
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-induced electrolyte improvement in cobalt tris(bipyridine)-mediated dye-sensitized solar cells†
Jiajia
Gao
a,
Wenxing
Yang
b,
Ahmed M.
El-Zohry
b,
Govind Kumar
Prajapati
a,
Yuan
Fang
a,
Jing
Dai
a,
Yan
Hao
a,
Valentina
Leandri
a,
Per H.
Svensson
ac,
István
Furó
a,
Gerrit
Boschloo
b,
Torben
Lund
d and
Lars
Kloo
 *a
*a
aDivision of Applied Physical Chemistry, Department of Chemistry, KTH Royal Institute of Technology, SE-100 44, Stockholm, Sweden. E-mail: larsa@kth.se
bDepartment of Chemistry, Ångström Laboratory, Uppsala University, Box 523, SE-75120, Uppsala, Sweden
cRISE Surface Process Formulation, Forskargatan 20j, SE-15136 Sodertalje, Sweden
dDepartment of Science and Environment, Roskilde University, DK-4000 Roskilde, Denmark
First published on 5th August 2019
Abstract
Lithium-ion-free tris(2,2′-bipyridine) Co(II/III)-mediated electrolytes have previously been proposed for long-term stable dye-sensitized solar cells (DSSCs). Such redox systems also offer an impressive DSSC performance improvement under light soaking exposure, manifested by an increase in photocurrent and fill factor without the expense of decreasing photovoltage. Kinetic studies show that charge transfer and ion diffusion at the electrode/electrolyte interface are improved due to the light exposure. Control experiments reveal that the light effect is unambiguously associated with electrolyte components, [Co(bpy)3]3+ and the Lewis-base additive tert-butylpyridine (TBP). Electrochemical and spectroscopic investigation of the [Co(bpy)3]3+/TBP mixtures points out that the presence of TBP, which retards the electrolyte diffusion, however causes an irreversible redox reaction of [Co(bpy)3]3+ upon light exposure that improves the overall conductivity. This discovery not only provides a new strategy to mitigate the typical Jsc–Voc trade-off in Co(II/III)-mediated DSSCs but also highlights the importance of investigating the photochemistry of a photoelectrochemical system.
Introduction
As a promising photovoltaic technology, dye-sensitized solar cells (DSSCs) have attracted much attention due to their expected advantages with respect to low-cost manufacturing and adaptive device structure based on the pioneering work of Grätzel and O'Regan in 1991.1 Most efforts have been devoted to the engineering of the dye structure and the electrolyte formulation for broader spectral absorption and more efficient photon-to-electron conversion. It must be borne in mind that in a multi-component DSSC system, molecular configuration and composition could be changed because of dynamic equilibria through interactions between the components. Molecular dynamics and interactions at the dye/TiO2 and TiO2/electrolyte interfaces (mostly for traditional I−/I3−-based electrolytes) that closely correlate with interfacial charge transfer kinetics, have been well studied and illustrated in past decades. However, less attention has been dedicated to the one-electron, outer-sphere transition-metal complexes recently qualified as attractive redox shuttle alternatives. Such redox complexes in particular offer a high photovoltage of DSSCs,4–7 and tunable redox potential and coordination sphere that can be used to control the driving forces and kinetics of the two rate-limiting electron transfer processes (including dye regeneration and electron recombination in the device8,9) via structural changes in the complex ligands.7,10–12 Paradoxically, one-electron redox couples of this type offer fast regeneration of the oxidized dye molecules, but at the same time suffer from fast recombination loss reactions.5,13–15 Co(II/III)-polypyridyl redox systems are the most studied and to date the most successfully employed in DSSCs, mainly due to the advantageous large inner-sphere reorganization energy necessarily for spin transition which reduces the interfacial charge-transfer loss.9,16–18 Nevertheless, the recombination issue at the electrode/electrolyte interface still prevails for such redox mediators. One reason is attributed to the sluggish transport of the bulky complex ions, especially so when high-boiling and typically viscous solvents are used.19 As a result, Co(III) ions attain a relatively high concentration at the interface that promotes recombination losses. In order to alleviate this issue, sensitizers with sterically bulky groups,7,20 atomic layer deposition (ALD)15,19 and potential-determining additives (PDAs), such as 4-tert-butylpyridine (TBP), are commonly employed to retard loss reactions at the TiO2/dye/electrolyte interface. The retardation of recombination reaction rates is macroscopically observed as an improvement in the open-circuit voltage.2,21 The inclusion of polarizing ions at or close to the semiconductor surface, e.g. Li+ or Mg2+ at the TiO2 surface, also cause similar effects by changing the local distribution of cobalt cations and in particular, reducing the concentration of [Co(bpy)3]3+ (or Co(III) for simplicity) in the electric double layer at the photoanode surface.18 Thus, the interaction between co-additives and the positively charged [Co(bpy)3]2+/3+ mediators, which by necessity must influence the function of [Co(bpy)3]2+/3+ in the device, calls for a more detailed study.Related interactions could include the involvement of the additive TBP, which has been commonly used in DSSCs. TBP as a strong Lewis base is a likely competitive ligand for coordination to the cobalt centres based on evidence regarding the instability of cobalt tris(bipyridine) complexes through ligand exchange.10,11,22,23 In addition, TBP was claimed by Salim et al. to exhibit a shielding effect with respect to the [Co(bpy)3]3+ ions via weak ion-dipole attraction accounting for the retarding role in the recombination loss reactions.21 This work will further explore the interaction between the two necessary electrolyte components in a lithium-ion-free [Co(bpy)3]2+/3+-based DSSC system, where a high concentration of [Co(bpy)3]3+ is used since it has been demonstrated beneficial for long-term device stability.2 In previous work, we have identified the role of TBP in promoting both the photovoltage and the photocurrent for this type of system. In this work, we will investigate the origin of a remarkable DSSC efficiency improvement after exposure to light.2,3 The beneficial effects of light soaking, on the short-circuit current in particular, can be traced to a lowering of the conduction band edge of the semiconductor substrate, or alternatively to an increase in the density of states (or photo-induced surface states) facilitating the electron injection process.24 Typically, such changes are considered to be caused by a photo-induced charge separation of the dye or semiconductor at open circuit changing the local electric field and thus causing a re-arrangement of charged species at the TiO2/dye/electrolyte interface.24–29 However, this work provides a new perspective, in which the electrolyte is involved in the light soaking effect. Control experiments based on the electrolyte components reveal that the light-induced change is associated with the combination of [Co(bpy)3]3+ and TBP. Changes in the electrochemical and spectroscopic properties have been studied and a new type of interaction between the two compounds is revealed.
Experimental section
Materials
All chemicals were purchased from Sigma Aldrich unless otherwise noted: fluorine-doped tin oxide (FTO; Pilkington, TEC 15 Ω cm−2 & TEC 7 Ω cm−2), TiO2 pastes (Dyesol Ltd., DSL 18NR-T and WER2-O), Surlyn frame (Solaronix). The dyes (D35, LEG4) and the tetracyanoborate or hexafluorophosphate salts of tris(2,2′-bipyridine-2N,N′) cobalt(II/III) were all obtained from Dyenamo AB, Sweden. All chemicals were of reagent grade and used without further purification.Solar cell fabrication and characterization
The details of solar cell fabrication have been reported previously.30 A transparent TiO2 layer (diluted paste: a mixture of 60 wt% TiO2 paste (DSL 18NR-T) with 36 wt% terpineol and 4 wt% ethyl cellulose; area: 0.5 cm × 0.5 cm, thickness: 5 μm) and a scattering layer (WER2-O, 3 μm) were in succession screen-printed on an FTO substrate (Pilkington, TEC 15 Ω cm−2) with a TiO2 blocking layer pre-deposited by a simple hydrothermal method. The working electrodes were sintered in the ambient atmosphere (325 °C for 25 min, thermostatic for 30 min, up to 500 °C for 25 min, thermostatic for 30 min and natural cooling), and post-treated with an aqueous TiCl4 solution (40 mM). The working electrodes were dipped into a 0.25 mM D35/ethanol dye bath in the dark overnight. The platinized counter electrodes were prepared by drop-casting 20 μl of a 4.8 mM H2PtCl6 isopropanol solution on a pre-drilled and cleaned FTO (Pilkington, TEC 7 Ω cm−2) glass substrate and then sintered in air at 400 °C for 30 min. The DSSC devices were fabricated by assembling the sensitized TiO2 electrodes with the Pt counter electrodes into a sandwich-type cell using a 25 μm thick hot-melt Surlyn frame (inner area: 0.6 cm × 0.6 cm) as sealant, introducing the electrolyte through pre-drilled holes under atmospheric pressure, and sealing by a 50 μm thermoplastic sheet and a glass coverslip. A metal contact was soldered on the edge of the FTO film to increase conductivity. Measurements of the thickness of the TiO2 layers were made by means of a profilometer (Veeco Dektak 150).Current density–voltage (J–V) characteristics were recorded with a light-shading metal mask (0.7 cm × 0.7 cm) on top of the cell under standard irradiation (AM 1.5G, 100 mW cm−2) supplied by a Newport solar simulator (model 91160-1000). Light illumination for incident photon-to-current conversion efficiency (IPCE) measurements was provided by a computer-controlled set-up assembled with a xenon arc lamp (300 W Cermax, ILC Technology), a monochromator (CVI Digikrom CM 110) and appropriate filters. Both J–V curves and IPCE spectra were recorded by a computerized Keithley 2400 source meter calibrated using a certified reference solar cell (Fraunhofer ISE).
