
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmorphous outperforms crystalline nanomaterials: surface modifications of molecularly derived CoP electro(pre)catalysts for efficient water-splitting†
Rodrigo
Beltrán-Suito
 ,
Prashanth W.
Menezes
* and
Matthias
Driess
*
,
Prashanth W.
Menezes
* and
Matthias
Driess
*
Department of Chemistry: Metalorganics and Inorganic Materials, Technische Universität Berlin, Straße des 17 Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de; prashanth.menezes@mailbox.tu-berlin.de
First published on 4th June 2019
Abstract
The single source precursor (SSP) approach was used to prepare highly active CoP bifunctional electro(pre)catalysts for the oxygen evolution reaction (OER), hydrogen evolution reaction (HER) and overall water splitting (OWS) reaction starting from a molecular β-diketiminato Co(I) cyclo-P4 complex. Crystalline or amorphous CoP particles were attained depending on the preparation route. Notably, the amorphous CoP displayed higher activity compared to the crystalline CoP on nickel foam (NF) and fluorinated tin oxide (FTO) substrates due to its unique electronic properties and surface characteristics. During the OER, severe oxidation to Co-oxy(hydroxides)/oxides by the loss of P was found to be crucial to increase the concentration of CoOx active sites. Interestingly, complete leaching of surface P from CoP and surface Co enrichment occurred during the HER. Finally, an OWS device was fabricated where the amorphous CoP outperformed the crystalline CoP with respect to low OWS cell voltage (with a difference of 130 mV) and enhanced stability for 5 days.
1. Introduction
The rapid growth of human population in the 21st century has increased the demand for energy and search for cleaner and sustainable energy sources.1 Hydrogen (H2) energy is a promising alternative to replace fossil fuels as it is contaminant free and has a high-energy density and zero CO2 release.2–4 In this respect, electrochemical water splitting can efficiently produce H2 and oxygen (O2).3,5 However, this already high energy consuming process requires the application of an overpotential (η).6,7 At present, the noble metal oxides of ruthenium or iridium for the OER and elemental platinum for the HER still represent the benchmark catalysts for practical applications of water splitting.8 Their high cost and scarce availability limit their use in large-scale applications. Consequently, it is imperative to use electrocatalysts that reduce the overpotentials and increase the energy-efficiency of the system based on low-cost, environmentally benign and earth-abundant transition metals,9 which is the ultimate focus of our research group.10–12Transition metal phosphides (TMPs) have emerged as high-performance catalysts for electrochemical water splitting because of their low hydrogen adsorption energies, high electrical conductivity and promising chemical resistance.3,13,14 Among them, cobalt phosphides (CoP or Co2P) have received attention recently due to their high HER electrocatalytic activity.15 In addition, follow-up studies revealed that negatively charged P atoms can trap protons and promote H2 liberation.16 Efforts have also been devoted to using these materials as OER catalysts for enabling OWS.15,17 In this case, the positively charged Coδ+ sites can act as hydroxyl acceptors, simultaneously facilitated by the negatively charged Pδ− centers, favouring O2 evolution by discharging and desorption.18
Recently, several novel synthetic strategies have been employed to prepare crystalline and amorphous CoP electrocatalysts and enhance their activity, such as aerosol spray from Co–P precursors,19 electrodeposition,20 MOF-derived TMPs21–23 and the combination with carbon nanostructures24,25 or other transition metals.26,27 The commonly applied synthetic routes are usually based on conventional solid-state syntheses which require highly reactive and pyrophoric reagents or high temperatures leading to a random distribution of aggregates and an infinite number of nanostructures.28 To prevent this, new synthetic strategies like the low-temperature molecular SSP approach are used, showing several advantages, foremost a better control of the composition and size distribution of the resulting nanomaterial, which can be varied depending on the experimental conditions.29 Recently, this synthetic method has been applied to access a broad range of high-performance electrocatalytic OER, HER and OWS materials, including chalcogenides30 and pnictides.31 Examples of preparation of crystalline cobalt phosphides by the SSP approach either for the HER or for the OER and/or OWS electrocatalysts are limited32–34 and access to amorphous cobalt phosphide phases by the SSP approach towards OWS remains unexplored. Moreover, the striking structural difference between amorphous and crystalline cobalt phosphides influencing the net catalytic activity and their structural transformation during bifunctional electrochemical OER and HER catalysis is currently unknown.
Herein, we present a novel molecular-based approach to synthesizing amorphous and crystalline CoP electro(pre)catalysts through hot injection and pyrolysis of a unique molecular β-diketiminato cyclo-P4 dicobalt(I) complex with a Co2P4 core. The substantial difference between amorphous and crystalline CoP structures with respect to electrocatalytic OER and HER activities has been systematically investigated on distinct electrode substrates under alkaline conditions. In addition, their surface structures and the nature of the active species have also been elucidated by means of advanced characterization techniques. Finally, a two-electrode alkaline electrolyser was fabricated to demonstrate the practical advantage of employing amorphous vs. crystalline CoP.
