
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcidic liquid-swollen polymer membranes exhibiting anhydrous proton conductivity higher than 100 mS cm−1 at around 100 °C†
Takato
Kajita‡
 a,
Haruka
Tanaka‡
a,
Atsushi
Noro‡
*a,
Yushu
Matsushita‡
a and
Naoki
Nakamura‡
b
a,
Haruka
Tanaka‡
a,
Atsushi
Noro‡
*a,
Yushu
Matsushita‡
a and
Naoki
Nakamura‡
b
aDepartment of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: noro@nagoya-u.jp
bAdvanced Material Engineering Division, Advanced R&D and Engineering Company, Higashifuji Technical Center, TOYOTA Motor Corporation, 1200 Mishuku, Susono, Shizuoka 410-1193, Japan
First published on 3rd May 2019
Abstract
Anhydrous proton-conductive membranes are prepared by incorporating sulfuric acid (H2SO4) into cross-linked poly(4-vinylpyridine) (CL-P), where H2SO4 mixes CL-P homogeneously at the molecular level, and therefore incorporated H2SO4 does not leach out from CL-P, even when the weight ratio of H2SO4 to CL-P is about four. The membranes are soft but self-standing and exhibit anhydrous conductivity higher than 100 mS cm−1 at around 100 °C. Furthermore, a membrane composed of polystyrene-b-poly(4-vinylpyridine)-b-polystyrene triblock copolymers and H2SO4 achieves not only high anhydrous conductivity but also good mechanical properties as a membrane.
Introduction
High-performance polymer membranes, such as lithium ion-conductive membranes1–7 and proton-conductive membranes,8–15 have been receiving attention in recent years. Proton conductive-membranes are useful in polymer electrolyte fuel cells (PEFCs) that generate electrical energy through a reaction of hydrogen and oxygen gases, producing water only as a byproduct. Other fuel cells, such as solid oxide fuel cells (SOFCs),16–18 molten carbonate fuel cells (MCFCs),19,20etc., must be typically operated at a higher temperature than at least 200 °C to attain high ionic conductivity of 100 mS cm−1 and they also require long time to start up.21 On the other hand, the proton-conductive membranes in PEFCs exhibit relatively high proton conductivity of 100 mS cm−1 even at 80 °C; hence, PEFCs are promising systems for use in clean power generation for vehicles. Nafion®, a perfluorosulfonic acid polymer membrane developed by DuPont, is the most well-known proton-conductive membrane since it exhibits a relatively high proton conductivity of 100 mS cm−1 or higher at 70–90 °C under hydrous conditions.8,22–29 However, the conductivity of Nafion is extremely low under dry conditions, especially over the boiling point of water (100 °C), where almost no water molecules exist.22–24,29 Hence, PEFCs with Nafion membranes cannot be operated without having humidification systems, especially over 100 °C. Note that a glass transition temperature (Tg) of Nafion is about 100 °C.23,26,29 Thus, Nafion cannot be used as a membrane over the Tg, i.e., about 100 °C.To overcome this drawback, membranes with moderate proton conductivity even under anhydrous conditions have been developed.30–40 One of the most well-known examples is polybenzimidazole (PBI; Fig. S1(a), ESI†) membranes doped with phosphoric acid (H3PO4) or sulfuric acid (H2SO4; Fig. S1(b), ESI†).30–32 These membranes exhibit a conductivity of a few tens of mS cm−1 at relatively high temperatures of about 200 °C; however, they exhibit the low conductivity of 5.6 mS cm−1 at a moderately high temperature of 130 °C. Moreover, the conductivity of the membranes spontaneously decreases over time because H3PO4 or H2SO4 in the membranes naturally leaches out. Thus, acid-doped PBI membranes are still insufficient for use in fuel cells, though various improvements have been made to the membranes to date.35,36,38,40 As another example, anhydrous proton-conductive metal organic framework (MOF) has also been developed,41–45 but the anhydrous conductivity was at most 10 mS cm−1 even at 150 °C,46 which is also inferior to the conductivity of humidified Nafion. Spontaneous leaching out of the acid from acid-doped PBI is probably attributed to the rigidness of PBI, which shows insufficient compatibility with even low-molecular-weight substances, such as H3PO4 or H2SO4. It should be noted that PBI has rigid aromatic rings on the backbone and hence it shows long Kuhn segment length of 3.0 nm,47 while a typical flexible vinyl polymer (e.g., polystyrene) has a Kuhn segment length around 0.70 nm.48 Therefore, if a flexible basic polymer without aromatic rings on the backbone chain is mixed with an acidic low-molecular-weight liquid medium that can be attached to a basic group on the side chain of flexible polymers via an attractive interaction, then spontaneous leaching out of acidic liquid would not occur because a large amount of acidic liquid can infiltrate the flexible polymer homogeneously at the molecular level. However, the flexible basic polymer mixed with a large amount of acidic liquid usually gives a homogeneous molten mixture that cannot be used as a membrane.
In order to prepare a soft-solid self-standing membrane, cross-linking of the polymer is typically required. In other words, there must be excellent utility in applying the molecular design concept of polymer gels49,50 that typically contain a large amount of solvents. Although the Tg of polymer gels is lower than ambient temperature due to the large amount of solvent, they do not flow and maintain their shape because the chains are cross-linked. If such polymer gels have adequate mechanical properties enabling self-standing abilities, then they can be used as soft membranes.
