
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh density graphene–carbon nanosphere films for capacitive energy storage†
Noel
Díez
 *ab,
Mo
Qiao
b,
Juan Luis
Gómez-Urbano
a,
Cristina
Botas
a,
Daniel
Carriazo
*ac and
Maria Magdalena
Titirici
b
*ab,
Mo
Qiao
b,
Juan Luis
Gómez-Urbano
a,
Cristina
Botas
a,
Daniel
Carriazo
*ac and
Maria Magdalena
Titirici
b
aCIC EnergiGUNE, Parque Tecnológico de Álava, Albert Einstein 48, 01510 Miñano, Álava, Spain. E-mail: noel@incar.csic.es; dcarriazo@cicenergigune.com
bSchool of Engineering and Materials Science, Materials Research Institute, Queen Mary University of London, London, UK
cIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 25th February 2019
Abstract
Highly packed films of reduced graphene oxide and sugar-based carbon nanospheres (CNSs) were prepared by a simple hydrothermal treatment. Under hydrothermal conditions, graphene oxide was partially reduced and self-assembled forming a monolith that effectively embedded the CNSs. The spheres were homogeneously distributed within the films, that had an apparent density of up to 1.40 g cm−3. The films thus synthesized were directly assembled into a cell and tested as free-standing electrodes for supercapacitors without using any binder or conductive additive. Electrodes with a mass loading similar to that of commercial devices showed very high values of volumetric capacitance (252 F cm−3) and also an excellent rate capability (64% at 10 A g−1) despite their highly packed microstructure. The homogeneous dispersion of the nanospheres was responsible for the improved ion diffusion when compared to the CNS-free counterpart. The use of a small CNS/graphene wt ratio is essential for achieving such good rate capability without compromising its performance in volumetric terms.
Introduction
Supercapacitors (SC) are, together with batteries, the major energy storage devices. While batteries are characterized by a higher energy density, SC are the preferred energy storage systems when rapid power delivery or fast recharging is required.1 In these systems, the main mechanism of energy storage comes from the electrostatic accumulation of electrolyte ions or fast redox reactions occurring on the electrode surface, which ensures fast charge–discharge and a long cycle life (>100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles). The most common electrode materials for SC are porous carbons due to their high specific surface area, good electronic conductivity, chemical inertness and low cost.2,3 Many carbons with optimized morphologies and porous architectures such as porous nanosheets4,5 or hierarchically porous carbons6,7 have shown excellent electrochemical performance in a gravimetric basis. In these electrode materials, mesopores and macropores act as electrolyte reservoirs close to the microporous surface and alleviate the diffusion resistance, allowing fast charge and discharge rates.8,9 However, the low density of activated carbons (generally lower than 0.7 g cm−3) translates into a moderate or bad performance in volumetric terms. Given that the growing market of portable electronics is conditioned by the development of more compact energy storage systems with high volumetric energy and power densities, it is a matter of special importance to search for new electrode materials that combine a compact structure and a good electrochemical response.10
000 cycles). The most common electrode materials for SC are porous carbons due to their high specific surface area, good electronic conductivity, chemical inertness and low cost.2,3 Many carbons with optimized morphologies and porous architectures such as porous nanosheets4,5 or hierarchically porous carbons6,7 have shown excellent electrochemical performance in a gravimetric basis. In these electrode materials, mesopores and macropores act as electrolyte reservoirs close to the microporous surface and alleviate the diffusion resistance, allowing fast charge and discharge rates.8,9 However, the low density of activated carbons (generally lower than 0.7 g cm−3) translates into a moderate or bad performance in volumetric terms. Given that the growing market of portable electronics is conditioned by the development of more compact energy storage systems with high volumetric energy and power densities, it is a matter of special importance to search for new electrode materials that combine a compact structure and a good electrochemical response.10
Graphene and graphene-derived materials have been repeatedly postulated as good candidates for supercapacitor electrode formulation.11–16 Due to its high theoretical surface-to-mass ratio (ca. 2600 m2 g−1) and outstanding electronic properties, graphene has been implemented as the electroactive material in high power micro-supercapacitors.17–19 Another important characteristic of graphene and graphene-based materials is that they can be processed into flexible self-standing films that can be directly used as electrodes without requiring any support, conductive additive and binder.20–23 Unfortunately, graphene and reduced graphene oxide sheets tend to pile up due to van der Waals interactions – especially during the drying process – ,24 which reduces their surface area accessible to electrolyte ions and causes a severe diffusion restriction.25,26 In a previous work we observed that highly packed graphene films with a high density of ca. 1.5 g cm−3 can retain a high capacitance value of 268 F cm−3 at a high current density of 10 A g−1 as long as the electrodes have an areal mass loading below 5 mg cm−2.26 In contrast, if the electrode mass loading is increased to values close to those of commercial devices (higher than 10 mg cm−2),27 the supercapacitors experienced a drastic capacitance decay with the increase of the current density. The hindered ion diffusion observed in the graphene-based films can be alleviated by the introduction of wrinkles/curvatures in the graphene layers.28,29 Another simple strategy to prevent restacking of graphene sheets and reduce the diffusion resistance is the intercalation of nanosized spacers. Thus, the use of nanofillers such as carbon black,24,30 carbon nanotubes,26,31,32 carbon spheres,33 carbide-derived carbon nanoparticles34 or carbon onions35 has been reported to improve the electrochemical properties of graphene films. In most of these cases, the mass fraction of the nanospacer required is so high (from 10 to 500 wt%) that it is not possible to achieve a self-standing film and the use of a binder additive is unavoidable.24,31–33 Importantly, the low packing density arising from the use of such high amounts of nanospacers results in a poor performance in volumetric terms, and sometimes also in gravimetric terms, given that the fillers have a lower (or negligible) electrochemical response. Moreover, the electrode mass loading used in these research studies is often very low,24,30–32 which brings along an overestimation of the material's performance.27
Herein we present a simple method for the preparation of a highly packed film composed of hydrothermally reduced graphene oxide (HrGO) and intercalated carbon nanospheres (CNSs) with an apparent density of 1.40 g cm−3. The nanospheres, that were prepared from sustainable precursors, were mixed with graphene oxide and homogeneously dispersed within the self-assembled graphene network during hydrothermal treatment. When tested as self-standing electrodes for supercapacitors, the composite films exhibited high capacitance and an outstanding rate capability, overcoming the high diffusion resistance of its CNS-free counterpart.