2. Experimental
2.1. General considerations and instrumentation
All synthetic procedures were done under inert conditions using standard Schlenk techniques or a M. Braun dry box containing an atmosphere of inert purified nitrogen. Solvents were dried by standard methods. 1H NMR spectra were recorded on a Bruker Spectrometer APX 200 at room temperature and the solvent residual signals were referenced to the internal standard. Fourier transform infrared (FTIR) spectra were recorded on a Thermo Fisher Nicolet iS5 IR spectrometer (ATR-Diamond) under inert conditions. Elemental analysis was carried out with a Thermo Flash EA 1112 Organic Elemental Analyzer by dynamic flash combustion at 1020 °C. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was carried out using a Thermo Jarrell Ash Trace Scan analyser. The samples were digested in aqua regia HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 3:1 v/v (nitric acid, SUPRA-Qualität ROTIPURAN® Supra 69% and hydrochloric acid, SUPRA-Qualität ROTIPURAN® Supra 30%) and the average of three reproducible independent experiments is reported. The digestion volume (2.5 mL) was diluted with Milli-Q water up to 15 mL. Calibration curves were recorded for both cobalt and phosphorus with concentrations between 1 mg L−1 and 100 mg L−1 from standard solutions (1000 mg L−1 Single-Element ICP-Standard Solution ROTI®STAR). Powder X-ray diffraction (PXRD) patterns were obtained on a Bruker AXS D8 advanced automatic diffractometer equipped with a position-sensitive detector (PSD) and a curved germanium (111) primary monochromator using Cu Kα radiation (λ = 1.5418 Å). The determination of the surface area was performed by nitrogen sorption using the BET method. Measurements were performed with a Nova 4000e from Quantachrome Instruments. Scanning electron microscopy (SEM) was carried out on a LEO DSM 982 microscope integrated with an EDX (EDAX, Apollo XPP). Data handling and analyses were performed with the software package EDAX. The most abundant elements were selected from the EDX spectra. Transmission electron microscopy (TEM) was accomplished on an FEI Tecnai G2 20 S-TWIN transmission electron microscope (FEI Company, Eindhoven, Netherlands) equipped with a LaB6 source at 200 kV acceleration voltage. Energy dispersive X-ray (EDX) analyses were performed with an EDAX r-TEM SUTW detector (Si (Li) detector), and the images were recorded with a GATAN MS794 P CCD camera. The SEM and TEM experiments were conducted at the Zentrum für Elektronenmikroskopie (ZELMI) of the TU Berlin.
:HNO3 3:1 v/v (nitric acid, SUPRA-Qualität ROTIPURAN® Supra 69% and hydrochloric acid, SUPRA-Qualität ROTIPURAN® Supra 30%) and the average of three reproducible independent experiments is reported. The digestion volume (2.5 mL) was diluted with Milli-Q water up to 15 mL. Calibration curves were recorded for both cobalt and phosphorus with concentrations between 1 mg L−1 and 100 mg L−1 from standard solutions (1000 mg L−1 Single-Element ICP-Standard Solution ROTI®STAR). Powder X-ray diffraction (PXRD) patterns were obtained on a Bruker AXS D8 advanced automatic diffractometer equipped with a position-sensitive detector (PSD) and a curved germanium (111) primary monochromator using Cu Kα radiation (λ = 1.5418 Å). The determination of the surface area was performed by nitrogen sorption using the BET method. Measurements were performed with a Nova 4000e from Quantachrome Instruments. Scanning electron microscopy (SEM) was carried out on a LEO DSM 982 microscope integrated with an EDX (EDAX, Apollo XPP). Data handling and analyses were performed with the software package EDAX. The most abundant elements were selected from the EDX spectra. Transmission electron microscopy (TEM) was accomplished on an FEI Tecnai G2 20 S-TWIN transmission electron microscope (FEI Company, Eindhoven, Netherlands) equipped with a LaB6 source at 200 kV acceleration voltage. Energy dispersive X-ray (EDX) analyses were performed with an EDAX r-TEM SUTW detector (Si (Li) detector), and the images were recorded with a GATAN MS794 P CCD camera. The SEM and TEM experiments were conducted at the Zentrum für Elektronenmikroskopie (ZELMI) of the TU Berlin.
2.2. X-ray photoelectron spectroscopy (XPS)
The X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Kratos Axis Ultra X-ray photoelectron spectrometer (Kratos Analytical Ltd., Manchester, U.K.) using an Al Kα monochromatic radiation source (1486.7 eV) with a 90° take off angle (normal to analyser). The vacuum pressure in the analysis chamber was kept at 2 × 10−9 Torr. The XPS spectra were collected for O 1s, Co 2p, and P 2p levels with a pass energy of 20 eV and a step of 0.1 eV. The binding energies were calibrated relative to the C 1s peak energy position at 285.0 eV. Data analyses were carried out using Casa XPS (Casa Software Ltd.) and the Vision data processing program (Kratos Analytical Ltd.).2.3. Synthesis of amorphous CoP by hot injection
