A1g modes at ∼265, ∼305 cm−1 and ∼380, respectively,102,103 while 3R-NbS2 shows the ones associated to E1, E2, A1 and A2, at ∼290, ∼330, ∼385, and ∼450 cm−1, respectively.102,104–106 The broad peaks observed at ∼160 and ∼180 cm−1 originate from two-phonon scattering processes in the presence of defects.86,91,102,104 Notably, NbS2 flakes show a prevalence of the characteristic Raman peaks assigned to 2H-NbS2. This suggests that the exfoliation of 3R-NbS2 crystals intrinsically causes a 3R-to-H phase conversion toward bilayer and monolayer flakes (i.e., 2H-NbS2 and 1H-NbS2), in agreement with previous observations.86 In addition, the A2 mode of 3R-NbS2 redshifts from ∼450 cm−1 in the NbS2 crystal to ∼435 cm−1 in the NbS2 flakes, as a consequence of the relaxation of the interlayer van der Waals forces with decreasing the NbS2 thickness.86,107 The prevalence of the 2H-phase in the NbS2 flakes was further confirmed by high resolution TEM (HRTEM) analysis. Fig. 2a shows a BF-TEM image of a partially suspended NbS2 flake. The corresponding HRTEM image of a portion of the suspended region is shown in Fig. 2b, which shows a single-crystal structure. Fig. 2c shows the corresponding fast Fourier transform (FFT), which matches with that of [001]-oriented 2H-NbS2 (ICSD card no. 250595 (ref. 108)). Fig. 2d shows a magnified portion of the HRTEM image, indicating lattice planes. Scanning transmission electron microscopy coupled with EDS (STEM-EDS) analyses were carried out to evaluate the chemical quality of the NbS2 flakes. Fig. 2e shows the high-angle annular dark-field STEM (HAADF-STEM) image of a partially suspended NbS2 flake. Fig. 2f and g show the corresponding STEM-EDS maps of Nb and S. The quantitative elemental analysis results in a S/Nb atomic ratio of ∼1.8 and a low atomic content of O (O/Nb atomic ratio ∼0.1), which excludes a predominant presence of oxide domains on the flakes. X-ray photoelectron spectroscopy (XPS) measurements (Fig. S1†) were performed to further analyse the element oxidation states on the surface of both the NbS2 crystal and flakes, revealing the presence multiple NbS2 phases, i.e., 2H (or 1H) and 3R ones. In particular, the Nb 3d spectra show the presence of three doublets. The first doublet (peaks at 203.4 ± 0.2 eV and 206.1 ± 0.2 eV) and the second doublet (peaks at 204.0 ± 0.2 and 206.7 ± 0.2 eV) may be both assigned to Nb4+ states in NbS2.107,109–111 The origin of two doublets for NbS2 could be ascribed to the presence of multiple NbS2 phases, i.e., 2H (or 1H) and 3R ones. The peaks located at binding energies of 207.7 ± 0.2 eV and 210.4 ± 0.2 eV are assigned to the Nb(5+) state in Nb2O5.112–116 Both the 3R phase and oxidization are attributed to the surface or the edges of the flakes because they were not detected using HRTEM and STEM-EDS (oxygen map not shown here, due to the low oxygen content), which are less surface-sensitive than XPS.
m1, derived under special deposition conditions in a monolayer or thin film form),94,95 or only theoretically predicted (Haeckelite 1S-NbS2; space group: P4/mbm;96 at pressure >20 GPa, and tetragonal NbS2; space group: I4/mmm (ref. 93)). The XRD data (Fig. 1k) show that the as-synthetized NbS2 crystals consist of a mixture of both 2H and 3R polytypes (indexed to PDF cards no. 04-005-8447 (ref. 97 and 98) and no. 04-004-7343,91,98 respectively). After exfoliation, the sample shows XRD peaks corresponding to the diffractions on NbS2 crystals aligned along the (001) plane99,100 (or to 1H-NbS2, i.e., the monolayer form of both 2H and 3R-NbS2). This indicates that the NbS2 flakes are arranged parallel to the c-axis perpendicular to the substrate and retain their native crystal structure.48,101 Raman spectroscopy analysis (Fig. 1l) further confirms the crystallinity of the exfoliated samples, well displaying the nondegenerate Raman active modes of the native crystals predicted by group theory for 2H-NbS2 (space group: D46h)102,103 and 3R-NbS2 (space group: C53v).102,104 In more detail, 2H-NbS2 exhibits the peaks attributed to E1g, E12g and A1g modes at ∼265, ∼305 cm−1 and ∼380, respectively,102,103 while 3R-NbS2 shows the ones associated to E1, E2, A1 and A2, at ∼290, ∼330, ∼385, and ∼450 cm−1, respectively.102,104–106 The broad peaks observed at ∼160 and ∼180 cm−1 originate from two-phonon scattering processes in the presence of defects.86,91,102,104 Notably, NbS2 flakes show a prevalence of the characteristic Raman peaks assigned to 2H-NbS2. This suggests that the exfoliation of 3R-NbS2 crystals intrinsically causes a 3R-to-H phase conversion toward bilayer and monolayer flakes (i.e., 2H-NbS2 and 1H-NbS2), in agreement with previous observations.86 In addition, the A2 mode of 3R-NbS2 redshifts from ∼450 cm−1 in the NbS2 crystal to ∼435 cm−1 in the NbS2 flakes, as a consequence of the relaxation of the interlayer van der Waals forces with decreasing the NbS2 thickness.86,107 The prevalence of the 2H-phase in the NbS2 flakes was further confirmed by high resolution TEM (HRTEM) analysis. Fig. 2a shows a BF-TEM image of a partially suspended NbS2 flake. The corresponding HRTEM image of a portion of the suspended region is shown in Fig. 2b, which shows a single-crystal structure. Fig. 2c shows the corresponding fast Fourier transform (FFT), which matches with that of [001]-oriented 2H-NbS2 (ICSD card no. 250595 (ref. 108)). Fig. 2d shows a magnified portion of the HRTEM image, indicating lattice planes. Scanning transmission electron microscopy coupled with EDS (STEM-EDS) analyses were carried out to evaluate the chemical quality of the NbS2 flakes. Fig. 2e shows the high-angle annular dark-field STEM (HAADF-STEM) image of a partially suspended NbS2 flake. Fig. 2f and g show the corresponding STEM-EDS maps of Nb and S. The quantitative elemental analysis results in a S/Nb atomic ratio of ∼1.8 and a low atomic content of O (O/Nb atomic ratio ∼0.1), which excludes a predominant presence of oxide domains on the flakes. X-ray photoelectron spectroscopy (XPS) measurements (Fig. S1†) were performed to further analyse the element oxidation states on the surface of both the NbS2 crystal and flakes, revealing the presence multiple NbS2 phases, i.e., 2H (or 1H) and 3R ones. In particular, the Nb 3d spectra show the presence of three doublets. The first doublet (peaks at 203.4 ± 0.2 eV and 206.1 ± 0.2 eV) and the second doublet (peaks at 204.0 ± 0.2 and 206.7 ± 0.2 eV) may be both assigned to Nb4+ states in NbS2.107,109–111 The origin of two doublets for NbS2 could be ascribed to the presence of multiple NbS2 phases, i.e., 2H (or 1H) and 3R ones. The peaks located at binding energies of 207.7 ± 0.2 eV and 210.4 ± 0.2 eV are assigned to the Nb(5+) state in Nb2O5.112–116 Both the 3R phase and oxidization are attributed to the surface or the edges of the flakes because they were not detected using HRTEM and STEM-EDS (oxygen map not shown here, due to the low oxygen content), which are less surface-sensitive than XPS.
 | ||
| Fig. 2 (a) Elastically filtered BF-TEM image of a NbS2 flake, partially suspended on a hole in the carbon support film. (b) HRTEM image of a portion of the suspended region of the NbS2 flake, with (c) the corresponding FFT matching with that of [001]-oriented 2H-NbS2. (d) Magnified portion of the HRTEM image. The lattice planes are also indicated. (e) HAADF-STEM image of a partially suspended NbS2 flake and (f and g) corresponding quantitative STEM-EDS maps of Nb (Kα, in green) and S (Kα, in blue), respectively. | ||
Overall, the morphological, structural and elemental analyses discussed above have shown that NbS2 flakes were successfully produced by LPE in IPA starting from synthetized NbS2 crystals. The NbS2 flakes optimally retained the crystallinity of the native crystal. However, their surface oxidation has to be considered for exploring effectively their electrocatalytic properties for the HER,78 in agreement with theoretical expectations.52–56
Fabrication and electrochemical characterization of NbS2-based electrodes
The direct exploitation of NbS2 as an electrocatalytic material for the HER has been reported only recently exclusively under acidic conditions.58,78,109,117,118 However, just two studies on 2H-NbS2 nanoplatelets54 and mechanically exfoliated tens of nm-thick 3R-NbS2 fakes57 have shown the possibility to approach Pt-like performance after thousands of electrochemical CV cycles, in agreement with theoretical predictions.52–57 The unusual self-optimizing/nanostructuring behaviour of NbS2 nanoplatelets inevitably raises questions about their reliability for a long-term HER (see the discussion in the Introduction section), pointing out the importance to provide new understanding for the fully exploitation of group-5 MX2.60The electrodes were advantageously produced through a room temperature vacuum filtration of the as-produced NbS2 flake dispersions (catalyst mass loading, ∼0.20 mg cm−2) on top of a single-walled carbon nanotube (SWCNT) buckypaper (SWCNT mass loading, ∼ 1.31 mg cm−2). In fact, by taking advantage of the material topologies, the NbS2 flakes cannot pass through the SWCNT tangle.119 Consequently, the SWCNTs effectively filter the NbS2 flakes, which then form a homogeneous film onto the buckypaper. The preparation of the SWCNT dispersion and the details of the fabrication of the electrodes are described in the Experimental section, following similar protocols adopted in our previous studies on LPE-produced transition metal (di or mono)chalcogenide-based electrocatalysts.21,28,48,50,51Fig. 3a shows a photograph of a representative NbS2 electrode, which was manually bent to show its flexibility. The surface morphology of the as-prepared electrodes was characterized by SEM measurements. The NbS2 flakes uniformly cover the mesoporous network of SWCNTs (Fig. 3b), which form a bundle-like morphology (Fig. S2†). The top-view SEM enlargement (Fig. 3c) shows a similar electrode surface to that displayed by NbS2 flakes directly filtered through a nylon membrane without SWCNTs (see Fig. 1i). To improve the catalytic properties of the NbS2 electrodes, a solution-based chemical treatment with hygroscopic Li-TFSI salt was used to enhance the electrolyte accessibility to the HER-active surface of the NbS2 flakes (Fig. 3d). In more detail, the salt ions penetrate into the porous electrodes, occupying inter-flakes sites. The Li+ can also intercalate into both surface niobium oxides (i.e., Nb2O5 and NbO)120–123 and NbS2,98,124–126 thus filling material interstitial sites. Concurrently, the Li-TFSI salt is highly hygroscopic, which means that it strongly adsorbs water, as shown in singular aqueous lithium-ion chemistry.127 The water uptake was confirmed by four-probe resistivity measurement of films of NbS2 flakes, showing a sheet resistance of ∼5.1 × 102 kΩ □−1 after Li-TFSI treatment, whereas the untreated films exhibited a sheet resistance higher than 103 kΩ □−1 due to the surface oxidation-induced passivation of the inter-flake electrical contact. During the air ambient exposure of the NbS2-based electrodes, Li-TFSI can also diffuse outward, as also observed in Li-TFSI-doped organic semiconducting films commonly exploited in perovskite solar cells.128,129 In addition, Li2O or LiOH can also be formed by exposing Li-TFSI-treated electrodes to air or water,130,131 resulting in H2 gas evolution. Therefore, the water dragging, the outward diffusion of Li-TFSI and the H2 evolution may drive the reorientation and nanostructuring (i.e., exfoliation and lateral dimension reduction) of the NbS2 flakes (Fig. S3†). These effects facilitate the water accessibility to the catalytic surface of the NbS2 flakes. Notably, the effect of the Li-TFSI treatment resembles the H2 evolution-aided nanostructuring of the group-5 MX2-based electrodes occurring during electrochemical treatments (e.g., CV scans).54,55 As reported in previous studies,54,55,57 the H2 evolution during CV scans causes a progressive self-optimizing evolution of the NbS2 morphology from thick-to-thin flakes. This change of the NbS2 morphology speeds up the electron transport towards the HER-active sites by shortening the interlayer electron-transfer pathways and facilitating the access of the aqueous protons (H3O+) to the catalytic films.54,55,57 Meanwhile, it increases the electrochemically accessible surface area of the electrode films.54,55,57 However, in contrast to our chemical approach, the electrochemical preconditioning of the electrodes is time-consuming and does not allow any reliable control of the electrode morphology from the start of the HER operation.