Experimental section
Synthesis of the CNSs
CNSs were synthesized by hydrothermal carbonization of xylose or glucose. In each case, 2 g of the saccharide was dissolved in 18 ml of distilled water and placed in a Teflon-lined autoclave. The reactors were heated at 5 °C min−1 to a final temperature of 180 °C. The holding time at the maximum temperature ranged from 1 to 24 h. Reaction products were repeatedly washed with distilled water and centrifuged. The purified product was finally washed with isopropanol and dried at 80 °C overnight. The spherical hydrochar particles were finally carbonized at 700 °C giving rise to the CNSs.Synthesis of the graphene-CNS films
HrGO-CNS monoliths were synthesized by hydrothermal reduction and self-assembly of a GO aqueous dispersion in the presence of a certain amount of carbonized nanospheres. 85 ml of the graphene oxide aqueous dispersion (2 mg ml−1) was mixed with appropriate amounts of xylose-derived CNSs to obtain composites with 10, 25 and 50 wt% of CNSs. During the hydrothermal treatment, graphene oxide suffers from a ca. 25 wt% loss, mainly due to CO2 evolution, as a consequence of the disproportionation and crosslinking reactions taking place.36,37 The amounts of CNSs added to the graphene oxide solution were calculated based on this assumption. The mixture of nanospheres and graphene oxide was first sonicated for 30 min and then transferred to a Teflon-lined autoclave. The reactor was heated up to 210 °C for 24 h. The monoliths obtained after the hydrothermal treatment were pressed at 8 tons using a hydraulic press without any previous drying step in order to preserve the solvated structure with certain plasticity that allowed its conformation into a highly packed film. The films thus obtained were denoted as HrGO@xCNS, where x is the wt% of CNSs in the composite. Composite films were also synthesized using the same procedure but using glucose-derived CNSs. The results presented refer to the xylose-based CNSs containing composites, unless otherwise specified. For comparison purposes, graphene films were prepared following the same procedure but without adding the CNSs. The rGO-xCNS electrodes were prepared by carbonization of the HrGO-xCNS films at 700 °C for 1 h under a nitrogen flow and using a heating rate of 5 °C min−1.Physico-chemical characterization
The parent graphene material (commercial graphene oxide from Graphenea) was characterized by AFM (NTegra, from NT-MDT Co.) after its dilution in distilled water (1:300) and deposition onto a mica substrate. X-ray diffraction (XRD) patterns were recorded for powdered samples in a Bruker D8 X-ray diffractometer; data were collected at 40 kV and 30 mA using CuKα radiation over 2θ within the range from 5 to 90° at a step of 0.02° and a residence time of 5 s. Raman spectra were recorded with a Renishaw spectrometer (Nanonics MultiView 2000) operating with an excitation wave-length of 532 nm. Spectra were acquired with 10 s of exposure time of the laser beam to the sample. X-ray photoelectron spectroscopy (XPS) measurements were carried out in a UHV spectrometer chamber with the base pressure below 10−10 mbar. The chamber features a hemispherical analyser PHOIBOS 150 with a 2D-DLD detector (SPECS) and a monochromatic X-ray source FOCUS 500 (SPECS) with two anodes: Al Kα (hν = 1486.74 eV) and Ag Lα (hν = 2984.3 eV). In order to calculate the apparent density of the electrodes (i.e. the density of the material including the porosity), their thickness was measured with the help of a micrometer. Nitrogen adsorption–desorption isotherms were recorded using an ASAP2020 instrument from Micromeritics. Samples were outgassed at 80 °C for 48 h prior to the analysis. Specific surface area values were determined using the BET equation within the 0.05–0.2 relative pressure range. Pore size distributions were determined by applying the Quenched-Solid Density Functional Theory (QSDFT) method to the nitrogen adsorption data assuming a slit pore model.
