
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Predicting the reactivity of energetic materials: an ab initio multi-phonon approach†
Adam A. L.
Michalchuk‡
 *ab,
Morris
Trestman§
a,
Svemir
Rudić
c,
Peter
Portius
d,
Peter T.
Fincham
e,
Colin R.
Pulham
*ab and
Carole A.
Morrison
*a
*ab,
Morris
Trestman§
a,
Svemir
Rudić
c,
Peter
Portius
d,
Peter T.
Fincham
e,
Colin R.
Pulham
*ab and
Carole A.
Morrison
*a
aEaStCHEM School of Chemistry, Centre for Science at Extreme Conditions, University of Edinburgh, Edinburgh, UK. E-mail: adam.michalchuk@bam.de; carole.morrison@ed.ac.uk; c.r.pulham@ed.ac.uk
bEPSRC Centre for Continuous Manufacturing and Crystallisation (CMAC), University of Edinburgh, Edinburgh, UK
cISIS Neutron and Muon Source, STFC, Rutherford Appleton Laboratory, Chilton, Didcot, OX11 0QX, UK
dDepartment of Chemistry, University of Sheffield, Western Bank, Sheffield S3 7HF, UK
eDefence Science and Technology Laboratory (DSTL), Porton Down, Salisbury SP4 0JQ, Wiltshire, UK
First published on 1st August 2019
Abstract
The ease with which an energetic material (explosives, propellants, and pyrotechnics) can be initiated is a critical parameter to assess their safety and application. Impact sensitivity parameters are traditionally derived experimentally, at great cost and risk to safety. In this work we explore a fully ab initio approach based on concepts of vibrational energy transfer to predict impact sensitivities for a series of chemically, structurally and energetically diverse molecular materials. The quality of DFT calculations is assessed for a subset of the materials by comparison with experimental inelastic neutron scattering spectra (INS). A variety of models are considered, including both qualitative and quantitative analysis of the vibrational spectra. Excellent agreement against experimental impact sensitivity is achieved by consideration of a multi-phonon ladder-type up-pumping mechanism that includes both overtone and combination pathways, and is improved further by the added consideration of temperature. This fully ab initio approach not only permits ranking of energetic materials in terms of their impact sensitivity but also provides a tool to guide the targeted design of advanced energetic compounds with tailored properties.
Introduction
Energetic materials (EMs; explosives, propellants and pyrotechnics) are characterized by their ability to convert large amounts of chemical potential energy rapidly into kinetic energy. This has made EMs central to the development of many technologies for both civilian and military application. Paramount to these technologies is the safe and controlled initiation of the EM, with accidental initiation being potentially life-threatening.1 The need for stringent safety and performance controls has greatly limited the number of compounds used in EMs. While many new EMs have been proposed,2 successful developments typically favour the preparation of composite materials that consist of a mixture of energetic compounds and additives. Amongst others, this approach has culminated in the formulation of dry mixtures, or more commonly, polymer-bonded explosives (PBXs) such as Semtex and C4. Recent paradigm changes in EM research have motivated focused studies towards development of insensitive munitions that exhibit minimal environmental impact.3 The development of new energetic molecules has been paramount to these efforts, accompanied by growing interest in the development of multi-component energetic solids.4The study of many important physical properties of EMs – including detonation velocity, enthalpy of formation, heat of explosion, and temperature of detonation – is amenable to computational approaches and quantitative predictions.5–7 The ability to predict properties quantitatively allows for powerful strategies to screen for new energetic molecules.8 Until recently, and despite a thorough understanding of materials chemistry and detonation physics, there remained very little understanding of the EM initiation mechanism. Therefore, the sensitivity of EMs could not be predicted reliably. Early work by Bowden and Yoffe9,10 showed that the bulk heating associated with mechanical impact is insufficient to describe the initiation processes. Thus, the concept of ‘hot-spots’ was developed to describe the localization of energy into highly concentrated regions capable of inducing bond dissociation. A number of more sophisticated mechanisms have since been proposed that typically describe the concentration of energy at interfaces (e.g. grain or particle boundaries) and the generation of high local temperatures or stresses. The former is particularly problematic in the description of processes in molecular materials, where melting occurs at temperatures considerably lower than those required to induce bond rupture.11 Regardless of the hot-spot origin, the mechanism by which energy leads to bond rupture has yet to be elucidated.
Considerable effort has been devoted to identifying a set of parameters capable of rationalizing the impact sensitivity of EMs.12 Noting that EM initiation necessarily involves bond rupture, models that are based on isolated molecules have often aimed at correlating impact sensitivity with the stability of bonds in the explosive molecule. This is done, for instance, by computing dissociation energies13 or analysing the topology of the electron density.14 However, these models are incapable of describing correctly the different sensitivity of polymorphic forms, i.e. crystalline phases that differ in the spatial arrangement of the energetic molecules. For example, δ-HMX is much more sensitive to impact than β-HMX.15 Furthermore, the impact sensitivity of an EM can be widely tuned by multi-component crystallization.4 In such systems, the structure of the energetic molecule itself again remains unaltered. This problem highlights that improved models of EM initiation must also account for characteristic parameters pertinent to the solid state. A number of such parameters have been suggested, including crystal void space and compressibility,16 intermolecular distances,17 and intermolecular interaction energies.18 In models that take account of these characteristics, the process of initial bond rupture has been identified as central. Authors have thus attempted to correlate bond rupture propensity to the electronic band gap19 and, in turn, impact sensitivity. However, as these methods offer only a qualitative rationale for impact sensitivity, they are generally restricted to chemically similar energetic molecules. In view of this restriction, other attempts have included a consideration of the dynamics of a mechanical impact, for example, by studying the effects of stress on band gaps and bond dissociation energies,20–22 or by considering the interlayer energies associated with imposing a structural distortion within the crystal.23,24 These more recent methods have typically been restricted to very small sample sets consisting of only a few EMs.
Coffey and Totton,25 and subsequently Zerilli and Totton,26 described a mechanism for the localisation of energy in crystalline materials that is based on vibrational energy transfer. This model assumes that mechanical energy, which excites low frequency phonon modes, undergoes rapid up-conversion (‘up-pumping’) through a process of phonon–phonon collisions. This ultimately increases the population (and hence amplitude) of internal molecular modes beyond covalent bond dissociation limits. The model was then developed further by Dlott and co-workers,27 who included a description of compression28 and localization of energy at crystal defects.29 Various authors have also developed predictive models of impact sensitivity which are based on the phonon up-pumping description of mechanically perturbed solids. The earliest attempts of this type used data from inelastic neutron scattering spectra,30,31 and were followed by work based on experimental Raman spectra.32 Some work was based on an analysis that considered the vibrational multi-phonon density of states,30,33,34 while other attempts have instead focused on up-pumping by high-order harmonic (e.g. overtone) coupling processes.32 Despite the promise offered by a model rooted in multi-phonon up-pumping to rationalize impact sensitivity of EMs, models based on experimental data alone do not offer predictive value. Correspondingly, the first results of vibrational up-pumping methods that rely on an ab initio input based on gas phase35 and periodic structure models34,36,37 have now appeared. Using zone-centre vibrational frequencies for molecular crystals, Bernstein36 was able to rank the relative impact sensitivity of six chemically similar organic EMs. In our previous work,37 we have investigated the vibrational up-pumping in nine azide-based crystals by calculating the complete vibrational dispersion curves. A correct ranking of these materials was achieved when both overtone and combination pathways were taken into account in the up-pumping process. Thus, while certain vibrational up-pumping models appear to be capable of describing impact sensitivity in EMs, it is not yet clear which models are applicable to a more chemically diverse set of complex organic EMs. This problem motivates the work in the present manuscript that sets out to develop further an ab initio model to describe the impact sensitivity for a set of chemically diverse EMs.
General theory
In this paper, the term phonon is used in reference to the delocalised, external vibrational modes of a crystal. Vibrational modes that are localised on molecules are denoted as internal vibrational modes.A mechanical perturbation can lead to the excitation of a crystalline material through various mechanisms. A high-energy impact, for instance, will lead to compression of a material and associated vibrational mode hardening. The magnitude of hardening depends on the average material Grüneisen parameter, Γ = (1/N)∑dln(ω)/dln(V), where ω and V denote frequency and volume, respectively, and N is the number of vibrational modes. Within the harmonic approximation, a material with energy E = E0 + ℏω(n + 1/2) will have a heat capacity at constant volume,
| CV ∝ ∫(exp(βε) − 1)−2g(ω)dω | (1) |
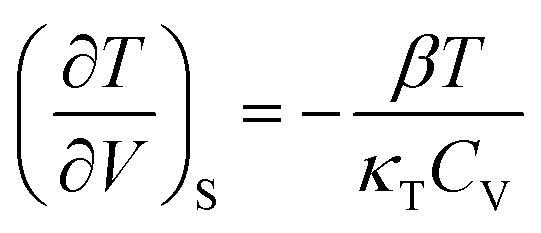 | (2) |
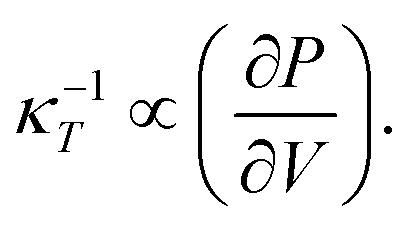 Here ∂P is derived from an increase in the temperature-dependent phonon pressure. Hence eqn (2) can also be regarded as a description of an increase in temperature that is proportional to dE/CV. This description leads to adiabatic heating of the bulk material according to:38
Here ∂P is derived from an increase in the temperature-dependent phonon pressure. Hence eqn (2) can also be regarded as a description of an increase in temperature that is proportional to dE/CV. This description leads to adiabatic heating of the bulk material according to:38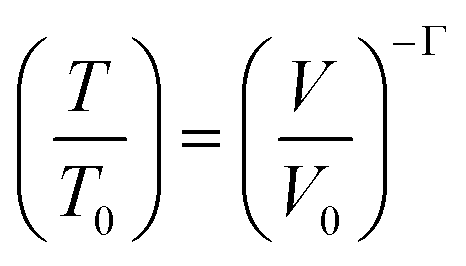 | (3) |
It follows that highly compressible materials undergo greater adiabatic heating. The Grüneisen parameters tend to be considerably larger for external vibrational modes than for internal states, and hence the initial adiabatic heating occurs predominantly in the external modes. As the heat capacity of the phonon modes is markedly lower than the total heat capacity, the phonon quasi-temperature greatly exceeds that of the final bulk temperature given by eqn (3).
Following initial excitation, the excess phonon energy dissipates through phonon–phonon collisions. By truncating the mathematical description of this process at the leading anharmonic term, the three-phonon collisions (i.e. the combination of two phonon states to create a third) dominate. This can be described by the general Hamiltonian
| H = Hph + Hv + Hph–v | (4) |
| Hph–v ∝ |V(3)|2δ[Ω(k) − ω1(k) − ω2(k)] | (5) |
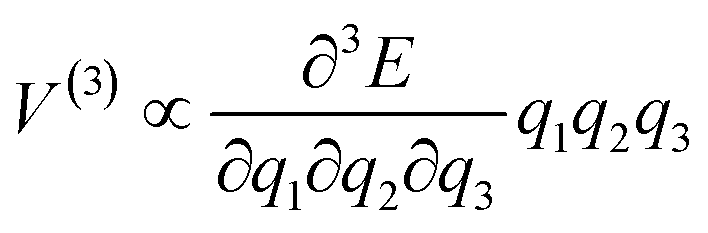 | (6) |
Although the initial up-pumped vibrational energy remains localized on molecules, it will eventually dissipate spatially at a rate that depends on the anharmonic coupling between molecules.42 In order for sufficient energy to be localized on an individual molecule, a large amount of energy must therefore up-pump at a rate that is faster than the dissipation rate. It follows that those materials which, according to eqn (4), exhibit faster vibrational up-pumping, can be expected to display greater susceptibility to initiation by mechanical impact.