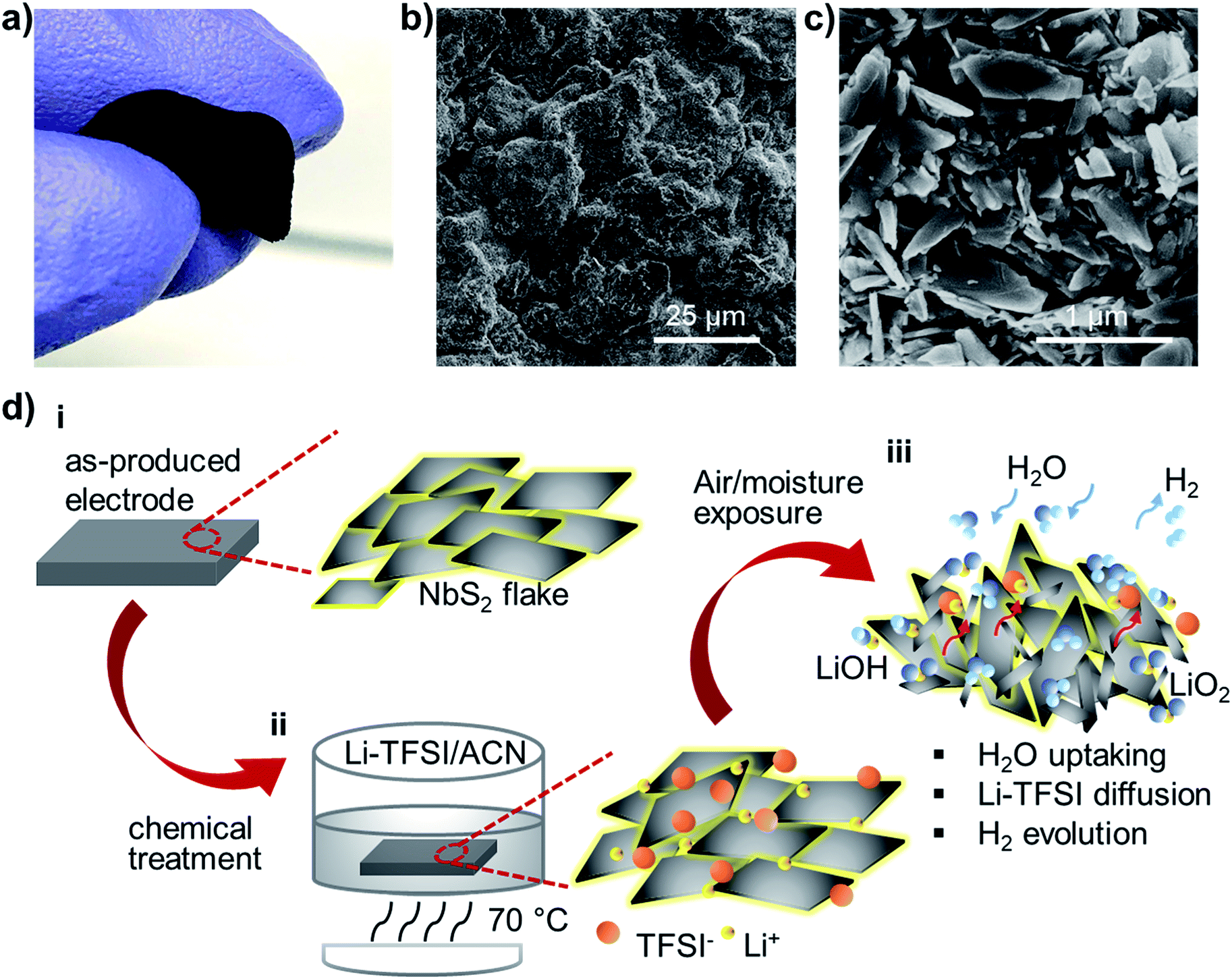 | ||
| Fig. 3 (a) A photograph of a flexible self-standing NbS2-based electrode produced by room-temperature sequential vacuum filtration of SWCNT and NbS2 flake dispersions onto a nylon membrane. (b and c) Top-view SEM images of a NbS2-based electrode. (d) Sketch of the chemical treatment of the NbS2-based electrodes: the as-produced NbS2-based electrode (panel i) and that immersed in a Li-TFSI solution in ACN and heated at 70 °C (panel ii). The salt ions (Li+ and TFSI−) penetrate between the flakes and/or intercalate into surface oxide or NbS2, forming a hygroscopic electrode surface. The latter takes up the water, Li-TFSI diffuses towards the surface, and Li+ species react with air and water to form Li2O and LiOH and evolve H2 (panel iii), reorienting the NbS2 flakes to increase the water accessibility to the electrode. | ||
The modification of the catalyst film morphology (deposited on flat glassy carbon substrates) induced by both chemical and electrochemical treatments was verified by measuring the double layer capacitances (Cdl) of the films (Fig. S4a†). The Cdl is proportional to the electrochemically accessible surface area of the films. These results show that the Cdl of the Li-TFSI-treated catalyst film (∼10.8 mF cm−2) is more than three orders of magnitude higher than that of the untreated film. After the electrochemical treatment (i.e., 1000 CV scans, see details in the Experimental section) in 0.5 M H2SO4, the catalyst film lost a significant amount of material, which was visible to the naked eye (Fig. S4b†). This effect could be a consequence of the catalyst film fragmentation originated from the H2 bubbling,54,55,57 as well as by the dissolution of possible surface oxides, which can form Nb(OH)4− in acidic media.132 However, the Cdl of the electrochemically treated NbS2 film is similar to that of the untreated film, indicating that the CV scans increase the specific electrochemically accessible surface area (defined by the ratio of the electrochemically accessible surface area and the mass loading) of the as-produced electrodes. X-ray photoelectron spectroscopy measurements (Fig. S5†) do not reveal significant changes in the chemical composition of the NbS2 flakes after both the chemical and the electrochemical treatments, indicating that the latter mainly affect the morphology of the catalysts. Beyond the aforementioned morphology changes, it is worth noticing that the presence of alkali-metal cations, including Li+, can control the catalytic activity of a catalyst in alkaline media.133–135 In fact, it has been suggested that Li+ induces steric and/or electronic effects on the interfacial water structure and reactivity, enhancing the HER activity of catalysts anchoring Li+ in the form of complexes.133 Noteworthily, the effective interaction between the Li-TFSI-treated electrode and the electrolyte is also supported by Cdl measurements (Fig. S4†), which show an efficient electrochemical double layer formation (Fig. S5†).
Electrochemical measurements of the electrodes
The HER activity of the as-produced electrodes was evaluated in both acidic (0.5 M H2SO4) and alkaline (1 M KOH) N2-purged solutions at room temperature. To the best of our knowledge, no previous study investigated the HER activity of NbS2 in alkaline solutions, either theoretically or experimentally.Fig. 4a and b show the iR-corrected linear sweep voltammetry (LSV) curves in 0.5 M H2SO4 and 1 M KOH, respectively, for the as-produced electrodes before and after Li-TFSI treatment (samples hereafter named NbS2 and Li-TFSI-treated NbS2, respectively). In addition, the LSV curves obtained for the NbS2 electrode after 1000 CV cycles (samples named NbS2-CV @ 1000 cycles), the commercial Pt/C benchmark and the SWCNTs (catalyst support) are also plotted. In 0.5 M H2SO4, Li-TFSI-treated NbS2 exhibits a HER activity (η10 = 0.31 V) significantly higher than that of as-produced NbS2 (η10 = 0.42 V). The electrochemical cycling also improves the HER activity of NbS2, and NbS2-CV @ 1000 cycles shows an η10 of 0.20 V. Similar results were also obtained in 1 M KOH, where Li-TFSI-treated NbS2 shows an η10 of 0.33 V, similar to that of NbS2-CV @ 1000 (0.31 V), whereas the untreated as-produced NbS2 displays an η10 of 0.49 V. Notably, the electrochemical treatment is also effective to improve the HER activity of Li-TFSI-treated NbS2 (see Fig. S6†) (η10 = 0.27 V in 0.5 M H2SO4; η10 = 0.28 V in 1 M KOH). As discussed in the previous section,54,55,57 the electrochemical treatments cause a progressive self-optimizing evolution of the NbS2 morphology from thick-to-thin flakes. This change of the flake morphology speeds up the electron transport towards the HER-active sites by shortening the interlayer electron-transfer pathways and facilitating the access of the aqueous protons (H3O+) to the catalytic films.54,55,57 Moreover, in the absence of catalyst film detachment/dissolution, this morphology change progressively increases the electrochemically accessible surface area of the electrodes, as demonstrated by Cdl measurement (Fig. S5a†). In this context, it is important to evidence that the SWCNT substrate is effective in maintaining the mechanical stability of the catalyst films during electrochemical tests.68,119 In fact, the electrode using SWCNTs as a support did not show any macroscopic material loss, while the catalyst film deposited onto glassy carbon partially precipitated on the bottom of the cell (see Fig. S4b†).