Light exposure treatments performed on open-circuit solar cells and electrolyte samples were performed under continuous irradiation (∼100 mW cm−2, 390 nm UV cut-off; ATLAS Suntest XLS) in a sample compartment maintaining a temperature of 60 °C. Samples aged in the dark were subjected to the same temperature conditions (60 °C). Two different approaches were used in this study, as schematically visualized in Scheme 1, where either model electrolytes with only selected components were pre-exposed to light (the pre strategy) and subsequently complemented with the remaining components to complete the electrolyte, according to the composition given below, and then introduced into a DSSC to be further characterized. Alternatively, the DSSC was made in the standard way, then exposed to light (the post strategy) and characterized. The post strategy is the typical way to perform ageing tests of DSSCs. In this work, the main insights have been obtained through the pre strategy.
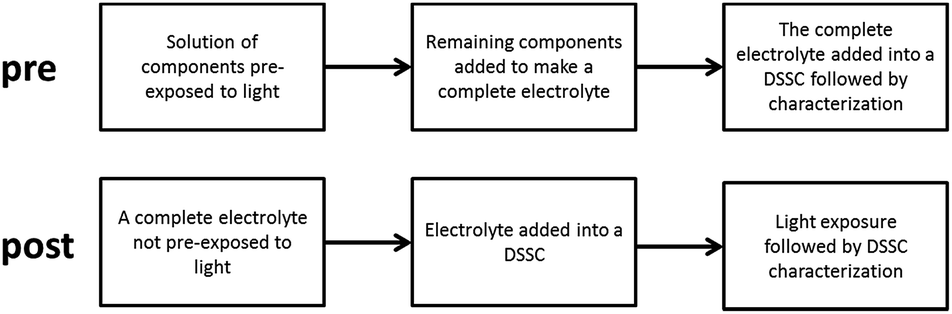 | ||
| Scheme 1 A visualization of the two main strategies, pre and post, used in this work. | ||
The electron lifetime (τn) and diffusion time (τd) in TiO2 films were estimated by first-order models of the transient voltage and current decay traces, respectively, following a small square-wave modulation to the base light intensity varied by controlling a white LED (Luxeon Star 1 W). The decay traces were recorded using a 16 bit resolution digital acquisition board (National Instruments) in combination with a current preamplifier (Stanford Research Systems SR570) and a custom-made system with electromagnetic switches. The methods of measurement have been reported previously.7,31 The relationship between voltage and extracted charge (Qoc) under open-circuit conditions was studied using a combined voltage decay/charge-extraction method.32 Relative data used in the figures were based on the best-performing cells for each type of electrolyte. Electrochemical impedance spectroscopy (EIS) measurements were conducted with an Autolab PGstat12 potentiostat with an impedance module. The frequency was swept from 105 Hz to 0.1 Hz using a 20 mV AC modulation amplitude. Electrochemical impedance spectra were recorded under dark conditions with an applied bias voltage. The curves were modeled using the Z-View software and an equivalent circuit model as shown in Scheme S1.†
Transient absorption measurements
Devices for transient absorption measurements were fabricated in the same way as the solar cells above, except that the photoanode contained a transparent TiO2 layer (no scattering layer was applied). Nanosecond transient absorption measurements were performed using an Edinburgh Instrument LP920 laser flash photolysis spectrometer with continuous wave xenon light as the probe light and a photomultiplier tube detector (system response time, ∼1 μs). Scattered light from the excitation was excluded with a 715 nm cut-on filter in front of the detector. Laser pulses were supplied by a Continuum Surelite II, Nd:YAG laser at a 10 Hz repetition rate in combination with an OPO (Continuum Surelite). The pulse intensity was attenuated to 0.2–3 mJ per pulse with the use of natural density filters. The pump light wavelength was selected to 530 nm. Kinetic traces of absorbance were detected at 760 nm, averaged over 50 to 100 pulses per sample. Three samples were prepared for each electrolyte composition, and the measurement error was estimated from the average deviation. The overlaid curves were modelled with a KWW function only for visualization. The kinetics of the system were characterized by the half time, t1/2, of the decay of the initial absorption difference.33 For the femtosecond transient absorption in the infrared region (IR), femtosecond laser pulses of 800 nm with a repetition rate of 3 kHz were used to generate an excitation wavelength of 520 nm with a power of 110–170 μW, and a probe pulse with a centered wavelength of ca. 5000 nm (∼2000 cm−1). More details about the femtosecond experimental setup can be found in previously published work.34Electrochemical measurements in a symmetrical dummy cell
Symmetrical dummy cells were fabricated by assembling two identical platinized FTO glass plates with the incomplete model electrolytes in between containing only 0.15 M [Co(bpy)3]3+ and TBP in various concentrations with exposure to light for different time periods. All electrochemical measurements were carried out under dark conditions. The same set-up and conditions as described above were used for the EIS measurements. Resistance and capacitances were analysed using an equivalent circuit model, and the diffusion time (τd) was calculated according to literature methods.35 Cyclic voltammograms (CVs) were recorded stepwise by increasing the scan rate from 10 to 250 mV s−1 with an interval waiting time of 100 s.In situ characterization of [Co(bpy)3]3+/TBP mixtures
UV-vis absorption spectra were recorded on a Cary 300 spectrophotometer in a quartz sample cell (1 cm path length). Cyclic voltammetry and differential pulse voltammetry (DPV) measurements were conducted using an Autolab potentiostat with a glassy carbon disk as the working electrode, a platinum wire as the counter electrode and Ag/AgNO3 as the reference electrode. 0.1 M [Bu4N]PF6 was added as conductive medium. The rest potential of the electrolyte was recorded by measuring the potential difference between the assembled electrode, composed of a Pt wire in contact with the target electrolyte in a plastic tube with a frit top allowing the free movement of ions, and an Ag/AgCl (1 M LiCl in ethanol) reference electrode in the supporting electrolytes (0.1 M [TBA]PF6 in acetonitrile) with the use of a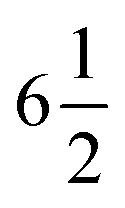 digit precision digital Keithley 2700 source meter. The reference electrode potential was calibrated by Fc/Fc+ in the same supporting electrolyte. The accuracy of the potentials determined was estimated to be around 5 mV.
digit precision digital Keithley 2700 source meter. The reference electrode potential was calibrated by Fc/Fc+ in the same supporting electrolyte. The accuracy of the potentials determined was estimated to be around 5 mV.