The probability of scattering is proportional to the number of phonons available to scatter. Thus, the initial up-pumping events are dominated by qqQ processes, in which the inequality ω(Q) < ω(q) + ω(q) applies. The ω(Q) states are known as the doorway modes, as they act as efficient conduits for vibrational energy transfer between extended phonon modes and localised internal vibrational modes. This efficiency is largely a consequence of their mixed nature. As energy and phase must be conserved, the energy transfer step is dominated by processes in which ω(q) = ω(Q)/2.29 Once the doorway modes are excited, they can subsequently scatter to excite higher frequency modes in a ‘ladder’-type approach.43 The overall-process can be summarized by Fig. 1.
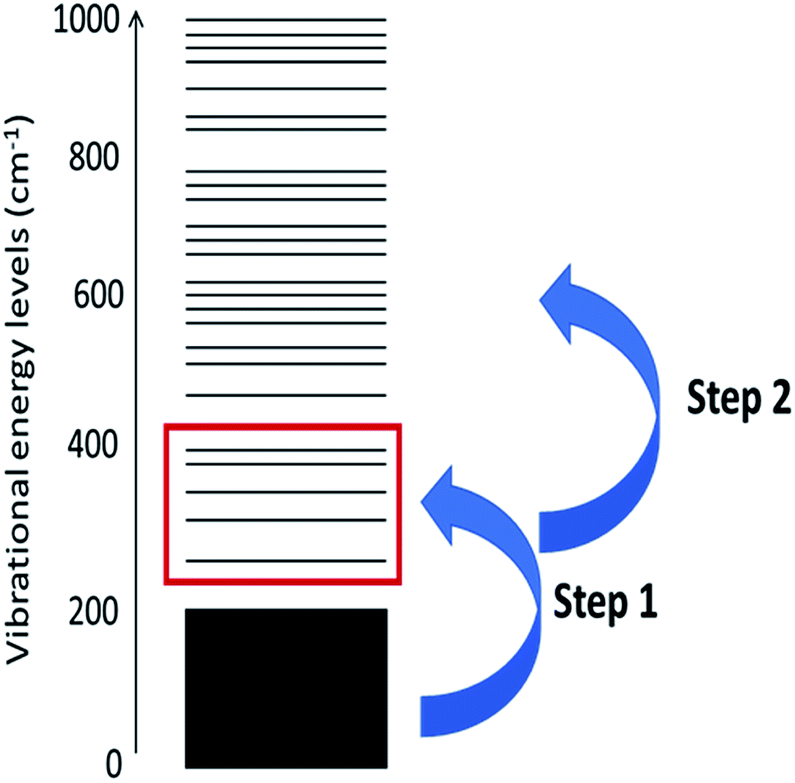 | ||
| Fig. 1 Schematic representation of the vibrational up-pumping model. The lowest frequency modes (black rectangle) absorb the initial mechanical energy, which then transfers to the ‘doorway’ modes (red rectangle). Finally, the doorway mode energy up-pumps into internal molecular modes at higher frequency to induce bond rupture. | ||
In order to induce the initiation of a chemical reaction, a dissociation barrier must be overcome. In our theory this requires that sufficient vibrational energy is transferred from the lattice to localised molecular modes. The (sub)set of vibrational modes that are capable of inducing a bond dissociation are known as the target modes. The chemical simplicity of the binary energetic azides, Mx(N3)y, permitted identification of a single vibrational normal coordinate that is responsible for initiating N−3.37 In such cases, a direct up-pumping model can be discussed.25 Such a mechanism is possible if either (1) the target mode is sufficiently isolated such that the multi-phonon density of states is negligible in its vicinity, or (2) the target mode has sufficiently strong coupling to the phonon bath to permit rapid, direct transfer. While condition (1) often applies to EMs composed of simple energetic molecules such as N−3, for other EMs, such as those consisting of cyclic molecules shown in Fig. 2, the decomposition pathways can be quite complex. Instead, an up-pumping model can be constructed that is based on an indirect (thermal) mechanism.41 In this case, vibrational energy rapidly up-pumps into the entire manifold of internal molecular modes which leads to the population of a subset of vibrational modes responsible for bond rupture and initiation. This is the approach used throughout this work. Sensitivity is thus correlated with the total up-pumping into internal vibrational modes. This correlation is consistent with RRKM theory, in which localisation on particular modes becomes more probable with higher amounts of localised energy. Furthermore, the 0 K limit previously employed in our earlier work on the azide materials37 is now extended to include temperature effects as well as the relative heating associated with adiabatic compression.
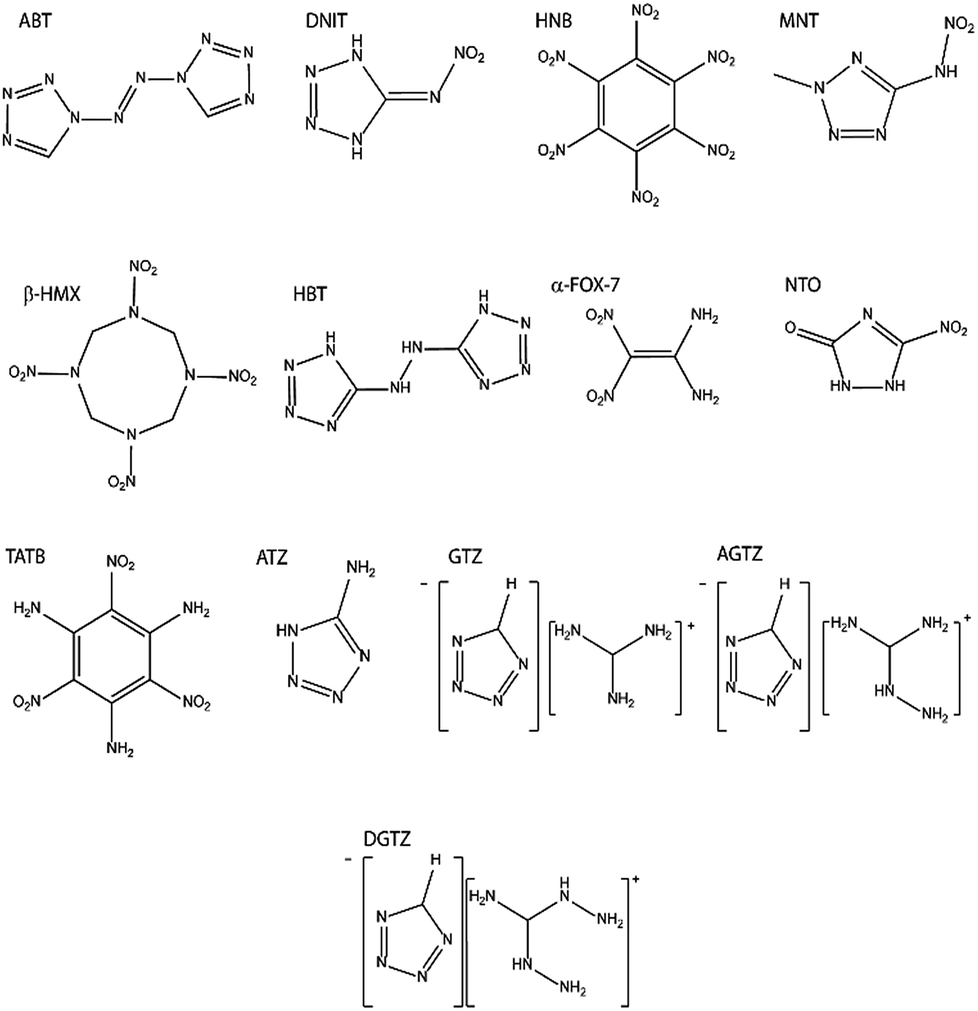 | ||
| Fig. 2 Lewis structures of the EMs investigated in this work. The corresponding crystal structures are given in the ESI S1.† | ||
Test set
Typical organic EMs consist of molecules with a large number of atoms and, common to other molecular materials, form crystals that adopt comparably large unit cells that possess low symmetry. Test candidates were therefore selected for which calculations were computationally tractable whilst ensuring that a broad range of impact sensitivities and structure types were represented. The test set was explicitly selected to include families of structurally related compounds, as well as a range of chemically diverse species. In this way, we aimed to test our model both within and between families of EMs, the latter having proved particularly challenging in the literature. Although many of these materials have been studied extensively, it is not uncommon for a range of impact sensitivities to be reported for the same compound. Documented factors influencing the reproducibility of impact sensitivity measurements for a given compound are the particle size, particle size distribution and phase purity.52 Three of the compounds, guanidinium tetrazolate (GTZ), aminoguanidinium tetrazolate (AGTZ) and diaminoguanidinium tetrazolate (DGTZ) are new materials, reported here for the first time. The test set used in our study is given in Fig. 2 (with crystal structures in the ESI S1†) and Table 1, alongside a summary of reported impact sensitivities. The output of the computational model will be compared against the trend in experimental impact sensitivities. This trend in impact sensitivity is based on the following collated observations based on the open literature and new measurements (for abbreviations see Table 1 and Fig. 2):| Material | Acronym | IS (h50)/cm | IS (E50)/J | IS (UTC)/J | Ref. |
|---|---|---|---|---|---|
| a Recalculated for a drop weight mass of 2.5 kg. b Assumed value. | |||||
| 1,1′-Azobistetrazole | ABT | — | — | <1 | 44 |
| 4,5-Dihydro-5-nitrimino-1H-tetrazole | DNIT | — | — | 1.5 | 45 |
| Hexanitrobenzene | HNB | 11 | 2.75 | — | 46 |
| 2-Methyl-5-nitramino-2H-tetrazole | MNT | — | — | 3 | 45 |
| 1,3,5,7-Tetranitro-1,3,5,7-tetrazocane | HMX | 32 | 8 | — | 47 |
| 29 | 7.25 | — | 48 | ||
| 26 | 6.5 | — | 48 | ||
| 5,5′-Hydrazinebistetrazole | HBT | — | — | >30 | 49 |
| 1,1-Diamino-2,2-dinitroethene | FOX-7 | 126 | 31.5 | — | 50 |
| Nitrotriazolone | NTO | 291 | 72.75 | — | 48 |
| 2,4,6-Triamino-1,3,5-trinitrobenzene | TATB | 490 | 122.5 | — | 48 |
| 5-Amino-1H-tetrazole | ATZ | — | >50b | — | See text |
| Guanidinium-1H-tetrazolate | GTZ | >204a | >50.1 | — | 51 |
| Aminoguanidinium-1H-tetrazolate | AGTZ | >31a | — | >8.2 | 51 |
| Diaminoguanidinium-1H-tetrazolate | DGTZ | >204a | >50.1 | — | 51 |
1. It is widely acknowledged that TATB is insensitive to impact.12 Most reports state its impact sensitivity to be ‘immeasurable’. Only a single source suggests that TATB has a sensitivity on the order of 50 J,52 although no indication was provided as to its origin or the experimental method used.