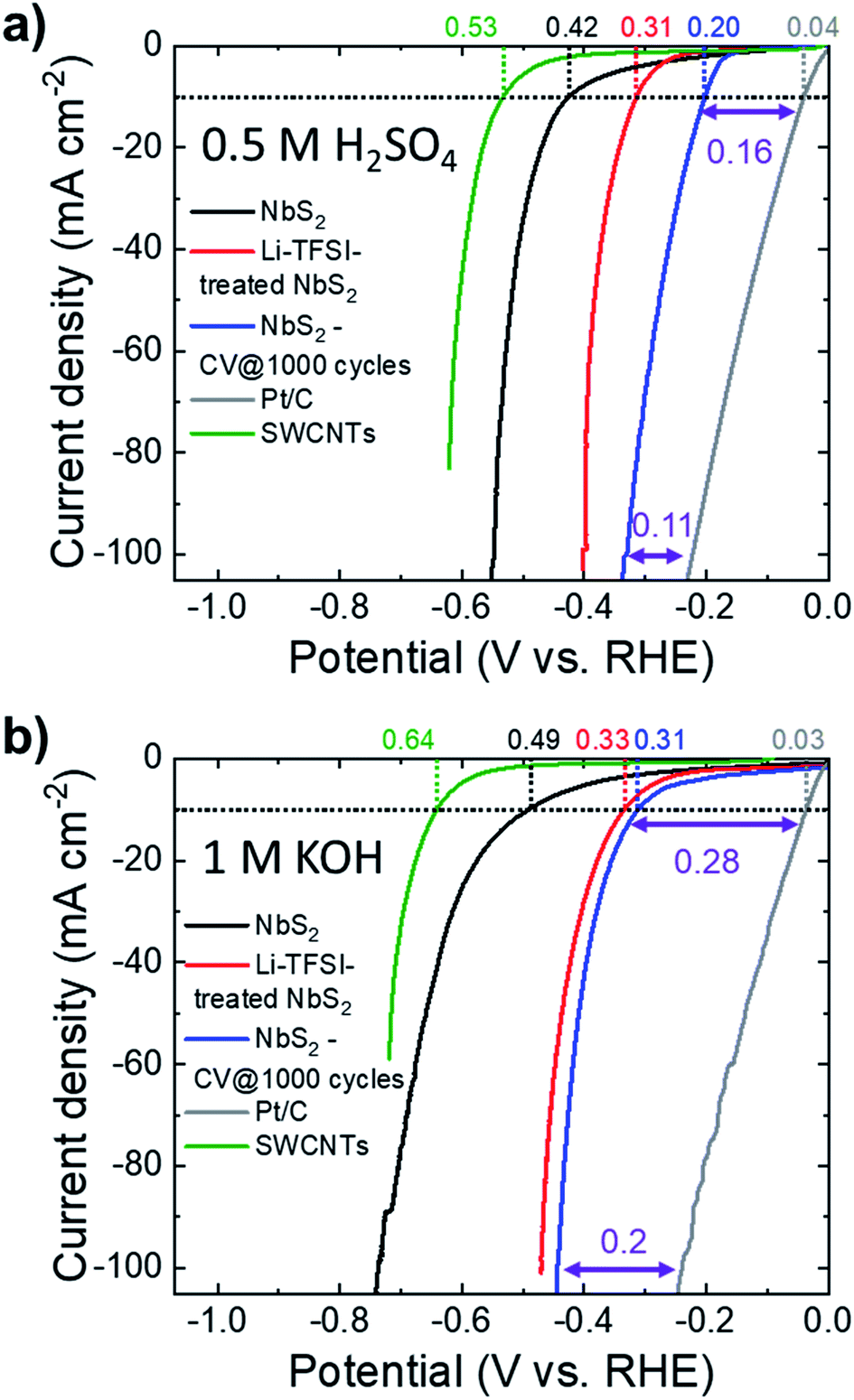 | ||
| Fig. 4 (a and b) iR-corrected LSV curves for NbS2, Li-TFSI-treated NbS2, NbS2-CV @ 1000 cycles in acidic (0.5 M H2SO4) and alkaline (1 M KOH) solutions, respectively. The LSV curves of the Pt/C benchmark and SWCNTs (catalyst support) are also shown for comparison. The η10 values measured for the electrodes are also shown. For NbS2-CV @ 1000 cycles, the HER overpotential vs. the Pt/C electrode at 10 mA cm−2 and 100 mA cm2 is also indicated. | ||
A rigorous analysis of the HER kinetics, including the extrapolation of the Tafel slope and the exchange current (j0), was not carried out in this work because ambiguous results can originate from the presence of the SWCNTs. In fact, SWCNTs have a high surface area that leads to a significant capacitive current density (in the range of mA cm−2) even at a low LSV sweep voltage rate (i.e., ≤5 mV s−1).51 This can be misleading during the interpretation of HER kinetic parameters.136,137 Interestingly, the overpotentials vs. Pt/C of our electrodes at 100 mA cm−2 are significantly lower (e.g., by ∼31% and 29% for NbS2-CV @ 1000 in 0.5 M H2SO4 and 1 M KOH, respectively) than those at a current density of 10 mA cm−2. This agrees with a HER activity also relying on highly abundant basal planes52–57 beyond that of the chalcogen and metal edges.55,56 Noteworthily, this is substantially different from group-6 MX2, in which the HER activity is restricted to only edge/defective sites.39–43
Heterogeneous NbS2:MoSe2 catalysts
To further exploit the potential of NbS2 flakes for the HER by taking advantage of the theoretically calculated database for the catalytic properties of 2D MX2 (ref. 39 and 56) (and combination thereof),56 heterogeneous electrocatalysts were produced by simply mixing LPE-produced NbS2 and MoSe2 flakes (electrocatalyst hereinafter referred to as NbS2:MoSe2). In fact, recent DFT calculations and AIMD simulations56 have shown that the heterogeneous stacking of NbS2 and (2H) MoSe2 flakes tunes the ΔG0H of the resulting heterogeneous configurations in acidic media to optimal thermo-neutral values (i.e., 0 eV) for both edge and basal sites. In more detail, the stacking process initiates an electron transfer from the MoSe2 to NbS2 flakes (Fig. 5a), decreasing the positive ΔG0H of the NbS2 basal plane (theoretical values: 0.061 eV for H-coverage = 8.3% and 0.11 for H-coverage = 16.6% from ref. 138; 0.31 eV from ref. 56; 0.12 eV from ref. 39; 0.15 eV from ref. 139) to nearly 0 eV (Fig. 5b).56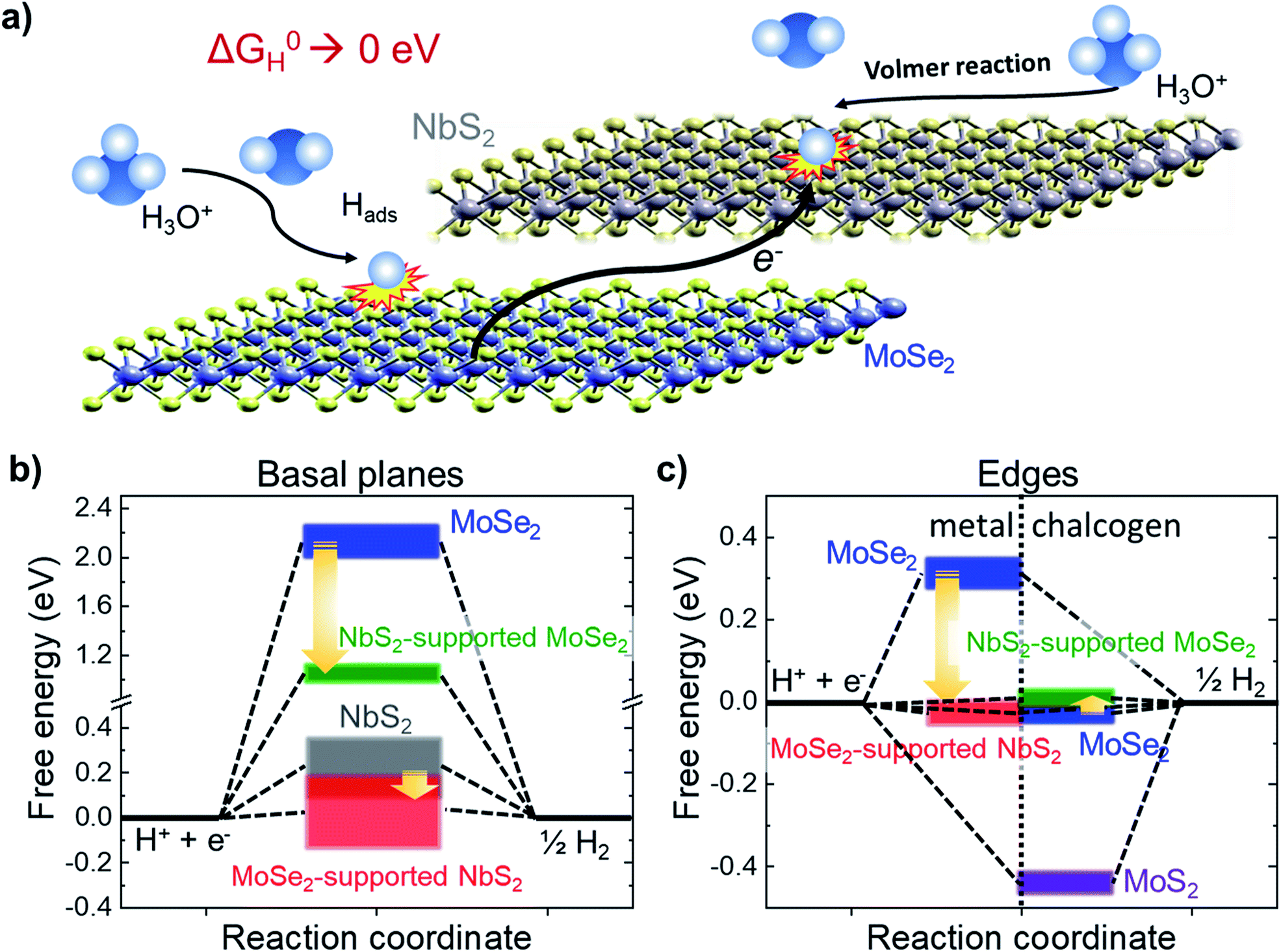 | ||
| Fig. 5 (a) Schematic illustration of the HER activation of the basal plane of the NbS2 flakes and metallic edges of MoSe2 through stacking the NbS2 and MoSe2 flakes. The interaction between the flakes promotes an electron transfer from the MoSe2 flakes towards the NbS2 flakes, decreasing the positive ΔG0H values of the basal plane of the NbS2 flakes and the metallic edges of MoSe2 toward 0 eV, promoting the Volmer reaction (H3O+ + e− ⇄ Hads + H2O in acidic condition), i.e., the first step of the HER. (b) Evolution of ΔG0H for the basal planes of the NbS2 and MoSe2 flakes, and the NbS2:MoSe2 hybrid catalysts. The data ranges have been extrapolated from literature databases (ref. 21, 39, 103 and 104). (c) Evolution of ΔG0H for the metallic (left side) and chalcogen (right side) edges of the MoSe2 flakes, and the NbS2:MoSe2 hybrid catalysts. The data ranges have been extrapolated from literature databases (ref. 21 and 39). | ||
In addition, we recently demonstrated that MoSe2 flakes are also efficient HER catalysts,28,50,51 showing (after appropriate physical or chemical treatments) an η10 < 0.10 V in both acidic and alkaline solutions.28,50,51 Theoretical calculations have proposed that the HER activity of MoSe2 flakes relies on edge sites (although it is still up for debate whether they are Mo or Se-edge sites, or both),39,56 whereas basal planes are inert (ΔG0H > 2 eV),39,56 also when stacked onto NbS2 flakes (ΔG0H ∼ 0.98 eV). As shown in Fig. 5c, the alleged HER activity of the Se-edge sites in MoSe2 flakes makes the latter advantageous over MoS2 flakes, whose S-edges strongly bind the H, resulting in negative ΔG0H (∼−0.45 eV).39,56 The ΔG0H of the chalcogen edges of MoSe2 flakes (∼−0.05 eV from ref. 39 and 56) is slightly varied around the thermoneutral value when MoSe2 flakes are stacked onto the NbS2 flakes (ΔG0H ∼ 0.06 eV). In addition, by stacking MoSe2 flakes onto the NbS2 flakes, the positive ΔG0H of metallic edges of MoSe2 (e.g., 0.31 eV from ref. 56) are low-shifted toward slightly negative near zero values (−0.01 eV from ref. 56). Similar effects have been also theoretically predicted and experimentally confirmed for MoS2-coated NbS2 catalysts.109 Noteworthily, the preparation of heterogeneous catalysts from LPE-produced catalysts is a scalable approach, which does not resort to any morphological/structural and/or chemical modifications of the LPE-produced flakes, such as the introduction of artificial defects, chemical doping of heteroatoms or strain constraints.56
The details regarding the production of MoSe2 flakes are reported in the Experimental section. The morphological, structural, and chemical characterization of the LPE-produced MoSe2 flakes has been recently reported by our studies.28,50,51 In particular, the MoSe2 flakes have a lateral size (measured by TEM) and thickness (measured by AFM) following lognormal distributions peaked at ∼30.0 nm and ∼2.9 nm, respectively.28,50,51 Optical absorbance, Raman spectroscopy and XRD measurements have shown that MoSe2 flakes optimally preserve the MoSe2 bulk crystallinity.28,50,51 In addition, the analysis of HRTEM images of the MoSe2 flakes and the corresponding FFTs confirmed that the flakes display a hexagonal phase,28 in agreement with XRD and Raman analyses.28,50,51