The NMR-spectroscopic experiments were performed on a Bruker Advance III 500 MHz spectrometer equipped with a 5 mm high-resolution probe, which can provide a maximum of 0.5 T m−1 gradient in the z-direction. In the diffusion experiments a stimulated echo pulse sequence was used with gradient pulse length of δ = 3.0 ms, a diffusion time Δ = 50 ms and allowing the gradient to increase linearly from 0.02 to 0.22–0.35 T m−1 in 16 steps. Each spectrum was obtained from 16 scans. The gradient strength g was calibrated with a sample of deuterated water at 298 K. For eNMR measurements, a double stimulated echo pulse sequence with a bipolar voltage pulse was used with gradient pulse length δ = 1.0 ms, drift time ΔE of 100 ms and gradient strength of 0.32 T m−1. The electric field E was incremented from 0 to 21 V cm−1 in at least 8 steps. All experiments were conducted at 293 K. The diffusion coefficients were obtained by modeling the signal attenuation with a conventional Stejskal–Tanner expression. The electrophoretic mobility was obtained from the phase modulation by the electric field strength according to
| ϕ = γgδΔEμE |
Computational details
Calculations were performed on [Co(bpy)3]3+ and various coordination species with partly or fully exchanged bpy ligands for TBP, with and without acetonitrile modelled as solvent. All calculations were performed using the Gaussian 09 program package,37 including a selection of different functionals and hybrid functionals. All results presented are based on results from B3LYP including the Coulomb-attenuating method improving long-range exchange interaction.38 A 10-electron effective-core pseudopotential (MDF10) was employed for Co including a (8s7p6d2f)/[6s5p3s2f] contracted valence space and basis sets of 6-311G type for all lighter elements.39,40 The effects of the acetonitrile solvent were implicitly introduced using the Polarizable Continuum Model (PCM) as implemented in Gaussian 09.Results and discussion
Light-induced efficiency enhancement
In this work, typical triphenylamine (TPA) based sensitizers with bulky chains (D35 and LEG4, see Fig. S1†) were employed, and cobalt-based electrolytes were prepared according to the same Li+-free recipe as previously reported: 0.30 M/0.15 M [Co(bpy)3]2+/3+ and 0.2 M TBP in acetonitrile.2 A higher than normal concentration of the Co(III) redox-system component (0.05 M) was used in this study to ensure efficient device performance in the absence of Li+. Fig. 1 shows the chemical structures of [Co(bpy)3]2+/3+ and TBP, where the Co2+/3+ centre adopts a quasi-octahedral coordination by N-atoms of the three bpy ligands. The acetonitrile solutions of systematically varied combinations of the three components: Co(II)/Co(III)/TBP, Co(II)/TBP, Co(III)/TBP, Co(III) only and TBP only were pre-exposed to light-soaking conditions (1 sun, 390 nm cut-off, 60 °C; denoted as the pre strategy in Scheme 1) before preparing the complete electrolyte and assembling the device; related DSSC efficiency (η) and short-circuit current (Jsc) were compared to non-light-treated complete electrolytes without and with assembled devices exposed to the same light treatment (denoted as the post strategy). By comparison, results in Fig. S2† show that the light-induced DSSC improvement in the post strategy can be achieved by the pre strategy based on the cobalt-based electrolyte and more specifically on the mixture of [Co(bpy)3]3+ and TBP only. This means that the beneficial light effect actually can be attributed to this mixture. For clarity, Fig. 2 and Table 1 show the performance of DSSCs assembled with different complete electrolytes: electrolyte A, non-treated; electrolyte A* and electrolyte AT: according to the pre strategy part of the electrolyte containing [Co(bpy)3]3+/TBP/acetonitrile exposed to one sun irradiation (390 nm cut-off)/60 °C and stored in the dark/60 °C, respectively. The electrolyte A-Li+ is the non-treated electrolyte A with additional 0.1 M LiClO4 included for comparison.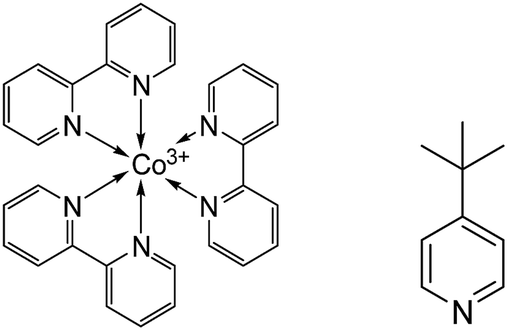 | ||
| Fig. 1 Molecular structures of the tris(2,2′-bipyridine) cobalt(III) complex (left) and TBP (right). | ||
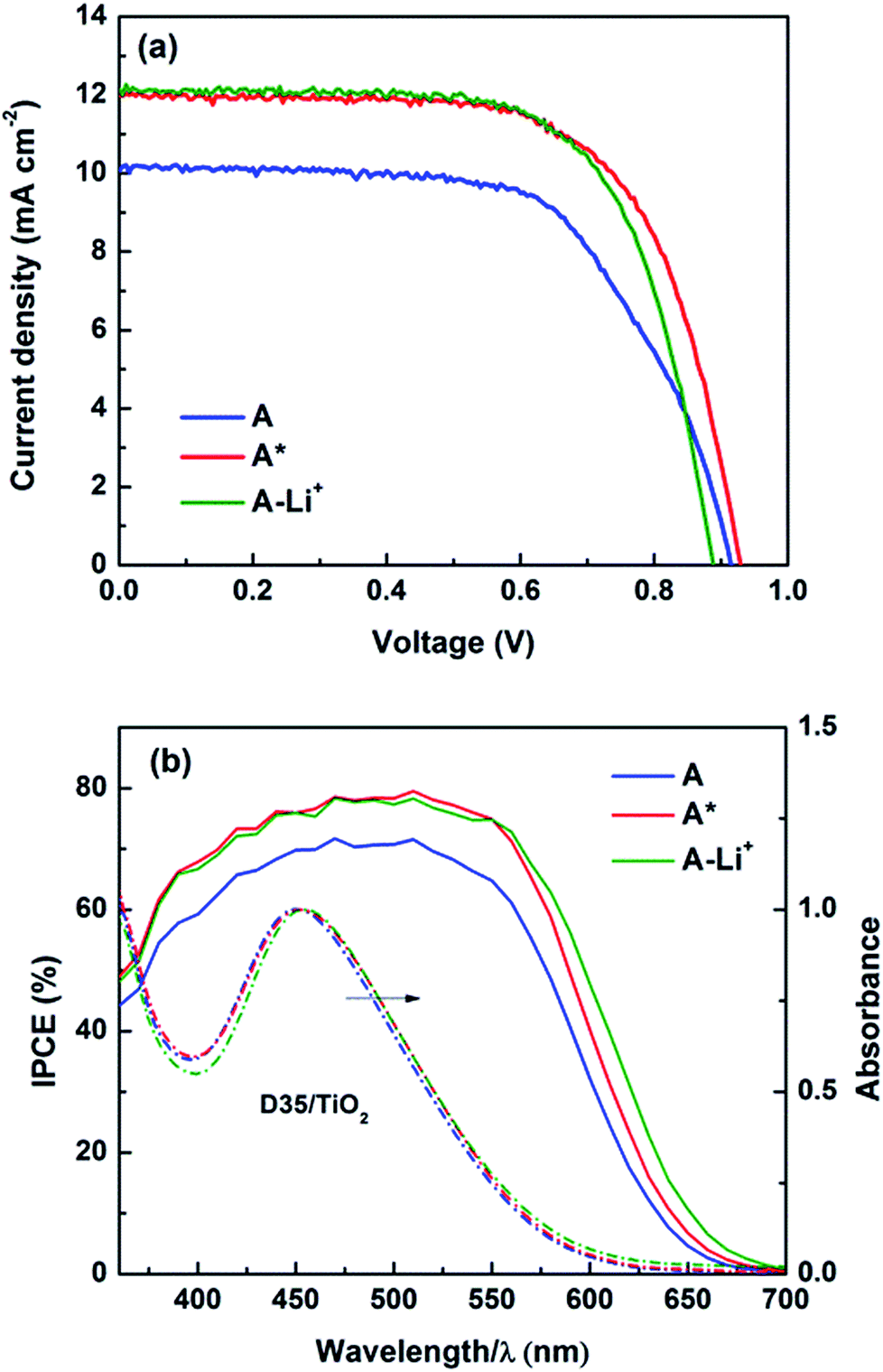 | ||
| Fig. 2 (a) The J–V characteristics and (b) incident photon-to-current conversion efficiency (IPCE) spectra (solid lines) of D35-sensitized DSSCs assembled with [Co(bpy)3]2+/3+-based electrolytes, untreated (A, blue; A-Li+, green) and treated by light as described in the main text (A*, red). In (b), normalized UV-vis absorption spectra (dashed lines) were recorded for D35 adsorbed onto the TiO2 film in contact with the Co(III)/TBP acetonitrile solution before (blue) and after (red) light exposure, and with Li+ salts added without light exposure (green). The absorption contribution of the contacting electrolyte was deducted. | ||
| Electrolytesa | V oc/mV | J sc/mA cm−2 | FF | η/% |
|---|---|---|---|---|
| a A* and AT were composed of a pre-exposed [Co(bpy)3]3+/TBP mixture, light/60 °C and dark/60 °C conditions for 4 hours, respectively. Each data in the table present the average of three independent cells. | ||||
| A | 900 ± 15 | 9.9 ± 0.2 | 0.63 ± 0.01 | 5.6 ± 0.3 |
| A* | 920 ± 10 | 11.9 ± 0.1 | 0.66 ± 0.01 | 7.2 ± 0.3 |
| AT | 871 ± 14 | 9.6 ± 0.2 | 0.65 ± 0.01 | 5.4 ± 0.4 |
| A-Li+ | 877 ± 13 | 12.0 ± 0.2 | 0.67 ± 0.01 | 7.1 ± 0.2 |
The results clearly demonstrate that the effect of light exposure, rather than that of thermal stress, on the electrolyte causes a notable increase in photocurrent (by ∼20%) and total efficiency (by ∼25%) of the resulting devices. Both parameters and fill factor (FF) are comparable to those of devices containing added Li+ salt, and notably the open-circuit voltage (Voc) is quite high mitigating the typical Jsc–Voc trade-off suffered by the addition of PDAs. The same results were obtained for devices based on other TPA-type dyes (LEG4), or cobalt redox couples with different counter ions (PF6−), see Table S1.† We also observed that the beneficial light effect is irreversible and remains stable after the illumination has been terminated. Consistently, the incident photon-to-current conversion efficiency (IPCE) within the spectral range investigated also increases and broadens towards the red, as shown in Fig. 2b and S3.† IPCE represents an integrated result of crucial processes occurring in DSSCs and is often expressed as