Based on the above consideration, we prepare an anhydrous proton-conductive polymer membrane that can be used even without humidification, while providing high conductivity of more than 100 mS cm−1 that is comparable to the performance of hydrous Nafion, by using both cross-linking of a flexible basic polymer and swelling with a large amount of nonvolatile strong acid, H2SO4, that is useful for attaining high proton conduction.51 First, we synthesize a cross-linked flexible basic polymer (CL-P; Fig. 1(a)) of poly(4-vinylpyridine) (Kuhn segment length: 0.797 nm)52 which can form acid–base complexes53 with nearly nonvolatile H2SO4via ionic interactions.54 Then, we prepare an anhydrous proton-conductive membrane by swelling the cross-linked polymer with a nearly nonvolatile H2SO4. Sulfuric acid molecules spontaneously and homogeneously infiltrate the cross-linked basic polymer via ionic interactions among the proton, sulfonate anion, and pyridinium cation. We also measure the anhydrous conductivity of the membranes.
 | ||
| Fig. 1 Synthetic schemes and schematic illustration. (a) Chemically cross-linked poly(4-vinylpyridine) (CL-P). (b) Uncross-linked poly(4-vinylpyridine) (P). | ||
Results and discussion
Preparation of mixtures and membranes
Before attempting our molecular design to preparing anhydrous proton-conductive membranes, we first prepared an H2SO4-doped PBI membrane (weight ratio H2SO4/PBI: 76/24) by infiltrating H2SO4 into a neat PBI membrane; which is a previously reported anhydrous proton-conductive membrane.30,31 Unfortunately, leaching out of H2SO4 was seen over several days after the preparation (Fig. S1(c) and (d), ESI†). This probably occurred due to poor solubility of PBI in H2SO4.To achieve high solubility of polymers in H2SO4, we used flexible poly(4-vinylpyridine) instead of rigid PBI. A membrane containing 20 wt% H2SO4 and 80 wt% uncross-linked poly(4-vinylpyridine) (P; Fig. 1(b)) was prepared (see also Table S1, ESI†) and resulted in a self-standing, homogeneous, hard membrane (Fig. 2(a)). However, this membrane exhibited almost no anhydrous conductivity. Narayanan and coworkers34 investigated a sample of uncross-linked poly(4-vinylpyridine) mixed with an equimolar amount of H2SO4 to the pyridyl groups on the polymer, and the mixture became a self-standing membrane and exhibited anhydrous conductivity; however, the conductivity was merely on the order of 0.1 mS cm−1, even at 140 °C, which was far below the 100 mS cm−1. Although the conductivity is insufficient, they did not further investigate a sample containing a larger amount of H2SO4. Therefore, to aim for a higher conductivity, we prepared an uncross-linked P/H2SO4 mixture containing much more H2SO4 (80 wt%) (see also Table S1, ESI†). Although the mixture had a homogeneous appearance as shown in Fig. 2(b) because H2SO4 in the mixture formed acid–base complexes with pyridyl groups on polymer P (see the Fourier transform infrared (FT-IR) spectra in Fig. S3, ESI†), the mixture showed fluidity. Thus, even if the mixture shows anhydrous high proton conductivity, it could not be used as a membrane.55–57 The reason for the liquidity was quite natural, namely the uncross-linked polymer was actually dissolved in a large quantity of a low-molecular-weight liquid.
 | ||
| Fig. 2 Schematic illustrations and photos of P/H2SO4 and CL-P/H2SO4. (a) A membrane containing 20 wt% H2SO4 and 80 wt% uncross-linked polymer P. The membrane was self-standing, homogeneous, and hard; however, its conductivity was very low under anhydrous conditions. (b) A mixture containing 20 wt% P and 80 wt% H2SO4. The mixture was homogeneous but could not be used as a membrane due to its fluidity. (c) A membrane prepared by swelling a cross-linked polymer CL-P membrane with the same weight of H2SO4. The membrane was self-standing, homogeneous, and hard, but its conductivity was very low (see Table 1). (d) A membrane containing 18 wt% CL-P and 82 wt% H2SO4. Since the polymer was cross-linked, the membrane was self-standing, homogeneous, and soft. Furthermore, it exhibited high conductivity even under anhydrous conditions (see also Fig. 3 and Table 1). In schematic, gray lines represent polymer chains with a higher Tg than ambient temperature, and pink lines represent chains with a lower Tg than ambient temperature. As shown in FT-IR spectra of Fig. S3,† a pyridyl group and H2SO4 interact with each other, but broken lines and dotted lines indicating the interaction are not shown here for simplicity. | ||
To overcome these difficulties, we prepared a cross-linked poly(4-vinylpyridine) (CL-P)/H2SO4 membranes by immersing the CL-P membrane in a solution of nearly nonvolatile H2SO4 dissolved in methanol, and then the volatile solvent was completely removed by heating at 50 °C. With this method, membranes can be prepared with good reproducibility (see also Fig. S4, ESI†). The sample containing 50 wt% H2SO4 was a self-standing, glassy, hard membrane and had a macroscopically homogeneous appearance (Fig. 2(c)). The sample containing 82 wt% H2SO4 was also self-standing but it formed a soft membrane (Fig. 2(d)). It should be noted that, unlike the H2SO4-doped PBI, the CL-P/H2SO4 membrane was macroscopically homogeneous and H2SO4 has never leached out, even it has been left for a long time (>100 days). This is because flexible CL-P is much more easily miscible with H2SO4 compared with rigid PBI. In fact, only the surface layer of PBI is likely to dissolve with H2SO4 in H2SO4-doped PBI. This also means that the number of contacts between the pyridyl groups on flexible CL-P and H2SO4 at the molecular level is much larger than that between imidazole groups on PBI and H2SO4. Thus, the Gibbs energy of mixing for the case, ΔmixG, represented by ΔmixH − TΔmixS (where ΔmixH, ΔmixS, and T are enthalpy of mixing, entropy of mixing, and absolute temperature, respectively), is mainly dominated by negative ΔmixH with a large absolute value. This prevents spontaneous leaching out of H2SO4 from the membrane. The above results show that it is effective to swell a cross-linked flexible base polymer with an acidic liquid in the preparation of an anhydrous proton-conductive membrane. It should be also noted that CL-P/H2SO4 containing 85 wt% H2SO4 was also prepared, but the sample was so soft and it was difficult to handle as a membrane. Therefore, swelling with 82 wt% acidic liquid could be the best result for the membrane preparation.