Following the protocols previously adopted for NbS2 electrodes, MoSe2 and NbS2:MoSe2 (material mass ratio of 1:1) electrodes were produced by vacuum filtration deposition of their dispersions on top of the SWNTs. Fig. S7† shows the top-view SEM images of the MoSe2 and NbS2:MoSe2 electrodes, respectively. The as-produced electrodes display a surface uniformly covered by the flakes. In particular, the heterogeneous electrode (i.e., NbS2:MoSe2) shows a morphology resembling those of the MoSe2 electrode, whose flakes are smaller than NbS2 flakes, in agreement with the characterization of the materials (see Fig. 1g for NbS2 flakes and ref. 28 and 51 for MoSe2 flakes). Although it is reasonable to assume that the vacuum filtration of a mixture of NbS2 and MoSe2 flakes in dispersion intrinsically promotes the stacking between flakes of different materials, SEM-EDS measurements were carried out to prove at least the absence of single material domains. The cross-sectional SEM-EDS analysis of the heterogeneous films composed of NbS2 and MoSe2 flakes (Fig. S8a†) shows homogeneous distributions for both Nb and Mo elements, which are compatible with an optimal mixing in the dispersion used for the preparation of the electrodes. The Li-TFSI treatment of the MoSe2 and NbS2:MoSe2 electrodes is expected to cause the same effects observed for the NbS2 electrodes (i.e., hygroscopicity and nanostructuring of the electrodes). The SEM-EDS analysis of the Li-TFSI-treated heterogeneous films (Fig. S8b†) leads to similar results to those of the untreated film (i.e., a homogeneous distribution of both Nb and Mo elements), suggesting a similar packing between NbS2 and MoSe2 flakes. The top-view SEM-EDS analyses of the untreated and the Li-TFSI heterogeneous films (Fig. S8c and d,† respectively) also show homogeneous distributions of the composing elements, further confirming the absence of single material domains. Fig. 6a and b show the LSV curves in 0.5 M H2SO4 and 1 M KOH, respectively, for the heterogeneous electrodes before and after the Li-TFSI treatment (electrodes named NbS2:MoSe2 and Li-TFSI-treated NbS2:MoSe2). In addition, we also plotted the LSV curves obtained for the untreated heterogeneous electrode after 1000 CV cycles (NbS2:MoSe2-CV @ 1000 cycles), the commercial Pt/C benchmark and the references NbS2 and MoSe2. The electrochemical characterization of the electrode based on solely MoSe2 before and after Li-TFSI treatment, as well as after 1000 CV cycles, is reported in Fig. S9,† showing an improvement of the initial HER activity (η10 = 0.28 and 0.35 V in 0.5 M H2SO4 and 1 M KOH, respectively) after both Li-TFSI treatment (η10 = 0.27 and 0.30 V in 0.5 M H2SO4 and 1 M KOH, respectively) and electrochemical cycling (η10 equal to 0.18 and 0.3 V in 0.5 M H2SO4 and 1 M KOH, respectively). However, the beneficial effects of the electrode treatments are less pronounced compared to the case of NbS2 electrodes. The heterogeneous NbS2:MoSe2 electrodes show a HER activity (η10 = 0.23 and 0.29 V in 0.5 M H2SO4 and 1 M KOH, respectively) which is higher than that of both NbS2 and MoSe2 electrodes. As reported for the individual counterpart-based electrodes, both the Li-TFSI treatment and the electrochemical cycling increase the HER activity of the heterogeneous electrodes. In 0.5 M H2SO4, the Li-TFSI-treated NbS2:MoSe2 electrode reaches an η10 of 0.14 V, approaching that of NbS2:MoSe2-CV @ 1000 cycles (η10 = 0.10 V). A similar trend was observed in 1 M KOH, in which the Li-TFSI-treated NbS2:MoSe2 electrode shows an η10 of 0.26 V, whereas NbS2:MoSe2-CV @ 1000 cycles achieves an η10 of 0.22 V. At a current density of 100 mA cm−2, NbS2:MoSe2-CV @ 1000 cycles displays a HER activity higher than that of the Pt/C benchmark in an acidic environment. Under alkaline conditions, the HER overpotential vs. the Pt/C benchmark (0.12 V) at 100 mA cm−2 is reduced by 0.03 V compared to that measured at 10 mA cm−2 (0.19 V).
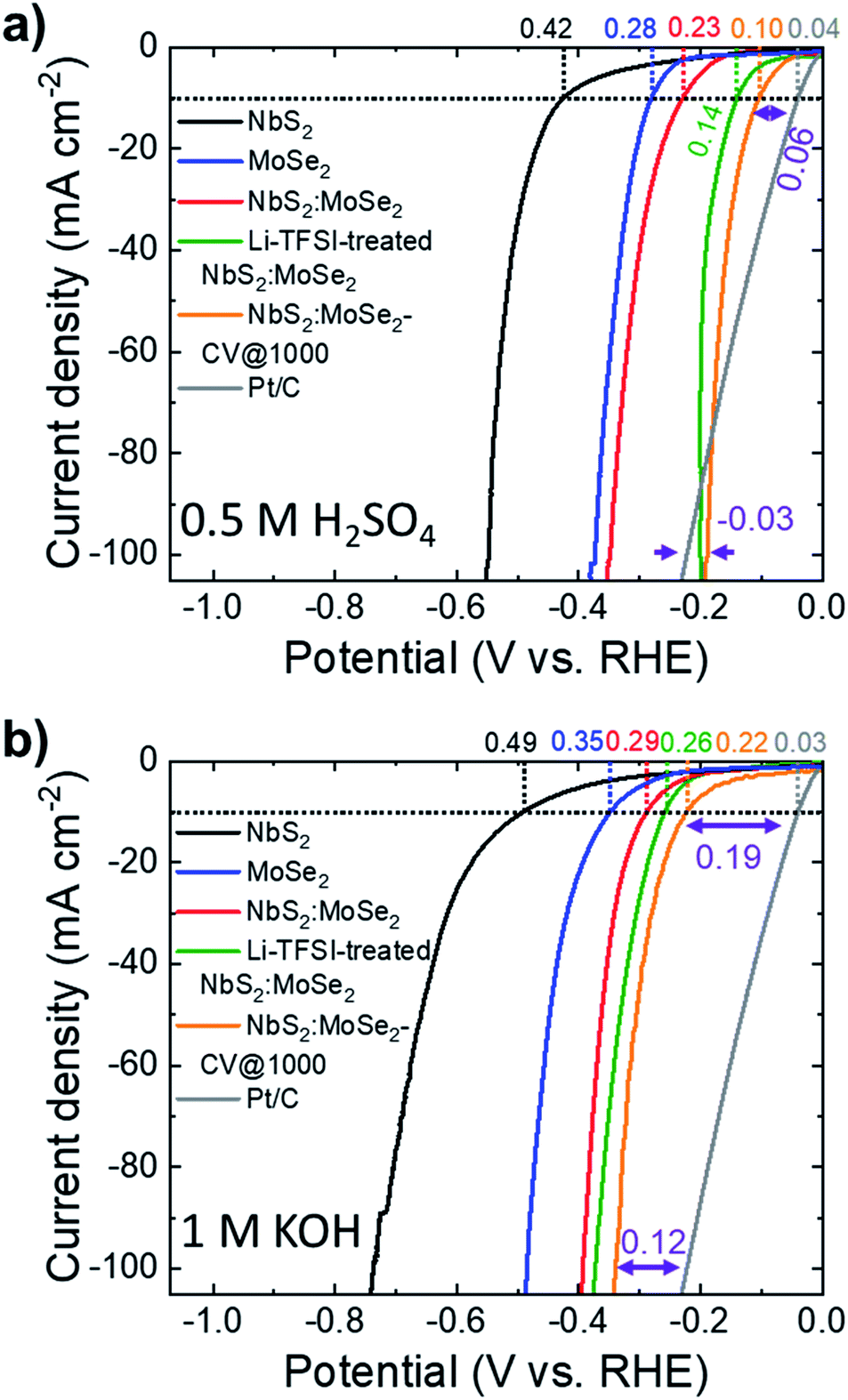 | ||
| Fig. 6 (a and b) iR-corrected LSV curves for NbS2:MoSe2, Li-TFSI-treated NbS2:MoSe2 and NbS2-CV @ 1000 cycles in acidic (0.5 M H2SO4) and alkaline (1 M KOH) solutions, respectively. The LSV curves of the Pt/C benchmark and NbS2 and MoSe2 references are also shown for comparison. The η10 values measured for the electrodes are also shown. For NbS2:MoSe2-CV @ 1000 cycles, the HER overpotential vs. the Pt/C electrode at 10 mA cm−2 and 100 mA cm2 is indicated. | ||
Beyond the electrocatalytic activity, the long-term steady-state durability of the catalysts is another important criterion for their practical utilization. Fig. 7a shows the chronoamperometric measurements for the Li-TFSI-treated NbS2:MoSe2 electrodes at a fixed potential corresponding to an initial cathodic current density of 80 mA cm−2. In 0.5 M H2SO4, the electrodes retained 97.3% of the initial current densities after 12 h. In 1 M KOH, the electrode improves the HER activity over time, increasing its initial current density by +22.2%. Fig. 7b shows the iR-corrected LSV curves of the electrodes measured after the stability test, showing an η10 of 0.11 and 0.12 V in 0.5 M H2SO4 and 1 M KOH, respectively.