2. NTO is generally regarded as a highly insensitive material.52
3.Despite discrepancies in the size of the sensitivity value (ranging from ∼20 J to 50 J), it is accepted that α-FOX-7 is less sensitive than β-HMX.52
4. HNB is well-known to be highly sensitive and exceeds the sensitivity of β-HMX.50
5. The sensitivity of 5-substituted 1H tetrazoles is ranked according to R groups and follows the series CH3 < NH2 < N2HO2 < Cl < NO2 < N3 < N+2.53,54 The Cl-substituted tetrazole is reported to have an impact sensitivity >40 J. It is reasonable to expect the NH2-substituted compound to be effectively insensitive. No data could be found to support the notion that 5-amino-1H-tetrazole exhibits any notable sensitivity. For the purpose of this study we have assumed a value of >50 J.
6. No sensitivity was detected for the new compounds GTZ or DGTZ at impact energies up to 50.1 J.51
7. No sensitivity was detected for the new compound AGTZ at impact energies up to 8.2 J (maximum tested energy).51
From these facts we derive the following series of sensitivity: ABT > HNB > HMX > HBT > FOX-7 > ATZ > NTO ≈ TATB ≈ GTZ ≈ AGTZ ≈ DGTZ.
Results and discussion
Electronic structure-band gap criterion
EM initiation requires rupture of a chemical bond, which itself requires population of an antibonding state. This has led to the so-called ‘band gap criterion’ for EM sensitivity prediction.55 The HSE06 screened hybrid functional is a recognised standard for band gap calculations, although it is often too expensive for large molecular systems. Based on results obtained for a series of inorganic materials, the less demanding hybrid GGA functional, B3PW91, has instead been shown to predict fundamental band gaps (Eg) in good agreement with experiment.56,57 For the study of the EMs considered here, it is therefore of interest to compare HSE06 and B3PW91 band gap energies alongside a standard GGA functional such as PBE.To the best of our knowledge, only limited experimental data regarding Eg values are available for the materials studied here. Experimental UV-Vis spectra have been documented for β-HMX.58 While the fundamental band gap (i.e. the difference between the ionization potential and electron affinities) is in principle different to the optical band gap (which is stabilized by electron–hole interactions, and may be restricted by symmetry), the discrepancy is often small in solid state organic materials.59 Hence, the UV-Vis spectra should offer a good indication when considering the validity of calculated Eg values. Furthermore, it should be noted that both α-FOX-7 and TATB are yellow powders, which indicates the presence of optical transitions in the region of about 2.7 eV.
The Eg values calculated for the series of molecular energetic compounds are given in Table 2. The B3PW91 functional predicts values of Eg that are consistently slightly higher than those obtained from HSE06, ranging from (Eg(HSE06)) +0.24 eV to (Eg(HSE06)) +0.34 eV. As expected, due to the absence of exact exchange in the PBE calculations, considerably lower Eg values are returned in comparison with the higher-level functionals. The values for PBE-based calculations available in the literature (see Table 2) differ only slightly from those calculated here. This is most notable for β-HMX, which may be attributed to other differences between the computational models such as the use of plane wave bases sets for the literature reports, which contrasts with the localised basis sets used in this work. In all cases, the G0W0 calculations found in the literature suggest a band gap that is larger than that calculated by either B3PW91 or HSE06. Given the limited availability of experimental data, and taking into account the colour of TATB and FOX-7, it can be inferred that G0W0 quasi-particle methods may overestimate the values of Eg. This has been demonstrated previously for inorganic systems.56
| Material | B3PW91 | PBE | HSE06 | Lit. Calc. | Lit. Exp. |
|---|---|---|---|---|---|
| a G0W0 from ref. 61. b G0W0 from ref. 62. c PBE from ref. 61. d PBE from ref. 63. e PBE from ref. 20. f From ref. 58 based on UV-Vis spectroscopy. | |||||
| ABT | 5.0317 (I) | 2.9982 (I) | 4.7882 (I) | — | — |
| DNIT | 5.1011 (I) | 3.4672 (D) | 4.8073 (I) | — | — |
| HNB | 3.9433 (I) | 2.1040 (I) | 3.6887 (D) | — | — |
| MNT | 5.7141 (D) | 3.9746 (D) | 5.4179 (D) | — | — |
| β-HMX | 5.4954 (D) | 3.6826 (I) | 5.2176 (D) | 7.21,a 4.66c | 5.32f |
| HBT | 5.9569 (I) | 4.2069 (I) | 5.6364 (I) | — | — |
| α-FOX-7 | 3.9833 (I) | 2.4483 (I) | 3.6719 (I) | 5.1,b 2.2,e 1.9d | — |
| NTO | 3.5024 (I) | 2.1027 (D) | 3.1892 (I) | — | — |
| TATB | 3.9824 (I) | 2.6334 (I) | 3.6599 (I) | 4.66,a 2.52c | — |
| ATZ | 5.3775 (I) | 3.6906 (I) | 5.0330 (D) | — | — |
| GTZ | 7.0430 (I) | 5.3301 (I) | 6.7056 (I) | — | — |
| AGTZ | 7.1283 (I) | 5.2721 (I) | 6.7922 (I) | — | — |
| DGTZ | 7.2912 (I) | 5.2183 (I) | 6.9785 (I) | — | — |
As has been noted for a set of energetic azide materials,37 there is no noticeable correlation between band gap values and the reported impact sensitivity of the molecular EMs. This also holds for the materials tested here, see Table 2. Based on the B3PW91 or HSE06 calculations, the predicted sensitivity ordering would be NTO > α-FOX-7 ≈ HNB ≈ TATB > ABT > DNIT > ATZ > β-HMX > MNT > HBT > GTZ > AGTZ > DGTZ. A comparison with the experimental ordering deduced above shows clearly that this is incorrect. Similar to observations for the azide materials,37 there is no evidence of any correlation between sensitivity and the nature of the band gap in a material (either direct or indirect band gap). The band gap criterion performs poorly even within classes of compounds. For example, within the series of tetrazole-based compounds, an ordering on the basis of Eg should deliver ABT > DNIT > ATZ > MNT > HBT > GTZ > AGTZ > DGTZ. While this ordering does capture the general trend of sensitive vs. insensitive, the accuracy of ordering remains poor compared to the data in Table 1. Thus, in order to find a correlation with experimental impact sensitivities, investigations must probe beyond electronic band gap energies.60
Vibrational up-pumping – sensitivity prediction
The simulated INS spectrum for one of the more sensitive test compounds, β-HMX (Fig. 3) shows, in general, a good agreement with experiment. The frequencies in the low-energy region of the INS spectrum are well-reproduced. The simulation underestimates the top level of the phonon bath (Ωmax) by only ca. 5 cm−1. There appears to be a systematic underestimation of the vibrational frequencies in the ω > 200 cm−1 region of the spectrum, amounting to ca. 20 cm−1. The intensities of the lowest frequency modes are not reproduced particularly well, which may be indicative of minor issues in generating the correct eigenvectors; we do not however expect this to make any significant difference to subsequent calculations in this work. A better agreement between simulation and experiment is achieved for α-FOX-7, Fig. 3. The simulated INS spectrum of NTO also exhibits excellent agreement with experiment, Fig. 3, despite a small instability (negative frequencies) that could not be converged out of the calculated phonon dispersion curves (ESI S3†). Both the intensities and frequencies appear well-reproduced across the spectrum. This strongly suggests that this minor instability has only a negligible influence on the vibrational structure. There appears to be some disagreement among the vibrational modes at higher frequency (ca. 800 cm−1) in the INS spectrum of TATB (Fig. 3); overall, however, the spectrum is reproduced very well in both frequencies and intensities. A description of the effects of q-vector sampling density on modelling the INS curves is given in ESI S4.†
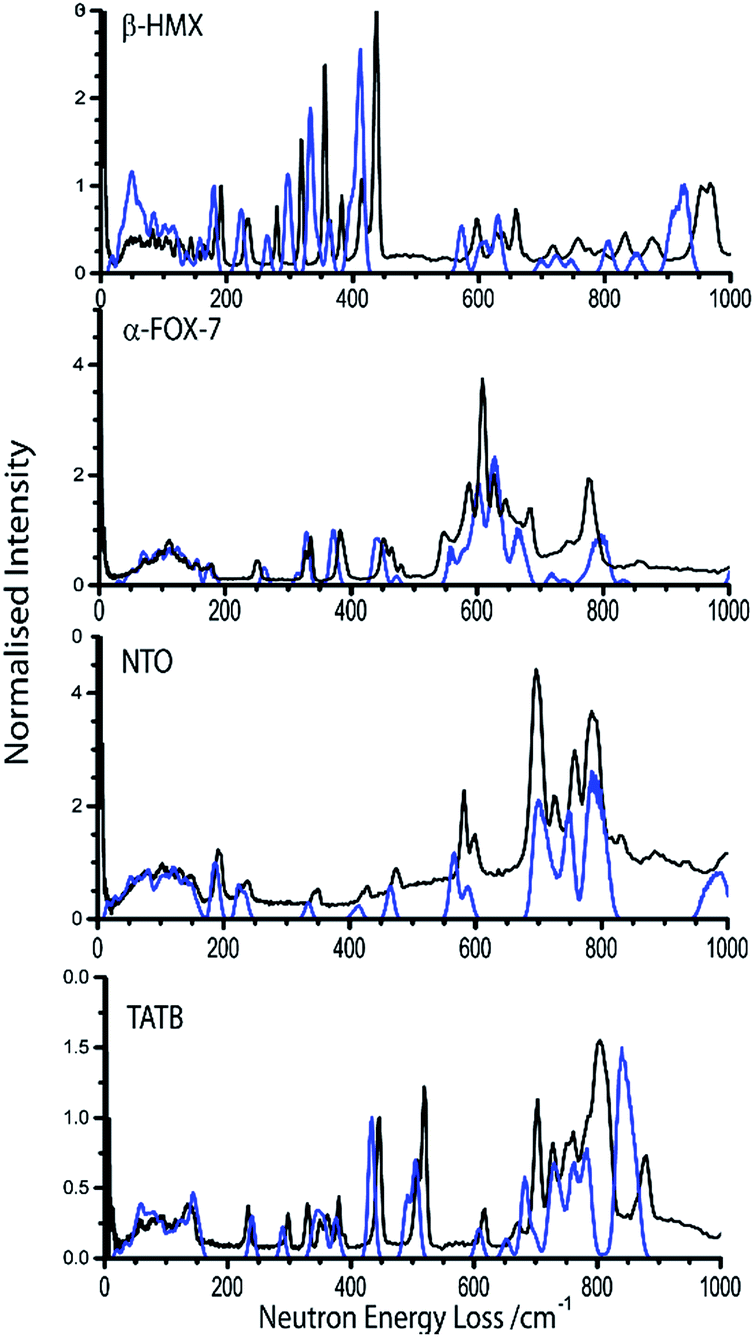 | ||
| Fig. 3 INS spectra for β-HMX, α-FOX-7, NTO, and TATB at 10 K. The experimental pattern (black lines) is shown in comparison to simulated INS spectra (blue lines) at sampling densities of the Brillouin zone 0.04 Å−1. See ESI S4† for discussion of sampling density. | ||
| τ ∝ V(3)Ω(2) | (7) |
| Ω max/cm−1 | ω d | Δωd | |
|---|---|---|---|
| ABT | 145 | 150 | 5 |
| DNIT | 145 | 158 | 13 |
| HNB | 135 | 160 | 25 |
| MNT | 175 | 180 | 5 |
| β-HMX | 193 | 210 | 17 |
| HBT | 200 | 225 | 25 |
| α-FOX-7 | 185 | 255 | 70 |
| NTO | 170 | 180 | 10 |
| NTO | 200 | 210 | 10 |
| TATB | 160 | 234 | 74 |
| ATZ | 220 | 285 | 65 |
| GTZ | 225 | 355 | 130 |
| AGTZ | 235 | 360 | 125 |
| DGTZ | 235 | 355 | 130 |
The data presented in Table 3 gives an initial qualitative insight into predicting the ordering of impact sensitivity, namely that an increase in the gap between Ωmax and the first doorway mode correlates with a decrease in impact sensitivity.