Electrochemical characterization
Electrodes were obtained by punching the film into discs with a diameter of 11 mm. These electrodes were dried at 80 °C overnight and then weighted. The mass loading of the electrodes (∼8 mg cm−2) was controlled by slicing the monoliths obtained from hydrothermal treatment to appropriate thicknesses. In each case, two electrodes of similar masses were placed in a symmetric Swagelok® cell using two stainless steel plungers as current collectors. Approximately 2 ml of 6 M KOH aqueous solution was added as the electrolyte and a glass fiber membrane was used as the separator. Good contact between the electrodes and current collectors was assured by applying pressure with the help of an F-clamp. Galvanostatic charge–discharge cycling, cyclic voltammetry and impedance spectroscopy measurements were conducted using a multichannel VSP-potentiostat/galvanostat (Biologic, France). The specific gravimetric and volumetric capacitances (Cg and Cv, expressed in F g−1 and F cm−3, respectively) were obtained from the discharge curve of the galvanostatic plots, recorded at different current densities, according to the following equations: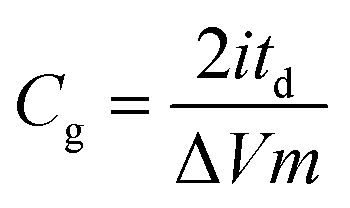
where i is the electrical current (A), V is the usable voltage range once the ohmic drop is subtracted (V), td is the discharge time (s), m is the mass of one electrode (g), ε is the thickness of one electrode (cm) and A corresponds to the area of the electrodes (cm2).
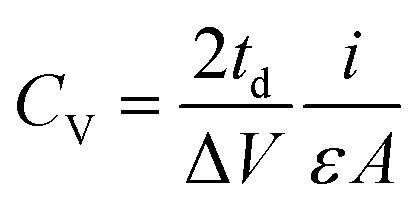
Results and discussion
Synthesis of the highly packed HrGO@CNS films
For the synthesis of the composites, as depicted in Scheme 1, carbon nanospheres were added to an aqueous GO dispersion which was then subjected to hydrothermal treatment. CNSs were previously synthesized by hydrothermal treatment of xylose, a low cost and naturally abundant saccharide. Optimization of the synthesis conditions are detailed in the ESI (Fig. S1 and ESI note 1).† Commercially available graphene oxide was used for the preparation of the graphene–carbon nanosphere composites. AFM observations revealed that the graphene oxide was composed exclusively of monolayers with a broad distribution of lateral sizes, the largest flakes having a length of around 2 microns (Fig. S2†). Hydrothermal treatment of the mixture of sugar-derived CNSs and the graphene oxide yielded a hybrid hydrogel consisting of a continuous network of interconnected hydrothermally reduced graphene oxide containing the carbon spheres.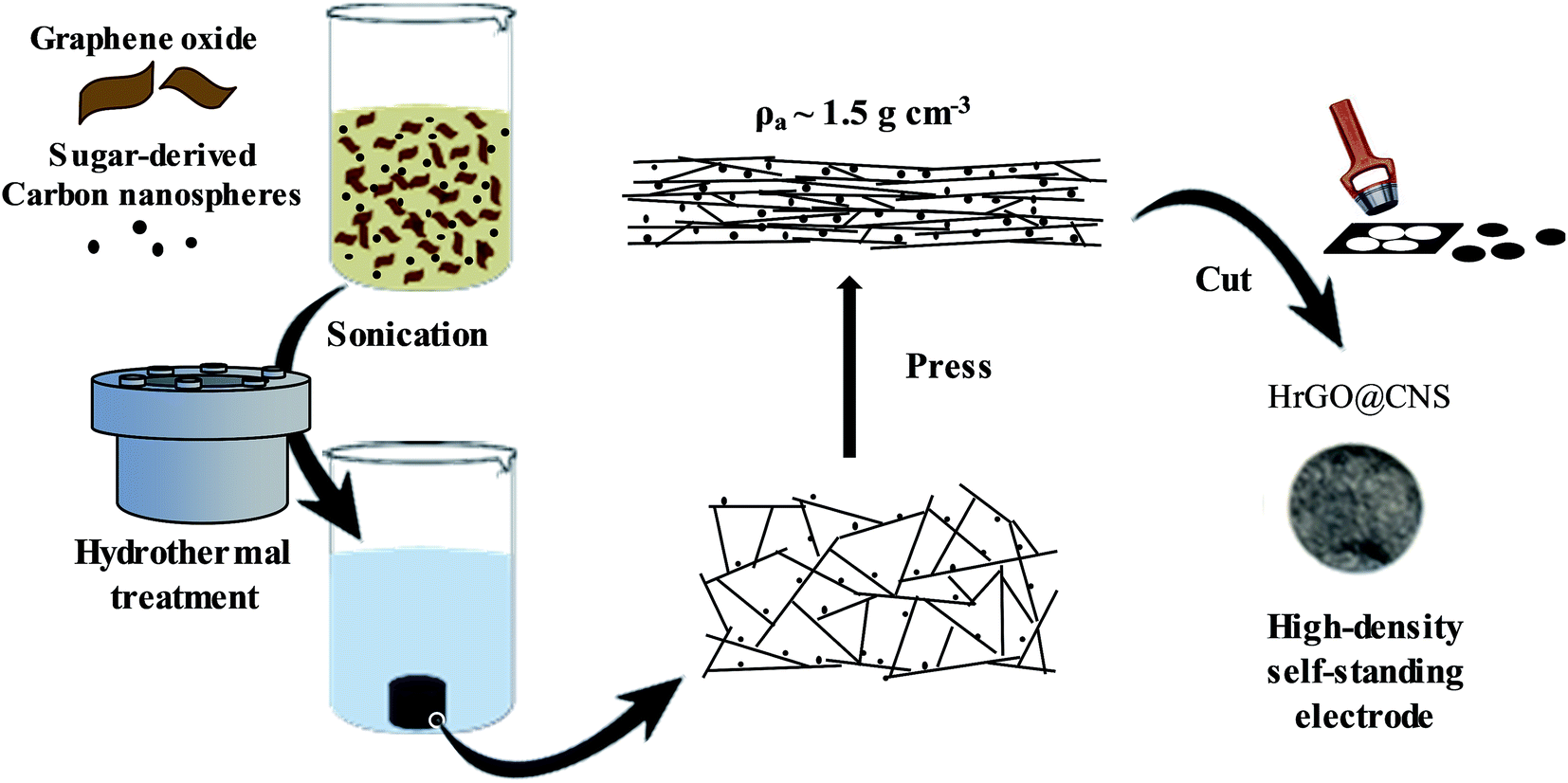 | ||
| Scheme 1 Scheme of the synthesis of high-density binder-free graphene-CNS electrodes. | ||
Digital pictures of the graphene-based monoliths with different contents of carbon nanospheres as well as the surrounding reaction media are shown in Fig. 1A and B. The liquid medium in which the monolith was immersed was clear, which indicates that all of the carbon nanospheres were successfully inserted into the structure of the self-assembled carbon monolith. The presence of CNSs modified the buoyancy of the hydrogels: the monoliths containing 10 wt% of CNSs sank in the water similarly to those obtained in the absence of CNSs, while the monolith with 50 wt% of CNSs floated in the reaction media. According to this, it can be envisaged that the