To a three-necked round bottom Schlenk flask fitted with a temperature sensor and a condenser, 25 mL oleic acid (Fisher Scientific) was added. The solvent was degassed by a 3-cycle freeze-pump method. The whole set-up was degassed using vacuum followed by refilling with nitrogen three times and then the flask was heated to 300 °C. The precursor (0.47 g, 0.46 mmol) was dissolved in 5 mL of dry oleic acid at 30 °C in another flask. The solution was transferred to the three-necked flask at 300 °C by injection under inert conditions. The reaction temperature was maintained at 300 °C for one more hour and then the mixture was allowed to cool down naturally to room temperature. The whole reaction mixture was transferred into a centrifuge tube and centrifuged along with an additional 20 mL methanol at 9000 rpm to produce a black solid. Washing with methanol was repeated thrice more to remove any excess ligand and oleic acid. The precipitate was then washed with acetone and dried to store for further use.2.4. Synthesis of crystalline CoP by pyrolysis
0.20 g (0.20 mmol) of the precursor was introduced into a quartz tube under an inert atmosphere and subsequently introduced into a tube furnace SR-A60-300/12 GERO (Neuhausen). The sample was then heated up to 600 °C at 10 °C min−1 from room temperature and held at that temperature for 2 h under Ar. After cooling to room temperature, a black powder was collected and washed with acetone several times. Furthermore, the use of a lower temperature (300 and 450 °C) leads to the formation of materials with a large amount of carbon due to the incomplete decomposition of the molecular precursor.2.5. Preparation of films
The synthesized materials films were prepared by electrophoretic deposition (EPD) on FTO and NF. 40 mg of the obtained material and 7 mg of I2 (Merck, double sublimated) were suspended in acetone by sonication for 1 h. In this way, generated protons get adsorbed onto the particles of the materials.35 A 10 V potential difference on a 1 × 1 cm2 area was applied on a two electrode system using a PS 303 Pro Conrad power supply. The applied potential forces the migration of the charged particles to the negative electrode (cathode). The deposition time was 5 min for each film and the respective films were washed with acetone. The sample loading was ∼0.4 mg cm−2, determined by weighing the FTO or NF substrate before and after EPD. A similar procedure was also followed to deposit commercial IrO2 (Alfa Aesar, 99%) and compare its activity with that of the synthesized materials.2.6. Electrochemical measurements
Several electrochemical measurements were performed on the as-prepared catalysts. A typical catalytic run was carried out using a three-electrode setup, consisting of a reference electrode (RE), a counter electrode (CE) and the catalyst-modified working electrode (WE). The three electrodes were immersed in an aqueous electrolyte (1 M KOH, Sigma Aldrich). The presence of Fe in it was ruled out by purifying the electrolyte before its use following a literature procedure.36 A potentiostat (SP-200, BioLogic Science Instruments) controlled with the EC-Lab v10.20 software package was used for all the experiments. The electrodes with samples deposited served as the WE, Pt wire (or a graphite rod for the HER) as the CE and Hg/Hg2SO4 (HER) or Hg/HgO (OER) as the RE. Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) experiments were carried out with an iR compensation of 85%, applied before each experiment. The potential ranges were 1.1 to 1.8 V vs. RHE for OER LSV and 0.0 to −0.5 V for HER LSV. The overpotential for the OER or HER was determined from the resulting polarization curves. CP measurements were done in 1 M KOH at a constant current density of ±10 mA cm−2, depending on OER or HER experiments. The overall water splitting LSV and CP experiments were performed in a two-electrode setup with the catalysts deposited on NF electrodes. The potentials were referenced to the reference hydrogen electrode (RHE) by using the following equations in 1 M KOH: E(RHE) = E(Hg/Hg2SO4, 1 M KOH) + 1.476 V and E(RHE) = E(Hg/HgO, 1 M KOH) + 0.924 V. The polarization curves were replotted as overpotential (η) vs. the logarithm of current density (logj) to obtain Tafel plots. The double layer capacitance (Cdl) was determined to calculate the active surface area of the materials and the substrate.37 From the already measured LSV, a potential range in which no faradaic process (no catalysis) occurs was selected. CVs were recorded at different scan rates (5 mV s−1 to 200 mV s−1). The half of the value of the slope of the plot of the capacitive current (the difference between anodic and cathodic current density) vs. the scan rate provides the double layer capacitances, Cdl, of the layer and the films. The ECSA can be calculated from the following equation: ECSA = Cdl/Cs, where Cs is the specific capacitance of the sample or the capacitance of an atomically smooth planar surface of the material per unit area under identical electrolyte conditions. The following value was used: Cs = 1.7 mF cm−2 for 1 M KOH on NF.38 EIS measurements were performed over a frequency range from 100 kHz to 10 mHz at 0.7 V vs. Hg/HgO for materials deposited on NF. A sinusoidal potential was applied, and the frequency-dependent complex impedance is measured. A Nyquist plot was constructed and the resistance of the electrolyte solution (Rs) and the resistance of the charge transfer (RCT) were calculated from the fit of the data to a Randles circuit.