Within the average anharmonic approximation, V(3) in eqn (7) depends on the anharmonic character of the scattering vibrational modes. Hence, the phonon bath (i.e. V(3) ∝ qqq) equilibrates rapidly within picoseconds.27 Following equilibration of the phonon states, vibrational up-conversion occurs via phonon–phonon coupling up to a maximum frequency of 2Ωmax within the first anharmonic approximation. This represents processes in which the relationship V(3) ∝ qqQ applies. Vibrational states at frequencies Ωmax < ω < 2Ωmax are defined as doorway modes. Subsequent excitation beyond the first anharmonic approximation requires scattering between a doorway mode and a phonon mode, for which V(3) ∝ qQQ applies and which therefore occurs somewhat more slowly.
The rate of phonon scattering in eqn (7) is proportional to n(ω,T). This is the Bose–Einstein population of a given phonon at a given temperature which can be used to predict the rate of energy transfer between the phonon bath and doorway modes via q(q)nQ processes. These occur at the rate kph → v (eqn (8))29
kp![[h with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/char_0068_20d1.gif) v ∝ V(n+1)Ω(n+1)[nph(θe) − nv(θe)]; v ∝ V(n+1)Ω(n+1)[nph(θe) − nv(θe)]; | (8) |
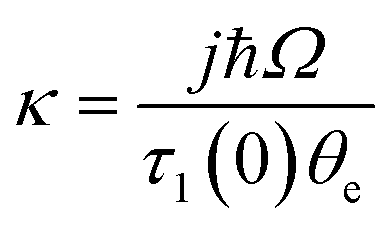 | (9) |
The probability of up-conversion into the doorway states therefore depends on the density of doorway states (i.e. the number of modes per unit cell volume), Fig. 4. This probability is calculated as the density of doorway modes. It is normalised by the magnitude of the doorway region and 3N (see ESI S5†). It is generally found that the more sensitive materials contain a higher density of vibrational states within the doorway region, whereas insensitive materials exhibit only minimal density in this part of the spectrum. Hence, despite yielding a rather poor ordering of relative impact sensitivities for the EMs investigated here, this qualitative model does appear capable of offering a broad classification (sensitive vs. insensitive) of each material. Its predictions agree well with previous studies that employed this model.35,36
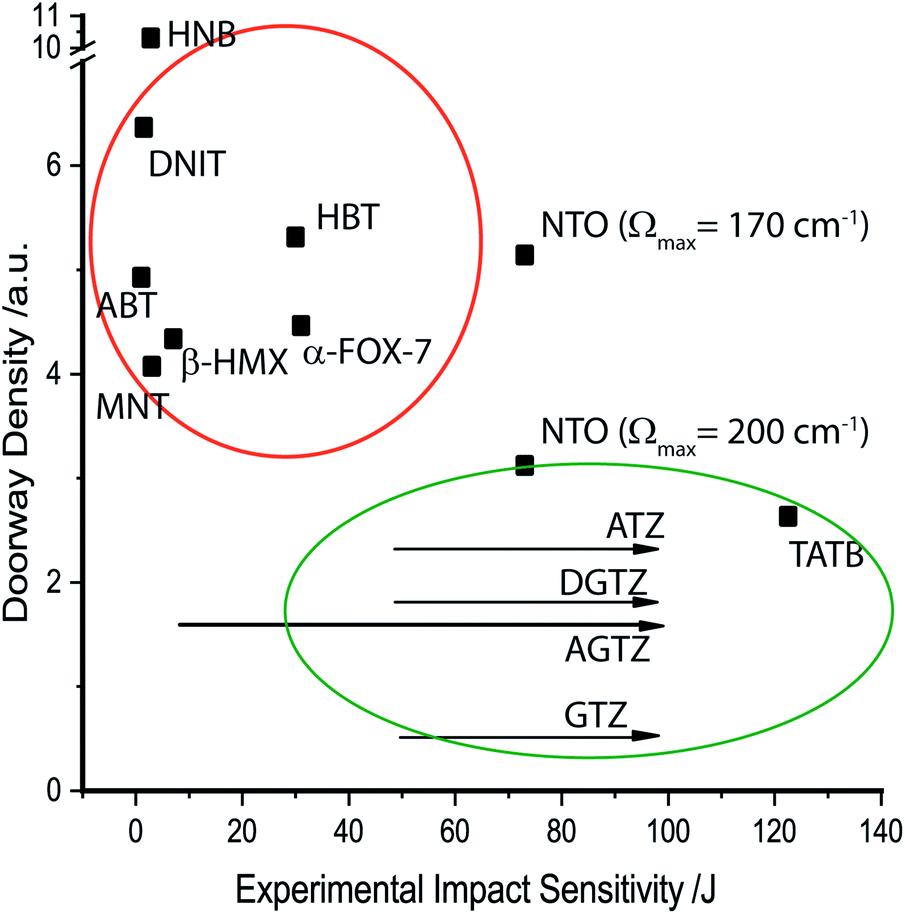 | ||
| Fig. 4 Correlating density of doorway states against experimental impact sensitivity, with the more sensitive materials clustered within the orange circle, and the less sensitive materials in the green circle. Arrows indicate that higher impact energies could not be measured, and so are approximate values only. | ||
To consider propagation of overtone up-pumping pathways, the phonon density of states, g(ω), is generated to the mth order,37
| g(ω)(m) = g(ω)/, m + 1. ; ω(m) = (m + 1) × ω | (10) |
Noting that as the rate of phonon–phonon scattering decreases with increasing order, we limit discussion to overtones of order m ≤ 2, i.e. the first two overtone pathways. The resulting trend is shown in Fig. 5 against available experimental impact sensitivity. It is generally observed that the sensitive materials are characterized by large values of ∫P(g(ω)(m)), while less sensitive materials exhibit increasingly smaller values in a nearly exponential fashion. Two materials are seemingly anomalous to this trend: DNIT and AGTZ. The former is predicted to be insensitive, while the latter is predicted to be sensitive (within a large uncertainty window). The opposite appears to be true in both cases.
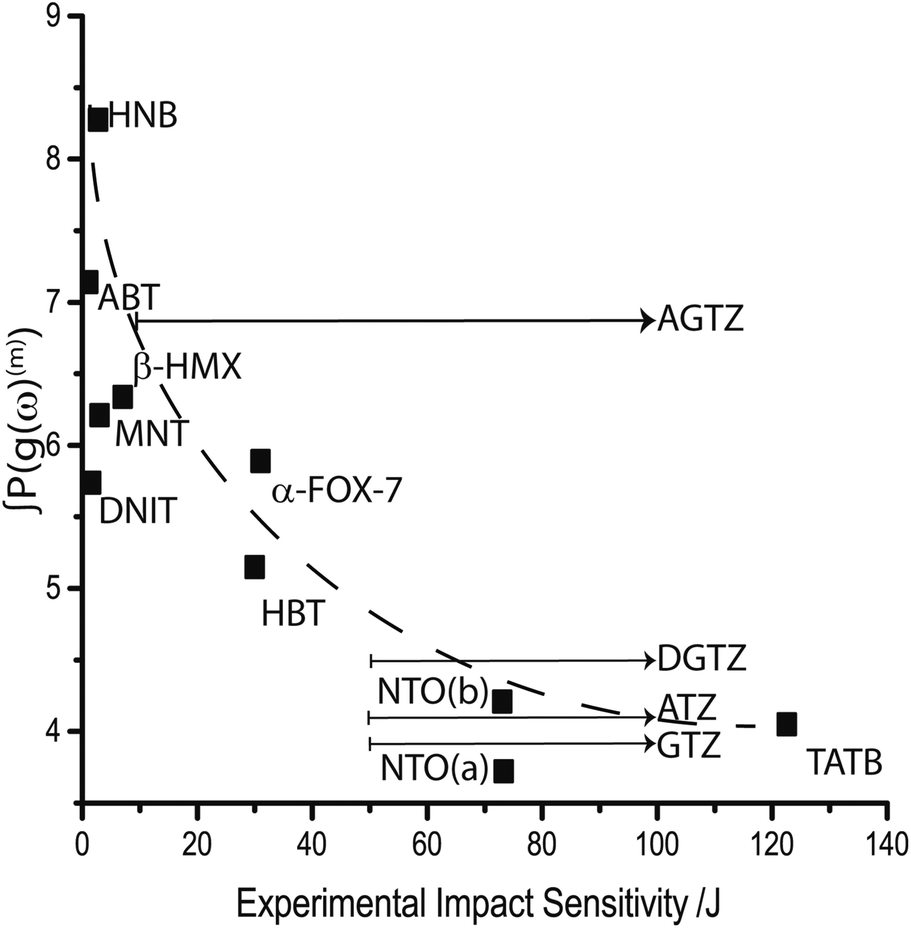 | ||
| Fig. 5 Correlating projected overtone pathways against experimental impact sensitivity. Only overtones of order m = 1, 2 are considered. A dashed line is included as visual aid. Arrows are added to indicate uncertainty in the measured sensitivity of DGTZ, ATZ, AGTZ and GTZ. NTO values are given for (a) Ωmax = 200 cm−1 and (b) Ωmax = 170 cm−1 | ||
Including overtone pathways of m > 2 leads to poorer correlation against experimental impact sensitivity (ESI S8†). This is in fact an encouraging observation, as a physical basis should not be found by including the low-probability higher-order scattering processes. It is important to note that this contrasts with earlier reports, which do include discussion of higher-order overtones.32,35,36 However, the data sets explored in these earlier reports have simplified the discussion to Brillouin zone centre (Γ only) frequencies only and did not consider population conservation between resonant states. These factors may well account for the differences observed.
| κOT ∝ Ω(2)[nph(T) − nd(T)] | (11) |
Time-dependent calculations by Kim and Dlott27 suggest that for a model molecular material, secondary up-pumping begins within picoseconds of the initial process of populating the doorway region. Hence, in step (2) of our up-pumping model we introduce a combination-based up-pumping process via30
| κC ∝ Ω(2)[nph(T)nd(T) − nω(T)] | (12) |
In accordance with eqn (11) and (12), this two-step model permits consideration of temperature. If the model is considered at an equilibrium temperature of 300 K, Fig. 6 is obtained. There is generally excellent agreement between the predicted ordering and the experimental sensitivity, with two notable exceptions: DNIT and NTO. It is promising, however, to find that the model corrects the ordering of the sensitive compounds, with the highly sensitive materials (HNB, ABT and MNT) all predicted to be higher than β-HMX. HNB is again predicted to be considerably more sensitive than ABT, and NTO (with Ωmax = 170 cm−1) is a clear outlier, although the simulated up-pumped density falls dramatically when Ωmax = 200 cm−1 (Fig. 6). While this higher value of Ωmax does not rigorously adhere to the definition of Ωmax stated above, the potential error in vibrational frequencies (particularly in light of the imaginary calculated frequency) warrants its consideration.