addition of the CNSs will have a negative impact on the density of the final self-standing electrodes. Indeed, the density of the highly packed films decreased with the increase of the CNS fraction (vide infra). The presence of CNSs also modified the appearance of the solvated monoliths. Fig. 1C shows a digital picture of a graphene-based monolith prepared in the absence of CNSs, which has a cylindrical shape and smooth surface. The addition of carbon spheres distorts the morphology of the monoliths, and this deformation is more evident when using high amounts of CNSs (Fig. 1B).
 | ||
| Fig. 1 Digital pictures of the as-received graphene hydrogels containing (A) 10 wt% and (B) 50 wt% of CNSs. (C) Digital picture of a CNS-free graphene hydrogel. Images of high-density electrodes prepared with (D) 10 wt% of carbon black and (E) 10 wt% of CNSs. | ||
The solvated state of the as-prepared monoliths endows them with certain plasticity, so that they can be easily processed into dense films by mechanical pressing.38Fig. 2A shows the image of a cross section of a HrGO electrode. The sheets of hydrothermally reduced graphene oxide self-assembled during the hydrothermal reduction form a continuous and interconnected network. Due to the mechanical pressing, these HrGO layers are preferentially oriented in the plane that is perpendicular to the pressing direction. A similar structure was obtained when the graphene oxide was hydrothermally reduced in the presence of the CNSs. As can be seen in Fig. 2B, the CNSs had a bimodal size distribution. The majority of the nanospheres had a small size of 60 to 120 nm, while a small portion of them reached diameters of ca. 500 nm. Fig. 2C and D show the images corresponding to the parallel sections of the HrGO-CNS films containing 10 wt% and 50 wt% of CNSs, respectively. As can be seen in these images, the carbonized nanospheres were homogeneously dispersed within the films. It should be remarked that, even in the film with the highest content of carbon spheres (50 wt%), these were well distributed along the material without aggregating. In contrast, when similar films were prepared using 10 wt% of carbon black as the nanofiller, macroscopic deposits of carbon black agglomerates, segregated from the graphene network, could be easily identified (Fig. 1D and E, the white arrows indicate the deposits of carbon black). The carbonized nanospheres were much better distributed in the hydrogel than the carbon black particles, presumably as a consequence of a higher affinity for the HrGO.
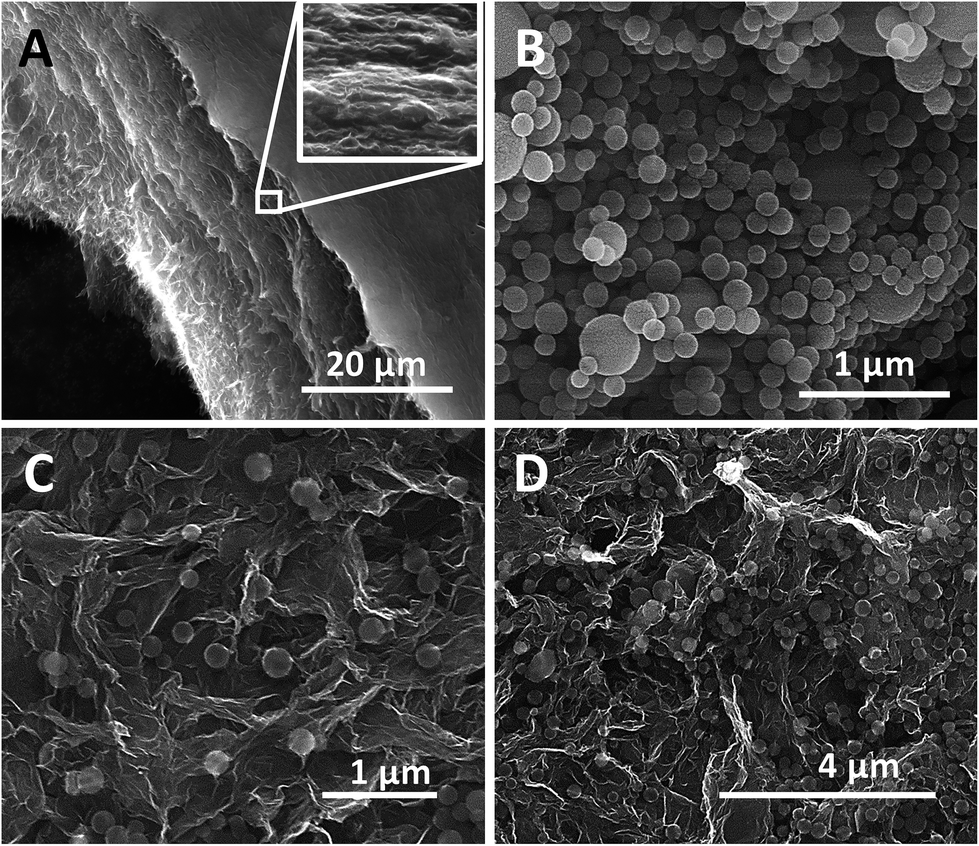 | ||
| Fig. 2 SEM images of (A) a cross section of a HrGO electrode, (B) the xylose-derived CNSs, and parallel sections of HrGO-CNS electrodes containing (C) 10 wt% and (D) 50 wt% of CNSs. | ||
The apparent density of the films was calculated by measuring the weight and volume of disc-like samples. Their apparent density decreased with the insertion of CNSs, from 1.52 g cm−3 in the HrGO film to 1.40, 1.33 and 1.22 g cm−3 in the films containing 10, 25 and 50 wt% of CNSs. This agrees with the aforementioned changes of buoyancy of the solvated hydrogels. We also prepared films out of CNSs and without graphene (90 wt% xylose-based CNSs and 10 wt% PTFE binder), that had a low apparent density of 0.59 g cm−3.