3. Results and discussion
The precursor was prepared according to the reported literature procedure and well characterized (Fig. S1–S7 and Tables S1 and S2, ESI†).39 Both hot injection and pyrolysis of the molecular precursor afforded black powders (Scheme 1). The powder X-ray diffraction (PXRD) analysis of the materials revealed an amorphous phase in the hot injection product and a crystalline CoP phase in the pyrolysis product (Fig. S8 and S9, ESI†). Scanning electron microscopy (SEM) revealed agglomerated materials, composed of smaller particles, and transmission electron microscopy (TEM) of the hot-injected product showed that the agglomerates were rather composed of particles of size ≤ 10 nm (Fig. 1a, b, S10 and S11b, ESI†) that do not show any crystalline fringes confirming the amorphous nature of the material, as supported by the selected area electron diffraction (SAED, Fig. 1c). The pyrolysis product consists of agglomerates of particles (Fig. 1d and S12a, ESI†) with clear crystalline fringes associated with the lattice spacing of 0.19 nm corresponding to the (211) plane of the CoP phase (Fig. 1e and S12b, ESI†). In addition, the SAED pattern displayed sharp diffraction rings representative of the crystalline CoP phase (JCPDS 29-0497, Fig. 1f). The phase composition and uniform distribution of the constituent elements were confirmed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) (Table S3†) and energy-dispersive X-ray spectroscopy (EDX) (Fig. S13–S16, ESI†). Surface-bonded oxygen (O) due to air exposure was verified by Fourier-transform infrared spectroscopy (FTIR) (Fig. S17†). Elemental analysis revealed the presence of a small amount of carbon (Table S4†), derived from the decomposition of the diketiminate ligand into unidentified organic species. Furthermore, the Brunauer–Emmett–Teller (BET) surface area determination using nitrogen (N2) adsorption–desorption isotherms evidenced almost similar specific surface areas for both materials (Table S5†). X-ray photoelectron spectroscopy (XPS) was performed to gain information on chemical composition and electronic states. The details of Co 2p, P 2p and O 1s XPS of both materials are given in Fig. S18 (ESI†).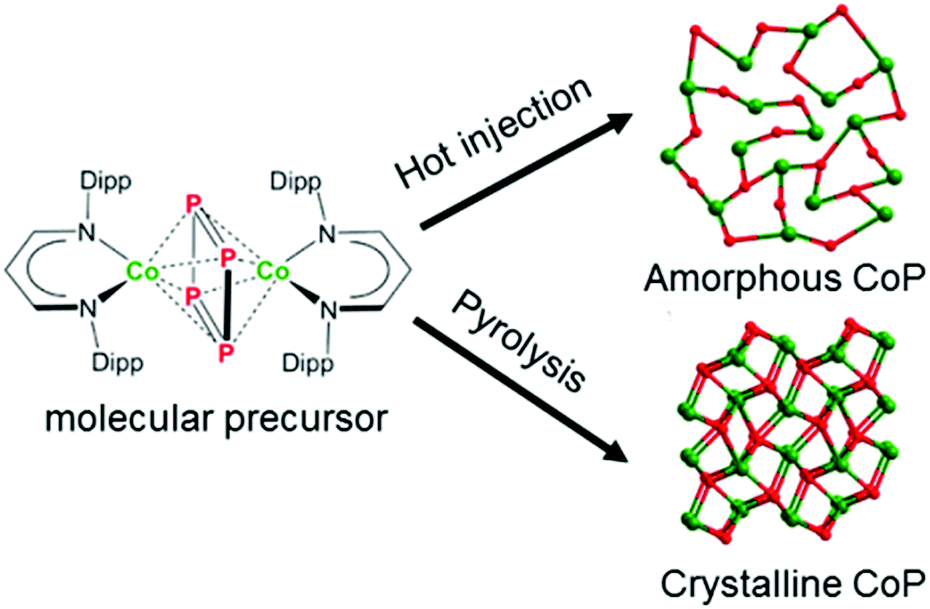 | ||
| Scheme 1 Schematic representation of synthetic routes applied to form amorphous and crystalline CoP by the molecular SSP approach. The crystal structure of crystalline CoP was determined being identical with that reported in JCPDS 29-0497, whereas the structure of amorphous CoP was drawn schematically only for a graphical differentiation. | ||
 | ||
| Fig. 1 TEM, HR-TEM and SAED of the as-prepared amorphous CoP (a–c) and crystalline CoP (d–f). The TEM and HR-TEM (a and b) of the amorphous CoP exhibited smaller agglomerated particles with no obvious diffraction rings in the SAED pattern (c). However, although the particle features were similar (d and e), the crystalline CoP displayed a lattice spacing of 0.19 nm corresponding to the (211) plane (inset e) and clear crystalline diffraction rings associated with CoP in the SAED pattern (f). | ||
The as-prepared materials were first electrophoretically deposited on NF to evaluate their OER activity by performing linear sweep voltammetry (LSV) in a 1 M KOH electrolyte and then compared to the state-of-the-art catalyst IrO2, Pt wire, and bare NF. The attained electrocatalytic activity of amorphous CoP for the OER (Fig. 2a) was very high, achieving an η10 mA = 284 mV, which surpassed the activity of the crystalline CoP (η10 mA = 305 mV), commercial IrO2 (η10 mA = 287 mV) and even the best performing TMP electrocatalysts reported to date in the literature (Tables S6–S8, ESI†). A low Tafel slope of 45 mV dec−1 was observed for the amorphous CoP which was significantly smaller than that of the crystalline CoP (82 mV dec−1) and IrO2 (118 mV dec−1), indicating a much faster reaction kinetics (Fig. 2c).40 A small reversible redox couple was detected prior to the catalytic water oxidation, which could be attributed to the presence of CoIII and CoIV (Fig. S19†). Both materials were found to be very stable at least for the period of 24 h by chronopotentiometric (CP) experiments at 10 mA cm−2 (Fig. S20b†). In addition to the NF, the catalysts were also deposited on a FTO substrate and measured under similar conditions, resulting in a similar trend of activity, long-term stability and Tafel slope (Fig. S21 and S22, ESI†).