| IPCE(λ) = ØA(λ) × Øinj × Øreg × ηcc | (1) |
Changes with respect to interfaces and kinetics in full devices
In order to elucidate the origin for the light-induced increase in IPCE and photocurrent, an investigation of the dye regeneration kinetics was performed; see the results in Fig. S5 and Table S3.† However, the difference in the half-time (τ1/2) of dye regeneration by the cobalt electrolyte is in the order of sub-microseconds. Previous studies on the D35 dye under similar conditions have revealed that the intrinsic charge recombination is in the order ∼10 to 100 μs.7 Thus, this small difference in the τ1/2 observed here would contribute negligibly to the dye regeneration efficiency, Øreg, and therefore cannot explain the large change in the IPCE nor in the Jsc observed in Fig. 2. Logically, it is instead expected that the photocurrent increase can be assigned to an increase in the product Øinjηcc. Fig. 3a and S6a† show the evolution of DSSC performance as a function of time of light-exposure treatment (the pre strategy). It is clear that the Jsc and FF increase dramatically to a steady-state situation reached after 4 hours of exposure, while the Voc stabilizes already after a slight increase in a considerably shorter time. In parallel, we monitored the change in the electron accumulation in the TiO2 film and the interfacial transfer kinetics. As shown in Fig. S6b,† by prolonging the Co(III)/TBP-solution light pre-exposure, the charge storage in the dye/TiO2 electrode at the same open-circuit voltage (or energy level) increases gradually, and the tail of the band-gap trap states is broadened. By exponential modelling of the distribution of trap states according to eqn (S1)–(S3),† it is notable that the difference in the TiO2 conduction band energy level (Ec) and the electrolyte redox potential (Eredox) decreases by pre-exposure of the electrolyte to light, indicating that Ec is positively shifted (∼0.1 eV) while Eredox remains almost constant; see Fig. S7 and Table S4.† As Ec is lowered, the density of electron-acceptor states in the TiO2 film iso-energetic with the dye excited states also increases, and it is expected that the band edge downshift promotes a higher Øinj, especially at longer wavelengths.24,28 From the electrolyte point of view, the origin of the Ec shift could be caused by electrolyte charging the TiO2 surface; the Ec downshift is either caused by an increase in the surface positive charge (e.g. protons) or through a decrease in the negative charge; the latter is linked to TBP concentrations in the systems studied. However, we have previously demonstrated that a mere reduction in initial concentration of TBP in the cobalt electrolyte shows little improvement.2 According to the results shown in Table S1 and Fig. S8,† by adding extra Lewis base (TBP or bipyridine) to the pre-exposed electrolytes, the downshift of the Ec is reduced and the improvement of the DSSC performance is also decreased. This means that the Ec shift constitutes a reason for the performance improvement, likely due to positive charges at the TiO2/electrolyte interface produced in the [Co(bpy)3]3+/TBP electrolyte during light exposure. | ||
| Fig. 3 (a) The photovoltaic performance improvement of DSSCs and (b) electron lifetime in D35-sensitized TiO2 films as a function of light exposure time of the [Co(bpy)3]3+/TBP component in the electrolyte. | ||
The electron collection efficiency (ηcc) is determined by two competing processes, the electron diffusion time (τd) and the electron recombination time, i.e. electron lifetime (τn) in TiO2, according to the relation of ηcc = 1/(1 + τd/τn). It was noted that τn is increased when pre-exposed electrolytes are used, while the change in τd is negligible, see Fig. 3b and S6c.† This indicates that ηcc increases after the electrolyte is pre-exposed to light, which is consistent with the increase in the IPCE and photocurrent. τn directly reflects the interfacial recombination kinetics of the injected electrons from TiO2 to the electrolyte; consistently, we observed a larger recombination resistance (the middle semicircle) in the light-exposed system in EIS results, see Fig. 4. In this perspective, two crucial factors need to be investigated: (1) Co(III) complexes, the concentration of which significantly influences the recombination loss rates operating as electron acceptors, and (2) interfacial recombination-suppressing additives, such as TBP or small cations; for the latter, Li+ has been demonstrated to effectively improve τn in our previous work,3 and in this work it could be protons being active in the Li+-free electrolyte. A loss of Co(III) likely due to light-induced degradation could be a reason for the increase in τn; however, by comparing the performance of DSSCs differing in the concentration of initially added Co(III) without light exposure treatment (Table S5 and Fig. S9†), the effect on increasing τn is significantly less even when the concentration is lowered to half, and little improvement occurs in cell performance. Moving to the other potential factor, the way for TBP to retard electron recombination is proposed to be either by passivating the TiO2 by surface adsorption, which promotes an increase in Ec as well, or by an impeding effect on the [Co(bpy)3]3+ complex, possibly in the form of ‘shielding’.21 The observed Ec downshift, as mentioned above, excludes the former TBP effect and also suggests adsorption of small cations, e.g. protons, probably as the reason. More noteworthy in Fig. 4 and Table S6† is that the pre-exposure treatment also causes a significant decrease in the diffusion resistance of the electrolyte from 10 Ω to 3 Ω, which may explain the increase in the current. In summary, the light-exposure strategy intrinsically solves the current-limiting issues of the classic cobalt-based electrolytes and irreversibly improves the performance of this type of DSSCs. Kinetics studies show a TiO2Ec downshift, retarded recombination and improved electrolyte diffusion, accounting for the significant increase in the photocurrent. The effects are directly tied to the combination of [Co(bpy)3]3+ and TBP, and an irreversible change induced by light exposure. Therefore, in the following section we focus on a more simple mixture, [Co(bpy)3]3+/TBP, to understand the chemistry involved.