The porous structure of the high-density films was analyzed by N2 physisorption experiments at −196 °C. The N2 adsorption isotherms of HrGO, HrGO-10CNS and HrGO-50CNS are shown in Fig. 3A. At very low relative pressures, the isotherms display a sharp increase in the amount of nitrogen adsorbed. This reveals the presence of microporosity (pores with a size < 2 nm) that can be due to defects or holes within the graphene sheets, as well as small slit-shaped cavities between different graphene layers. The curves corresponding to the CNS-containing samples have a more pronounced slope within the 0.1–0.8 relative pressure range, which is indicative of higher mesopore content in these samples. The pore size distributions, plotted in the inset of Fig. 3A, corroborate that HrGO-10CNS and HrGO-50CNS contain a larger number of mesopores compared to HrGO, and a mesopore size distribution extended to larger pore sizes. At high relative pressures (p/p0 > 0.9), these curves also show a sharp nitrogen uptake due to the existence of large mesopores and/or macropores. Since this feature is not present in the isotherm corresponding to HrGO, the presence of such textural porosity must be due to the voids and channels created between the CNSs and graphene layers. This textural porosity, together with the low density of the CNSs, counts against the packing density of the material. However, as will be explained later, its presence is crucial for their good electrochemical behavior when used as electrodes in supercapacitors. The BET surface area calculated from the isotherm data is 160, 190 and 160 m2 g−1 for HrGO, HrGO-10CNS and HrGO-50CNS, respectively, but these values should be simply taken as an indicator because the films are partially solvated even during the electrochemical testing, and part of the microporosity of these materials might have collapsed due to the evolution of trapped water during the sample degassing.
 | ||
| Fig. 3 (A) N2 adsorption isotherms and QSDFT pore size distributions (inset) of the high-density films. XRD patterns (B) and Raman spectra (C) obtained for the composite films. | ||
The microstructure of the composite films was also examined by X-ray diffraction and Raman spectroscopy. The XRD patterns of HrGO-10CNS and HrGO-50CNS are shown in Fig. 3B. Both spectra exhibit a broad and low intensity (002) diffraction peak at 2θ values of ca. 24° and 43°, which indicates that the restacking degree of the graphene sheets achieved during the hydrothermal reduction and subsequent pressing/drying was very low. The Raman spectra of these materials, shown in Fig. 3C, show two characteristic G and D bands at 1593 cm−1 and 1352 cm−1, originating from the first-order scattering of E2g phonons by the graphitic planes and the breathing mode of k-point phonons of the A1g symmetry. These bands relate to the presence of ordered graphitic domains and structural defects in the graphene nanosheets, respectively. The areal ratio of these peaks (AD/AG) was 1.5 for HrGO-10CNS and 1.3 for HrGO-50CNS, respectively. This is consistent with the higher content of nanospheres carbonized at a high temperature, that present small graphitic domains, in HrGO-50CNS.
Electrochemical performance of the high-density HrGO-CNS electrodes
According to previous studies,26 the capacitance retention of high-density HrGO electrodes (without intercalated nanofillers) is greatly influenced by the mass/thickness of the electrodes. For this reason, only electrodes with mass loadings close to those used in commercial devices (higher than 7.5 mg cm−2) were assembled in symmetric two-electrode cells and electrochemically tested in 6 M KOH using a cell voltage of 1.2 V.The values of gravimetric and volumetric capacitance, calculated from the discharge branches recorded at different current densities, are plotted in Fig. 4A and B, respectively. At low current densities, the HrGO electrode exhibited a very high capacitance of 343 F cm−3, that corresponds to a gravimetric capacitance of 225 F g−1. Given the moderate specific surface area calculated for this material, these high capacitance values are attributable to a substantial contribution of pseudocapacitance due to fast redox reactions in the highly functionalized surface of HrGO (oxygen content of 24.7 wt% according to XPS measurements, see Fig. S3†). These electrodes experience a sharp capacitance decay with the increase of the current density (37 F cm−3 at 15.2 A cm−3). The poor conductivity derived from the relatively high oxygen content of the hydrothermally reduced graphene oxide sheets, together with diffusion restrictions within the highly packed carbon matrix, seems to be the reason for this behavior.