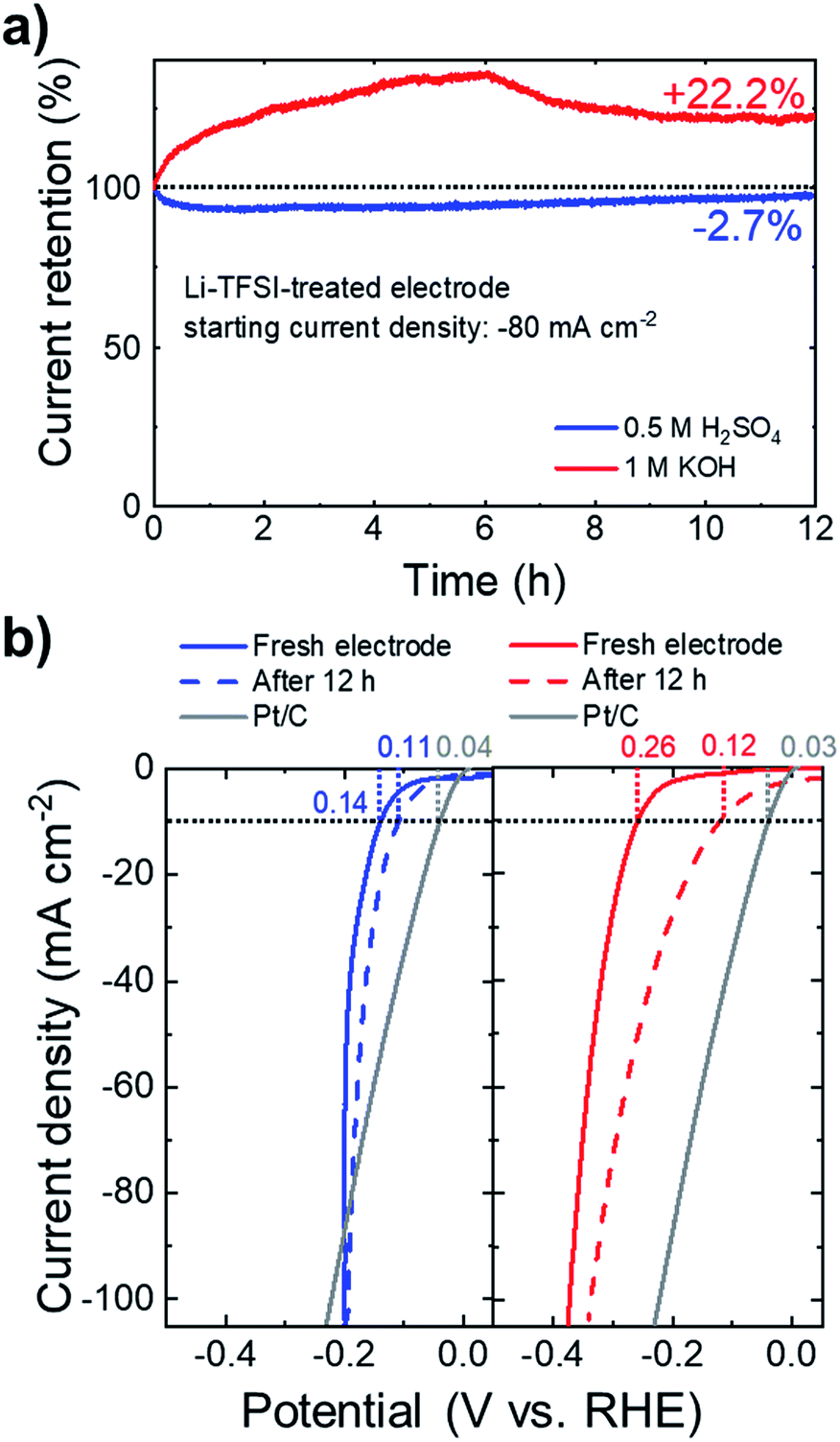 | ||
| Fig. 7 (a) Chronoamperometry measurement of Li-TFSI-treated NbS2:MoSe2 in 0.5 M H2SO4 (blue line) and 1 M KOH (red line). The percentage increase of the starting cathodic current density (80 mA cm−2) after 12 h is also indicated. (b) The LSV curves of Li-TFSI-treated NbS2:MoSe2 in 0.5 M H2SO4 (left panel) and 1 M KOH (red panel). The LSV curves of the PT/C benchmark are shown for comparison. The η10 values measured for the electrodes are also indicated. | ||
Under acidic conditions, the stability of the electrode is further verified by XPS analysis (Fig. S10a†), which does not show relevant chemical alterations of the NbS2 and MoSe2 flakes after the long-term test. Noteworthily, these results have been achieved without using any binder, such as Nafion, which could prospectively improve further the mechanical stability of the electrodes during H2 evolution.140,141 In fact, although the gas evolution, causing the nanostructuring of the electrodes, has been correlated with the self-optimization behaviour of catalytic group-5 MX2 in acidic media,54,55,57,77 it may imply a loss of catalytic materials, which should be controlled for practical purposes. In this context, the Li-TFSI-treated electrodes perform similarly to the electrochemically cycled electrodes, but do not require in operando pre-conditioning of the electrodes. Therefore, our approach to chemically treat the NbS2-based electrodes could be beneficial for controlling the design of the group-5 MX2 catalyst, as well as for their long-term durability. Under alkaline conditions, the remarkable decrease of η10 (from 0.26 to 0.12 V) is still under investigation. In fact, the XPS (Fig. S10†) data reveal a significant decrease of the NbS2-related components (i.e., those attributed to Nb(4+) and S 2p). These observations indicate a progressive oxidation and a possible dissolution of the NbS2 flakes. The decrease of the Nb atomic content relative to that of Mo is further verified by SEM-EDS analysis with a clear intensity decrease of the Nb (Kα) peak compared to the Mo (Kα) peak (Fig. S11†). Moreover, the peaks of the doublet assigned to such Nb(5+) (∼207.4 and ∼210.1 eV) are located at binding energy slightly lower than those of the doublet assigned to Nb2O5 in the as-produced NbS2 flakes (typically at ∼207.7 eV and ∼210.4 eV,112–116 see Fig. S1†). Therefore, such XPS peaks could be ascribed to either sub-stoichiometric Nb2O5−x or hydr(oxy)oxide species. Noteworthily, the latter can synergistically interact with MoSe2 flakes (which are chemically stable, see Fig. S10† and ref. 28 and 51), increasing the HER activity compared to that of the initial electrode (see Fig. 7a). In fact, it has been recently demonstrated that the presence of transition metal oxides (or hydroxides) on an MX2 surface can also increase the HER activity of the pristine MX2 under alkaline conditions,28,51,142 similarly to what was observed in noble metal-based electrocatalysts.143–145
Conclusion
In summary, we have produced phase-mixed NbS2 flakes (i.e., 1H-, 2H- and 3R-NbS2 flakes) by an environmentally friendly liquid phase exfoliation (LPE) of synthetized NbS2 crystals in 2-propanol (IPA). On the basis of a literature database for theoretically calculated Gibbs free energy of adsorbed atomic H (ΔG0H), NbS2 flakes have been investigated as efficient HER catalyst candidates. Vacuum filtration has been used as a scalable approach to manufacture electrodes based on NbS2 flakes. The as-produced electrodes were also chemically treated with a Li-TFSI bath in order to vertically orient the NbS2 flakes and to induce the nanostructuring of their morphology, increasing the water accessibility to their surface. The full potential of NbS2 flakes was exploited in a heterogeneous electrode obtained by mixing NbS2 and MoSe2 flakes. In fact, the flake stacking increases the HER activity of both the basal plane of the NbS2 flakes and the metallic edge of MoSe2, without affecting that of the catalytic sites of the single counterparts. In acidic solution (0.5 M H2SO4), the designed NbS2-based electrocatalysts reach an overpotential at a cathodic current density of 10 mA cm−2 (η10) as low as 0.14 and 0.10 V after chemical Li-TFSI treatment and electrochemical cycling, respectively. At current density ≥100 mA cm−2 the HER activity of the electrodes is higher than that of the Pt/C benchmark (overpotential reduced by 0.03 V). For the first time, the HER activity of NbS2-based electrocatalysts is investigated in alkaline solutions (1 M KOH), showing a promising η10 of 0.26 and 0.22 V after chemical Li-TFSI treatment and electrochemical cycling, respectively. The durability of the electrodes is validated over 12 h of continuous operation at a fixed potential corresponding to an initial cathodic current density of 80 mA cm−2. The results show an optimal performance retention of the chemically treated electrodes in 0.5 M H2SO4 (97.3%). In 1 M KOH, the electrodes significantly improve their initial cathodic current density (+22.2%), displaying an η10 of only 0.12 V after 12 h. However, chemical changes affect the NbS2 flakes during HER operation, and further studies are needed to validate practical applications. Our results provide new insight into the exploitation of metallic group-5 TMDs for the HER under pH-universal conditions via scalable material preparation and electrode processing. The chemical treatment of the electrodes may be advantageous to their in operando electrochemical cycling, because it can be prospectively coupled with the use of proton conducting binders to avoid catalyst dissolution/loss.Experimental
Materials
Nb (99.9%, <100 μm) and S (99.999%, <6 mm) powders were purchased from Strem, USA. H2SO4 (99.999%), KOH (≥85% purity, ACS reagent, pellets), Pt/C (10 wt% loading) and Nafion solution (5 wt%) were purchased from Sigma Aldrich. The SWCNTs (>90% purity) were purchased from Cheap Tubes. The MoSe2 crystal was purchased from HQ graphene.Crystals' synthesis and exfoliation
NbS2 crystals were made by direct synthesis from elements. An amount of Nb and S powders with a Nb:S stoichiometry of ca. 1:2 (1 wt% excess of S) and corresponding to a total mass of 10 g of NbS2 was placed in a quartz glass ampoule (20 mm × 120 mm) and sealed under high vacuum (1 × 10−3 Pa). The ampoule was heated at 450 °C for 12 h and subsequently at 600 °C for 48 h. Finally the derived product was treated at 900 °C for 48 h and cooled down to room temperature over a period of 24 h, obtaining NbS2 crystal powder. The NbS2 and MoSe2 flakes were produced by LPE in IPA from the as-synthetized NbS2 crystal powder and purchased MoSe2 crystal,63,80 followed by SBS to remove the unexfoliated material by ultracentrifugation.81,82 Experimentally, 50 mg of bulk crystals were added to 50 mL of anhydrous IPA and ultrasonicated in a bath sonicator (Branson® 5800 cleaner, Branson Ultrasonics) for 6 h. The resulting dispersions were ultracentrifuged at 2700 g (Optima™ XE-90 with a SW32Ti rotor, Beckman Coulter) for 20 min at 15 °C in order to separate un-exfoliated bulk crystals (collected as sediment) from the exfoliated materials that remained in the supernatant. Then, 80% of the supernatant was collected by pipetting, obtaining an exfoliated material dispersion. The concentration of the NbS2 and MoSe2 flake dispersions was 0.30 g L−1 and 0.25 g L−1, respectively.