Anhydrous conductivity of CL-P/H2SO4
The conductivity of the CL-P/H2SO4 membranes in the temperature range from 50 °C to 120 °C without humidification was determined by the AC impedance method, where the overall ionic conductivity contributed by all ion species including protons was measured. Contributions of proton conductivity to the overall ionic conductivity can be expressed by the proton transference number, and the proton transference number of concentrated sulfuric acid is probably very close to unity as phosphoric acid,58 since the proton is the smallest ion in sulfuric acid and move much more easily than other ions such as a sulfate ion. Therefore, the overall ionic conductivity measured by AC impedance method is considered to be equivalent to the proton conductivity in this study. Table 1 summarizes the anhydrous conductivity and Fig. 3 shows a plot of the anhydrous conductivity of the CL-P/H2SO4 membranes against the reciprocal of the absolute temperature. In the membrane with an H2SO4 content of 50 wt% (Fig. 2(c)), we could not obtain an impedance spectrum because the resistance was extremely large; i.e., the anhydrous conductivity was extremely low. However, for the membrane in which the content of H2SO4 increased by 5 wt% from 50 wt%, i.e., H2SO4 content was set to be 55 wt% (Fig. S4(a), ESI†), the membrane exhibited a conductivity of 0.077 mS cm−1 at 80 °C. Moreover, the anhydrous conductivity was 13 mS cm−1 at 80 °C for the membrane with an H2SO4 content of 60 wt% (Fig. S4(b), ESI†); the conductivity increased drastically by 2 to 3 orders of magnitude due to a mere 5 wt% increase in the H2SO4 content. Furthermore, the conductivity increased as the H2SO4 content increased; notably, the membrane with an H2SO4 content of 82 wt% (Fig. 2(d)) exhibited a very high conductivity of more than 100 mS cm−1 (120 mS cm−1 at 80 °C), even under anhydrous conditions. At temperatures above 100 °C, this membrane showed even higher conductivity (e.g., 160 mS cm−1 at 120 °C). The present molecular design which enables to maintain the membrane shape by homogeneous and spontaneous swelling with a large amount of H2SO4, probably resulted in much higher conductivity than that in previously reported H2SO4-doped PBI membranes (the H2SO4 content: 44 wt%), i.e., ca. 0.35 mS cm−1 at 85 °C.31 The molecular design for membrane preparation can also be applied to another cross-linked flexible basic polymer, cross-linked poly(1-vinylimidazole) (CL-I, see Scheme S1, Table S2 and Fig. S6, ESI†). Note that the leaching out of the H2SO4 from the membranes did not occur, either.| Polymer | H2SO4 weight contenta [wt%] | n Acid/nBaseb [mol mol−1] | H2SO4 mole contentc [mol%] | σ DC [mS cm−1] | ||||
|---|---|---|---|---|---|---|---|---|
| 50 °C | 80 °C | 95 °C | 110 °C | 120 °C | ||||
| a The weight content of H2SO4 in polymer membranes. b The molar ratio of H2SO4 to the pyridyl group on the polymer. c The mole content of H2SO4, which can be calculated according to (1 + nBase/nAcid)−1 × 100. d The DC conductivity estimated from the |Z| value of the plateau in the AC impedance spectrum, i.e., the bulk resistance. Here, the proton transference number is assumed to be unity. Therefore, the overall ionic conductivity is regarded as the proton conductivity. e The conductivity cannot be determined because it is very low. f The conductivity was not measured because the temperature is higher than the Tg of polystyrene. | ||||||||
| CL-P | 50 | 1.1 | 52 | —e | —e | —e | —e | —e |
| 55 | 1.3 | 57 | 0.022 | 0.077 | 0.11 | 0.14 | 0.15 | |
| 60 | 1.6 | 62 | 7.4 | 13 | 15 | 18 | 19 | |
| 70 | 2.6 | 72 | 25 | 36 | 40 | 49 | 59 | |