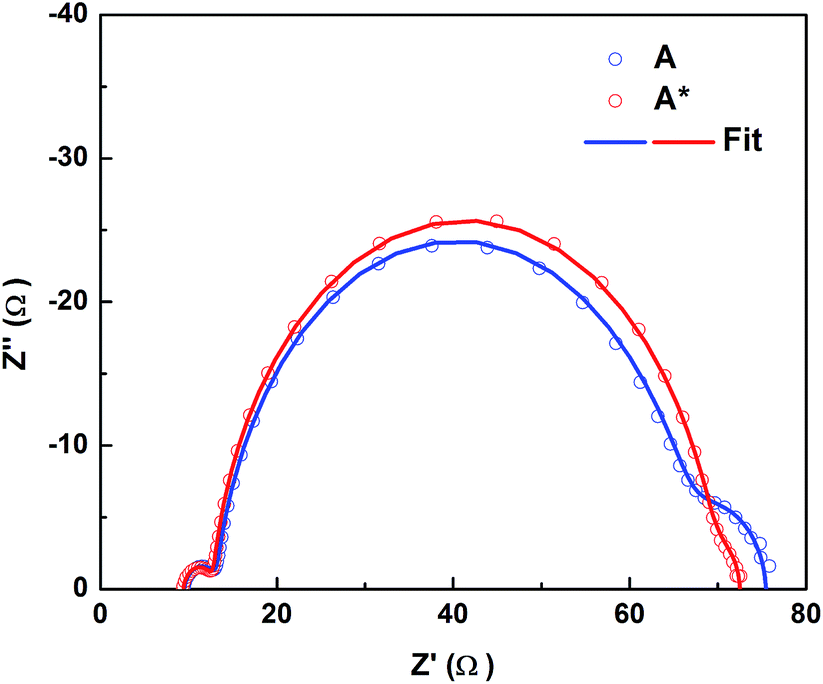 | ||
| Fig. 4 Nyquist plots of D35-sensitized solar cells containing Co(III)/TBP electrolytes without (A) and with (A*) pre-exposure to light. The results were obtained from EIS measurements under dark conditions and at −0.9 V bias. Three semicircles in the plots represent: from left to right, the charge transfer resistances at the electrolyte/counter electrode interface, the charge-transfer resistance at the photoanode/electrolyte interface and the diffusion resistance of the electrolyte, respectively. Parameters obtained from the models are shown in Table S6.† | ||
Electrochemical studies on [Co(bpy)3]3+/TBP mixtures
The changes in the electrochemical properties of the [Co(bpy)3]3+/TBP acetonitrile solution before and after light exposure were investigated. Electrochemical studies of the mixtures were conducted in a symmetrical dummy cell with platinized FTO glass electrodes. In Fig. 5, cyclic voltammetry (CV) results show that when TBP present, the current response is lower compared to solutions containing Co(III) alone (Fig. S10a†), and the peak current (ip) shows a clear dependence on the scan rate (v). The linearity in ip as function of the square root of scan rate (v1/2) (Fig. S11†) indicates that the charge transfer is controlled by ion diffusion. As shown in Fig. S12,† by comparing EIS results of the solution containing [Co(bpy)3]3+ alone with those also containing different concentrations of TBP, it is clear that introduction of TBP to the [Co(bpy)3]3+ electrolyte causes an increase in both diffusion resistance (Rd) and diffusion time (τd). In principle, τd = L2/D, where L is the electrolyte layer thickness and D is the effective diffusion coefficient of the limiting species. Thus, the diffusion limit originates from the retarding effect of TBP and probably a lower diffusion coefficient of the limiting species. Previous work has attributed this effect to the high viscosity of TBP.47 This effect has also been noted by Kirner and Elliot suggesting a Co(III) ligand exchange involving TBP.48 Based on our previous work,2 the adsorption of TBP molecules could influence the surface property of the Pt electrode and thus the charge transport kinetics. After light exposure, CVs of the TBP-containing system show no scan-rate dependence and a 3-fold limiting current. This indicates that the charge transfer process turns to redox kinetic control, and the mass transport in the system is greatly improved. We added the reduced cobalt component ([Co(bpy)3]2+, 0.3 M) to the exposed system to complete the electrolyte. A higher I–V slope was observed but almost the same limiting current, see Fig. S10b.† Such a comparison demonstrates that increasing the concentration of Co(II) (the rate-limiting species) improves the redox kinetics and reversibility in the symmetric cell system, but it does not influence the diffusion. Consistently, τd decreases to 1/3, indicating that the diffusion kinetics at the electrode/electrolyte interface is improved, either through an increase in the diffusion coefficient of the limiting species or a change in the electrode surface environment. These results explain the decrease in the diffusion resistance to one third for a complete cell after pre-exposure treatment as depicted in previous section.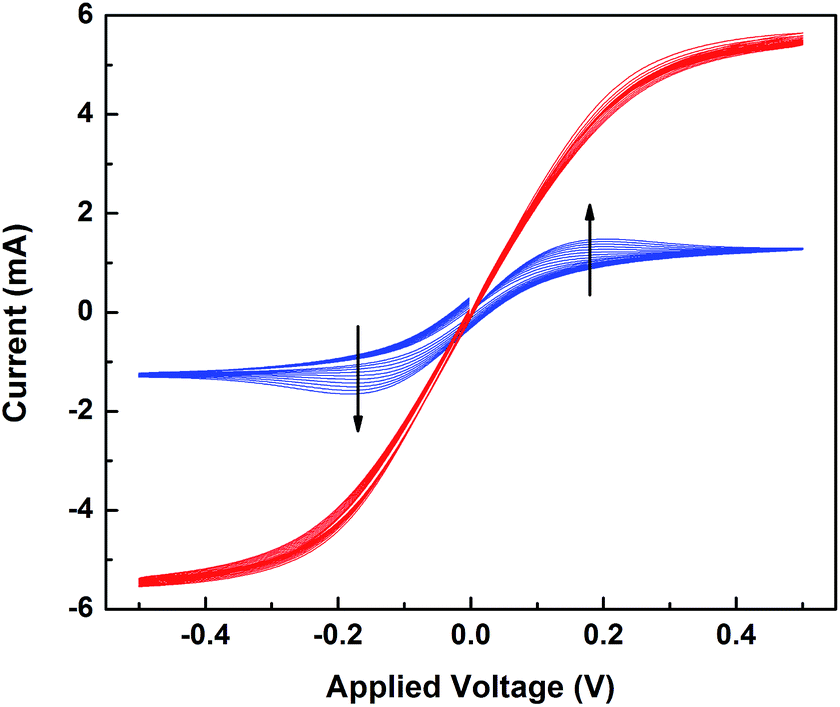 | ||
| Fig. 5 Cyclic voltammograms of 0.15 M/0.2 M [Co(bpy)3]3+/TBP electrolytes in acetonitrile; fresh (blue) and after light exposure (red), symmetrical dummy cells at scan rates from 25 mV s−1 to 250 mV s−1, interspace 25 mV s−1. The arrows show the direction of change with increasing scan rate. | ||
In summary, the higher charge-transport ability of light-exposed electrolytes in the presence of [Co(bpy)3]3+ and TBP is notable, and the link to the better photovoltaic properties is clear. However, the reasons at a molecular level are less obvious.