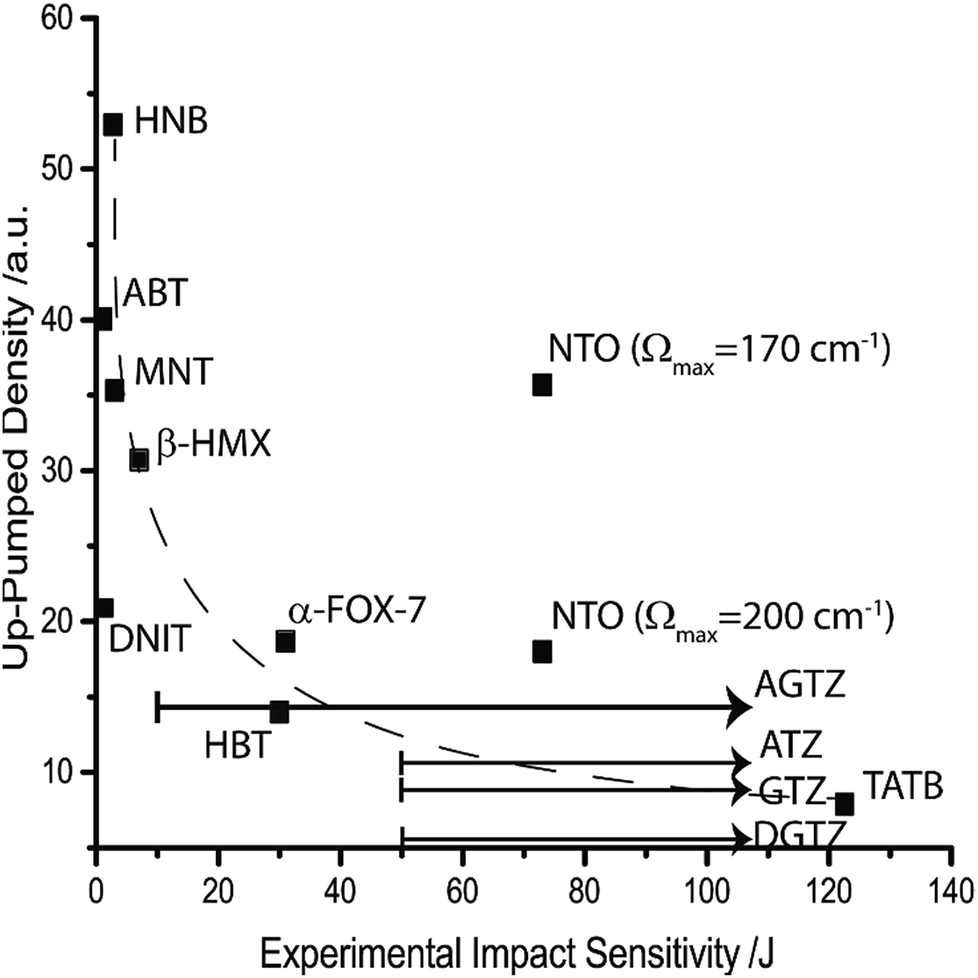 | ||
| Fig. 6 Correlation of the two-layered up-pumping model at 300 K against experimentally determined impact sensitivity. A dashed line is added to aid visualisation. | ||
As has been discussed earlier, the increase in equilibrium temperature will depend on both compressibility and anharmonicity of the material through eqn (3). Upon instantaneous compression, the shock energy is deposited into the phonon bath and, as the phonon heat capacity, Cph, is considerably lower than the total heat capacity, the quasi-equilibrium temperature achieved by the phonon bath, θph, will greatly exceed that achievable by the bulk solid. The phonon heat capacity is given by the Einstein model as roughly 6kB per molecule in the primitive unit cell. Due to the amalgamation of some molecular modes into the phonon bath, the approximation Cph ≈ 6kBZ + Y for a primitive cell of Z molecules and Y amalgamated modes holds. This therefore reflects the number of modes within the phonon bath, Table 4. It is reasonable to assume that the compressibility and anharmonicity of the molecular materials studied here will be similar in this respect. Therefore, the respective values of Cph scale as T = q/Cph ⇒ T = q/N, where N is the total number of vibrational modes below Ωmax.
| Z | Y | C ph | scaled θph/K | |
|---|---|---|---|---|
| ABT | 4 | 12 | 36 ⇒ 0.72 | 4166 |
| DNIT | 4 | 2 | 26 ⇒ 0.52 | 5769 |
| HNB | 2 | 18 | 30 ⇒ 0.60 | 5000 |
| MNT | 4 | 12 | 36 ⇒ 0.72 | 4166 |
| β-HMX | 4 | 10 | 34 ⇒ 0.68 | 4410 |
| HBT | 2 | 4 | 16 ⇒ 0.32 | 9375 |
| α-FOX-7 | 4 | 10 | 36 ⇒ 0.72 | 4166 |
| NTO (170) | 8 | 2 | 50 ⇒ 1.00 | 3000 |
| NTO (200) | — | — | 64 ⇒ 1.28 | 2343 |
| TATB | 2 | 12 | 24 ⇒ 0.48 | 6250 |
| ATZ | 4 | 0 | 24 ⇒ 0.48 | 6250 |
| GTZ | 6 | 0 | 48 ⇒ 0.96 | 3125 |
| AGTZ | 8 | 4 | 52 ⇒ 1.04 | 2884 |
| DGTZ | 6 | 4 | 28 ⇒ 0.56 | 5357 |
As a roughly representative value for shock temperature a value of θph = 3000 K was chosen. When Fig. 6 is re-examined with the addition of a shock temperature, the correlation with experimental impact sensitivity improves substantially, see Fig. 7. At this stage, the sensitive EMs are, for the first time, correctly ordered, with a steep descent towards the insensitive materials. Importantly, NTO is placed correctly, but only if the higher-lying –NO2 wagging vibrational modes are included in the phonon bath, i.e. when the higher value of Ωmax is used. Furthermore, it becomes possible to predict an ordering for the tetrazolate salts, whose experimental sensitivities have not been measured above 50 J.
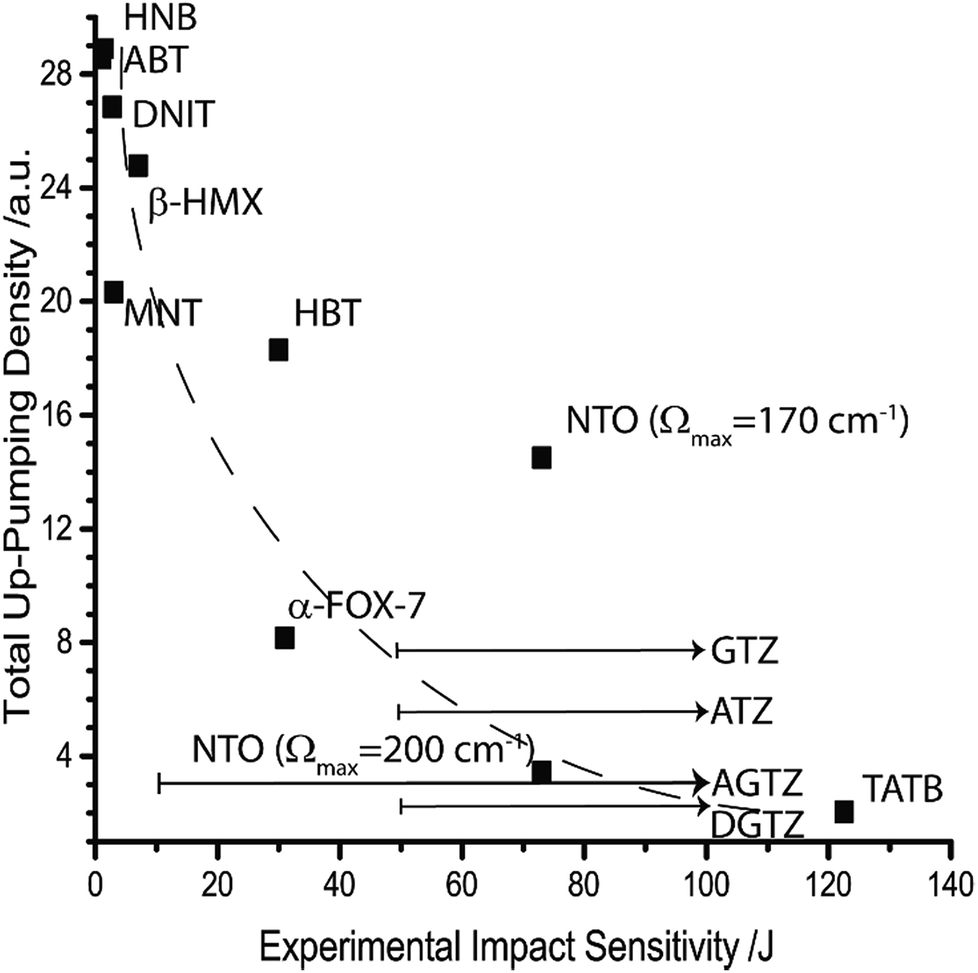 | ||
| Fig. 7 Up-pumping densities according to the two-layered multi-phonon approach for predicting impact sensitivity. The value of θph is set according to Table 4, with all modes ω > Ωmax set at T = 300 K. The dashed line is added as a visual aid. Arrows indicate uncertainty in experimental sensitivity measurement based on the experimentally determined lower limits. | ||
It is worth highlighting that the correlation based on a multi-phonon up-pumping model given here is the strongest one established to date. Moreover, this model appears capable of ranking EMs across a chemically diverse set of energetic molecules. This finding suggests that all of the information regarding bond strength (i.e. dissociation energies) is captured within the relative vibrational structures of these materials and can thus be taken account of in a multi-phonon model. While many of the data points in Fig. 7 appear to fit a decay line, it should be noted that, within the model proposed here, a completely insensitive material must display an up-pumping density of zero. Hence, it is reasonable to expect an asymptotic function to fit the data points and this has been explored in Fig. 5 through 7. The validity of this type of correlation strengthens the suggestion that the Ωmax = 200 cm−1 value found for NTO is consistent across all datasets in this work. This finding is consistent with earlier works on HMX, which assume amalgamation of low-lying NO2 waging modes into the phonon bath.28
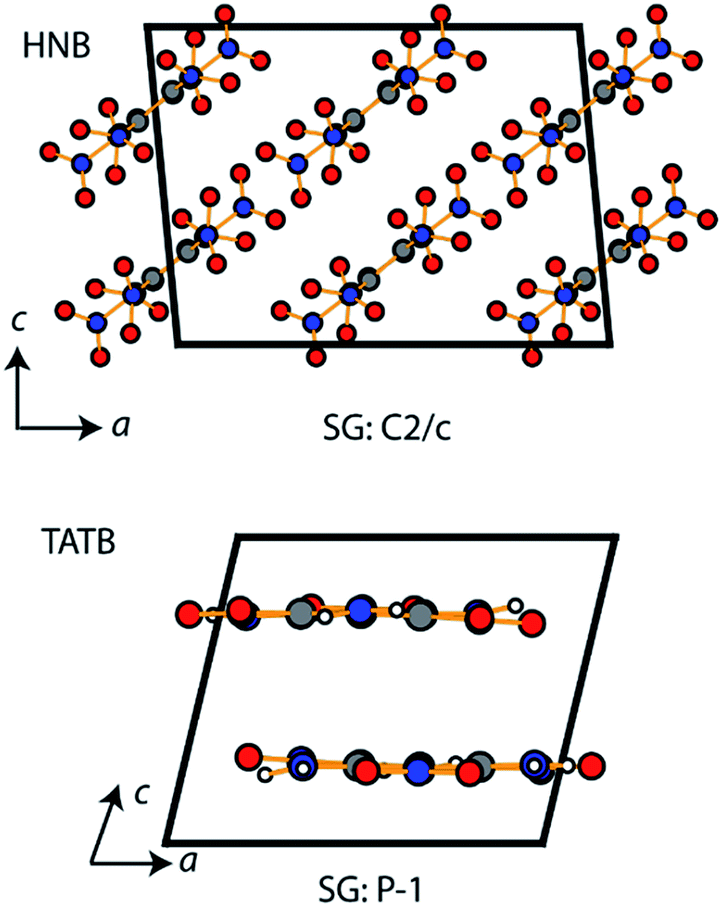 | ||
| Fig. 8 Crystal packing of TATB and HNB primitive unit cells. The space group (SG) is given for each material and the unit cell axes indicated. Atoms are labelled as O – red, N – blue, C – grey, and H – white. | ||
It is a commonly assumed observation that layered materials are generally insensitive to mechanical impact. HNB is however known experimentally to be substantially more sensitive to impact than TATB. Interestingly, all of the up-pumping models presented in this work correctly predict the impact sensitivity ordering of these two compounds, thereby presenting an opportunity to determine why two structurally similar compounds exhibit such different impact sensitivity behaviour. Firstly, we note that the doorway density of HNB is considerably higher than that of TATB (Fig. 4), which suggests that the former is less rigid than the latter. This region is dominated by the –NO2 waging motions and ring deformation modes. The same motions in TATB occur at higher frequencies as the –NO2 groups are held in the plane of the ring by internal hydrogen bond interactions with adjacent NH2 groups. They are therefore outside this crucial vibrational region (see ESI S9†). Secondly, we observe that the localised vibrational modes for HNB appear at lower wavenumbers than for TATB (see ESI S9†), such that more modes fall below the 3Ωmax upper limit for vibrational up-pumping for the more sensitive compound. This again suggests that HNB is less rigid than TATB. The weakening in ring vibrational modes also correlates with a weakening in ring aromaticity in HNB when comparing the electron density distribution with that for TATB (Fig. 9).