Material dispersions' preparation
The dispersions of NbS2 and MoSe2 flakes were used as-produced. The heterogeneous dispersions of the NbS2 and MoSe2 flakes were obtained by mixing the single material component dispersions with a material weight ratio (w/w) of 1:1. The SWCNT dispersions were produced by dispersing SWCNTs in N-methyl-2-pyrrolidone (NMP) with a concentration of 0.2 g L−1 by means of ultrasonication-based de-bundling.146–148 In particular, 10 mg of SWCNT powder was added to 50 mL of NMP. The dispersion was then ultrasonicated for 30 min by using a sonic tip (Vibra-cell 75185, Sonics) with the vibration amplitude set to 45%. The sonic tip was pulsed for 5 s on and 2 s off to reduce the solvent heating, which was also mitigated by using an ice bath around the vessel. The dispersion of Pt/C was produced by dissolving 4 mg of Pt/C and 80 μL of Nafion solution in 1 mL of 1:4 v/v ethanol/water.
Materials' characterization
The SEM analysis of the as-synthesized crystals was carried out using a JEOL JSM-7500FA equipped with a cold FEG, operated at 10 kV accelerating voltage. The EDS analyses were performed using an Oxford X-Max 80 system equipped with an 80 mm2 silicon drift detector (SDD) and using Oxford's AZtec TruMap software. The displayed SEM-EDS maps were obtained by background subtraction and peak deconvolution in order to correct for the overlapping S (Kα peak) and Nb (Lβ peak) features in the EDS spectrum. The surface of the samples was cleaned by mechanical exfoliation and imaged without any conductive coating. The BF-TEM images of the NbS2 flakes were acquired with a JEM 1011 (JEOL) TEM (thermionic W filament), operated at 100 kV. The morphological and statistical analysis was performed by using ImageJ software (NIH) and OriginPro 9.1 software (OriginLab), respectively. The samples for the TEM measurements were prepared by drop casting the as-prepared exfoliated material dispersions onto ultrathin C-on-holey C-coated Cu grids and rinsed with deionized water and subsequently dried overnight under vacuum. The HRTEM, HAADF-STEM imaging and STEM-EDS analyses were carried out on an image-Cs-corrected JEOL JEM-2200FS TEM (Schottky emitter), operated at 200 kV, equipped with an in-column image filter (Ω-type) and a Bruker XFlash 5060 EDS detector. For these analyses, the NbS2 flake dispersion was drop cast onto a holey-carbon-coated Cu grid. The displayed STEM-EDS maps are “quantitative”, in analogy to what was done for SEM-EDS maps of the crystals.The AFM images were acquired with a Nanowizard III (JPK Instruments, Germany), mounted on an Axio Observer D1 (Carl Zeiss, Germany) inverted optical microscope. The AFM measurements were carried out by means of PPP-NCHR cantilevers (Nanosensors, USA) having a nominal tip diameter of 10 nm. We used a drive frequency of ∼295 kHz. We collected intermittent contact mode AFM images (512 × 512 data points) of 2.5 × 2.5 μm2 by keeping the working set point above 70% of the free oscillation amplitude. We used a scan rate of 0.7 Hz for the acquisition of the images. JPK Data Processing software (JPK Instruments, Germany) was exploited to process the height profiles, while the data were analysed by using OriginPro 9.1 software. The latter was also used to carry out the statistical analysis on multiple AFM images for all the tested samples. The samples were prepared by drop-casting the as-prepared exfoliated material dispersions onto mica sheets (G250-1, Agar Scientific Ltd.) and dried overnight under vacuum. The XRD measurements were acquired with a PANalytical Empyrean using Cu Kα radiation. The samples for XRD were prepared by drop-casting the as-prepared exfoliated material dispersions onto Si/SiO2 substrates and dried overnight under vacuum. Raman spectroscopy measurements were performed by using a Renishaw microRaman inVia 1000 mounted with a 50× objective, with an excitation wavelength of 532 nm and an incident power of 1 mW on the samples. For each sample, we collected 50 spectra. The samples were prepared by drop casting the as-prepared exfoliated material dispersions onto Si/SiO2 substrates and subsequently dried under vacuum. The XPS characterization was performed with a Kratos Axis UltraDLD spectrometer, having a monochromatic Al Kα source (15 kV, 20 mA). The spectra were acquired on a 300 × 700 μm2 area. A constant pass energy of 160 eV and an energy step of 1 eV were used to collect wide scans. High-resolution spectra were acquired at a constant pass energy of 10 eV with an energy step of 0.1 eV. The binding energy scale was referenced to the C 1s peak at 284.8 eV. The spectra were analysed using CasaXPS software (version 2.3.17). The samples were prepared by drop casting the NbS2 flake dispersions onto a Si/SiO2 substrate (LDB Technologies Ltd), followed by a drying process under vacuum.
Electrodes' fabrication
The electrodes were produced by sequentially depositing the SWCNT and exfoliated catalytic material (NbS2, MoSe2 and heterogeneous NbS2:MoSe2) dispersions onto nylon membranes (Whatman® nylon membrane filters, 0.2 μm pore size) through a vacuum filtration process.48 The material mass loading was 1.31 mg cm−2 for SWCNTs and 0.2 mg cm−2 for the exfoliated catalytic materials. The electrode area was 3.8 cm2. Additional electrodes were produced by depositing the as-produced NbS2 flake dispersion onto glassy carbon by a drop casting method (NbS2 flake mass loading = 0.5 mg cm−2). Before the electrochemical characterization, the as-prepared electrodes were dried overnight at room temperature. The electrodes were chemically treated with Li-TFSI inside a glove box. Experimentally, 2 mg of Li-TFSI (Sigma-Aldrich) were dissolved in 10 mL of anhydrous acetonitrile (ACN) (Sigma-Aldrich) and stirred for 15 min at room temperature to make a 0.2 g L−1 solution. Subsequently, the electrodes were immersed in the Li-TFSI solution in a closed vial for 20 min at a temperature of 70 °C. The electrodes were removed (without rinsing) and subsequently annealed at 100 °C for 5 min in air in order to remove the residuals of ACN. Electrodes made entirely of SWCNTs were also produced as a reference with a mass loading of 1.31 mg cm−2. A Pt/C electrode was produced as a benchmark for the HER by depositing the corresponding dispersion onto glassy carbon substrates (Sigma Aldrich). The active material mass loading of the electrodes was 0.262 mg cm−2 for Pt/C, in agreement with previously reported protocols.28,51Electrode characterization
The XPS analyses of the as-produced electrodes and the electrodes after chemical and electrochemical treatments were performed with the same set-up and parameters used for the characterization of the materials. The detailed SEM images of the electrodes were collected on a Helios Nanolab® 600 DualBeam microscope (FEI Company) and 5 kV and 0.2 nA as measurement conditions. The SEM-EDS measurements were performed on a JEOL JSM-6490LA SEM at 30 kV. The electrochemical measurements were performed at room temperature in a flat-bottom fused silica cell using the three-electrode configuration of a potentiostat/galvanostat station (VMP3, Biologic), controlled via own software. A glassy carbon rod and saturated KCl Ag/AgCl were employed as the counter electrode and reference electrode, respectively. The measurements were carried out in 200 mL of 0.5 M H2SO4 or 1 M KOH. Before starting the measurements, the oxygen was purged from the electrolyte by flowing N2 gas throughout the liquid volume using a porous frit. The Nernst equation: ERHE = EAg/AgCl + 0.059 × pH + E0Ag/AgCl, where ERHE is the converted potential vs. RHE, EAg/AgCl is the experimental potential measured against the Ag/AgCl reference electrode, and E0Ag/AgCl is the standard potential of Ag/AgCl at 25 °C (0.1976 V vs. RHE), was used to convert the potential difference between the working electrode and the Ag/AgCl reference electrode to the reversible hydrogen electrode (RHE) scale. The double-layer capacitances (Cdl) of the untreated and treated NbS2 films deposited onto glassy carbon were estimated by cyclic voltammetry (CV) measurements in a non-faradaic region of potential (between 0.2 and 0.4 V vs. RHE) at various potential scan rates (ranging from 20 to 600 mV s−1). Cyclic voltammetry was also exploited for the electrochemical treatment of the electrodes. Experimentally, 1000 CV scans were carried out between 0.25 and 0.75 V vs. RHE at a potential scan rate of 100 mV s−1. The LSV curves of the electrodes using a SWCNT substrate were acquired at a 5 mV s−1 scan rate and were iR-corrected by considering i as the measured working electrode current and R as the series resistance arising from the working electrode substrate and electrolyte resistances. Electrochemical impedance spectroscopy (EIS) measurements were performed on the electrodes at open circuit potential and at a frequency of 10 kHz to measure R (in agreement with methods reported in previous studies).48,50,51 The stability tests were performed by chronoamperometry measurements, i.e., by measuring the current in potentiostatic mode over time, at a fixed overpotential corresponding to a cathodic current density of 80 mA cm−2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 785219—GrapheneCore2. We thank Lea Pasquale and Marco Salerno (Materials Characterization Facility – Istituto Italiano di Tecnologia) for support in XRD and AFM data acquisition; the Electron Microscopy Facility – Istituto Italiano di Tecnologia for support in TEM and SEM/EDS data acquisition; and the Clean Room facility – Istituto Italiano di Tecnologia for the access to carry out part of the SEM characterization. This work was supported by the project Advanced Functional Nanorobots (reg. No. CZ.02.1.01/0.0/0.0/15_003/0000444 financed by the EFRR). Z. S and V. M. were supported by the Czech Science Foundation (GACR No. 17-11456S).Notes and references
- B. Johnston, M. C. Mayo and A. Khare, Technovation, 2005, 25, 569–585 CrossRef.
- M. Momirlan and T. N. Veziroglu, Int. J. Hydrogen Energy, 2005, 30, 795–802 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- W. E. Winsche, K. C. Hoffman and F. J. Salzano, Science, 1973, 180, 1325–1332 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- K. E. Ayers, J. N. Renner, N. Danilovic, J. X. Wang, Y. Zhang, R. Maric and H. Yu, Catal. Today, 2016, 262, 121–132 CrossRef CAS.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- D. A. Singer, Econ. Geol., 1995, 90, 88–104 CrossRef CAS.
- C. Sealy, Mater. Today, 2008, 12, 65–68 CrossRef.
- M. Ball and M. Weeda, Int. J. Hydrogen Energy, 1995, 90, 88–104 Search PubMed.
- P. Wang, K. Jiang, G. Wang, J. Yao and X. Huang, Angew. Chem., Int. Ed., 2016, 55, 12859–12863 CrossRef CAS PubMed.
- Z. Cao, Q. Chen, J. Zhang, H. Li, Y. Jiang, S. Shen, G. Fu, B. Lu, Z. Xie and L. Zheng, Nat. Commun., 2017, 8, 15131 CrossRef PubMed.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- L. Yu, S. Song, B. McElhenny, F. Ding, D. Luo, Y. Yu, S. Chen and Z. Ren, J. Mater. Chem. A, 2019, 7, 19728–19732 RSC.
- J. X. Feng, J. Q. Wu, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2018, 140, 610–617 CrossRef CAS PubMed.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 RSC.