Spectroscopic studies on [Co(bpy)3]3+/TBP mixtures
In order to identify the origin of the light effect at a molecular level, we investigated the [Co(bpy)3]3+/TBP mixture using different spectroscopic techniques. From a coordination chemistry perspective, the [Co(bpy)3]3+ complex can be regarded as a quasi-octahedral d6 system, and using a simple ligand-field-inspired model it is expected that the [Co(bpy)3]3+ complex is diamagnetic (low spin) and that d–d absorption bands are weak, consistent with the UV-vis absorption features in Fig. 6, because the 6 (mainly) d-type electrons are located in a (near)degenerate orbital of t2g symmetry. Upon mixing with TBP and before light exposure, the absorption spectrum of [Co(bpy)3]3+ in the visible region shows a notable change: a new absorption band at ∼425 nm appears and the total absorbance is considerably increased. This change increases when adding more TBP, see Fig. S13a.† The TBP-induced spectral changes implies that TBP present in the solution must influence the inner coordination sphere of the Co(III) complex and in some way lower the symmetry of the coordination. 1H-NMR spectroscopy can be used to monitor the TBP. A slight downfield shift was found for the TBP 2H-pyridine (peak a, at ∼7.35 ppm) upon mixing and by comparing 1H NMR spectra of TBP alone to that of a mixture with [Co(bpy)3]3+, see Fig. 7. Furthermore, peak a shifts back (hidden in the 2H-bpy peak at ∼7.40 ppm) when extra ligand, bipyridine (bpy), is added to the solution, while the characteristic peaks and the d–d absorption spectra of [Co(bpy)3]3+ remain unchanged, see Fig. S14.† It is here notable that the NMR spectra show a significant difference in the dynamics of ligand exchange between bpy and TBP, where TBP clearly is involved in a fast exchange with Co(III) and bpy is characterized by a slow exchange. Nevertheless, the NMR spectra show that some sort of ligand exchange affecting the chemical environment of TBP must take place. The effects of adding TBP to a solution/electrolyte containing [Co(bpy)3]3+ must involve a change in the coordination geometry of [Co(bpy)3]3+.48 There are however no clues in the spectroscopic results to what type of coordination that may take place. Considering the fast TBP exchange, a general expression of ligand effect can be formulated (eqn (2)). Such an in-binding of TBP may or may not cause a dissociation of bpy, and thus x and y are unknown in eqn (2). Considering that the TBP exchange is fast and the bpy exchange is slow, it seems unlikely that a Co(III) complex of lower coordination number than 6 is formed. Quantum-chemical calculations on initial 7-coordinate [Co(bpy)3(TBP)]3+ complexes converge to quasi-octahedral, 6-coordinate species, where bpy ligands placed trans to TBP are shifted sideways to release the coordination of one of its two pyridine N-atoms. Thus, also higher coordination numbers than 6 appear less feasible. Nevertheless, a lowering of the Co(III) coordination symmetry, possibly via effects from TBP in the second coordination shell of Co(III), as proposed by Salim and co-workers,21 could cause a splitting of the molecular orbitals dominated by the Co d-orbitals. Such a splitting may both change the magnetic properties of the complex and certainly affect the strength of the d–d-transitions, which is thus consistent with the new UV-vis spectral feature observed.| [Co(bpy)3]3+ + xTBP ⇌ [Co(bpy)y(TBP)x]3+ + (3 − y)bpy | (2) |
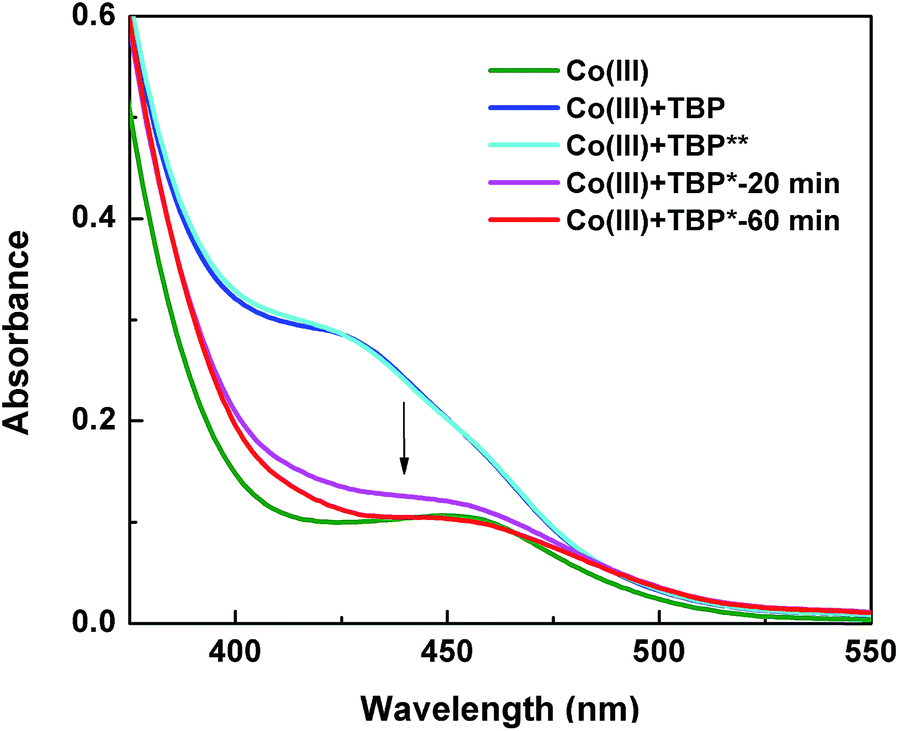 | ||
| Fig. 6 UV-vis absorption spectra of [Co(bpy)3]3+ only (green), [Co(bpy)3]3+/TBP in acetonitrile before (blue) and after storage in the dark (light blue) and after light illumination for 20 min (pink) and 60 min (red). | ||
Using the NMR-spectroscopic data focusing on the exchange of TBP, the exchange equilibrium constant can be estimated (K = 3.7 × 10−3, see Table S8†). In such an estimate changes in bpy coordination are neglected. Consequently, we can deduce that only one to a few percent of the [Co(bpy)3]3+ will be converted according to eqn (2) in the normal DSSC electrolyte, making it hard to identify the exact structure of the resulting Co(III) complex of lower symmetry. Such effects are drowned by the predominantly present and symmetrical [Co(bpy)3]3+ ions. From the results in Table 2, the most dramatic change in diffusive properties is observed for TBP, decreasing from about 24 × 10−10 m2 s−1 to 15 × 10−10 m2 s−1 when mixed with the [Co(bpy)3]3+ ions in acetonitrile. This indicates that in a dilute solution for 1H-NMR spectroscopic investigations, at least 40% of the added TBP in some way interacts with the [Co(bpy)3]3+ complexes and slightly reduces the mobility of the Co(III) ions (9.6 × 10−10 m2 s−1 to 8.7 × 10−10 m2 s−1) in the bulk solution. In summary, the spectroscopic results demonstrate an interaction between [Co(bpy)3]3+ and TBP, which irrespective if involving a ligand exchange or only a secondary interaction, could be the nature of the favourably shielding effect of TBP on electron recombination to Co(III) acceptors as reported and the adverse retarding effect on the electrolyte diffusion as discussed above.
| Samples | D TBP (×10−10 m2 s−1) | D CoB3 (×10−10 m2 s−1) | D Co′ (×10−10 m2 s−1) | Z eff |
|---|---|---|---|---|
| a Derived from the peak a. | ||||
| [Co(bpy)33+] | — | 9.6 | — | 1.38 |
| TBP | 23.9 | — | — | 0 |
| [Co(bpy)33+]/TBP-fresh | 15.4 | 8.7 | — | 1.34 |
| [Co(bpy)33+]/TBP-light | 18.2 | 8.5 | 9.0 | 1.20 |
After the [Co(bpy)3]3+/TBP mixture has been exposed to light, the d–d absorption intensity decreases with time and end up close to the spectrum of the pure and untreated solution of [Co(bpy)3]3+. 1H NMR spectra show that the TBP peak (a) is broadened (Fig. S15†) and changed back to a sharp peak when extra bpy is added from the start (Fig. 7) or added afterwards (Fig. S16†). In Fig. 7, it can be clearly observed that the peak a further and gradually shifts downfield with light exposure time, as also observed for the water peak (∼2.25 ppm). A concomitant new peak (henceforth identified as Co′) arises at ∼14.5 ppm. These phenomena are not observed for the same mixture stored in dark (Fig. 6 and S17†). The spectral changes are irreversible after turning off the light. Since TBP exchange is fast, the original state of the [Co(bpy)3]3+/TBP system will not be re-formed (otherwise it would again partially go back to the asymmetric species with strong d–d absorption, as described above, immediately after turning off the light). The decrease in the Co(III) d–d absorption intensity as function of light exposure time was studied by varying the initial concentrations of [Co(bpy)3]3+ and TBP, see Fig. S13b,† highlighting that the change rate is related to the concentrations of the two compounds. Thus, an irreversible chemical reaction is indicated to take place between the two components upon light exposure. The spectral change is much more difficult to model, but combined with the 1H-NMR-spectroscopic results shown in Fig. 7, it is clear that [Co(bpy)3]3+ is still the main species present in solution, and it must return to a pseudo-octahedral configuration (weak d–d transitions) after light exposure. The Raman spectra in Fig. S18a† show that a new peak arises with a 27 cm−1 shift with respect to the TBP ring breathing frequency at ∼997 cm−1. IR-spectroscopic results shown in Fig. S18b† display a similar qualitative effect, where the pyridine ring vibration modes (δ(C–H), 765 cm−1 and 819 cm−1) decrease in intensity after light exposure. The Raman downshift in wavenumbers, which indicates a lower ring binding strength or alternatively a higher effective mass affecting the ring vibration, could be attributed to TBP either becoming coordinated to the cobalt centre or protonated.49–51 However, a persistent coordination to Co(III) is not a feasible explanation because of the observed fast TBP ligand exchange. The shift of the TBP peak a is reduced either when adding fresh TBP to the exposed mixture (the integral intensity increases as well, Fig. 7) or when more TBP is added from the start, see Fig. S19.† This demonstrates that the peak corresponds to the weighted average of a free/modified TBP system in the fast exchange regime with respect to the frequency difference between the spin systems (peaks) involved. In this context we also note that the diffusion coefficient of TBP after light exposure is still lower than that of free TBP.