 | ||
| Fig. 4 Evolution of the gravimetric (A) and volumetric (B) capacitance calculated for the high density films vs. current density. Galvanostatic charge/discharge profiles recorded at a current density of (C) 1 A g−1 and (D) 10 A g−1. | ||
Among the HrGO-CNS films, the film containing 10 wt% of CNSs was selected for its electrochemical testing taking into consideration the results previously presented. This low concentration of CNSs seems enough to provide efficient diffusion channels while minimizing the content of carbon spheres, whose electrochemical response is negligible39 (vide infra). Moreover, this film maintains a density of 1.40 g cm−3, which is among the highest reported in the literature for carbon electrode materials.26 As shown before, higher CNS contents lead to lower values of apparent density that would negatively affect the electrochemical behavior of the films in volumetric terms. As can be seen in Fig. 4A and B, the electrochemical response of the composite electrodes differs from that of the HrGO film. The film containing 10 wt% of CNSs exhibited a capacitance of 252 F cm−3 at low current densities, which is lower than that of HrGO but still among the highest reported for carbon materials. The lower capacitance values of HrGO-10CNS at very low current densities are due to the presence of the CNSs that have a poor electrochemical activity due to their low specific area and oxygen content. Galvanostatic charge–discharge experiments using electrodes containing 90 wt% of CNSs and 10 wt% of PTFE binder confirmed the very low capacitance expected for this material (Fig. 4A and B). The HrGO-10CNS electrodes experienced a high capacitance retention of 64% (162 F cm−3) at a high current density of 14 A cm−3. It should be emphasized that this excellent rate capability was achieved despite the use of electrodes with a mass loading typical of commercial devices and their high packing density. Under these testing conditions, the volumetric capacitance of the HrGO-10CNS electrode is ∼5 times higher than that of its CNS-free counterpart.
Fig. 4C and D show the galvanostatic charge–discharge plots at 1 and 10 A g−1, respectively. At 1 A g−1 all of the profiles overlap and have a triangular shape. At the highest current density, the HrGO electrode experiences an abrupt voltage drop at the beginning of the discharge. This voltage drop is notably smaller in the case of the HrGO-CNS film. The ESR values calculated for HrGO and HrGO-10CNS are 2.02 and 0.33 ohm, respectively. As can be deduced from the same plots, the composite electrodes possess a smaller equivalent distributed resistance (EDR). The charge propagation restrictions in the HrGO electrodes arisen from the poor conductivity and impeded ion diffusion are therefore alleviated by the insertion of the nanospacers.26 The improved charge transfer properties and rate capability of the HrGO-CNS electrodes were corroborated by cyclic voltammetry experiments. The voltammograms of the HrGO-10CNS electrodes have a more rectangular shape, especially at a high scan rate of 100 mV s−1 (Fig. 5). Electrochemical impedance spectroscopy measurements were also performed on the symmetric cells (see Fig. S4†). The smaller Warburg regions in the Nyquist plots of the HrGO-CNS electrodes ratify the lower resistance to ion diffusion in these materials.
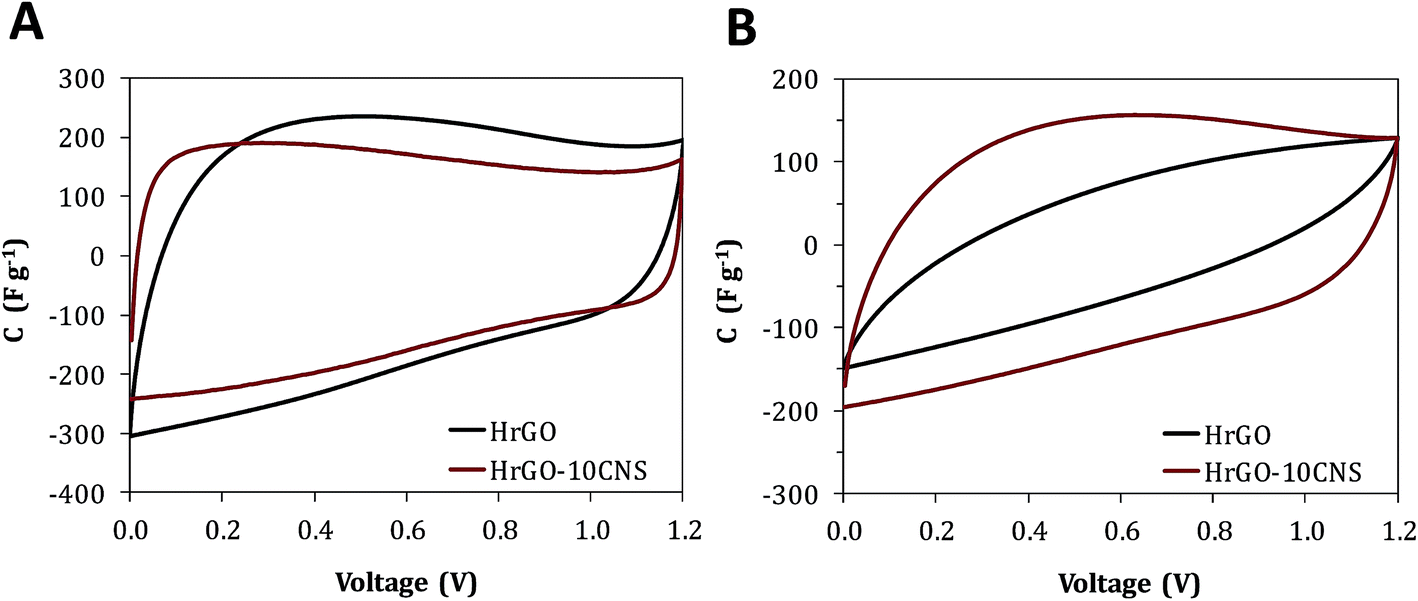 | ||
| Fig. 5 Cyclic voltammograms recorded at a scan rate of (A) 10 mV s−1 and (B) 100 mV s−1 for the symmetric cells built with HrGO and HrGO-10CNS films. | ||
All these results demonstrate the improved electrochemical performance of the highly packed HrGO films when a small amount of CNSs is homogeneously inserted within the graphene network in situ during the hydrothermal reduction of graphene oxide. A low fraction of CNSs of 10 wt% seems enough to boost the ion diffusion during fast charge/discharge while maintaining high values of volumetric capacitance.