 | ||
| Fig. 2 Polarization curves of the (a) OER and (b) HER of different CoP materials and commercial noble metal-based catalysts deposited on NF with a scan rate of 10 mV s−1 in 1 M KOH (iR compensation: 85%) and corresponding Tafel slopes obtained for the (c) OER and (d) HER. Selected regions of the polarization curves are shown in Fig. S20a and S23a (ESI†). | ||
Similar experiments were carried out to explore the catalytic activity towards the HER. The overpotential of the amorphous CoP was 143 mV at a current density of −10 mA cm−2 when deposited on NF (Fig. 2b). The crystalline CoP and commercial IrO2 were clearly less active (η−10 mA = 261 mV and 209 mV, respectively). However, the lowest overpotential was achieved with Pt (η−10 mA = 39 mV). In addition, Tafel plots for the HER were determined (Fig. 2d) from which a Tafel slope of 63 mV dec−1 was obtained for the amorphous CoP which was lower than that of the crystalline CoP (80 mV dec−1) and even Pt wire (73 mV dec−1). The attained values of Tafel slopes fall in the range of 40–120 mV dec−1 which indicates that the HER reaction proceeds via the Volmer–Heyrovsky mechanism on the surface.41 The long-term experiments (Fig. S23b†) showed that both materials have a stable activity under operating conditions over 24 h. When FTO was used instead of NF, a similar trend was observed in activity, stability and the Tafel slope (Fig. S24 and S25, ESI†). A detailed comparison of the activity of the prepared materials to that of other non-noble metal-based catalysts, non-noble TMP catalysts and Co–P-based catalysts is given in Tables S9–S11,† respectively (ESI†). Throughout the electrochemical OER and HER measurements, the amorphous CoP activity was found to be superior to that of the crystalline phase. This difference arises from the surface and electronic characteristics of the materials. As the BET surface areas were similar (Table S5†), the electrochemically active surface area (ECSA) was used to compare the active area available for catalysis.42 The ECSAs of the materials were determined from the calculation of the double layer capacitance (Cdl) (see details in the ESI†). The ECSA of the amorphous CoP is about ∼2.7 times larger than that of the crystalline CoP (Table S12†). After the OER and HER, the ECSA increases ∼1.5 times in the amorphous CoP (∼0.49 cm2) and ∼2.1 times in the crystalline CoP (∼0.24 cm2) owing to their increased surface transformation.43 This proves that the amorphous CoP has a higher density of surface defects, randomly oriented bonds, higher structural flexibility,44 and coordinative unsaturated surface metal sites available for reaction.16,45 In addition, the ECSA normalized LSV curves also confirmed the higher electrocatalytic activity of the amorphous CoP (Fig. S28†). Electrochemical impedance spectroscopy (EIS) measurements were carried out to probe the charge transfer properties of the materials, which determine their activity. A lower resistance to charge transfer (RCT) was found on the amorphous CoP relative to the crystalline CoP from the Nyquist plots of the materials (Fig. S29†). Moreover, the RCT decreases on both materials after the OER and HER, indicating a highly efficient electron transfer during catalysis (Table S13†). The use of carbon nanomaterials to improve the charge transfer efficiency and conductivity has been shown to be a common strategy for structure and electronic tuning of nanomaterials.46 However, we assume that the amount of carbon species attained was very low to influence the catalytic performance of the prepared CoP, as demonstrated for metal–organic framework (MOF) derived TMP electrocatalysts.47–50
The post-catalysis characterization revealed the transformation in the structure and composition. After the OER, the amorphous CoP did not show any drastic changes in its overall structure as well as in the SAED pattern (Fig. S30†). In the case of the crystalline CoP, an amorphous layer encapsulating the crystalline core is formed, which was also demonstrated by the SAED pattern (Fig. S30†). Elemental mapping shows that Co, O, and P are present in both materials (Fig. S31 and S32, ESI†). Phosphate formation also enhances the OER activity because it can act as a promoting ligand in the CoII/CoIII/CoIV redox process and also facilitate the four-electron proton-coupled transfer steps during the OER.51 The structural transformations at the near-surface were further confirmed by the XPS analysis (Fig. 3). The peaks corresponding to Coδ+ (778.7 eV 2p3/2 and 793.9 eV 2p1/2) completely disappeared during the OER forming new peaks: the ones associated with Co2+ and Co3+ which indicate the formation of cobalt oxidized species (Fig. 3a, S33a and b, ESI†).52 Additionally, the P 2p spectra displayed the diminishing Pδ− signal at 129.0 eV and the oxidised P5+ (phosphate) after LSV and CP experiments (133.7 eV) (Fig. 3b, S33c and d, ESI†).52,53 In the O 1s spectra (Fig. 3c, S33e and f, ESI†), the deconvolution resulted in one major (largely hydroxylated) and two minor peaks (formation of an oxide and adsorbed water molecules at the surface).54,55 Similar transformations were observed for the crystalline CoP (Fig. 3d–f) and the detailed information on its deconvoluted spectra is given in Fig. S34 (ESI†). The FTIR spectra (Fig. S35†) additionally confirmed the surface hydroxylation. Therefore, it can be deduced that during the OER in alkaline media, the surface of CoP undergoes oxidation to polyphosphate and oxo(hydroxo) containing species (CoOx(OH)y).56–58 Since the polyphosphates are highly soluble in alkaline solution,58,59 the surface becomes enriched with CoOx(OH)y which is the true active electrocatalyst for the OER.15 This oxidation process is likely to go deeper beyond the nanoparticle surface under further prolonged electrolysis and may completely transform the original CoP structure to CoOx(OH)y. Thus, the higher leaching of P as polyphosphate (85%) anions from the amorphous CoP confirms greater structural rearrangements in the defect-enriched (CoOx(OH)y) surface.60,61 On the other hand, the P dissolution rate in the crystalline CoP is only moderate (11%) (Table S14†), which confirms the larger number of arbitrarily oriented bonds and higher structural flexibility of the amorphous CoP relative to its crystalline counterpart, which intensifies the structural modification and translates into the formation of a larger number of surface unsaturated sites for facile reactant adsorption, as confirmed by the ECSA difference of the materials and its change during catalysis.43
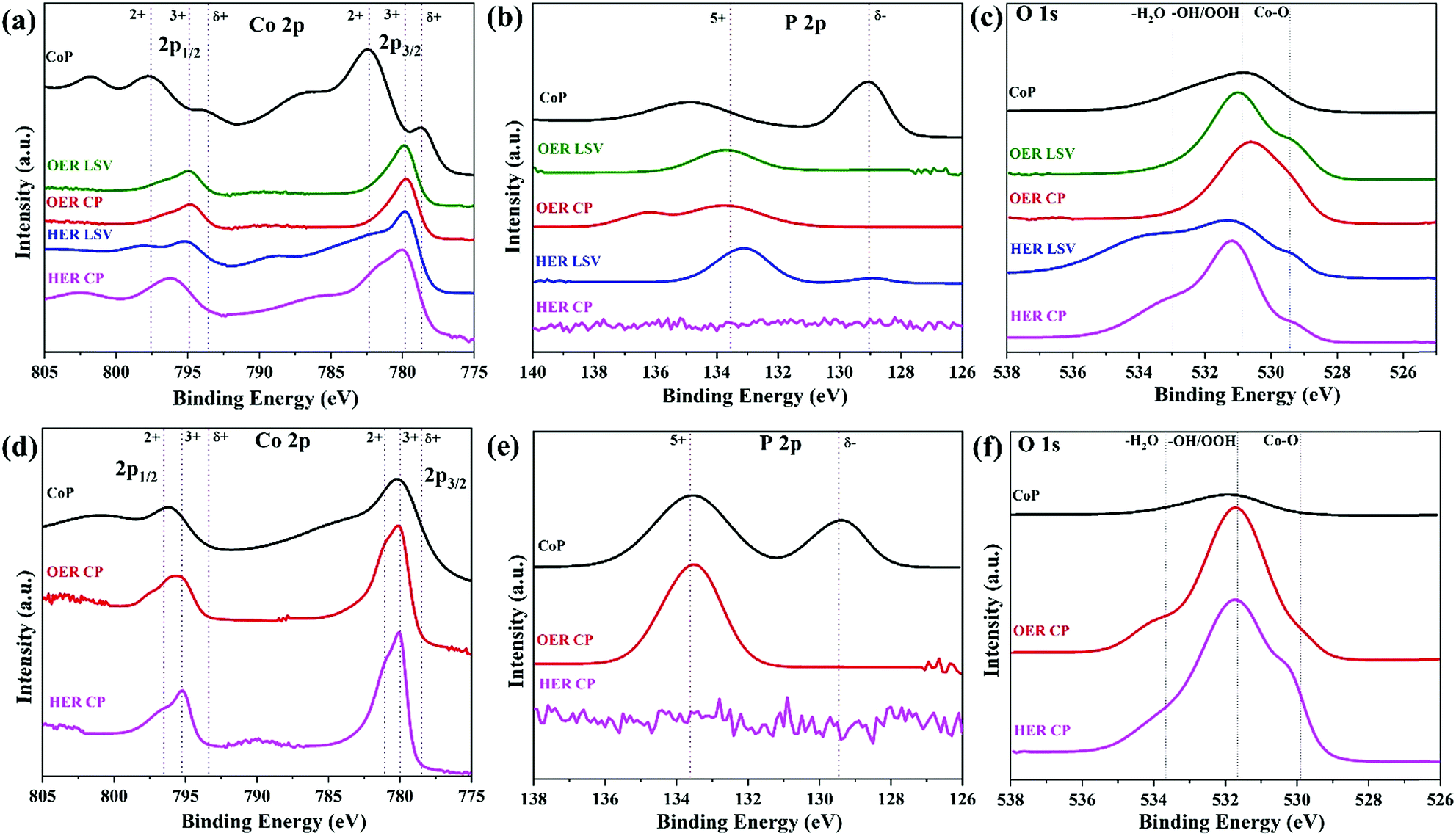 | ||
| Fig. 3 XPS spectra of the amorphous CoP: (a) Co 2p, (b) P 2p and O 1s (c) and the crystalline CoP: (d) Co 2p, (e) P 2p and (f) O 1s before and after catalysis. Deconvoluted spectra and details on assignments are shown in the ESI (Fig. S18, S32, S33, S38 and S39, ESI†). | ||
Likewise, the characterization after the HER was also carried out. TEM and SAED of the amorphous CoP (Fig. S36a and b, ESI†) showed no apparent change in the phase after catalysis. An amorphous layer was formed over the crystalline CoP but retaining its crystalline core (Fig. S36c and d, ESI†). Elemental mapping and EDX of the amorphous material and crystalline CoP (Fig. S37 and S38, ESI†) revealed that Co, O, and P are present. In both cases, the HER gave rise to the loss of P into solution by 13% and 5% for the amorphous and crystalline CoP, respectively (Table S14†). Post-HER XPS spectra of the amorphous CoP revealed oxidation of Co to Co2+/3+ (Fig. 3a and S39d, ESI†) and the absence of P signals (Fig. 3b). O 1s deconvolution after LSV and CP results in three deconvoluted peaks with assignments as in the case after the OER (Fig. 3c, S39e and f, ESI†). Similar assignments were observed in the XPS spectra of the crystalline CoP and FTIR after the HER (Fig. 3d–f, S40 and S41, ESI†). Two important phenomena contribute to the surface transformation during the HER. Initially, a similar phenomenon as in the OER occurs and results in the outer surface transformation to oxy(hydroxide) species induced by the phosphorus dissolution. At the same time, the application of a negative potential leads to the electrochemical reduction of the oxidized species generated in the surface.58 In consequence, phosphorus remains as Pδ− after catalysis in the XPS spectrum (Fig. S39c†). However, after the CP 24 h experiment, a Co-enriched surface is produced since all surface phosphorus is dissolved in solution, and hence no signal in the P 2p spectrum appears. During the application of the negative potential, in situ generated thermodynamically favourable Co0 species are formed (Fig. S42†) which can act as binding sites for protons. Similar surface transformations and formation of in situ Co0 at the surface under reductive conditions have been shown before for several phosphorus-based materials.33,43 Although phosphorus was lost on the outer surface of CoP, the presence of P in the bulk also contributes to catalysis. It has been shown previously that more electronegative P atoms can withdraw electron density from the metal by acting as a base to trap protons, whereas metal atoms can behave as a hydride acceptor.62,63 The metallic-character CoP core also contributes to catalysis by accelerating the charge transfer from the active catalyst site on the surface to the electrode substrate to efficiently accomplish the HER, which was verified by EIS measurements (Table S13†).