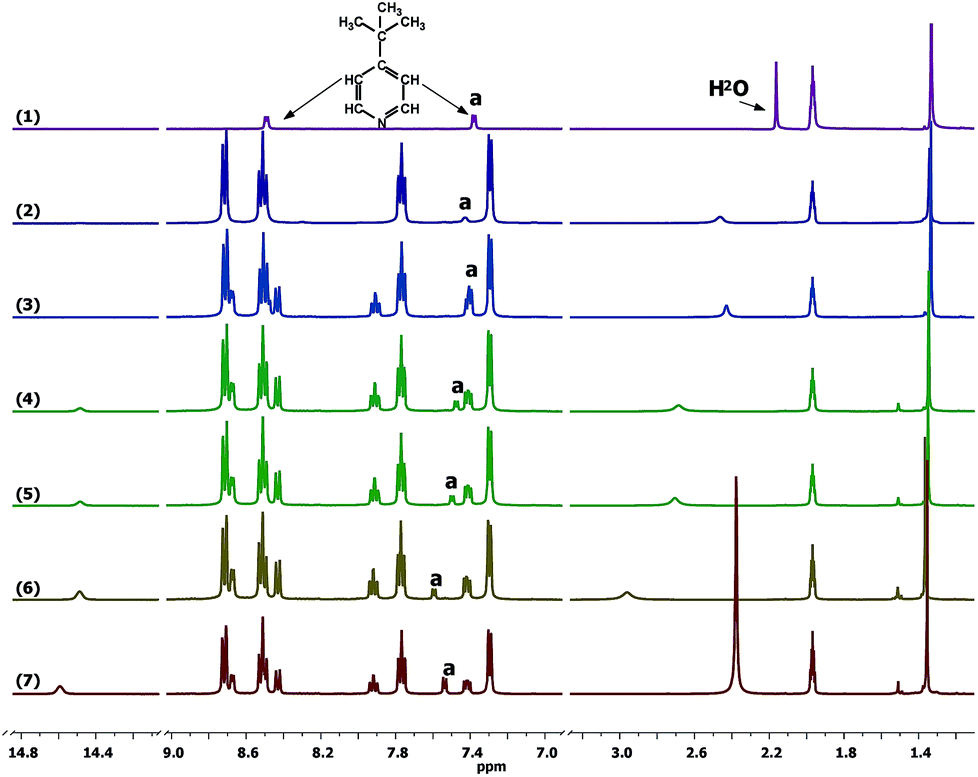 | ||
| Fig. 7 1H NMR spectra of (1) TBP only, (2) [Co(bpy)3]3+/TBP, (3) [Co(bpy)3]3+/TBP/bpy when fresh and after light exposure for (4) 2 h, (5) 4 h and (6) 24 h, and (7) with one more equivalent of fresh TBP added in CD3CN. The characteristic peak of the TBP pyridine hydrogen atoms is labelled a. The initial component concentrations were: TBP 0.008 M, [Co(bpy)3]3+ 0.05 M and bpy 0.05 M. | ||
It is also worth noting that the Co′ 1H NMR-spectroscopic peak with a chemical shift at around 14.5 ppm, alluded to above, emerges and increases with time of light exposure. The peak areas indicate that part of Co(III) has been converted into this new species. The peak shows a short relaxation time (T1 = 0.082 s) and stronger temperature dependence (Fig. S20†), indicating the formation of a paramagnetic complex. Possible explanations to this new peak include (1) the formation of paramagnetic Co(II) (d7 configuration) involving a redox reaction;52 (2) a light-induced spin-crossover of Co(III) to a high-spin state driven by TBP.53,54 A third alternative could involve the generation of a binuclear complex coordinated by TBP, but the essentially unaffected diffusion coefficients of the Co-containing species speak against such an explanation.55,56 These hypothesized complexes cannot be detected by QC-MS, see Fig. S21.† In fact, all attempts to isolate any new species from the [Co(bpy)3]3+–TBP mixtures, light-exposed or not, have resulted in solid compounds containing the pure [Co(bpy)3]3+ complex only. We have observed that in the presence of [Co(bpy)3]3+, addition of the [Co(bpy)3]2+ complex to a TBP solution leads to a broadened TBP peak a in 1H-NMR spectra (Fig. S22†), which is consistent with that in the presence of [Co(bpy)3]3+ after light exposure. The reason could be fast ligand exchange between TBP and the labile paramagnetic Co(II) species. Thus, the fact that extra addition of bpy to the light-exposed system leads to sharper TBP peaks is understandable, as a TBP/bpy ligand exchange equilibrium at the Co(II) centre is reformed. Hence, scenario (1) is more reasonable; that is, a redox reaction occurs in the [Co(bpy)3]3+/TBP system under light with [Co(bpy)3]2+ being formed. Redox reaction products of TBP or bpy are unlikely to explain the spectral changes, but more likely reaction with some unintentional electrolyte component, such as water.57 A possible reaction is proposed in eqn (3).
| 4[Co(bpy)3]3+ + 2H2O + TBP → 4[Co(bpy)3]2+ + 4TBP–H+ + O2 | (3) |
The production of protons binding to the base TBP could be inferred by the gradual downfield shift of the TBP peak a and water peak in Fig. 7.50,51 The hard acidity of protons is similar to that of Li+ ions, and so are the effects on DSSCs, including those on the photoanode, such as the positively shifted Ec, the red-shifted absorption spectra of the dye/TiO2 film, the suppressed recombination loss and for the other part, reduced TBP activity in interacting with [Co(bpy)3]3+ due to protonation, allowing [Co(bpy)3]3+ to go back to a symmetric configuration with weak d–d light absorption. Since no significant increase is observed for the diffusion coefficients of Co(III) and Co′ after light exposure, the enhancement of diffusion/charge transport is likely to be attributed to the reduction of available TBP at the cathode surface due to protonation. Thus, by the strategy of light exposure on [Co(bpy)3]3+/TBP mixture with a possible production of protons as a result, the DSSC performance reaches the same levels as when Li+ salts have been added. In addition, the presence of Li+ salts shows worse dye photostability than the presence of a Brønsted acid (Fig. S23†). This result explains the benefits of the Li+-ion-free cobalt electrolytes with respect to the long-term photostability of DSSCs.
Conclusions
This work has identified an irreversible light-induced reaction in the commonly used cobalt tris-(bipyridine)-mediated electrolyte for DSSCs. This effect causes a significant improvement of cell performance, mainly in the photocurrent. After prior light exposure treatment of a Co(III)/TBP mixture, solar cells outperform typical lithium-ion-containing ones both regarding initial conversion efficiency and long-term stability. Detailed characterization based on fully operational devices, as well as model systems containing electrolyte mixtures, points to an interaction between Co(III) and TBP, which macroscopically influences the kinetics at the electrode/electrolyte interfaces. The microscopic picture remains a bit unclear, although we can present a plausible explanation to a [Co(bpy)3]3+–TBP interaction upon mixing, based on secondary coordination, and to a proposed redox reaction between Co(III) with trace water in the presence of TBP upon light exposure leading to the formation of protons or TBP protonation. All in all, the work points to the importance of investigating the detailed molecular dynamics in multi-component DSSC systems, and particularly regarding the electrolyte and its interface reactions.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the Swedish Research Council, the Swedish Energy Agency as well as the China Scholarship Council (CSC) for financial support. The authors also thank Dr Lei Wang for his helpful assistance in 1H-NMR spectroscopic studies.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- J. Gao, M. Bhagavathi Achari and L. Kloo, Chem. Commun., 2014, 50, 6249–6251 RSC.
- J. Gao, W. Yang, M. Pazoki, G. Boschloo and L. Kloo, J. Phys. Chem. C, 2015, 119, 24704–24713 CrossRef CAS.
- T. C. Li, A. M. Spokoyny, C. She, O. K. Farha, C. A. Mirkin, T. J. Marks and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 4580–4582 CrossRef CAS PubMed.
- B. A. Gregg, F. Pichot, S. Ferrere and C. L. Fields, J. Phys. Chem. B, 2001, 105, 1422–1429 CrossRef CAS.
- T. W. Hamann, Dalton Trans., 2012, 41, 3111–3115 RSC.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724 CrossRef CAS PubMed.
- S. M. Feldt, P. W. Lohse, F. Kessler, M. K. Nazeeruddin, M. Gratzel, G. Boschloo and A. Hagfeldt, Phys. Chem. Chem. Phys., 2013, 15, 7087–7097 RSC.