- Y. Hou, M. Qiu, T. Zhang, X. Zhuang, C.-S. Kim, C. Yuan and X. Feng, Adv. Mater., 2017, 29, 1701589 CrossRef PubMed.
- E. Petroni, E. Lago, S. Bellani, D. W. Boukhvalov, A. Politano, B. Gürbulak, S. Duman, M. Prato, S. Gentiluomo, R. Oropesa-Nuñez, J.-K. Panda, P. S. Toth, A. E. Del Rio Castillo, V. Pellegrini and F. Bonaccorso, Small, 2018, 14, 1800749 CrossRef PubMed.
- Y. Hou, M. Qiu, G. Nam, M. G. Kim, T. Zhang, K. Liu, X. Zhuang, J. Cho, C. Yuan and X. Feng, Nano Lett., 2017, 17, 4202–4209 CrossRef CAS PubMed.
- Y. Hou, M. Qiu, M. G. Kim, P. Liu, G. Nam, T. Zhang, X. Zhuang, B. Yang, J. Cho, M. Chen, C. Yuan, L. Lei and X. Feng, Nat. Commun., 2019, 10, 1392 CrossRef PubMed.
- F. H. Saadi, A. I. Carim, W. S. Drisdell, S. Gul, J. H. Baricuatro, J. Yano, M. P. Soriaga and N. S. Lewis, J. Am. Chem. Soc., 2017, 139, 12927–12930 CrossRef CAS PubMed.
- P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J.-Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 RSC.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., 2014, 126, 6525–6528 CrossRef.
- L. Liao, S. Wang, J. Xiao, X. Bian, Y. Zhang, M. D. Scanlon, X. Hu, Y. Tang, B. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 RSC.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, M. Prato, B. Martín-García, R. Brescia and F. Bonaccorso, ACS Nano, 2019, 13, 3162–3176 CrossRef CAS PubMed.
- J. Lai, S. Li, F. Wu, M. Saqib, R. Luque and G. Xu, Energy Environ. Sci., 2016, 9, 1210–1214 RSC.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, B. Martín-García, M. Prato and F. Bonaccorso, ACS Appl. Energy Mater., 2019, 2, 5373–5379 CrossRef CAS.
- C. Lei, Y. Wang, Y. Hou, P. Liu, J. Yang, T. Zhang, X. Zhuang, M. Chen, B. Yang, L. Lei, C. Yuan, M. Qiu and X. Feng, Energy Environ. Sci., 2019, 12, 149–156 RSC.
- M. Chhowalla, Z. Liu and H. Zhang, Chem. Soc. Rev., 2015, 44, 2584–2586 RSC.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 RSC.
- J. Yang and H. S. Shin, J. Mater. Chem. A, 2014, 2, 5979–5985 RSC.
- D. Merki and X. Hu, Energy Environ. Sci., 2011, 4, 3878–3888 RSC.
- M. Pumera, Z. Sofer and A. Ambrosi, J. Mater. Chem. A, 2014, 2, 8981–8987 RSC.
- H. Li, X. Jia, Q. Zhang and X. Wang, Chem, 2018, 4, 1510–1537 CrossRef CAS PubMed.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Surf. Sci., 2015, 640, 133–140 CrossRef CAS.
- D. Er, H. Ye, N. C. Frey, H. Kumar, J. Lou and V. B. Shenoy, Nano Lett., 2018, 18, 3943–3949 CrossRef CAS PubMed.
- C. Tsai, H. Li, S. Park, J. Park, H. S. Han, J. K. Nørskov, X. Zheng and F. Abild-Pedersen, Nat. Commun., 2017, 8, 15113 CrossRef PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963 CrossRef CAS PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- D. McAteer, Z. Gholamvand, N. McEvoy, A. Harvey, E. O'Malley, G. S. Duesberg and J. N. Coleman, ACS Nano, 2016, 10, 672–683 CrossRef CAS PubMed.
- Y. Yu, S.-Y. Huang, Y. Li, S. N. Steinmann, W. Yang and L. Cao, Nano Lett., 2014, 14, 553–558 CrossRef CAS PubMed.
- Y. Huang, Y.-E. Miao, J. Fu, S. Mo, C. Wei and T. Liu, J. Mater. Chem. A, 2015, 3, 16263–16271 RSC.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed.
- L. Najafi, S. Bellani, B. Martín-García, R. Oropesa-Nuñez, A. E. Del Rio Castillo, M. Prato, I. Moreels and F. Bonaccorso, Chem. Mater., 2017, 29, 5782–5786 CrossRef CAS.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, A. Ansaldo, M. Prato, A. E. Del Rio Castillo and F. Bonaccorso, Adv. Energy Mater., 2018, 8, 1703212 CrossRef.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, A. Ansaldo, M. Prato, A. E. Del Rio Castillo and F. Bonaccorso, Adv. Energy Mater., 2018, 8, 1801764 CrossRef.
- C. Zhu, D. Gao, J. Ding, D. Chao and J. Wang, Chem. Soc. Rev., 2018, 47, 4332–4356 RSC.
- Y. Huan, J. Shi, X. Zou, Y. Gong, Z. Zhang, M. Li, L. Zhao, R. Xu, S. Jiang, X. Zhou, M. Hong, C. Xie, H. Li, X. Lang, Q. Zhang, L. Gu, X. Yan and Y. Zhang, Adv. Mater., 2018, 30, 1705916 CrossRef PubMed.
- Y. Liu, J. Wu, K. P. Hackenberg, J. Zhang, Y. M. Wang, Y. Yang, K. Keyshar, J. Gu, T. Ogitsu, R. Vajtai, J. Lou, P. M. Ajayan, B. C. Wood and B. I. Yakobson, Nat. Energy, 2017, 2, 17127 CrossRef CAS.
- J. Shi, X. Wang, S. Zhang, L. Xiao, Y. Huan, Y. Gong, Z. Zhang, Y. Li, X. Zhou, M. Hong, Q. Fang, Q. Zhang, X. Liu, L. Gu, Z. Liu and Y. Zhang, Nat. Commun., 2017, 8, 958 CrossRef.
- B. Han, S. H. Noh, D. Choi, M. H. Seo, J. Kang and J. Hwang, J. Mater. Chem. A, 2018, 6, 20005–20014 RSC.
- J. Zhang, J. Wu, X. Zou, K. Hackenberg, W. Zhou, W. Chen, J. Yuan, K. Keyshar, G. Gupta, A. Mohite, P. M. Ajayan and J. Lou, Mater. Today, 2019, 25, 28–34 CrossRef CAS.
- C. Huang, X. Wang, D. Wang, W. Zhao, K. Bu, J. Xu, X. Huang, Q. Bi, J. Huang and F. Huang, Chem. Mater., 2019, 31, 4726–4731 CrossRef CAS.
- J. Hwang, S. H. Noh and B. Han, Appl. Surf. Sci., 2019, 471, 545–552 CrossRef CAS.
- D. N. Chirdon and Y. Wu, Nat. Energy, 2017, 2, 17132 CrossRef.
- Z. Cai, B. Liu, X. Zou and H. M. Cheng, Chem. Rev., 2018, 118, 6091–6133 CrossRef CAS PubMed.
- Y. Huang, E. Sutter, N. N. Shi, J. Zheng, T. Yang, D. Englund, H. J. Gao and P. Sutter, ACS Nano, 2015, 9, 10612–10620 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- A. E. Del Rio Castillo, V. Pellegrini, A. Ansaldo, F. Ricciardella, H. Sun, L. Marasco, J. Buha, Z. Dang, L. Gagliani, E. Lago, N. Curreli, S. Gentiluomo, F. Palazon, M. Prato, R. Oropesa-Nuñez, P. S. Toth, E. Mantero, M. Crugliano, A. Gamucci, A. Tomadin, M. Polini and F. Bonaccorso, Mater. Horiz., 2018, 5, 890–904 RSC.
- F. Bonaccorso, A. Lombardo, T. Hasan, Z. Sun, L. Colombo and A. C. Ferrari, Mater. Today, 2012, 15, 564–589 CrossRef CAS.
- S. Bellani, F. Wang, G. Longoni, L. Najafi, R. Oropesa-Nuñez, A. E. Del Rio Castillo, M. Prato, X. Zhuang, V. Pellegrini, X. Feng and F. Bonaccorso, Nano Lett., 2018, 18, 7155–7164 CrossRef CAS PubMed.
- S. Bellani, B. Martín-García, R. Oropesa-Nuñez, V. Romano, L. Najafi, C. Demirci, M. Prato, A. E. Del Rio Castillo, L. Marasco, E. Mantero, G. D'Angelo and F. Bonaccorso, Nanoscale Horiz., 2019, 4, 1077–1091 RSC.
- S. Bellani, E. Petroni, A. E. Del Rio Castillo, N. Curreli, B. Martín-García, R. Oropesa-Nuñez, M. Prato and F. Bonaccorso, Adv. Funct. Mater., 2019, 29, 1807659 CrossRef.
- A. Agresti, S. Pescetelli, A. L. Palma, B. Martin-Garcia, L. Najafi, S. Bellani, I. Moreels, M. Prato, F. Bonaccorso and A. Di Carlo, ACS Energy Lett., 2019, 4, 1862–1871 CrossRef CAS.
- Y. L. Zhong, Z. Tian, G. P. Simon and D. Li, Mater. Today, 2015, 18, 73–78 CrossRef CAS.
- K. Parvez, S. Yang, X. Feng and K. Müllen, Synth. Met., 2015, 210, 123–132 CrossRef CAS.
- S. Bellani, L. Najafi, A. Capasso, A. E. Del Rio Castillo, M. R. Antognazza and F. Bonaccorso, J. Mater. Chem. A, 2017, 5, 4384–4396 RSC.
- S. Yang, M. R. Lohe, K. Müllen and X. Feng, Adv. Mater., 2016, 28, 6213–6221 CrossRef CAS PubMed.
- P. Yu, S. E. Lowe, G. P. Simon and Y. L. Zhong, Curr. Opin. Colloid Interface Sci., 2015, 20, 329–338 CrossRef CAS.
- R. Yan, G. Khalsa, B. T. Schaefer, A. Jarjour, S. Rouvimov, K. C. Nowack, H. G. Xing and D. Jena, Appl. Phys. Express, 2019, 12, 23008 CrossRef.
- S. K. Basu and M. Taniguchi, J. Therm. Anal., 1987, 32, 1105–1113 CrossRef CAS.
- K. Hackenberg, et al., Self-improving electrocatalysists for gas evolution reactions, US Pat. 20160153098A1, 2016.
- X. Chia, A. Ambrosi, P. Lazar, Z. Sofer and M. Pumera, J. Mater. Chem. A, 2016, 4, 4241–14253 RSC.
- M. Leroux, L. Cario, A. Bosak and P. Rodière, Phys. Rev. B, 2018, 97, 195140 CrossRef CAS.
- F. Bonaccorso, A. Bartolotta, J. N. Coleman and C. Backes, Adv. Mater., 2016, 28, 6136–6166 CrossRef CAS PubMed.
- A. Capasso, A. E. Del Rio Castillo, H. Sun, A. Ansaldo, V. Pellegrini and F. Bonaccorso, Solid State Commun., 2015, 224, 53–63 CrossRef CAS.