Inspired by the outstanding OER and HER activities of amorphous CoP on NF, we assembled an OWS device in a two electrode configuration using both CoP/NF as both the anode and cathode (CoP/NF‖CoP/NF) in 1 M KOH (Fig. S43a†). For comparison, crystalline CoP and bare NF electrodes were also measured under similar conditions. A cell voltage of 1.65 V is needed to reach a current density of 10 mA cm−2 for the amorphous CoP, whereas for the crystalline CoP, a potential of 1.79 V was required (LSV, Fig. 4a), which were superior to the bare NF. Finally, the long-term CP of both CoP for OWS was measured resulting in exceptional stability over 5 days for both materials (Fig. 4b). Moreover, an inverted electrochemical cell (graduated) was used in which H2 and O2 could be collected separately at atmospheric pressure (Fig. S44†) and the H2:O2 volume ratio was ∼2:1, showing an efficient selectivity of the catalysts for each half-cell reaction (Fig. S45†). The calculated faradaic efficiency (FE) also was nearly 100% for each half-cell reaction (see Table S15†) with amorphous CoP.
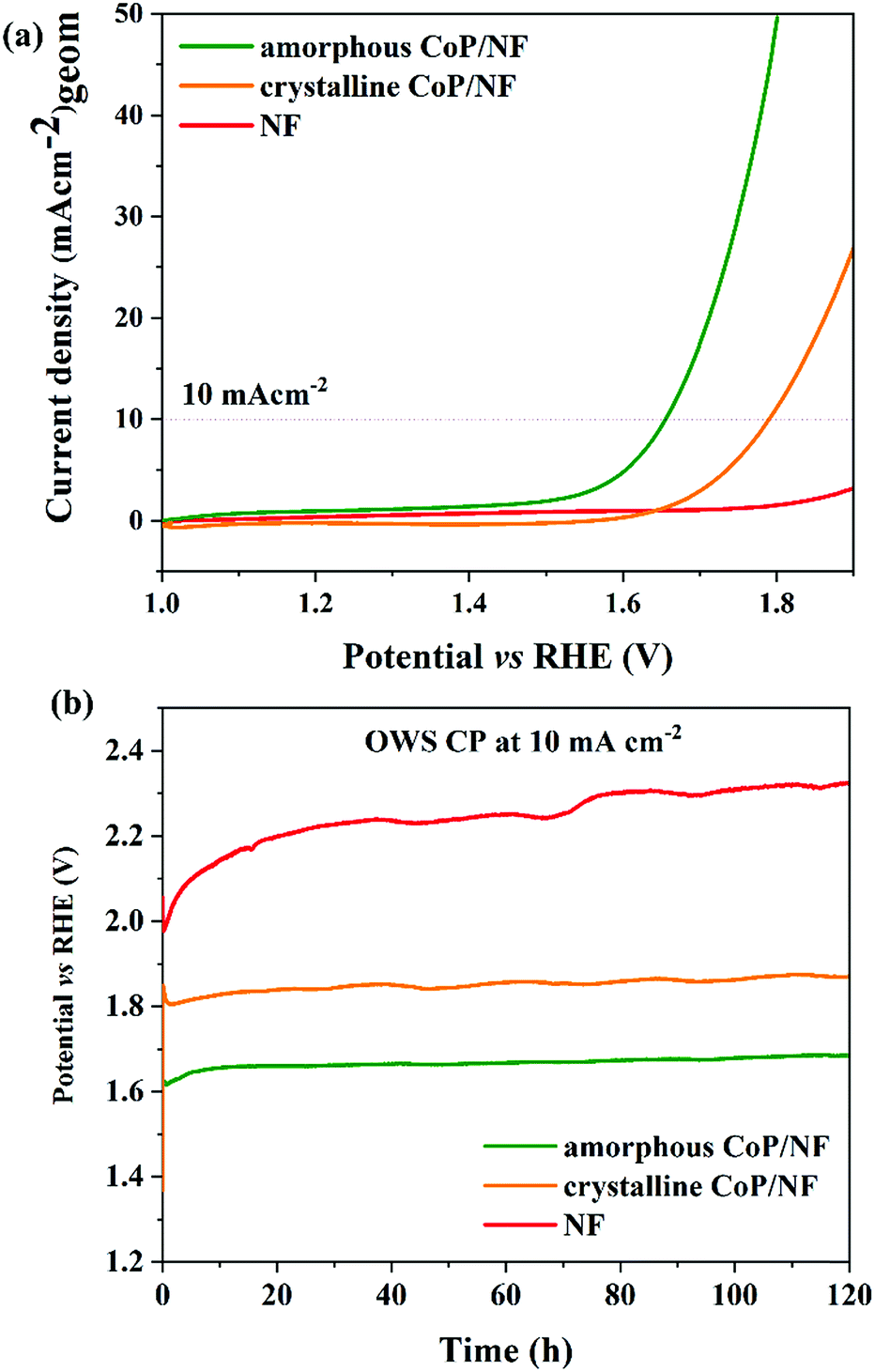 | ||
| Fig. 4 (a) LSV curves of the amorphous and crystalline CoP (CoP/NF‖CoP/NF) along with bare NF (NF‖NF) for alkaline OWS and (b) OWS durability tests (CP at 10 mA cm−2) over 5 days. | ||
4. Conclusions
In summary, structurally different amorphous and crystalline CoP have been prepared through two different thermolytic approaches and utilized as electro(pre)catalysts for the OER, HER and OWS in alkaline media. The amorphous CoP material exhibits remarkable catalytic activity for both the OER and the HER with long-term stability and a considerably small Tafel slope in comparison to the crystalline CoP. During the OER in alkaline solution, Co oxy(hydroxide)/oxide enrichment occurs on the surface of both catalysts as a result of P dissolution from the pre-catalyst forming polyphosphate species that dissolve in the electrolyte. Two simultaneous processes occur during the HER: the initial oxidation of CoP by the contact with the strongly alkaline electrolyte and concomitant phosphorus loss into the electrolyte, forming a Co-rich surface that encapsulates the CoP conductive core. Second, the reduction of the oxidized Co species to in situ generated Co0 active sites, which serve for proton reduction. Moreover, the metallic character of the CoP core also contributes to catalysis by its high conductivity to accomplish charge transfer from the active catalyst surface to the electrode substrate, as determined by the EIS experiments. The observed difference in activity is attributed to different dissolution rates of P as phosphate, diverse morphological, surface and electronic characteristics and higher active surface area of amorphous CoP than that of crystalline, which is also consistent with the lower RCT of the amorphous CoP. Moreover, the overpotentials attained for amorphous CoP in OER and HER catalysis are far better than those of most of the reported Co–P catalyst films to date. Finally, the bifunctional catalytic activity of both materials was tested using a two-electrode cell. As anticipated, the amorphous CoP exhibited a low voltage and strikingly over 100 h stability for OWS. Furthermore, the presented preparation method could be easily extended to prepare other bifunctional electrocatalysts starting from molecular single-source organometallic precursors.Conflicts of interest
The authors declare no competing interests or other interests that might be perceived to influence the results and discussion reported in this paper.Acknowledgements
The authors are indebted to Dr Vitaly Gutkin (Hebrew University, Jerusalem) for XPS, Mr Christoph Fahrenson (ZELMI, TU Berlin) for SEM, Jan Niklas Haussmann (TU Berlin) for TEM, and the group of Prof. Martin Lerch (TU Berlin) for PXRD measurements. We thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038 (UniSysCat) for financial support.References
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed.
- P. W. Menezes, A. Indra, C. Das, C. Walter, C. Göbel, V. Gutkin, D. Schmeiβer and M. Driess, ACS Catal., 2017, 7, 103–109 CrossRef CAS.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. Wiechen, M. M. Najafpour, S. I. Allakhverdiev and L. Spiccia, Energy Environ. Sci., 2014, 7, 2203–2212 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- B. You and Y. Sun, Adv. Energy Mater., 2016, 6, 1502333 CrossRef.
- P. W. Menezes, C. Panda, S. Loos, F. Bunschei-Bruns, C. Walter, M. Schwarze, X. Deng, H. Dau and M. Driess, Energy Environ. Sci., 2018, 11, 1287–1298 RSC.
- P. W. Menezes, A. Indra, I. Zaharieva, C. Walter, S. Loos, S. Hoffmann, R. Schlögl, H. Dau and M. Driess, Energy Environ. Sci., 2019, 12, 988–999 RSC.
- P. W. Menezes, C. Panda, S. Garai, C. Walter, A. Guiet and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 15237–15242 CrossRef CAS PubMed.
- Y. Pan, Y. Lin, Y. Chen, Y. Liu and C. Liu, J. Mater. Chem. A, 2016, 4, 4745–4754 RSC.
- J. A. Vigil, T. N. Lambert and B. T. Christensen, J. Mater. Chem. A, 2016, 4, 7549–7554 RSC.
- J. Wang, Z. Liu, Y. Zheng, L. Cui, W. Yang and J. Liu, J. Mater. Chem. A, 2017, 5, 22913–22932 RSC.
- J. Yu, Y. Zhong, X. Wu, J. Sunarso, M. Ni, W. Zhou and Z. Shao, Adv. Sci., 2018, 5, 1800514–1800522 CrossRef.