The use of glucose as the precursor for the synthesis of CNSs was also explored. Thus, electrodes with 10 wt% of glucose-derived CNSs were prepared following the same synthetic procedure. As can be seen in Fig. S5†, the capacitance values at a low current density are similar for both films, but the retention of the capacitance at a high current rate is slightly superior in the case of the film containing the xylose-based carbonized spheres. This can be due to the fact that, contrary to the glucose-derived spheres, xylose-based CNSs have a bimodal particle size distribution (see Fig. S1G†) that can be beneficial for fast ion transport. The apparent density measured for the films containing 10 wt% of glucose derived-CNSs is similar to that of HrGO-10CNS: 1.38 and 1.40 g cm−3, respectively. Another aspect to take into account is that xylose degrades under hydrothermal conditions to more aromatic condensed structures than glucose,40 which might enhance the electron transfer properties of the nanofillers.
The high-density electrodes were subjected to thermal treatment in an inert atmosphere in order to evaluate the influence of the exposure to high temperatures on the characteristics and electrochemical response of the electrodes. After the heat treatment at 700 °C, the capacitive behavior of the rGO film strongly worsens compared to that of HrGO (Fig. S6A†). This can be ascribed to the loss of pseudocapacitance and wettability provided by the oxygen functionalities of HrGO, and also to the loss of trapped water molecules intercalated between graphene layers.16 The loss of this solvated state facilitates a high degree of restacking upon thermal treatment at high temperatures. SEM observations on the carbonized electrodes revealed that the CNSs migrated during the high temperature treatment and formed clusters of agglomerated spheres, leaving graphene domains devoid of carbon spheres (Fig. S6B†). Insertion of a small amount of CNSs into the highly packed solvated HrGO network is therefore a good strategy to improve the charge transfer properties of the HrGO films and to reduce the ion diffusion resistance maintaining its self-standing nature and allowing the preparation of binder-free electrodes.
Ragone plots comparing the gravimetric and volumetric energy and power densities of the HrGO and HrGO-10CNS highly packed films are shown in Fig. 6. At slow discharge, HrGO achieved an energy density value of 14.2 W h dm−3, but this value decays rapidly when high power is demanded. Thus, at a power of 3.07 kW dm−3, the energy density provided by this material was 1.02 W h dm−3. A much higher energy was obtained at a high power demand for the HrGO-10CNS film, which supplied 6.67 W h dm−3 at 3.81 kW dm−3, around 7 times higher than that of the CNS-free film. These values are higher than those of other aqueous supercapacitors based on commercial porous carbon,16 MOF-derived porous carbons,41 hierarchical porous carbons42 and N-doped carbon nanofibers,43 being slightly surpassed by the porous monolithic graphene reported by Tao et al.16 and by the high density CNT-reduced graphene oxide films.26
 | ||
| Fig. 6 Ragone plot showing the gravimetric (filled dots) and volumetric (empty dots) energy density versus power density of the HrGO (black) and HrGO-10CNS (red) films, calculated taking into account the mass of the two electrodes. Volumetric energy and power densities of other carbon materials reported in the literature are plotted as empty circles. | ||
Finally, the electrochemical stability of the HrGO-10CNS electrodes upon long cycling at 5 A g−1 was confirmed, and the cell retained 92% of its initial capacitance after 5000 cycles of charge–discharge (Fig. 7). The long term stability of this composite film was better than that of the CNS-free HrGO film, which, when cycled under the same conditions, retained 84.4% of its initial capacitance after 5000 charge–discharge cycles.
 | ||
| Fig. 7 Long cycling stability of the symmetric supercapacitors built with HrGO-10CNS and HrGO electrodes. Charge–discharge cycles were run at a current density of 5 A g−1. | ||
Conclusions
Highly packed films based on reduced graphene oxide and sugar-derived carbon nanospheres were prepared by a simple hydrothermal treatment. The CNSs were homogeneously distributed within the reduced graphene oxide network, and the composite films achieved high apparent densities up to 1.4 g cm−3. Self-standing electrodes prepared out of these films were successfully tested as electrodes for supercapacitors. Despite their dense microstructure and their high mass loading, they showed very high capacitance values (252 F cm−3) and excellent rate capabilities (64% at 14 A cm−3). The addition of a small amount of CNSs (10 wt%) is the key to achieve such an excellent response at high current rates as well as to maintain a good electrochemical performance in volumetric terms. The electrodes also showed an excellent electrochemical stability upon 5000 charge–discharge cycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Union (Graphene Flagship, Grant number 785219) and Spanish Ministry of Economy and Competitiveness (MINECO/FEDER) (MAT2015-64617-C2-2-R). Noel Díez acknowledges the Spanish Ministry of Education and Sports (MECD) for a Jose Castillejo mobility grant. Authors also thank the GRAPHENEA Company for supplying the graphene oxide used in this study.References
- J. M. Miller, Ultracapacitor applications, The Institution of Engineering and Technology, 2011 Search PubMed.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- Z. Fan, Y. Liu, J. Yan, G. Ning, Q. Wang, T. Wei, L. Zhi and F. Wei, Adv. Energy Mater., 2012, 2, 419–424 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, ACS Nano, 2014, 8, 5069–5078 CrossRef CAS PubMed.