- Y. Xie, J. Baillargeon and T. W. Hamann, J. Phys. Chem. C, 2015, 119, 28155–28166 CrossRef CAS.
- M. K. Kashif, J. C. Axelson, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, J. Am. Chem. Soc., 2012, 134, 16646–16653 CrossRef CAS PubMed.
- M. K. Kashif, M. Nippe, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, Angew. Chem., Int. Ed., 2013, 52, 5527–5531 CrossRef CAS PubMed.
- Y. Xie and T. W. Hamann, J. Phys. Chem. Lett., 2013, 4, 328–332 CrossRef CAS PubMed.
- B. M. Klahr and T. W. Hamann, J. Phys. Chem. C, 2009, 113, 14040–14045 CrossRef CAS.
- H. Nusbaumer, S. M. Zakeeruddin, J.-E. Moser and M. Grätzel, Chem.–Eur. J., 2003, 9, 3756–3763 CrossRef CAS PubMed.
- S. M. Feldt, U. B. Cappel, E. M. J. Johansson, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2010, 114, 10551–10558 CrossRef CAS.
- J.-H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J.-E. Moser, C. Yi, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2012, 3, 631 CrossRef PubMed.
- E. Mosconi, J.-H. Yum, F. Kessler, C. J. Gómez García, C. Zuccaccia, A. Cinti, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, J. Am. Chem. Soc., 2012, 134, 19438–19453 CrossRef CAS PubMed.
- S. Nakade, Y. Makimoto, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2005, 109, 3488–3493 CrossRef CAS PubMed.
- J. J. Nelson, T. J. Amick and C. M. Elliott, J. Phys. Chem. C, 2008, 112, 18255–18263 CrossRef CAS.
- T. N. Murakami, N. Koumura, T. Uchiyama, Y. Uemura, K. Obuchi, N. Masaki, M. Kimura and S. Mori, J. Mater. Chem. A, 2013, 1, 792–798 RSC.
- N. T. Salim, X. Yang, S. Zhang, J. Liu, A. Islam and L. Han, J. Mater. Chem. A, 2014, 2, 10532–10539 RSC.
- W. Yang, Y. Hao, P. Ghamgosar and G. Boschloo, Electrochim. Acta, 2016, 213, 879–886 CrossRef CAS.
- J. T. Kirner and C. M. Elliott, J. Phys. Chem. C, 2015, 119, 17502–17514 CrossRef CAS.
- A. Listorti, C. Creager, P. Sommeling, J. Kroon, E. Palomares, A. Fornelli, B. Breen, P. R. F. Barnes, J. R. Durrant, C. Law and B. O'Regan, Energy Environ. Sci., 2011, 4, 3494–3501 RSC.
- L. Yang, B. Xu, D. Bi, H. Tian, G. Boschloo, L. Sun, A. Hagfeldt and E. M. J. Johansson, J. Am. Chem. Soc., 2013, 135, 7378–7385 CrossRef CAS PubMed.
- P. Tiwana, P. Docampo, M. B. Johnston, L. M. Herz and H. J. Snaith, Energy Environ. Sci., 2012, 5, 9566–9573 RSC.
- B. O'Regan and D. T. Schwartz, Chem. Mater., 1998, 10, 1501–1509 CrossRef.
- S. Ferrere and B. A. Gregg, J. Phys. Chem. B, 2001, 105, 7602–7605 CrossRef CAS.
- B. A. Gregg, S.-G. Chen and S. Ferrere, J. Phys. Chem. B, 2003, 107, 3019–3029 CrossRef CAS.
- M. Bhagavathi Achari, V. Elumalai, N. Vlachopoulos, M. Safdari, J. Gao, J. M. Gardner and L. Kloo, Phys. Chem. Chem. Phys., 2013, 15, 17419–17425 RSC.
- J. Nissfolk, K. Fredin, A. Hagfeldt and G. Boschloo, J. Phys. Chem. B, 2006, 110, 17715–17718 CrossRef CAS PubMed.
- M. Bailes, P. J. Cameron, K. Lobato and L. M. Peter, J. Phys. Chem. B, 2005, 109, 15429–15435 CrossRef CAS PubMed.
- K. C. D. Robson, K. Hu, G. J. Meyer and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1961–1971 CrossRef CAS PubMed.
- L. J. Antila, F. G. Santomauro, L. Hammarström, D. L. Fernandes and J. Sá, Chem. Commun., 2015, 51, 10914–10916 RSC.
- S. Sarker, A. J. S. Ahammad, H. W. Seo and D. M. Kim, Int. J. Photoenergy, 2014, 2014, 17 CrossRef.
- F. Hallberg, I. Furó, P. V. Yushmanov and P. Stilbs, J. Magn. Reson., 2008, 192, 69–77 CrossRef CAS PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408–3420 CrossRef CAS.
- S. Agrawal, T. Leijtens, E. Ronca, M. Pastore, H. Snaith and F. De Angelis, J. Mater. Chem. A, 2013, 1, 14675–14685 RSC.
- W. Yang, Y. Hao, N. Vlachopoulos, A. I. K. Eriksson and G. Boschloo, J. Phys. Chem. C, 2016, 120, 22215–22224 CrossRef CAS.
- R. M. O'Donnell, R. N. Sampaio, T. J. Barr and G. J. Meyer, J. Phys. Chem. C, 2014, 18, 16976–16986 CrossRef.
- J. Wiberg, T. Marinado, D. P. Hagberg, L. Sun, A. Hagfeldt and B. Albinsson, J. Phys. Chem. B, 2010, 114, 14358–14363 CrossRef CAS PubMed.
- R. Gao, L. Wang, Y. Geng, B. Ma, Y. Zhu, H. Dong and Y. Qiu, Phys. Chem. Chem. Phys., 2011, 13, 10635–10640 RSC.
- J. Teuscher, J.-D. Décoppet, A. Punzi, S. M. Zakeeruddin, J.-E. Moser and M. Grätzel, J. Phys. Chem. Lett., 2012, 3, 3786–3790 CrossRef CAS PubMed.
- T. M. Koh, K. Nonomura, N. Mathews, A. Hagfeldt, M. Grätzel, S. G. Mhaisalkar and A. C. Grimsdale, J. Phys. Chem. C, 2013, 117, 15515–15522 CrossRef CAS.
- J. T. Kirner and C. M. Elliot, J. Phys. Chem. C, 2015, 119, 17502–17514 CrossRef CAS.
- L. Kavan, Y. Saygili, M. Freitag, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Electrochim. Acta, 2017, 227, 194–202 CrossRef CAS.
- G. Roy, J. F. Miravet, B. Escuder, C. Sanchez and M. Llusar, J. Mater. Chem., 2006, 16, 1817–1824 RSC.
- N. Wattanavichean, E. Casey, R. J. Nicholsa and H. Arnolds, Phys. Chem. Chem. Phys., 2018, 20, 866–871 RSC.
- J. K. Beattie and H. Elsbernd, Inorg. Chim. Acta, 1995, 240, 641–644 CrossRef CAS.
- M. Khalil, M. A. Marcus, A. L. Smeigh, J. K. McCusker, H. H. W. Chong and R. W. Schoenlein, J. Phys. Chem. A, 2006, 110, 38–44 CrossRef CAS PubMed.
- S. Thies, H. Sell, C. Schütt, C. Bornholdt, C. Näther, F. Tuczek and R. Herges, J. Am. Chem. Soc., 2011, 133, 16243–16250 CrossRef CAS PubMed.
- Ş. Yurdakul and M. Banat, J. Inclusion Phenom. Macrocyclic Chem., 1996, 26, 127–131 CrossRef.
- M. Fondo, N. Ocampo, A. M. Garcia-Deibe, M. Corbella, M. S. El Fallah, J. Cano, J. Sanmartin and M. R. Bermejo, Dalton Trans., 2006, 4905–4913, 10.1039/b609961k.
- J. Gao, A. Fischer, P. H. Svensson and L. Kloo, ChemistrySelect, 2017, 2, 1675–1680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Dye structures, photovoltaic performance of DSSCs, IPCE, Toolbox, electron injection and dye regeneration kinetic data, 1H NMR and infrared absorption spectra. See DOI: 10.1039/c9ta07198a |
| This journal is © The Royal Society of Chemistry 2019 |