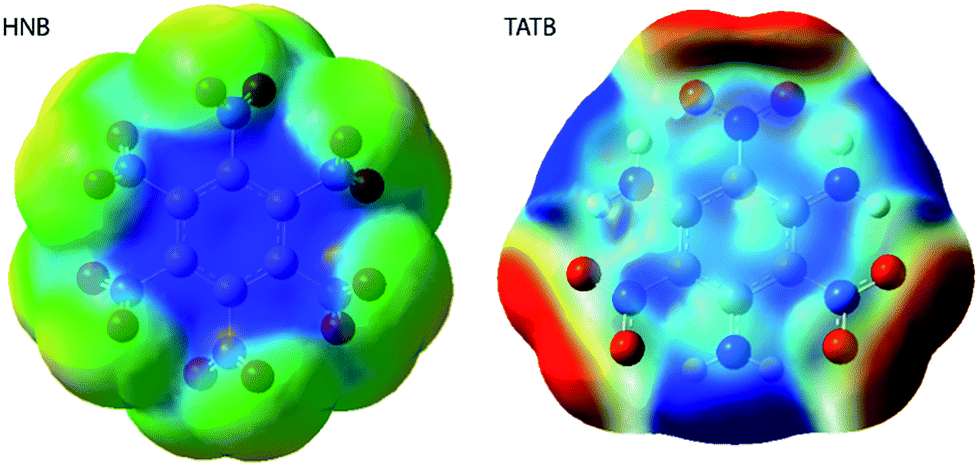 | ||
| Fig. 9 Electrostatic potential surfaces of HNB and TATB. Surfaces are generated with isovalue 2 × 10−4, scaled from −0.02 to 0.02. Blue represent electron deficient regions, and red are regions of high electron density. | ||
Thus, the vibrational up-pumping model presents the first indication of a physical mechanism that is able to rationalise the enormous difference in the sensitivity of two layered EMs. This finding has two important implications for rationalising the design of new insensitive EMs. Firstly, in order to reduce the density of states in the doorway region and shift the localised vibrational modes that receive the up-pumped vibrational energy, rigid, electron dense molecules are required. This is characteristic of molecules such as FOX-7, NTO and TATB. Secondly, the value of Ωmax should be minimised in order to minimise the upper bound up-pumping limit of 3Ωmax, as defined by the second overtone population limit. This can be achieved by increasing the degree of layering in the crystallographic packing. The present contribution makes clear that the value of Ωmax is not the crucial design parameter, but it can assist to reduce sensitivity, provided that the density of the doorway states remains low.
Conclusions
In the present work, a vibrational up-pumping model is developed to rationalise the sensitivity of a chemically and crystallographically diverse range of molecular energetic materials (EMs). The fully ab initio vibrational structures for a subset of these EMs are in excellent agreement with experimental inelastic neutron scattering spectra (INS). This confirms the validity of the theoretically predicted vibrational structures. A qualitative analysis of the vibrational doorway mode density appears to offer an excellent means for ready assignment of sensitivity. However, these assignments are, at best, limited to ‘sensitive’ vs. ‘insensitive’. Subsequently, vibrational energy up-pumping was explored using an overtone mechanism, where Ω(2) is restricted to the coupling of two counterpropagating phonons. As suggested in previous works,36 this type of model does offer good agreement with experimental sensitivities, despite failing in some cases. However, the correlation can be improved considerably when a two-layer up-pumping mechanism is adopted. This mechanism implies an initial excitation of the doorway states that occurs by overtone pathways. Subsequently, the overtone pathways are pumped higher via combination pathways with the underlying vibrational density of states. This produces excellent agreement between predicted up-pumping rates and experimental sensitivities. This is particularly striking when differences in the relative shock (adiabatic heating) temperatures are considered. Despite its ability to predict the relative impact sensitivity of a broad range of EMs, it must be remembered that the model remains limited to ideal crystal structures. The model predicts the intrinsic sensitivity of EMs, but cannot yet describe the influence of crystallographic defects, grain size, distribution and grain boundary effects. Nevertheless, the new model is still of considerable benefit for screening new materials, and indicative of their inherent potential towards initiation, once the crystal structure is known.Furthermore, it is important to highlight that the work presented here is based on the internal flow of energy which follows an external perturbation. For the purposes of this work, we have assumed that the excitation occurs via a conserved adiabatic heating process. Alternative excitation processes, including fracture and contact stresses (e.g. with grit) have also been suggested. Importantly, the nature of the excitation primarily affects the magnitude and spatial distribution of the excitation. The model presented here is largely independent of these parameters. Further work is instead required to understand the magnitude of excitation imposed by these alternative mechanisms and will be the subject of follow-up investigation.
As a result of this work it is now possible to suggest design targets for new EMs. Foremost in the search for insensitive materials appears to be the need for a low density of doorway vibrational modes and for the localised molecular vibrations to be shifted to higher wavenumbers. Both of these features, which are molecular design targets, suggest that increasing structural rigidity will suppress vibrational up-pumping. An excellent example of this is provided in the comparison of the structurally very similar compounds TATB and HNB. The value of Ωmax also appears to be crucial for suppressing rapid up-pumping into the highest levels of the vibrational structure. This design parameter falls within the realm of crystal engineering, and suggests a rationalisation for the differing sensitivities of polymorphic and multi-component crystal forms encountered previously.
The ab initio model developed here offers excellent predictive capacity across a chemically and crystallographically diverse range of EMs. By introducing consideration of temperature and heat capacity, it allows further exploration of temperature dependence, and dynamic effects associated with material compressibility upon impact. Further development of this model therefore offers immense potential to rationalise EM property behaviour in terms of experimentally tuneable parameters.
Experimental and computational methods
Preparative procedures and analytical chemistry
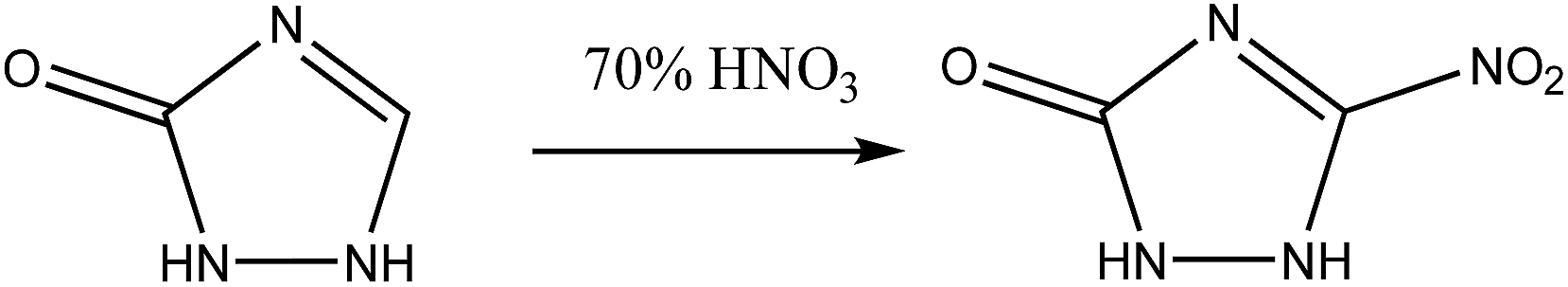 | ||
| Scheme 1 Synthetic approach for the synthesis of NTO. | ||
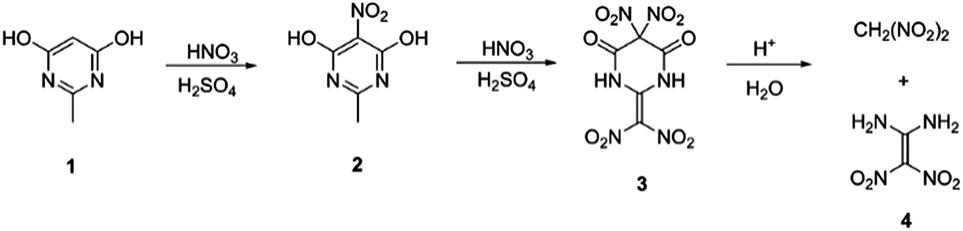 | ||
| Scheme 2 Synthetic approach for preparation of FOX-7. | ||
The crystallographic information files for compounds GTZ (CCDC 1900007), AGTZ (CCDC 1900008) and DGTZ (CCDC 1900009) are available free of charge from the Cambridge Crystallographic Data Centre (CCDC).51 A structure discussion of the crystals of the compounds GTZ, AGTZ, DGTZ (reported here for the first time) as well as triaminoguanidinium tetrazolate (TGTZ) will be provided in a forthcoming paper.