| 82 | 5.0 | 83 | 87 | 120 | 140 | 150 | 160 | |
| Post-CL-P | 80 | 4.3 | 81 | 60 | 94 | 120 | 140 | 150 |
| S–P–S | 80 | 4.5 | 82 | 63 | 100 | 130 | —f | —f |
 | ||
| Fig. 3 Anhydrous conductivity versus reciprocal absolute temperature for several CL-P/H2SO4 membranes. Solid lines are drawn as guide for eyes. | ||
To further investigate the dependence of the anhydrous conductivity on the H2SO4 content in CL-P/H2SO4 membranes, Tgs of uncross-linked P/H2SO4 mixtures were measured by differential scanning calorimetry (DSC). DSC thermograms are shown in Fig. S5 (ESI†). Fig. 4 shows a plot of Tg (the left side of vertical axis) against the molar ratio of H2SO4 to the pyridyl group on the polymer P, nAcid/nBase (see also Table S1, ESI†). Here we used uncross-linked P because it can be easily synthesized and its molecular characterization is also easy (see also ESI†). Note that the Tg dependence of uncross-linked P/H2SO4 and CL-P/H2SO4 should be almost the same, since the molar amount of cross-links in CL-P is very small. When the weight contents of H2SO4 were 10 wt% (nAcid/nBase = 0.12), 20 wt% (nAcid/nBase = 0.27), 30 wt% (nAcid/nBase = 0.46), and 40 wt% (nAcid/nBase = 0.71), where the molar ratio of H2SO4 to the pyridyl group is below unity, the Tg values were high: 141 °C, 158 °C, 170 °C, and 171 °C, respectively. This is presumably because most of H2SO4 in the mixtures formed acid–base complexes with pyridyl groups on polymer P (see the FT-IR spectra in Fig. S3, ESI†), and the segmental motions of the polymer were strongly restricted. On the other hand, when the weight contents of H2SO4 were 50 wt% (nAcid/nBase = 1.1), 55 wt% (nAcid/nBase = 1.3), and 60 wt% (nAcid/nBase = 1.6), where the molar ratio of H2SO4 to the pyridyl group on the polymer P exceeds unity, the Tg values suddenly dropped and became 141 °C, 43 °C, and −52 °C, respectively. This is probably because an excessive molar amount of H2SO4 to the pyridyl groups served as a plasticizer, causing activated segmental motions of the polymer chain. When the H2SO4 weight content was further increased, the Tg gradually decreased; the Tgs at 70 wt% (nAcid/nBase = 2.5) and 80 wt% (nAcid/nBase = 4.3) are very low at −86 °C and about −95 °C, respectively.
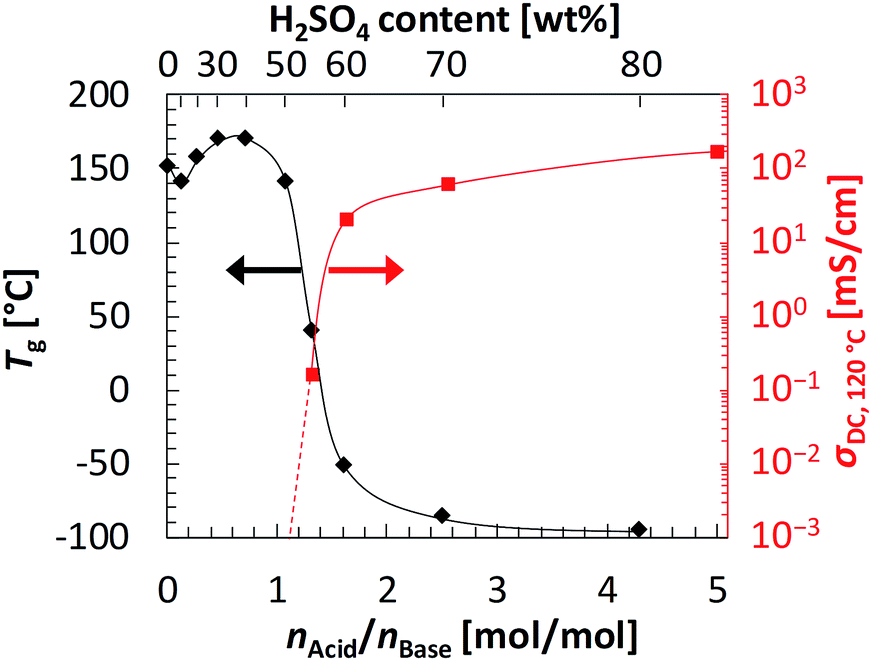 | ||
| Fig. 4 T g of P/H2SO4 mixtures (left axis) and anhydrous conductivity at 120 °C of CL-P/H2SO4 membranes (right axis) against H2SO4/pyridyl group molar ratio (nAcid/nBase) and the content of H2SO4 in mixtures. Solid and broken lines are drawn as guide for eyes. | ||
In the region where nAcid/nBase is larger than unity, the conductivity also varied remarkably. Fig. 4 also shows the dependence of the conductivity of CL-P/H2SO4 at 120 °C (the right side of vertical axis) against nAcid/nBase. When nAcid/nBase was 1.1 or less, i.e., the H2SO4 content was 50 wt% or less, the conductivity was not able to be estimated by impedance measurements because of high resistance. However, when nAcid/nBase was varied slightly from 1.3 (H2SO4 content: 55 wt%) to 1.6 (H2SO4 content: 60 wt%), the conductivity increased drastically by 2 to 3 orders of magnitude. This drastic increase in the conductivity was attributed to both an increase in the number of free protons derived from an excessive molar amount of H2SO4 that is not used for acid–base complexation and a drastic decrease in Tg of the membrane. Furthermore, the membrane of CL-P/H2SO4 containing even 82 wt% H2SO4 maintained its shape due to cross-linking, despite of the low Tg, thereby attaining the very high conductivity of more than 100 mS cm−1 (160 mS cm−1 at 120 °C) even under anhydrous conditions.