- S. Zhu, J. Li, C. He, N. Zhao, E. Liu, C. Shi and M. Zhang, J. Mater. Chem. A, 2015, 3, 22266–22273 RSC.
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- T. Liu, F. Zhang, Y. Song and Y. Li, J. Mater. Chem. A, 2017, 5, 17705–17733 RSC.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- Q. Wang, J. Yan and Z. Fan, Energy Environ. Sci., 2016, 9, 729–762 RSC.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- A. Śliwak, B. Grzyb, N. Díez and G. Gryglewicz, Appl. Surf. Sci., 2017, 399, 265–271 CrossRef.
- Y. Xu, G. Shi and X. Duan, Acc. Chem. Res., 2015, 48, 1666–1675 CrossRef CAS PubMed.
- Y. Sun, Q. Wu and G. Shi, Phys. Chem. Chem. Phys., 2011, 13, 17249–17254 RSC.
- Y. Tao, X. Xie, W. Lv, D.-M. Tang, D. Kong, Z. Huang, H. Nishihara, T. Ishii, B. Li and D. Golberg, Sci. Rep., 2013, 3, 2975 CrossRef PubMed.
- Z. S. Wu, K. Parvez, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 2487 CrossRef PubMed.
- J. R. Miller, R. Outlaw and B. Holloway, Science, 2010, 329, 1637–1639 CrossRef CAS PubMed.
- D. Aradilla, M. Delaunay, S. Sadki, J.-M. Gérard and G. Bidan, J. Mater. Chem. A, 2015, 3, 19254–19262 RSC.
- A. Yu, I. Roes, A. Davies and Z. Chen, Appl. Phys. Lett., 2010, 96, 253105 CrossRef.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457 CrossRef CAS PubMed.
- H. F. Wang, R. Chen, J. Feng, M. Qiao, S. Doszczeczko, Q. Zhang, A. B. Jorge and M. M. Titirici, ChemElectroChem, 2018, 5, 1786–1804 CrossRef CAS.
- J. Yan, T. Wei, B. Shao, F. Ma, Z. Fan, M. Zhang, C. Zheng, Y. Shang, W. Qian and F. Wei, Carbon, 2010, 48, 1731–1737 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- N. Díez, C. Botas, R. Mysyk, E. Goikolea, T. Rojo and D. Carriazo, J. Mater. Chem. A, 2018, 6, 3667–3673 RSC.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 RSC.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- J. Yan, J. Liu, Z. Fan, T. Wei and L. Zhang, Carbon, 2012, 50, 2179–2188 CrossRef CAS.
- Y. Wang, J. Chen, J. Cao, Y. Liu, Y. Zhou, J.-H. Ouyang and D. Jia, J. Power Sources, 2014, 271, 269–277 CrossRef CAS.
- S.-Y. Yang, K.-H. Chang, H.-W. Tien, Y.-F. Lee, S.-M. Li, Y.-S. Wang, J.-Y. Wang, C.-C. M. Ma and C.-C. Hu, J. Mater. Chem., 2011, 21, 2374–2380 RSC.
- D. Yu and L. Dai, J. Phys. Chem. Lett., 2009, 1, 467–470 CrossRef.
- C. X. Guo and C. M. Li, Energy Environ. Sci., 2011, 4, 4504–4507 RSC.
- M. Alhabeb, M. Beidaghi, K. L. Van Aken, B. Dyatkin and Y. Gogotsi, Carbon, 2017, 118, 642–649 CrossRef CAS.
- Y. Sun, Q. Wu, Y. Xu, H. Bai, C. Li and G. Shi, J. Mater. Chem., 2011, 21, 7154–7160 RSC.
- K. Hu, X. Xie, T. Szkopek and M. Cerruti, Chem. Mater., 2016, 28, 1756–1768 CrossRef CAS.
- N. Díez, A. Śliwak, S. Gryglewicz, B. Grzyb and G. Gryglewicz, RSC Adv., 2015, 5, 81831–81837 RSC.
- M. Sevilla, G. A. Ferrero, T. T. Vu and A. B. Fuertes, ChemNanoMat, 2016, 2, 33–36 CrossRef CAS.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Energy Mater., 2011, 1, 356–361 CrossRef CAS.
- M.-M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- J. Hu, H. Wang, Q. Gao and H. Guo, Carbon, 2010, 48, 3599–3606 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., 2008, 120, 379–382 CrossRef.
- L.-F. Chen, X.-D. Zhang, H.-W. Liang, M. Kong, Q.-F. Guan, P. Chen, Z.-Y. Wu and S.-H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta12050a |
| This journal is © The Royal Society of Chemistry 2019 |