Condensed matter calculations
The PBE-D2 scheme has been previously demonstrated to work well for molecular energetic materials.66–68 It was therefore chosen as a starting point for the calculations presented here. All input structures were taken from the Cambridge Crystallographic Database (CCDC): β-HMX (Ref: OCHTET15 (ref. 69)), α-FOX-7 (Ref: SEDTUQ03 (ref. 70)), α-NTO (Ref: QOYJOD06 (ref. 71)), HNB (Ref: HNOBEN72), ABT (EWEYEL44), HBT (TIPZAU49), and TATB (TATNBZ73) or were recently determined (AGTZ, GTZ, DGTZ). Optimisation was performed using plane wave DFT as implemented in CASTEP v16.74 The electronic wavefunction was expanded in plane waves to a kinetic energy of 1800 eV for all systems, except HBT (cut-off 1600 eV), and β-HMX and TATB (cut-off 1300 eV). Forces were converged to <5 × 10−4 eV Å−1, and stresses to <5 × 10−4 GPa. The energy change per atom was converged to <1 × 10−9 eV per atom. The resulting unit cell parameters are given in Table S3.1.† In all cases, norm-conserving pseudopotentials were used, as available within the CASTEP v16 software package. All phonon calculations were based on primitive unit cells. Phonon calculations were performed using the linear response method to calculate the dynamical matrices on a regular grid of wave vectors, and Fourier interpolated to a fine grid of >150 points for generation of phonon density of states (DOS, g(ω)). Phonon dispersion curves were generated by Fourier interpolation of the computed dynamical matrices along high symmetry paths as proposed by SeeKPath75 and labelled by IUPAC convention.Electronic band structures were calculated in CRYSTAL17 (ref. 76) using localised basis sets, available from the CRYSTAL17 database. Their selection was based on previous success with similar materials and DFT functionals (H–H_pob_TZVP_2012;77 C–C_m-6-311G(d)_Heyd_2005; N–N_m-6-311G(d)_Heyd_2005;78 O–O_m-6-311G(2d)_Heyd_2005.78 In order to ensure closest reproduction of experimental results, all calculations were performed on the crystallographically determined geometries. The band structures were calculated using the HSE06,78 B3PW91 (ref. 79) and PBE80 functionals. For all materials, the tolerances (TOLINTEG) were set at 7 7 7 9 30 (as recommended for use with these basis sets78). The electronic structure was sampled across a regular grid of points, with ca. 120 points sampled in each material.
Inelastic neutron scattering spectroscopy
All INS spectra were collected using the TOSCA spectrometer at the ISIS Neutron and Muon source.81,82 Samples (ca. 1.5 g) were placed in aluminium sample holders. Samples were cooled to ca. 10 K and data collected for a total of ca. 400 μA h. The sample temperature was subsequently increased in steps of 50 K up to a maximum of 200 K for β-HMX and TATB, and 150 K for α-FOX-7. Data were collected at each 50 K interval. Both forward and back-scattered data were accumulated and corrected for scattering from the sample holder and background. All data processing was done using Mantid.83 INS spectra were simulated using ABINS,84 as implemented in Mantid. Only first-order quantum events (i.e. the fundamentals) are considered in the simulation of INS spectra.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
INS Spectra were collected on the TOSCA instrument at the ISIS Neutron and Muon Source (RB 1710382). The authors thank an Edinburgh Global Research Scholarship (AALM), EPSRC CMAC EP/I033459/1 (AALM). We are grateful to the UK Materials and Molecular Modelling Hub for computational resources, which is partially funded by EPSRC (EP/P020194/1). Further thanks to the Edinburgh Computing and Data Facility (ECDF) for additional computational resources. Dr D. Williamson (University of Cambridge) and Dr H. Lloyd (University of Edinburgh) are thanked for their assistance in the drop weight testing. Final thanks to Prof Adam Cumming for helpful discussion.References
- L. A. Medard, Accidental Explosions Volume 2: Types of Explosive Substances, ed. L. Medard, Wiley & Sons, Ltd., New York, USA, 1989 Search PubMed.
- J. J. Sabatini and K. D. Oyler, Recent Advances in the Synthesis of High Explosive Materials, Crystals, 2016, 6, 5 CrossRef.
- I. J. Powell, Insensitive Munitions – Design Principles and Technology Developments, Propellants, Explos., Pyrotech., 2016, 41(3), 409–413 CrossRef CAS.
- S. R. Kennedy and C. R. Pulham, Co-Crystallization of Energetic Materials, in Co-crystals: Preparation, Characterization and Applications, ed. C. B. Aakeröy, A. S. Sinha, Royal Society of Chemistry, Cambridge, UK, 2018, pp. 231–266 Search PubMed.
- S. V. Bondarchuk and N. A. Yefimenko, An Algorithm for Evaluation of Potential Hazards in Research and Development of New Energetic Materials in Terms of Their Detonation and Ballistic Profiles, Propellants, Explos., Pyrotech., 2018, 43, 818 CrossRef CAS.
- E. Koch, L. Energetic and V. Weiser, Review on Thermochemical Codes, in 4th Workshop on Pyrotechnic Combustion Mechanism, 2009 Search PubMed.
- E. F. C. Byrd and B. M. Rice, Improved Prediction of Heats of Formation of Energetic Materials Using Quantum Mechanical Calculations, J. Phys. Chem. A, 2006, 110(3), 1005–1013 ( J. Phys. Chem. A , 2009 , 113 , 5813 ) CrossRef CAS PubMed.
- Y. Wang, Y. Liu, S. Song, Z. Yang, X. Qi, K. Wang, Y. Liu, Q. Zhang and Y. Tian, Accelerating the Discovery of Insensitive High-Energy-Density Materials by a Materials Genome Approach, Nat. Commun., 2018, 9, 2444 CrossRef PubMed.
- F. P. Bowden and A. Yoffe, Hot Spots and the Initiation of Explosion, Symp. Combust. Flame Explos. Phenom., 1949, 3(1), 551–560 CrossRef.
- F. P. Bowden and A. D. Yoffe, Initiation and Growth of Explosion in Liquids and Solids, Cambridge University Press, Cambridge, UK, 1952 Search PubMed.
- P. M. Dickson, G. R. Parker, L. B. Smilowitz, J. M. Zucker and B. W. Asay, Frictional Heating and Ignition of Energetic Materials, AIP Conf. Proc., 2006, 845(II), 1057–1060 CrossRef CAS.
- R. V. Tsyshevsky, O. Sharia and M. M. Kuklja, Molecular Theory of Detonation Initiation: Insight from First Principles Modeling of the Decomposition Mechanisms of Organic Nitro Energetic Materials, Molecules, 2016, 21(2), 236 CrossRef PubMed.
- B. M. Rice, S. Sahu and F. J. Owens, Density Functional Calculations of Bond Dissociation Energies for NO2 Scission in Some Nitroaromatic Molecules, J. Mol. Struct.: THEOCHEM, 2002, 583(1), 69–72 CrossRef CAS.
- J. S. Murray, M. C. Concha and P. Politzer, Links between Surface Electrostatic Potentials of Energetic Molecules, Impact Sensitivities and C–NO2/N–NO2 bond Dissociation Energies, Mol. Phys., 2009, 107(1), 89–97 CrossRef CAS.
- B. W. Asay, B. F. Henson, L. B. Smilowitz and P. M. Dickson, On the Difference in Impact Sensitivity of Beta and Delta HMX, J. Energ. Mater., 2003, 21(4), 223–235 CrossRef CAS.
- P. Politzer and J. S. Murray, Impact Sensitivity and Crystal Lattice Compressibility/Free Space, J. Mol. Model., 2014, 20(5), 2233 CrossRef PubMed.
- M. Cartwright and J. Wilkinson, Correlation of Structure and Sensitivity in Inorganic Azides I Effect of Non-Bonded Nitrogen Nitrogen Distances, Propellants, Explos., Pyrotech., 2010, 35(4), 326–332 CrossRef CAS.
- D. E. Taylor, Prediction of the Impact Sensitivity of Energetic Molecules Using Symmetry Adapted Perturbation Theory, U.S. Army Research Laboratory, Aberdeen Proving Grounds, MD, 2011 Search PubMed.
- H. Zhang, F. Cheung, F. Zhao and X. Cheng, Band Gaps and the Possible Effect on Impact Sensitivity for Some Nitro Aromatic Explosive Materials, Int. J. Quantum Chem., 2009, 109, 1547–1552 CrossRef CAS.
- M. M. Kuklja and S. N. Rashkeev, Shear-Strain-Induced Structural and Electronic Modifications of the Molecular Crystal 1,1-Diamino-2,2-Dinitroethylene: Slip-Plane Flow and Band Gap Relaxation, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(10), 104111 CrossRef.
- M. M. Kuklja and S. N. Rashkeev, Shear-Strain-Induced Chemical Reactivity of Layered Molecular Crystals, Appl. Phys. Lett., 2007, 90(15), 2005–2008 CrossRef.
- S. V. Bondarchuk, Quantification of Impact Sensitivity Based on Solid-State Derived Criteria, J. Phys. Chem. A, 2018, 122, 5455–5463 CrossRef CAS PubMed.
- J. Zhang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, Enforced Layer-by-Layer Stacking of Energetic Salts towards High-Performance Insensitive Energetic Materials, J. Am. Chem. Soc., 2015, 137(33), 10532–10535 CrossRef CAS PubMed.
- J. Zhang and J. M. Shreeve, Time for Pairing: Cocrystals as Advanced Energetic Materials, CrystEngComm, 2016, 18(33), 6124–6133 RSC.
- C. S. Coffey and E. T. Totton, A Microscopic Theory of Compressive Wave-Induced Reactions in Solid Explosives, J. Chem. Phys., 1982, 76(2), 949–954 CrossRef CAS.
- F. J. Zerilli and E. T. Totton, Shock-Induced Molecular Excitation in Solids, Phys. Rev. B, 1984, 29(10), 5891–5902 CrossRef CAS.
- H. Kim and D. D. Dlott, Theory of Ultrahot Molecular Solids: Vibrational Cooling and Shock-Induced Multiphonon up Pumping in Crystalline Naphthalene, J. Chem. Phys., 1990, 93(3), 1695–1709 CrossRef CAS.
- A. Tokmakoff, M. D. Fayer and D. D. Dlott, Chemical Reaction Initiation and Hot-Spot Formation in Shocked Energetic Molecular Materials, J. Phys. Chem., 1993, 97(9), 1901–1913 CrossRef CAS.
- D. D. Dlott and M. D. Fayer, Shocked Molecular Solids: Vibrational up Pumping, Defect Hot Spot Formation, and the Onset of Chemistry, J. Chem. Phys., 1990, 92(6), 3798–3812 CrossRef CAS.
- L. E. Fried and J. A. Ruggiero, Energy-Transfer Rates in Primary, Secondary, and Insensitive Explosives, J. Phys. Chem., 1994, 98(39), 9786–9791 CrossRef CAS.
- J. R. Hill, E. L. Chronister, T. Chang, H. Kim, J. C. Postlewaite and D. D. Dlott, Vibrational Relaxation and Vibrational Cooling in Low Temperature Molecular Crystals, J. Chem. Phys., 1988, 88(2), 949–967 CrossRef CAS.
- K. L. McNesby and C. S. Coffey, Spectroscopic Determination of Impact Sensitivities of Explosives, J. Phys. Chem. B, 1997, 101(16), 3097–3104 CrossRef CAS.
- S. D. McGrane, J. Barber and J. Quenneville, Anharmonic Vibrational Properties of Explosives from Temperature-Dependent Raman, J. Phys. Chem. A, 2005, 109(44), 9919–9927 CrossRef CAS PubMed.
- S. Ye and M. Koshi, Theoretical Studies of Energy Transfer Rates of Secondary Explosives, J. Phys. Chem. B, 2006, 110(37), 18515–18520 CrossRef CAS PubMed.
- S. Ye, K. Tonokura and M. Koshi, Energy Transfer Rates and Impact Sensitivities of Crystalline Explosives, Combust. Flame, 2003, 132(1–2), 240–246 CrossRef CAS.
- J. Bernstein, Ab Initio Study of Energy Transfer Rates and Impact Sensitivities of Crystalline Explosives, J. Chem. Phys., 2018, 148(8), 084502 CrossRef PubMed.
- A. A. L. Michalchuk, P. T. Fincham, P. Portius, C. R. Pulham and C. A. Morrison, A Pathway to the Athermal Impact Initiation of Energetic Azides, J. Phys. Chem. C, 2018, 122, 19395–19408 CrossRef CAS.