Properties of post-cross-linked polymer/H2SO4 and triblock copolymer/H2SO4
Although CL-P described above was prepared by cross-linking a polymer during polymerization, a post-cross-linked base polymer (Post-CL-P) can also be prepared by mixing 1,4-dibromobutane (BrC4Br) as a cross-linking agent with the uncross-linked homopolymer P (Fig. 5(a); see also Scheme S2, ESI†), so that a quaternization reaction of the pyridyl group occurs (see FT-IR spectra in Fig. S7, ESI†).59 Thus, anhydrous proton-conductive membranes can also be easily prepared by infiltrating H2SO4 into the Post-CL-P membranes. Fig. 5(b) and (c) show a schematic illustration and a photograph of Post-CL-P/H2SO4 containing 80 wt% H2SO4. The Post-CL-P/H2SO4 membrane was soft and homogeneous. As shown in Fig. 5(d), the conductivity of Post-CL-P/H2SO4 was also very high, for example, 94 mS cm−1 at 80 °C, 150 mS cm−1 at 120 °C, and 220 mS cm−1 at 160 °C (see also Tables 1 and S3, ESI†) under anhydrous conditions. It should be noted that the conductivity of Post-CL-P/H2SO4 at 95 °C decreased slightly over time, but the conductivity in approximately 300 hours after keeping at 95 °C was still around 94% of the initial conductivity, 120 mS cm−1 (Fig. S8, ESI†). Leaching out of the acid did not occur, either, even after heating at 95 °C for approximately 300 hours. This also means fatal sulfate poisoning to catalyst does not occur at this temperature. | ||
| Fig. 5 (a) Chemical structure of a post-cross-linked P (Post-CL-P) with base groups on the P chain. (b) Schematic illustration and (c) a photo of a proton-conductive membrane of Post-CL-P/H2SO4 containing 80 wt% H2SO4. (d) Plots of anhydrous conductivity against reciprocal absolute temperature for Post-CL-P/H2SO4 and S–P–S/H2SO4, where the weight content of H2SO4 is 0.8. Solid lines are drawn just as guide for eyes. (e) Chemical structure of an ABA triblock copolymer (S–P–S) comprising glassy polystyrene (S) and glassy poly(4-vinylpyridine) (P) at ambient temperature. (f) Schematic illustration and (g) a photo of a proton-conductive membrane of S–P–S/H2SO4 containing 80 wt% H2SO4. (h) Stress–strain curves for Post-CL-P/H2SO4 and S–P–S/H2SO4, where the weight content of H2SO4 is 0.8. In schematic, as shown in the FT-IR spectra of Fig. S3,† a pyridyl group and H2SO4 interact with each other, but broken lines and dotted lines indicating the interaction are not shown here for simplicity. | ||
Proton-conductive membranes can also be prepared not only by chemical cross-linking but also by physical cross-linking.60,61 One example is self-assembly of an ABA triblock copolymer62 that forms a nanophase-separated structure63 consisting of incompatible polymer blocks, where the middle block B is bridged between the glassy domains of the end blocks A. As a pioneering study of anhydrous proton-conductive membranes using a block copolymer, Park and coworkers prepared a membrane exhibiting the anhydrous conductivities of 20 mS cm−1 at 115 °C and 45 mS cm−1 at 165 °C by infiltrating a neutral aprotic ionic liquid into a poly(styrenesulfonate)-b-polymethylbutylene diblock copolymer, which can form a nanophase-separated structure, but cannot adopt a bridging conformation because of the nature of diblock copolymer.64–66 Segalman and coworkers also prepared a membrane exhibiting the anhydrous conductivities of 3 mS cm−1 at 65 °C and 10 mS cm−1 at 140 °C by infiltrating a protic ionic liquid into a poly(styrene)-b-poly(2-vinylpyridine) diblock copolymer.67,68 More recently, Lodge, Hillmyer, and coworkers prepared an anhydrous proton-conductive membrane (4 mS cm−1 at 120 °C and 14 mS cm−1 at 180 °C) by incorporating a protic ionic liquid of 1-ethylimidazolium N,N-bis(trifluoromethylsulfonyl)imidide into a poly(ethylene oxide)-b-polystyrene diblock copolymer where the polystyrene domain was chemically cross-linked.69 In our study, we prepared an anhydrous proton-conductive membrane by simply infiltrating H2SO4 into the polystyrene-b-poly(4-vinylpyridine)-b-polystyrene (S–P–S) triblock copolymer (Fig. 5(e); see also Scheme S3, ESI†) membrane via acid–base complexation and ionic interaction. Note that H2SO4 can selectively dissolve the middle block P only in S–P–S. As expected, even the S–P–S/H2SO4 containing 80 wt% H2SO4 was a self-standing soft membrane (Fig. 5(g)), because the middle chains P are bridging the glassy S domains, probably forming the nanophase-separated structure (Fig. 5(f)).60 The S–P–S/H2SO4 membrane also exhibited very high conductivity (for example, 100 mS cm−1 at 80 °C and 130 mS cm−1 at 95 °C; see also Table 1), whose value is comparable to that of the humidified Nafion membrane. To the best of our knowledge, this is the first report of a proton-conductive membrane comprising a self-assembled block copolymer that exhibits a quite high conductivity of higher than 100 mS cm−1 at approximately 100 °C under almost no humidity; furthermore, it can be favorably processed with very little thickness and a large area since these membranes show thermoplastic property, which is a very useful feature for practical applications.