- S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- M. Liu and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 2158–2165 CrossRef CAS PubMed.
- H. Jia, R. Jiang, W. Lu, Q. Ruan, J. Wang and J. C. Yu, J. Mater. Chem. A, 2018, 6, 4783–4792 RSC.
- N. Bai, Q. Li, D. Mao, D. Li and H. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 29400–29407 CrossRef CAS PubMed.
- M. Xu, L. Han, Y. Han, Y. Yu, J. Zhai and S. Dong, J. Mater. Chem. A, 2015, 3, 21471–21477 RSC.
- X. Li, X. Wang, J. Zhou, L. Han, C. Sun, Q. Wang and Z. Su, J. Mater. Chem. A, 2018, 6, 5789–5796 RSC.
- B. You, N. Jiang, M. Sheng, S. Gul, J. Yano and Y. Sun, Chem. Mater., 2015, 27, 7636–7642 CrossRef CAS.
- J. Wang, W. Yang and J. Liu, J. Mater. Chem. A, 2016, 4, 4686–4690 RSC.
- X. Li, Y. Fang, F. Li, M. Tian, X. Long, J. Jin and J. Ma, J. Mater. Chem. A, 2016, 4, 15501–15510 RSC.
- C.-Z. Yuan, S.-L. Zhong, Y.-F. Jiang, Z. K. Yang, Z.-W. Zhao, S.-J. Zhao, N. Jiang and A.-W. Xu, J. Mater. Chem. A, 2017, 5, 10561–10566 RSC.
- J. Li, G. Wei, Y. Zhu, Y. Xi, X. Pan, Y. Ji, I. V. Zatovsky and W. Han, J. Mater. Chem. A, 2017, 5, 14828–14837 RSC.
- J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- C. Panda, P. W. Menezes and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 11130–11139 ( Angew. Chem. , 2018 , 130 , 11298–11308 ) CrossRef CAS.
- C. Panda, P. W. Menezes, C. Walter, S. Yao, M. E. Miehlich, V. Gutkin, K. Meyer and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 10506–10510 CrossRef CAS.
- C. Walter, P. W. Menezes, S. Orthmann, J. Schuch, P. Connor, B. Kaiser, M. Lerch and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 698–702 CrossRef CAS PubMed.
- J. A. Vigil and T. N. Lambert, RSC Adv., 2015, 5, 105814–105819 RSC.
- S. Anantharaj, P. N. Reddy and S. Kundu, Inorg. Chem., 2017, 56, 1742–1756 CrossRef CAS PubMed.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., Int. Ed., 2015, 54, 6251–6254 CrossRef CAS PubMed.
- L. Besra and M. Liu, Prog. Mater. Sci., 2007, 52, 1–61 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlögl and H. N. Alshareef, Nano Lett., 2016, 16, 7718–7725 CrossRef CAS PubMed.
- S. Yao, N. Lindenmaier, Y. Xiong, S. Inoue, T. Szilvási, M. Adelhardt, J. Sutter, K. Meyer and M. Driess, Angew. Chem., Int. Ed., 2015, 54, 1250–1254 CrossRef CAS PubMed.
- F. Zhou, A. Izgorodin, R. K. Hocking, L. Spiccia and D. R. MacFarlane, Adv. Energy Mater., 2012, 2, 1013–1021 CrossRef CAS.
- C. Du, M. Shang, J. Mao and W. Song, J. Mater. Chem. A, 2017, 5, 15940–15949 RSC.
- J. S. Lee, G. S. Park, H. Il Lee, S. T. Kim, R. Cao, M. Liu and J. Cho, Nano Lett., 2011, 11, 5362–5366 CrossRef CAS PubMed.
- Y. Zhang, L. Gao, E. J. M. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- C. Zhang, S. Trudel and C. P. Berlinguette, Eur. J. Inorg. Chem., 2014, 2014, 660–664 CAS.
- P. He, X.-Y. Yu and X. W. D. Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef CAS.
- Q. Wang, Z. Liu, H. Zhao, H. Huang, H. Jiao and Y. Du, J. Mater. Chem. A, 2018, 6, 18720–18727 RSC.
- L. Yan, L. Cao, P. Dai, X. Gu, D. Liu, L. Li, Y. Wang and X. Zhao, Adv. Funct. Mater., 2017, 27, 1–10 Search PubMed.
- S. He, S. He, X. Bo, Q. Wang, F. Zhan, Q. Wang and C. Zhao, Mater. Lett., 2018, 231, 94–97 CrossRef CAS.
- P. He, X.-Y. Yu and X. W. D. Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef CAS PubMed.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- L. Ai, Z. Niu and J. Jiang, Electrochim. Acta, 2017, 242, 355–363 CrossRef CAS.
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874–6878 CrossRef CAS.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- J.-K. Chang, C.-M. Wu and I.-W. Sun, J. Mater. Chem., 2010, 20, 3729–3735 RSC.
- B. You, N. Jiang, M. Sheng, M. W. Bhushan and Y. Sun, ACS Catal., 2016, 6, 714–721 CrossRef CAS.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- Z. Wu, Q. Gan, X. Li, Y. Zhong and H. Wang, J. Phys. Chem. C, 2018, 122, 2848–2853 CrossRef CAS.
- A. Indra, P. W. Menezes, N. R. Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS.
- X. Zhou, H. Gao, Y. Wang, Z. Liu, J. Lin and Y. Ding, J. Mater. Chem. A, 2018, 6, 14939–14948 RSC.
- Q. He, H. Xie, Z. ur Rehman, C. Wang, P. Wan, H. Jiang, W. Chu and L. Song, ACS Energy Lett., 2018, 3, 861–868 CrossRef CAS.
- P. Liu and J. A. Rodriguez, J. Am. Chem. Soc., 2005, 127, 14871–14878 CrossRef CAS PubMed.
- S. Yao, V. Forstner, P. W. Menezes, C. Panda, S. Mebs, E. M. Zolnhofer, M. E. Miehlich, T. Szilvási, N. Ashok Kumar, M. Haumann, K. Meyer, H. Grützmacher and M. Driess, Chem. Sci., 2018, 9, 8590–8597 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta04583j |
| This journal is © The Royal Society of Chemistry 2019 |