- Y. B. Zel’dovich and Y. P. Raizer, Physics of Shock Waves and High-Temperature Hydrodynamic Phenomena, Academic, New York, 1967, vol. II Search PubMed.
- L. Paulatto, F. Mauri and M. Lazzeri, Anharmonic Properties from a Generalized Third-Order Ab Initio Approach: Theory and Applications to Graphite and Graphene, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(21), 1–18 CrossRef.
- C. L. Schosser and D. D. Dlott, A Picosecond CARS Study of Vibron Dynamics in Molecular Crystals: Temperature Dependence of Homogeneous and Inhomogeneous Linewidths, J. Chem. Phys., 1984, 80(4), 1394–1406 CrossRef CAS.
- D. D. Dlott, Multi-Phonon up-Pumping in Energetic Materials, in Overview of Recent Research on Energetic Materials, ed. R. W. Shaw, T. B. Brill, and D. L. Thompson, World Scientific, 2005, pp. 303–333 Search PubMed.
- J. R. Hill, E. L. Chronister, T. C. Chang, H. Kim, J. C. Postlewaite and D. D. Dlott, Vibrational Relaxation of Guest and Host in Mixed Molecular Crystals, J. Chem. Phys., 1988, 88(4), 2361–2371 CrossRef CAS.
- S. Chen, W. A. Tolbert and D. D. Dlott, Direct Measurement of Ultrafast Multiphonon Up-Pumping in High Explosives, J. Phys. Chem., 1994, 98(32), 7759–7766 CrossRef CAS.
- T. M. Klapötke and D. G. Piercey, 1,1′-Azobis(Tetrazole): A Highly Energetic Nitrogen-Rich Compound with a N10 Chain, Inorg. Chem., 2011, 50(7), 2732–2734 CrossRef PubMed.
- T. M. Klapötke and J. Stierstorfer, Nitration Products of 5-Amino-1H-Tetrazole and Methyl-5-Amino-1H-Tetrazoles – Structures and Properties of Promising Energetic Materials, Helv. Chim. Acta, 2007, 90, 2132–2150 CrossRef.
- W. S. Wilson, D. E. Bliss, S. L. Christian, and D. J. Knight, Explosive Properties of Polynitroaromatics, China Lake, CA, USA, 1990 Search PubMed.
- R. L. Simpson, P. A. Urtiew, D. L. Ornellas, G. L. Moody, K. J. Scribner and D. M. Hoffman, CL-20 Performance Exceeds That of HMX and Its Sensitivity Is Moderate, Propellants, Explos., Pyrotech., 1997, 22, 249–255 CrossRef CAS.
- C. B. Storm, J. R. Stine, J. F. Kramer, Sensitivity Relationships in Energetic Materials, in Chemistry and Physics of Energetic Materials, ed. S. N. Bulusu, Springer, Dordrecht, 1990, pp. 605–639 Search PubMed.
- T. M. Klapötke and C. M. Sabaté, 5,5′-Hydrazinebistetrazole: An Oxidation-Stable Nitrogen-Rich Compound and Starting Material for the Synthesis of 5,5′-Azobistetrazolates, Z. Anorg. Allg. Chem., 2007, 633(15), 2671–2677 CrossRef.
- B. M. Rice and J. J. Hare, A Quantum Mechanical Investigation of the Relation between Impact Sensitivity and the Charge Distribution in Energetic Molecules, J. Phys. Chem. A, 2002, 106(9), 1770–1783 CrossRef CAS.
- P. Portius, Personal Communication, Sheffield, UK, 2019.
- R. Meyer, J. Köhler, A. Homburg, Explosives, ed. Meyer, R., Köhler, J., and Homburg, A., Wiley-VCH, Weinheim, 6th edn, 2007 Search PubMed.
- Z. X. Chen and H. Xiao, Impact Sensitivity and Activation Energy of Pyrolysis for Tetrazole Compounds, Int. J. Quantum Chem., 2000, 79(6), 350–357 CrossRef CAS.
- T. M. Klapötke, M. Stein and J. Stierstorfer, Salts of 1H-Tetrazole – Synthesis, Characterization and Properties, Z. Anorg. Allg. Chem., 2008, 634(10), 1711–1723 CrossRef.
- W. Zhu and H. Xiao, First-Principles Band Gap Criterion for Impact Sensitivity of Energetic Crystals: A Review, Struct. Chem., 2010, 21(3), 657–665 CrossRef CAS.
- J. M. Crowley, J. Tahir-Kheli and W. A. Goddard, Resolution of the Band Gap Prediction Problem for Materials Design, J. Phys. Chem. Lett., 2016, 7(7), 1198–1203 CrossRef CAS PubMed.
- A. J. Garza and G. E. Scuseria, Predicting Band Gaps with Hybrid Density Functionals, J. Phys. Chem. Lett., 2016, 7(20), 4165–4170 CrossRef CAS PubMed.
- J. K. Cooper, C. D. Grant and J. Z. Zhang, Experimental and TD-DFT Study of Optical Absorption of Six Explosive Molecules: RDX, HMX, PETN, TNT, TATP, and HMTD, J. Phys. Chem. A, 2013, 117(29), 6043–6051 CrossRef CAS PubMed.
- J.-L. Bredas, Mind the Gap!, Mater. Horiz., 2014, 1(1), 17–19 RSC.
- S. V. A. Bondarchuk, Unified Model of Impact Sensitivity of Metal Azides, New J. Chem., 2019, 43(3), 1459–1468 RSC.
- S. Appalakondaiah, G. Vaitheeswaran and S. Lebègue, Dispersion Corrected Structural Properties and Quasiparticle Band Gaps of Several Organic Energetic Solids, J. Phys. Chem. A, 2015, 119(24), 6574–6581 CrossRef CAS PubMed.
- S. Appalakondaiah, G. Vaitheeswaran and S. Lebègue, Structural, Vibrational, and Quasiparticle Band Structure of 1,1-Diamino-2,2-Dinitroethelene from Ab Initio Calculations, J. Chem. Phys., 2014, 140(1), 014105-1–014105-7 CrossRef PubMed.
- A. Hu, B. Larade, H. Abou-Rachid, L. S. Lussier and H. Guo, A First Principles Density Functional Study of Crystalline FOX-7 Chemical Decomposition Process under External Pressure, Propellants, Explos., Pyrotech., 2006, 31(5), 355–360 CrossRef CAS.
- A. A. L. Michalchuk, S. Rudić, C. R. Pulham and C. A. Morrison, Vibrationally Induced Metallisation of the Energetic Azide α-NaN3, Phys. Chem. Chem. Phys., 2018, 20(46), 29061–29069 RSC.
- J. Ledgard. The Preparatory Manual of Explosives, Jared Ledgard, 3rd edn, 2007 Search PubMed.
- S. Hunter, A. J. Davidson, C. A. Morrison, C. R. Pulham, P. Richardson, M. J. Farrow, W. G. Marshall, A. R. Lennie and P. J. Gould, Combined Experimental and Computational Hydrostatic Compression Study of Crystalline Ammonium Perchlorate, J. Phys. Chem. C, 2011, 115(38), 18782–18788 CrossRef CAS.
- S. Hunter, T. Sutinen, S. F. Parker, C. A. Morrison, D. M. Williamson, S. Thompson, P. J. Gould and C. R. Pulham, Experimental and DFT-D Studies of the Molecular Organic Energetic Material RDX, J. Phys. Chem. C, 2013, 117(16), 8062–8071 CrossRef CAS.
- S. Hunter, P. L. Coster, A. J. Davidson, D. I. A. Millar, S. F. Parker, W. G. Marshall, R. I. Smith, C. A. Morrison and C. R. Pulham, High-Pressure Experimental and DFT-D Structural Studies of the Energetic Material FOX-7, J. Phys. Chem. C, 2015, 119(5), 2322–2334 CrossRef CAS.
- J. R. Deschamps, M. Frisch and D. Parrish, Thermal Expansion of HMX, J. Chem. Crystallogr., 2011, 41(7), 966–970 CrossRef CAS.
- J. Evers, T. M. Klapötke, P. Mayer, G. Oehlinger and J. Welch, α- and β-FOX-7, Polymorphs of a High Energy Density Material, Studied by X-Ray Single Crystal and Powder Investigations in the Temperature Range from 200 to 423 K, Inorg. Chem., 2006, 45(13), 4996–5007 CrossRef CAS PubMed.
- N. B. Bolotina, M. J. Hardie, R. L. Speer Jr and A. A. Pinkerton, Energetic Materials: Variable-Temperature Crystal Structures of {γ}- and {ε}-HNIW Polymorphs, J. Appl. Crystallogr., 2004, 37, 808–814 CrossRef CAS.
- Z. A. Akopyan, Y. T. Struchkov and V. G. Dashevskii, Crystal And Molecular Structure of Hexanitrobenzene, Zh. Strukt. Khim., 1966, 7, 408 CAS.
- H. H. Cady and A. C. Larson, The Crystal Structure of 1,3,5-Triamino-2,4,6-Trinitrobenzene, Acta Crystallogr., 1965, 18(3), 485–496 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First Principles Methods Using CASTEP, Z. Krist., 2005, 220, 567–570 CAS.
- Y. Hinuma, G. Pizzi, Y. Kumagai, F. Oba and I. Tanaka, Band Structure Diagram Paths Based on Crystallography, Comput. Mater. Sci., 2017, 128, 140–184 CrossRef CAS.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre and P. D'Arco, et al., CRYSTAL14: A Program for the Ab Initio Investigation of Crystalline Solids, Int. J. Quantum Chem., 2014, 114(19), 1287–1317 CrossRef CAS.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, Consistent Gaussian Basis Sets of Triple-Zeta Valence with Polarization Quality for Solid-State Calculations, J. Comput. Chem., 2013, 34(6), 451–459 CrossRef CAS PubMed.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, Energy Band Gaps and Lattice Parameters Evaluated with the Heyd–Scuseria–Ernzerhof Screened Hybrid Functional, J. Chem. Phys., 2005, 123(17), 174101 CrossRef PubMed.
- F. Tran, P. Blaha, K. Schwarz and P. Novák, Hybrid Exchange-Correlation Energy Functionals for Strongly Correlated Electrons: Applications to Transition-Metal Monoxides, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74(15), 155108 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- D. Colognesi, M. Celli, F. Cilloco, R. J. Newport, S. F. Parker, V. Rossi-Albertini, F. Sacchetti, J. Tomkinson and M. Zoppi, TOSCA Neutron Spectrometer: The Final Configuration, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 64–66 CrossRef.
- R. S. Pinna, S. Rudić, S. F. Parker, J. Armstrong, M. Zanetti, G. Škoro, S. P. Waller, D. Zacek, C. A. Smith and M. J. Capstick, et al., The Neutron Guide Upgrade of the TOSCA Spectrometer, Nucl. Instrum. Methods Phys. Res., Sect. A, 2018, 896, 68–74 CrossRef CAS.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal and M. A. Gigg, et al., Mantid - Data Analysis and Visualization Package for Neutron Scattering and μ SR Experiments, Nucl. Instruments Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- K. Dymkowski, S. F. Parker, F. Fernandez-Alonso and S. Mukhopadhyay, AbINS: The Modern Software for INS Interpretation, Phys. B: Condens. Matter, 2018, 1–6 Search PubMed in press.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta06209b |
| ‡ Current address: BAM Federal Institute for Materials Research and Testing, Richard-Willstatter-Strasse 11, 12489 Berlin, Germany. |
| § Current affiliation: Department of Physical Chemistry, University of Basel, Basel, Switzerland. |
| This journal is © The Royal Society of Chemistry 2019 |