The mechanical properties were also evaluated by tensile tests of the above proton-conductive membranes. Fig. S9(a) and (b) (ESI†) show photographs of the samples before and after/during elongation for Post-CL-P/H2SO4 and S–P–S/H2SO4, respectively. Fig. 5(h) exhibits stress–strain curves acquired, and Table S4 (ESI†) summarizes the following mechanical properties: Young's modulus (E), elongation at break (εb), maximum stress (σmax), and inner area of the stress–strain curve (S). E is the initial slope of the stress–strain curve estimated from the low strain region between 0% and 10% by the least squares method. S corresponds to the energy used until break, which is an index representing the robustness of the material. The E, εb, σmax, and S of the Post-CL-P/H2SO4 membrane are 0.10 MPa, 60%, 0.054 MPa, and 0.021 MJ m−3, respectively. From these results, Post-CL-P/H2SO4 is found to be a soft solid membrane, although it cannot be largely deformed. In contrast, the E, εb, σmax, and S of the S–P–S/H2SO4 membrane are 0.10 MPa, 880%, 0.56 MPa, and 2.1 MJ m−3, respectively. Here, E is the same value as that of the Post-CL-P/H2SO4 membrane, while both εb, and σmax have 10 times larger values. Therefore, S is 100 times larger than that of the Post-CL-P/H2SO4 membrane, revealing that the S–P–S/H2SO4 membrane has far superior mechanical properties than Post-CL-P/H2SO4 membrane. Since Post-CL-P is chemically cross-linked and the cross-linking points are generated nonuniformly (Fig. 5(b)), the stress concentration at certain cross-linking points tends to occur during elongation, which probably induces easy fracture of the membrane. On the other hand, the S hard domains formed by the self-assembly of the S–P–S block copolymer are located uniformly (Fig. 5(f)), which reduces the stress concentration on certain domains that behave as physical cross-links, resulting in the large εb of S–P–S/H2SO4.
Conclusions
In conclusion, we report the preparation of anhydrous proton-conductive polymer membranes exhibiting high conductivity of higher than 100 mS cm−1, comparable to that of humidified Nafion. The preparation procedure adopted in this study includes the spontaneous and homogeneous infiltration of a strong acidic liquid into the cross-linked flexible basic polymer. Even if the membranes contain a large amount of H2SO4 (4 times the weight of the polymer), no spontaneous leaching out was observed. In addition, even if Tg is far below the ambient temperature due to the incorporation of a large amount of H2SO4, the membranes are self-standing because the polymers are cross-linked. When the molar ratio of the acid to the base group (nAcid/nBase) exceeds unity, the excess molar amount of H2SO4 acts as a plasticizer and the concentration of free protons increases, resulting in a drastic decrease in Tg and a remarkable increase in anhydrous conductivity. Moreover, by incorporating about 4 times the weight of H2SO4 in CL-P, the membrane, despite anhydrous conditions, exhibits the very high conductivity of more than 100 mS cm−1, comparable to humidified Nafion. Furthermore, by swelling an S–P–S triblock copolymer membrane with H2SO4, it exhibits not only anhydrous high conductivity, but also excellent robustness, especially to a large breaking elongation of 800% or more, since the hard domains serving as physical cross-links are located uniformly. In the future, we will investigate effects of humidity on the conductivity of membranes and the performance of assembled fuel cells under anhydrous conditions (see also Fig. S10 (ESI†) of the preliminary result on performance of a fuel cell assembled with the anhydrous proton-conductive membranes under anhydrous conditions, though the performance was not good; this is probably because preparation and measurements of the assembled fuel cell have not been optimized yet. If catalyst layer preparation by infiltrating ion species is optimized, the much better fuel cell performance will be attained even under anhydrous conditions). Further development of higher-performance anhydrous proton-conductive polymer membranes based on a similar molecular design are desired to achieve practical applications of next generation fuel cells.Conflicts of interest
Two patent applications covering this work have been filed by Nagoya University and Toyota Motor Corporation, naming N. N., A. N., T. K., H. T., and Y. M. as inventors.Acknowledgements
The authors thank Mr Masashi Suzuki and Mr Naoki Inukai at the Aichi Center for Industry and Science Technology, Japan, for their kind help for AC impedance measurements. This work was partially supported through KAKENHI grant number 15K13785 (A. N.) from JSPS, Japan. A. N. is also grateful for Toyota Most Advanced Collaborative Research Grant.Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Yoshizawa, M. Hirao, K. Ito-Akita and H. Ohno, J. Mater. Chem., 2001, 11, 1057–1062 RSC.
- M. Singh, O. Odusanya, G. M. Wilmes, H. B. Eitouni, E. D. Gomez, A. J. Patel, V. L. Chen, M. J. Park, P. Fragouli, H. Iatrou, N. Hadjichristidis, D. Cookson and N. P. Balsara, Macromolecules, 2007, 40, 4578–4585 CrossRef CAS.
- K. P. Barteau, M. Wolffs, N. A. Lynd, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2013, 46, 8988–8994 CrossRef CAS.
- Y. Kitazawa, K. Iwata, S. Imaizumi, H. Ahn, S. Y. Kim, K. Ueno, M. J. Park and M. Watanabe, Macromolecules, 2014, 47, 6009–6016 CrossRef CAS.
- S. A. Chopade, J. G. Au, Z. Li, P. W. Schmidt, M. A. Hillmyer and T. P. Lodge, ACS Appl. Mater. Interfaces, 2017, 9, 14561–14565 CrossRef CAS PubMed.
- Y. Kitazawa, K. Iwata, R. Kido, S. Imaizumi, S. Tsuzuki, W. Shinoda, K. Ueno, T. Mandai, H. Kokubo, K. Dokko and M. Watanabe, Chem. Mater., 2018, 30, 252–261 CrossRef CAS.
- T. A. Zawodzinski, C. Derouin, S. Radzinski, R. J. Sherman, V. T. Smith, T. E. Springer and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 1041–1047 CrossRef CAS.
- M. Rikukawa and K. Sanui, Prog. Polym. Sci., 2000, 25, 1463–1502 CrossRef CAS.
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS PubMed.
- J. Qiao, T. Hamaya and T. Okada, Chem. Mater., 2005, 17, 2413–2421 CrossRef CAS.
- M. J. Park and N. P. Balsara, Macromolecules, 2010, 43, 292–298 CrossRef CAS.
- H. Zhang and P. K. Shen, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- A. Kraytsberg and Y. Ein-Eli, Energy Fuels, 2014, 28, 7303–7330 CrossRef CAS.
- M. Zakeri, E. Abouzari-Lotf, M. M. Nasef, A. Ahmad, M. Miyake, T. M. Ting and P. Sithambaranathan, Arabian J. Chem., 2018 DOI:10.1016/j.arabjc.2018.05.010.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- S. M. Haile, Acta Mater., 2003, 51, 5981–6000 CrossRef CAS.
- D. J. L. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Chem. Soc. Rev., 2008, 37, 1568–1578 RSC.
- C. Y. Yuh and J. R. Selman, J. Electrochem. Soc., 1991, 138, 3642–3648 CrossRef CAS.
- N. J. Cherepy, R. Krueger, K. J. Fiet, A. F. Jankowski and J. F. Cooper, J. Electrochem. Soc., 2005, 152, A80–A87 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- Y. Sone, P. Ekdunge and D. Simonsson, J. Electrochem. Soc., 1996, 143, 1254–1259 CrossRef CAS.
- F. Bauer, S. Denneler and M. Willert-Porada, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 786–795 CrossRef CAS.
- J. Xie, D. L. Wood, D. M. Wayne, T. A. Zawodzinski, P. Atanassov and R. L. Borup, J. Electrochem. Soc., 2005, 152, A104–A113 CrossRef CAS.
- R. Souzy and B. Ameduri, Prog. Polym. Sci., 2005, 30, 644–687 CrossRef CAS.
- S. J. Osborn, M. K. Hassan, G. M. Divoux, D. W. Rhoades, K. A. Mauritz and R. B. Moore, Macromolecules, 2007, 40, 3886–3890 CrossRef CAS.
- K. Schmidt-Rohr and Q. Chen, Nat. Mater., 2008, 7, 75–83 CrossRef CAS PubMed.
- X. Ling, M. Bonn, S. H. Parekh and K. F. Domke, Angew. Chem., Int. Ed., 2016, 55, 4011–4015 CrossRef CAS PubMed.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- J. S. Wainright, J. T. Wang, D. Weng, R. F. Savinell and M. Litt, J. Electrochem. Soc., 1995, 142, L121–L123 CrossRef CAS.
- R. Bouchet and E. Siebert, Solid State Ionics, 1999, 118, 287–299 CrossRef CAS.
- Y.-L. Ma, J. S. Wainright, M. H. Litt and R. F. Savinell, J. Electrochem. Soc., 2004, 151, A8–A16 CrossRef CAS.
- M. Yamada and I. Honma, Polymer, 2005, 46, 2986–2992 CrossRef CAS.
- S. R. Narayanan, S. P. Yen, L. Liu and S. G. Greenbaum, J. Phys. Chem. B, 2006, 110, 3942–3948 CrossRef CAS PubMed.
- Q. Li, J. O. Jensen, R. F. Savinell and N. J. Bjerrum, Prog. Polym. Sci., 2009, 34, 449–477 CrossRef CAS.
- A. Verma and K. Scott, J. Solid State Electrochem., 2010, 14, 213–219 CrossRef CAS.
- T. Yasuda, S. Nakamura, Y. Honda, K. Kinugawa, S.-Y. Lee and M. Watanabe, ACS Appl. Mater. Interfaces, 2012, 4, 1783–1790 CrossRef CAS PubMed.
- D. Aili, J. Zhang, M. T. D. Jakobsen, H. Zhu, T. Yang, J. Liu, M. Forsyth, C. Pan, J. O. Jensen, L. N. Cleemann, S. P. Jiang and Q. Li, J. Mater. Chem. A, 2016, 4, 4019–4024 RSC.
- K.-S. Lee, J. S. Spendelow, Y.-K. Choe, C. Fujimoto and Y. S. Kim, Nat. Energy, 2016, 1, 16120 CrossRef CAS.
- S. S. Araya, F. Zhou, V. Liso, S. L. Sahlin, J. R. Vang, S. Thomas, X. Gao, C. Jeppesen and S. K. Kær, Int. J. Hydrogen Energy, 2016, 41, 21310–21344 CrossRef CAS.
- S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, Nat. Mater., 2009, 8, 831–836 CrossRef CAS PubMed.
- J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. L. Moudrakovski and G. H. K. Shimizu, Nat. Chem., 2009, 1, 705–710 CrossRef CAS PubMed.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. H. K. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- Y.-S. Wei, X.-P. Hu, Z. Han, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2017, 139, 3505–3512 CrossRef CAS PubMed.
- V. G. Ponomareva, K. A. Kovalenko, A. P. Chupakhin, D. N. Dybtsev, E. S. Shutova and V. P. Fedin, J. Am. Chem. Soc., 2012, 134, 15640–15643 CrossRef CAS PubMed.
- O. V. Okatova, L. N. Andreeva, I. A. Strelina, N. N. Ul'yanova, A. Y. Leykin and A. L. Rusanov, Polym. Sci., Ser. C, 2010, 52, 17–23 CrossRef.
- F. S. Bates, C. V. Berney and R. E. Cohen, Macromolecules, 1983, 16, 1101–1108 CrossRef CAS.
- P. J. Flory and J. Rehner Jr, J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1950, 18, 108–111 CrossRef CAS.
- H. E. Darling, J. Chem. Eng. Data, 1964, 9, 421–426 CrossRef CAS.
- S. W. Yeh, K. H. Wei, Y. S. Sun, U. S. Jeng and K. S. Liang, Macromolecules, 2005, 38, 6559–6565 CrossRef CAS.
- E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1997, 119, 6345–6359 CrossRef CAS.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- A. Noda, M. A. B. H. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- W. Ogihara, H. Kosukegawa and H. Ohno, Chem. Commun., 2006, 34, 3637–3639 RSC.
- S.-Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda and M. Watanabe, J. Am. Chem. Soc., 2010, 132, 9764–9773 CrossRef CAS PubMed.
- T. Dippel, K. D. Kreuer, J. C. Lassègues and D. Rodriguez, Solid State Ionics, 1993, 61, 41–46 CrossRef CAS.
- H. Nishide, J. Deguchi and E. Tsuchida, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 3023–3029 CrossRef CAS.
- T. P. Lodge, Science, 2008, 321, 50–51 CrossRef CAS PubMed.
- A. Noro, M. Hayashi and Y. Matsushita, Soft Matter, 2012, 8, 6416–6429 RSC.
- Y. He, P. G. Boswell, P. Bühlmann and T. P. Lodge, J. Phys. Chem. B, 2007, 111, 4645–4652 CrossRef CAS PubMed.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- S. Y. Kim, S. Kim and M. J. Park, Nat. Commun., 2010, 1, 88 Search PubMed.
- S. Y. Kim, E. Yoon, T. Joo and M. J. Park, Macromolecules, 2011, 44, 5289–5298 CrossRef CAS.
- O. Kim, K. Kim, U. H. Choi and M. J. Park, Nat. Commun., 2018, 9, 5029 CrossRef PubMed.
- M. L. Hoarfrost and R. A. Segalman, Macromolecules, 2011, 44, 5281–5288 CrossRef CAS.
- M. L. Hoarfrost, M. S. Tyagi, R. A. Segalman and J. A. Reimer, Macromolecules, 2012, 45, 3112–3120 CrossRef CAS.
- S. A. Chopade, S. So, M. A. Hillmyer and T. P. Lodge, ACS Appl. Mater. Interfaces, 2016, 8, 6200–6210 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supplementary figures and supplementary tables. See DOI: 10.1039/c9ta01890e |
| ‡ Author contributions: A. N. conceived the original idea and designed the study with T. K. and N. N. T. K. and H. T. carried out the experiments, and A. N., T. K., Y. M., and N. N. analyzed data. A. N., T. K., and Y. M. co-wrote the paper with input from all the authors. |
| This journal is © The Royal Society of Chemistry 2019 |