- O. M. Maragó, F. Bonaccorso, R. Saija, G. Privitera, P. G. Gucciardi, M. A. Iatì, G. Calogero, P. H. Jones, F. Borghese, P. Denti, V. Nicolosi and A. C. Ferrari, ACS Nano, 2010, 4, 7515–7523 CrossRef PubMed.
- D. Zheng, X. Zhang, C. Li, M. E. McKinnon, R. G. Sadok, D. Qu, X. Yu, H. S. Lee, X. Q. Yang and D. Qu, J. Electrochem. Soc., 2015, 162, A203–A206 CrossRef CAS.
- F. Bonaccorso, A. Lombardo, T. Hasan, Z. Sun, L. Colombo and A. C. Ferrari, Mater. Today, 2012, 15, 564–589 CrossRef CAS.
- S. Zhao, T. Hotta, T. Koretsune, K. Watanabe, T. Taniguchi, K. Sugawara, T. Takahashi, H. Shinohara and R. Kitaura, 2D Mater., 2016, 3, 025027 CrossRef.
- X. Wang, J. Lin, Y. Zhu, C. Luo, K. Suenaga, C. Cai and L. Xie, Nanoscale, 2017, 9, 16607–16611 RSC.
- Z. Li, W. Yang, Y. Losovyj, J. Chen, E. Xu, H. Liu, M. Werbianskyj, H. A. Fertig, X. Ye and S. Zhang, Nano Res., 2018, 11, 5978–5988 CrossRef CAS.
- J. A. Wilson and A. D. Yoffe, Adv. Phys., 2015, 6, 5763 Search PubMed.
- S. Jeong, D. Yoo, M. Ahn, P. Miro, T. Heine and J. Cheon, Nat. Commun., 2015, 6, 5763 CrossRef CAS.
- P. Rabu, A. Meerschaut, J. Rouxel and G. A. Wiegers, J. Solid State Chem., 1990, 88, 451–458 CrossRef CAS.
- W. Ge, K. Kawahara, M. Tsuji and H. Ago, Nanoscale, 2013, 5, 5773–5778 RSC.
- F. Jellinek, G. Brauers and H. Muller, Nature, 1960, 185, 376–377 CrossRef CAS.
- Z.-L. Liu, L.-C. Cai and X.-L. Zhang, J. Alloys Compd., 2014, 610, 472–477 CrossRef CAS.
- C. J. Carmalt, T. D. Manning, I. P. Parkin, E. S. Peters and A. L. Hector, J. Mater. Chem., 2004, 14, 290–291 RSC.
- C. J. Carmalt, T. D. Manning, I. P. Parkin, E. S. Peters and A. L. Hector, Thin Solid Films, 2004, 469–470, 495–499 CrossRef CAS.
- Y. Ma, A. Kuc, Y. Jing, P. Philipsen and T. Heine, Angew. Chem., Int. Ed., 2017, 56, 10214–10218 CrossRef CAS PubMed.
- Y. Liao, K. S. Park, P. Xiao, G. Henkelman, W. Li and J. B. Goodenough, Chem. Mater., 2013, 25, 1699–1705 CrossRef CAS.
- Y. Liao, K. S. Park, P. Singh, W. Li and J. B. Goodenough, J. Power Sources, 2014, 245, 27–32 CrossRef CAS.
- http://www.hqgraphene.com/NbS2.php, accessed on 28 August 2019.
- http://www.hqgraphene.com/3R-NbS2.php, accessed on 28 August 2019.
- N. Yue, J. Weicheng, W. Rongguo, D. Guomin and H. Yifan, J. Mater. Chem. A, 2016, 4, 8198–8203 RSC.
- W. G. McMullan and J. C. Irwin, Solid State Commun., 1983, 45, 557–560 CrossRef CAS.
- S. Nakashima, Y. Tokuda, A. Mitsuishi, R. Aoki and Y. Hamaue, Solid State Commun., 1982, 42, 601–604 CrossRef CAS.
- S. Onari, T. Arai, R. Aoki and S. Nakamura, Solid State Commun., 1979, 31, 577–579 CrossRef CAS.
- D. Gopalakrishnan, A. Lee, N. K. Thangavel and L. M. Reddy Arava, Sustainable Energy Fuels, 2017, 2, 96–102 RSC.
- Y. Ding, Y. Wang, J. Ni, L. Shi, S. Shi and W. Tang, Phys. B, 2011, 406, 2254–2260 CrossRef CAS.
- J. K. Dash, L. Chen, P. H. Dinolfo, T. M. Lu and G. C. Wang, J. Phys. Chem. C, 2015, 119, 19763–19771 CrossRef CAS.
- https://materialsproject.org/materials/mp-10033/, accessed on 28 August 2019.
- X. Zhou, S.-H. Lin, X. Yang, H. Li, M. N. Hedhili, L.-J. Li, W. Zhang and Y. Shi, Nanoscale, 2018, 10, 3444–3450 RSC.
- H. Bark, Y. Choi, J. Jung, J. H. Kim, H. Kwon, J. Lee, Z. Lee, J. H. Cho and C. Lee, Nanoscale, 2018, 10, 1056–1062 RSC.
- J. Zhang, C. Du, Z. Dai, W. Chen, Y. Zheng, B. Li, Y. Zong, X. Wang, J. Zhu and Q. Yan, ACS Nano, 2017, 11, 10599–10607 CrossRef CAS PubMed.
- M. K. Bahl, J. Phys. Chem. Solids, 1975, 36, 485–491 CrossRef CAS.
- K. Kim, M. S. Kim, P. R. Cha, S. H. Kang and J. H. Kim, Chem. Mater., 2016, 28, 1453–1461 CrossRef CAS.
- S. Ramakrishna, A. Le Viet, M. V. Reddy, R. Jose and B. V. R. Chowdari, J. Phys. Chem. C, 2010, 114, 664–671 CrossRef.
- R. Romero, J. R. Ramos-Barrado, F. Martin and D. Leinen, Surf. Interface Anal., 2004, 36, 888–891 CrossRef CAS.
- M. Aufray, S. Menuel, Y. Fort, J. Eschbach, D. Rouxel and B. Vincent, J. Nanosci. Nanotechnol., 2009, 9, 4780–4785 CrossRef CAS PubMed.
- D. Gopalakrishnan, A. Lee, N. K. Thangavel and L. M. Reddy Arava, Sustainable Energy Fuels, 2018, 2, 96–102 RSC.
- J. Si, Q. Zheng, H. Chen, C. Lei, Y. Suo, B. Yang, Z. Zhang, Z. Li, L. Lei, Y. Hou and K. Ostrikov, ACS Appl. Mater. Interfaces, 2019, 11, 13205–13213 CrossRef CAS.
- V. Romano, B. Martín-García, S. Bellani, L. Marasco, J. Kumar Panda, R. Oropesa-Nuñez, L. Najafi, A. E. Del Rio Castillo, M. Prato, E. Mantero, V. Pellegrini, G. D'Angelo and F. Bonaccorso, ChemPlusChem, 2019, 84, 882–892 CrossRef CAS.
- J. Li, W.-W. Liu, H.-M. Zhou, Z.-Z. Liu, B.-R. Chen and W.-J. Sun, Rare Met., 2018, 37, 118–122 CrossRef CAS.
- A. A. Lubimtsev, P. R. C. Kent, B. G. Sumpter and P. Ganesh, J. Mater. Chem. A, 2013, 1, 14951–14956 RSC.
- K. J. Griffith, A. C. Forse, J. M. Griffin and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 8888–8899 CrossRef CAS PubMed.
- N. Kumagai, Y. Koishikawa, S. Komaba and N. Koshiba, J. Electrochem. Soc., 1999, 146, 3203–3210 CrossRef CAS.
- X. Ou, X. Xiong, F. Zheng, C. Yang, Z. Lin, R. Hu, C. Jin, Y. Chen and M. Liu, J. Power Sources, 2016, 325, 410–416 CrossRef CAS.
- J. Zhang, C. Du, Z. Dai, W. Chen, Y. Zheng, B. Li, Y. Zong, X. Wang, J. Zhu and Q. Yan, ACS Nano, 2017, 11, 10599–10607 CrossRef CAS PubMed.
- B. Stanje, V. Epp, S. Nakhal, M. Lerch and M. Wilkening, ACS Appl. Mater. Interfaces, 2015, 7, 4089–4099 CrossRef CAS PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- Z. Hawash, L. K. Ono, S. R. Raga, M. V. Lee and Y. Qi, Chem. Mater., 2015, 27, 562–569 CrossRef CAS.
- Z. Hawash, L. K. Ono and Y. Qi, Adv. Mater. Interfaces, 2016, 3, 1600117 CrossRef.
- M. Otero, G. Lener, J. Trincavelli, D. Barraco, M. S. Nazzarro, O. Furlong and E. P. M. Leiva, Appl. Surf. Sci., 2016, 383, 64–70 CrossRef CAS.
- J. Phillips and J. Tanski, Int. Mater. Rev., 2005, 50, 265–286 CrossRef CAS.
- E. Asselin, T. M. Ahmed and A. Alfantazi, Corros. Sci., 2007, 49, 694–710 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- D. Strmcnik, K. Kodama, D. Van Der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466 CrossRef CAS PubMed.
- D. Strmcnik, M. Escudero-Escribano, K. Kodama, V. R. Stamenkovic, A. Cuesta and N. M. Markovií, Nat. Chem., 2018, 2, 880 CrossRef.
- J. Staszak-Jirkovský, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K.-C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2015, 15, 197 CrossRef PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- H. Pan, Sci. Rep., 2014, 4, 5348 CrossRef CAS PubMed.
- J. Lee, S. Kang, K. Yim, K. Y. Kim, H. W. Jang, Y. Kang and S. Han, J. Phys. Chem. Lett., 2018, 9, 2049–2055 CrossRef CAS PubMed.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, A. Ansaldo, M. Prato, A. E. Del Rio Castillo and F. Bonaccorso, Adv. Energy Mater., 2018, 8, 1703212 CrossRef.
- S. Ma Andersen and E. Skou, ACS Appl. Mater. Interfaces, 2014, 61, 16565–16576 CrossRef PubMed.
- O. J. Curnick, P. M. Mendes and B. G. Pollet, Electrochem. Commun., 2010, 12, 1017–1020 CrossRef CAS.
- Z. Zhu, H. Yin, C.-T. He, M. Al-Mamun, P. Liu, L. Jiang, Y. Zhao, Y. Wang, H.-G. Yang, Z. Tang, D. Wang, X.-M. Chen and H. Zhao, Adv. Mater., 2018, 30, 1801171 CrossRef PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K.-C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., 2012, 124, 12663–12666 CrossRef.
- F. Bonaccorso, T. Hasan, P. H. Tan, C. Sciascia, G. Privitera, G. Di Marco, P. G. Gucciardi and A. C. Ferrari, J. Phys. Chem. C, 2010, 114, 17267–17285 CrossRef CAS.
- T. Hasan, P. H. Tan, F. Bonaccorso, A. G. Rozhin, V. Scardaci, W. I. Milne and A. C. Ferrari, J. Phys. Chem. C, 2008, 112, 20227–20232 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta07210a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |