
Selective separation and preconcentration of Th(IV) using organo-functionalized, hierarchically porous silica monoliths†
Yimu
Hu
abc,
Simon
Giret
abc,
Rafael
Meinusch
d,
Jongho
Han
ef,
Frédéric-Georges
Fontaine
 *acg,
Freddy
Kleitz
*h and
Dominic
Larivière
*ac
*acg,
Freddy
Kleitz
*h and
Dominic
Larivière
*ac
aDepartment of Chemistry, Université Laval, Québec, G1V 0A6, QC, Canada. E-mail: frederic.fontaine@chm.ulaval.ca; dominic.larriviere@chm.ulaval.ca
bCentre de Recherche sur les Matériaux Avancés (CERMA), Université Laval, Québec, G1V 0A6, QC, Canada
cCentre en Catalyse et Chimie Verte (C3V), Université Laval, Québec, G1V 0A6, QC, Canada
dInstitute of Physical Chemistry, Justus-Liebig-University Giessen, 35392 Giessen, Germany
eDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Republic of Korea
fCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science, Daejeon 305-701, Republic of Korea
gCanada Research Chair in Green Catalysis and Metal-Free Processes, Canada
hDepartment of Inorganic Chemistry – Functional Materials, Faculty of Chemistry, University of Vienna, 1090 Vienna, Austria. E-mail: freddy.kleitz@univie.ac.at
First published on 26th November 2018
Abstract
The potential application of thorium (Th) as nuclear fuel, as well as the environmental and public health concerns associated with it, promotes the development of economic and sustainable materials for the separation and removal of Th(IV) from minerals and environmental samples. In this work, centimeter-size, porous silica monoliths exhibiting hierarchical macroporosity–mesoporosity and a robust silica skeleton were prepared using a sol–gel process combined with post-synthetic hydrothermal treatment in ammonium hydroxide. Upon functionalization with diglycolamide (DGA), the resulting monolithic hybrid material was used as a column-type fixed bed sorbent for continuous flow extraction. An enhanced Th(IV) uptake from aqueous solution was achieved with a high enrichment factor and selectivity in the presence of competitive ions such as rare earth elements (REEs) and uranium (U). Systematic mechanistic studies show that the hierarchical pore system is crucial for enhanced adsorption kinetics and capacity. Two mineral leachates were used to assess the performances of the hybrid material, and despite the complex ion matrix and high ionic composition, the sorbent shows highly efficient recovery of Th(IV). The material was able to undergo 10 extraction–stripping–regeneration cycles, which bodes well for potential industrial applications.
Introduction
As globalization and rapid population growth drastically increase global energy needs, the demand for carbon-free and environmentally sustainable energy sources is high. As such, the construction and expansion of infrastructures designed for low-emission energy technologies (e.g., nuclear power plants and wind turbines) have boosted the demand for valuable technological materials, such as actinides (Ac) and rare earth elements (REEs).1 However, several ecological, social and safety issues have to be addressed before these “clean energy” technologies reach their full potential. For example, the nuclear fuel reprocessing and waste management are among the biggest limitations of nuclear energy, especially from the viewpoint of environmental protection and public health. Because of its potential usage in nuclear energy and inherent safety concerns, much attention has recently been drawn toward thorium (typical as 232Th(IV)).2 Also related is the increasing demand for rare earth elements that has risen drastically due to their important applications in green technologies such as hybrid vehicles, permanent magnets, and high efficiency lightings. The large amount of thorium commonly found in rare earth minerals (e.g., monazite and bastnasite),3 and the concomitant radiologic pollution is a severe concern during rare earth production. Due to their similar physical and chemical properties, the partitioning of thorium and REEs remains one of the most challenging hydrometallurgical separations.4 In this regard, there is a need for the development of highly efficient methods for the separation and recovery of thorium from sources such as mining and industrial wastes.Various methods including extraction, adsorption, ion-exchange, and chemical precipitation have been developed to recover actinides from aqueous solutions, among which the multiple-step liquid–liquid extraction (LLE) is the most widely applied technique on the industrial level, owing to its good selectivity and versatility in regard to solvents and organic ligands.4 However, the large amount of organic solvent consumed and the radioactive waste generated during repetitive extraction steps are the main drawbacks in the process. In comparison, alternative methods based on solid-phase extraction (SPE), which features high enrichment factor, fast kinetics, and significant decrease in solvent consumption, have recently emerged.5–7 A remarkable number of materials and nanomaterials have been developed for actinide and nuclide removal from aqueous solution. Traditional porous materials used for this purpose include clay, bare silica gel, active carbon, and zeolites, but they all suffer from low adsorption capacities and slow kinetics.8–11 Advanced functional porous materials, such as amorphous porous organic polymers (POP),12,13 porous aromatic frameworks (PAF),14–16 ion-exchangers,17,18 metal–organic frameworks (MOF),19,20 and covalent organic frameworks (COF)21,22 have been designed to adapt to various conditions (e.g., pH, ionic strength, and presence of interfering metal ions) for nuclear waste removal. Other promising candidates for stationary phase materials in SPE systems include mesoporous silica and carbon, with a pore size between 2–50 nm. These materials have found wide applications for the capture of important radionuclides (e.g., U and Th) and REEs.6 These materials exhibit a remarkably high specific surface area allowing for high contact efficiency and enhanced mass transfer kinetics between the two hydrophilic phases (sorbent and analyte), as well as easily functionalizable surface properties through post-synthetic grafting23–32 or ion-imprinting technique.33–35
In general, mesoporous materials are suitable as stationary phases in chromatographic applications, as the nano-size pores can significantly enhance the extraction efficiency and reduce the analysis time. However, the use of small size particles as packing materials is often associated with a high backpressure of the column, thus reducing mass transport kinetics and limiting their industrial applicability, especially in high flow-rate chromatography analysis. Under this context, bimodal, hierarchically structured silica monoliths that contain both macropores (pore size > 50 nm) and mesopores are highly desirable. The macroporosity will enable the rapid permeation of the aqueous phase, whereas the mesopores provide high specific surface area and active sites. Ever since the first porous silica monolith prepared by Nakanishi et al. in 1991,36 monolithic silica materials with hierarchical porosity have been synthesized based on the similar sol–gel process using a myriad of templates, including Pluronic-type triblock co-polymers,37 cationic,38 and anionic39 surfactants. Among various commonly known methods relying on the mechanism of sol–gel chemistry (e.g., phase separation, emulsions and foams, and ice templating),40 polymerization induced phase separation is a versatile approach towards hierarchically macroporous–mesoporous networks. During the process, the mesoporosity, the size and shape of the macropores, and the degree of macroscopic phase separation can be easily adjusted by factors such as pH, concentration of the polymer, and gelation time of the sol. Another benefit associated with the use of sol–gel route is that the resulting monoliths can be prepared in a variety of morphologies or shapes (e.g., columns, disks, and capillaries), which is particularly interesting for industrial applications.41,42
Novel extraction chromatographic sorbents would greatly improve the ability to separate and recover actinides, and the overall performance of these sorbents is intimately reliant on functional groups. Nowadays, the most commonly used functional groups are phosphorus-, amine- or sulfoxide-based ligands, inspired by organic extracting agents utilized in LLE.4,6 In particular, derivatives of the diglycolyl amide (DGA) ligands have recently been studied for extraction of REEs;6,32,43 however, studies focusing on their ability for actinide extraction in SPE remain scarce.26 In this work, monolithic silica exhibiting a bimodal, hierarchical macroporous–mesoporous structure was successfully prepared via sol–gel processing. A highly stable silica skeleton was obtained through post-gelation treatment in mildly basic ammonia solution using Ostwald ripening inside the silica backbone. The centimeter-size monolithic body exhibits interconnected and adjustable macropores in the micrometer range in addition to disordered mesopores with pore diameters in the 2–4 nm range. Upon functionalization by DGA, the monolithic sorbent with a high specific surface area demonstrated significant selectivity and extraction capacity toward Th(IV) from aqueous solution in both continuous column extraction and batch conditions. The hierarchical nanostructures considerably accelerate the mass transfer, significantly reduce the backpressure, efficiently promote the full utilization of active sites, and markedly improve the kinetic performance. The applicability of sorbents was demonstrated by the removal of Th(IV) from two mineral leachates: rare earth elements ore (OKA-2) and bauxite residue (red mud). The possibility of regenerating the porous sorbents was demonstrated over up to 10 cycles with no significant loss in Th(IV) extraction capacity, suggesting their potential for industrial applications.
Results and discussion
Synthesis and characterization of the materials
The centimeter-size, hierarchically macroporous–mesoporous silica monoliths were prepared using a sol–gel process accompanied by phase separation from tetraethoxysilane (TEOS) in acidic conditions in the presence of water soluble homopolymer (PEG 35000) and a surfactant (C16TAB), as described in the literature.38 Since the procedure employed results in the formation of a silica gel, the size and shape of the monolith are determined by the size and shape of its initial mold. In this study, polyvinylchloride (PVC) tubes with an internal diameter of 1.2 cm and a length of 6 cm were used as synthesis vessels. After the aging step, the wet monoliths were submitted to an ammonia hydrothermal post-treatment (solvent exchange). The increase in pH or temperature enhances Ostwald ripening inside the silica skeleton, leading to a stabilization of the monolith skeleton, and generates a disordered mesoporosity at the same time.44 In the present study, an efficient procedure for obtaining large size materials was to keep the wet monoliths in 200 mL of NH4OH at 90 °C for 8 h. The use of NH4OH at lower concentration (0 to 1 mol L−1) at a given temperature (90 °C), or at lower temperature (75 °C) at a given ammonia concentration (2 mol L−1), induced fragile silica skeleton that could not resist the drying process, and eventually lead to the destruction of the monolithic structure. The monoliths after solvothermal treatment were named as M0.1, M0.5, M1, and M2 for treatment with 0.1, 0.5, 1, and 2 mol L−1 of ammonia solution at 90 °C, respectively; M0 for monoliths without solvothermal treatment; and M2_75 for material treated with a 2 mol L−1 ammonia solution at 75 °C. For M2, white, monolithic rods were obtained after calcination, with an averaged diameter of 1 cm and length of 5 cm, corresponding to total size shrinkage of 8.3% compared to initial gelled silica body in the molds (Fig. 1a). | ||
| Fig. 1 Pictures of M2 after calcination (a) and DGA-functionalized monolith M-DGA (e) (the scale is in cm), HRSEM images of M2 (b and c) and M-DGA (f and g) showing the macroporous skeletons, and TEM images of M2 (d) and M-DGA (h) showing the hybrid macroporous–mesoporous structure. | ||
During the synthesis, the size of the mesopores was controlled by the pH (i.e., the concentration of ammonia) and the temperature of the post-synthesis treatment. This behavior arises from the formation of larger nanoparticles inside the skeleton at higher ammonia concentration or higher temperature, which leads to larger pore sizes. After calcination, the mesoporosity features of the monoliths under various solvothermal treatment conditions were thoroughly investigated by N2 physisorption. The silica skeleton of materials without solvothermal treatment (M0) was weakly condensed and shattered during the drying process. Furthermore, M0 exhibits only a small size mesoporosity after drying and calcination (a pore size of 2.6 nm calculated via the NLDFT adsorption branch analysis, Table S1, ESI†), and thus no obvious hysteresis loop associated with capillary condensation in large mesopores was observed on the N2 physisorption isotherm (Fig. S2†). For all of the monoliths treated with ammonia, the size of mesopores increases from 3.7 nm to 4.1 nm at higher ammonia concentration and temperature due to Ostwald ripening process (Fig. S2 and Table S1, ESI†). For M2, for example, a typical type IV N2 adsorption isotherm was obtained with capillary condensation reflecting the structural mesoporosity in the relative pressure range (P/P0) of 0.3–0.4, and the pore size distribution curve showed a sharp peak centered at 4.1 nm with high uniformity (Fig. 2). A slight decrease in specific surface area and pore volume was observed for materials treated with higher temperature or pH, as expected, since harsher solvothermal treatment generates larger pore sizes. The apparent density (i.e., the density of the material excluding the gas-accessible pore volume) was measured from the standard N2 physisorption data as described by Weinberger et al.,45 and was determined to be 1.06 g cm−3. Compared to the values for standard mesoporous silica (e.g., MCM-41, MCM-48, SBA-15, and KIT-6), which vary between 2.37 g cm−3 to 2.61 g cm−3, the significantly lower apparent density of monoliths could be explained by the presence of macropores in the structure (vide infra).
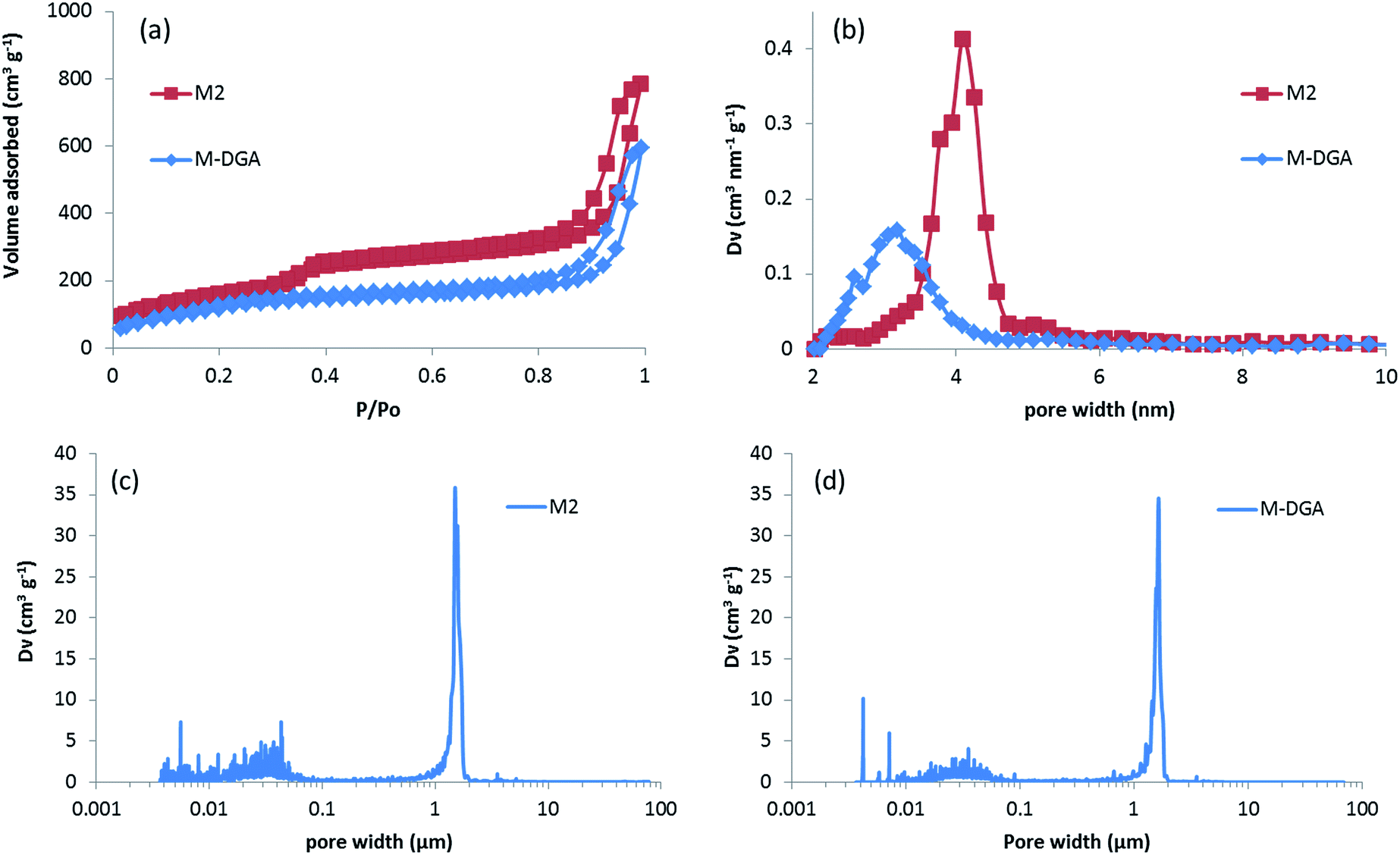 | ||
| Fig. 2 N2 physisorption isotherms measured at – 196 °C (a) and the respective NLDFT mesopore size distributions (b) of M2 and M-DGA, and pore size distribution (PSD) calculated from mercury intrusion porosimetry for M2 (c) and M-DGA (d). | ||
The high-resolution scanning electron microscopy (HRSEM) images highlight the macropores that extend throughout the silica skeleton, and the mesopores that are highly accessible through the macropores (Fig. 1b and c). The transmission electron microscopy (TEM) image of pristine silica monolith M2 shows that the mesopores are disordered and buried in the skeleton (Fig. 1d). The macroporosity parameters of the materials were determined using mercury intrusion porosimetry, and the derived pore size distribution (PSD), pore volume, and apparent and bulk density are compiled in Tables 1 and S1 (ESI†). Mercury porosimetry of the monoliths reveals two intrusion steps: one at lower pressure (<1 MPa) in the domain of micrometer-range macropores and the second one at higher pressure (>30 MPa) in the domain of nanometer-range mesopores (Fig. S4, ESI†). Moreover, for M2, the mercury extrusion branch parallels the intrusion branch, suggesting their good mechanical properties. Well-defined and uniform macropores were obtained through concurrent phase separation and sol–gel transition induced by the polymerization reaction, as confirmed by the very sharp step in the mercury intrusion branch in the micrometer domains and the narrow PSD (Fig. 2c and S4, ESI†). The bimodal porosity of the monoliths was clearly observable from mercury porosimetry analysis; however, the sizes of mesopores obtained from mercury intrusion branch are slightly higher compared to the pore sizes calculated via NLDFT method from N2 physisorption. In general, the pore size derived from mercury porosimetry significantly underestimates the real pore size of narrow mesopores (e.g., for ordered mesoporous silica KIT-6 with 3-dimensional structure).46,47 In this case, although solvothermal treatment significantly enhanced the overall mechanical property and robustness of the silica skeleton, part of the 2-dimensional, MCM-41-type mesopore structure collapsed at high pressure during the mercury intrusion,48 and the pore size is therefore overestimated in the nanometer-range. The apparent density obtained from mercury porosimetry for M2 is 1.32 g cm−3, which is in good accordance with the value measured using N2 physisorption. The significant difference between apparent density and bulk density (0.22 g cm−3) again confirms the highly porous nature of the material.
| Samples | N2 physisorption | Hg porosimetry | Bulk density (g cm−3) | |||||
|---|---|---|---|---|---|---|---|---|
| S BET (m2 g−1) | d meso (nm) | V mesopore (cm3 g−1) | Apparent density (g cm−3) | d macro (μm) | V macropore (cm3 g−1) | Apparent density (g cm−3) | ||
| M2 | 621 | 4.1 | 1.36 | 1.06 | 1.65 | 3.87 | 1.32 | 0.22 |
| M-DGA | 485 | 3.3 | 0.89 | 1.61 | 1.47 | 2.91 | 1.89 | 0.32 |
The DGA-APTS ligand (see Experimental section) was introduced onto the silica monolith piece using a standard grafting procedure in toluene at 120 °C. Prior to adding the DGA-APTS, the pristine monolith was immersed in dry toluene to ensure the penetration of the ligand into the monolith body and homogeneous grafting inside the pores. After the grafting procedure, the monolithic body became slightly yellow (Fig. 1e). The efficiency of the grafting was evaluated using thermogravimetric analysis–differential thermal analysis (TGA–DTA), which showed a total mass loss of 14.3% (Fig. S5, ESI†). Elemental analysis showed 10.4 wt% of carbon and 1.52 wt% of nitrogen in the functionalized monolith (denoted as M-DGA), corresponding to an apparent surface ligand density of 0.45 nm−2. After anchoring of the organic ligands on the silica surface, the shape of the hysteresis loop was well-maintained compared to the pristine monolithic support, and the loop was shifted to the lower relative pressure range (P/P0) of 0.2–0.3, indicating a decrease in pore size. A surface area of 485 m2 g−1 and a pore volume of 0.89 cm3 g−1 were obtained, and a narrow PSD with a mean value of 3.3 nm could be calculated from the adsorption branch based on the NLDFT model (Fig. 2a and b). The overall hierarchically macroporous–mesoporous structure was also well maintained after functionalization, as shown in the mercury intrusion/extrusion curves (Fig. S4, ESI†) and narrow macropore size distribution of M-DGA (Fig. 2d). Furthermore, the HRSEM images (Fig. 1f and g) and TEM (Fig. 1h) of M-DGA also confirm that the hierarchical structure remains essentially intact and that the mesopores, as well as the functional groups, are easily accessible.
The grafting through covalent bonds is further confirmed by solid state NMR spectroscopy. The peaks corresponding to the DGA ligand can be clearly attributed in the 13C CP/MAS NMR spectrum, as reported previously (Fig. S6, ESI†).43,49 From the 29Si MAS NMR spectra, peaks representing trifunctional (T) silicon between −50 to −65 ppm are observed in addition to the Q3 and Q4 peaks of the pristine silica framework between −90 and −120 ppm (Fig. S7, ESI†). The DGA has been anchored to the silica support mostly through T1((SiO)(OR)2Si–R) and T2((SiO)2(OR)Si–R) species appearing at −52 and −60 ppm, respectively, whereas a portion of the ligands is attached to the monolith surface through T3((SiO)3Si–R) at −65 ppm. In addition, the FTIR spectrum of M-DGA shows new peaks at 1672 and 1550 cm−1, which correspond to amide I (CO stretching) and amide II bands (NH deformation, CN stretching), which are not observed in the pristine monolith (Fig. S8, ESI†).
Continuous column studies
The M-DGA (0.89 g) and pristine monolith M2 (0.70 g) were evaluated as stationary phases for the chromatographic extraction and preconcentration of Th(IV) under a continuous flow of standard feed solution containing 300 μg L−1 of REEs, Al(III), Fe(III), Th(IV) and U(VI) at pH 3.0, with a flow rate of 1 mL min−1 controlled by an HPLC pump. The outlet of the column was connected to the ICP-MS/MS for online elemental analysis. For clarity, only the simplified extraction chromatograms showing certain elements of interest (e.g., Al(III), Fe(III), Sc(III), Th(IV), and U(VI)) are shown in Fig. 3 (for the full chromatograms, see Fig. S9, ESI†). For M-DGA and M2, the elution time for all REEs was 3.45 and 4.00 min, respectively, which corresponds approximately to the total pore volume of the monoliths obtained from N2 physisorption and Hg porosimetry (3.30 and 3.50 cm3 for M-DGA and M2, respectively). The M-DGA showed an elution time of 14 min for thorium, indicating significant selectivity toward Th(IV). After 14 min, the Th(IV) signal gradually increased and reached only less than 80% of its original concentration even after 90 min, which suggests that the column has not yet been saturated with Th(IV). After 90 min, the elements trapped in the solid phase were recovered using an ammonium oxalate solution ((NH4)2C2O4, 0.05 M). All the metals were recovered at approximately the same time, i.e., 3 min. The concentration of thorium was significantly enriched by the column during the extraction process, with an enrichment factor of more than 3200% compared to its original concentration (Fig. 3b). The sum of the Th(IV) quantities recovered by ammonium oxalate was estimated to be 80.1% of the total amount of Th(IV) initially introduced in the column. Slight enrichment for Fe(III) and Sc(III) was also observed (320% and 240% for Fe(III) and Sc(III), respectively), while only trace amounts of REEs, Al(III), and U(VI) were recovered (Fig. S9, ESI†).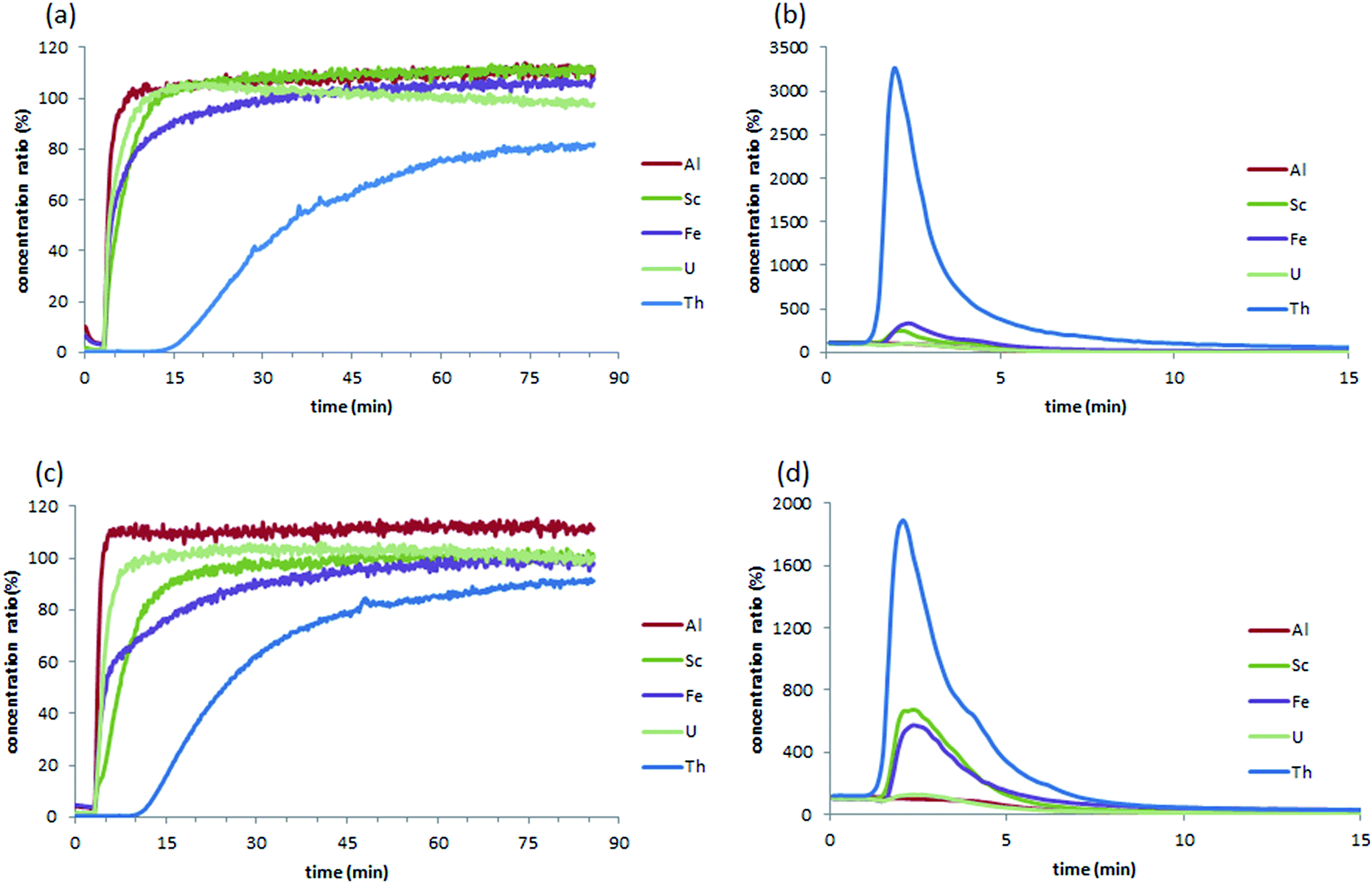 | ||
| Fig. 3 Extraction chromatograms of the feeding solution containing REEs, Al, Fe, Th and U (concentration of 300 μg L−1) with M-DGA (a) and M2 (c), and the corresponding recovery chromatograms using a 0.05 M ammonium oxalate solution (b and d). For clarity, only chromatograms for Al(III), Sc(III), Fe(III), Th(IV), and U(VI) are shown here. | ||
This extraction pattern is intriguing, since the efficient separation of Th(IV) from U(VI) or REE matrices is a difficult task.50–53 In the solution, the species distribution of actinides largely depends on the pH, and the speciation of Th(IV) and U(VI) as a function of pH has been previously studied in detail. For example, at pH 2–4 and Th(IV) concentration < 5 mmol L−1, Th4+, Th(OH)22+ and Th(OH)3+ are the predominant species in solution,24 whereas similar conditions lead to the formation of UO22+, which is in general less favorable for the sorbents compared to its multinuclear hydroxide complexes species formed at higher pH (e.g., (UO2)3(OH)5+ and (UO2)3(OH)7−).25 The size of UO22+ is significantly larger than that of Th(IV) species present in the medium, which renders the chelation between DGA ligand and the U(VI) species less efficient during the dynamic extraction process. On the other hand, although the DGA-based ligands showed high affinity towards middle-size lanthanides (i.e., Sm(III), Eu(III) and Gd(III)) when grafted on ordered mesoporous silica,43,49,54 strikingly high selectivity was observed here for Th(IV) over REEs (Fig. S9, ESI†), possibly because the spatial arrangement of DGA ligands grafted on the silica surface in M-DGA is optimal for Th(IV) extraction.
The substantial amount of radioactive uranium and thorium associated with rare earth deposits has caused considerable concern in rare earth industry, and the separation of these elements from the matrices is therefore of paramount importance for efficient rare earth production and radioactive nuclides management. A recent study by Hopkins et al. demonstrated high selectivity of DGA covalently bonded to mesoporous silica (KIT-6-N-DGA) for radioactive thorium and protactinium over uranium.26 In the present work, the system based on hierarchically structured silica monoliths functionalized by DGA shows superior uptake capacity and specific selectivity towards Th(IV) under dynamic flow-through conditions over the competitive ions that commonly co-exist with the element, suggesting promising potential for applications in rare earth or nuclear industries.
For comparison, the pristine monolith M2 showed an elution time of 8.90 min for Th(IV), with an enrichment factor of 1820%, which corresponds to 54.4% of the total amount of Th(IV) initially introduced into the column. Furthermore, Sc(III) was also enriched by 660%, while only trace amounts of REEs were retained. Indeed, the high selectivity of M2 toward Th(IV) and Sc(III) can be rationalized by the large surface area of the pristine material (SBET = 621 m2 g−1) as well as the abundant amount of accessible silanol groups. It is well-known that the exposed silanol groups on the mesoporous silica surface serve as O-donors for the complexation with actinides ions.23,24,55 Recently, unmodified mesoporous silica materials (e.g., KIT-6 and SBA-15) were used as an extracting medium in a SPE process for the selective separation and preconcentration of scandium, in which the specific binding sites for Sc(III) was seemingly related to silanol groups on the surface of the material, although the binding mechanism has yet to be understood.56 In this work, the grafting of DGA on monolith not only increases the adsorption capacity but also provides a better selectivity for Th(IV) over Sc(III). This is quite important especially for certain types of real-world samples (such as the bauxite residue in this work), in which large amount of Sc(III) could be easily recovered by pure silica material after Th(IV) is eliminated from the solution. Further, it is suggested that the macropores in the bimodal hierarchically structured silica monoliths facilitate mass transport, and expose more silanol groups to the metal ions, leading to an enhanced and selective sorption of Th(IV) and Sc(III). In the case of M-DGA, after functionalization with the DGA ligand, the oxygen in the carbonyl group readily binds Th(IV), further increasing the uptake capacity of the material.
To investigate the impact of accessible silanol groups on the Th(IV) uptake, a surface passivation using 1,1,3,3-tetramethyldisilazane (TMDS) was carried out for M2 and M-DGA, yielding a surface covered by –SiH(Me)2 groups.57 The resulting monoliths were denoted as M2-TMDS and M-DGA-TMDS, respectively. An additional mass loss of 3% for M2-TMDS and 1.6% for M-DGA-TMDS was observed on TGA after surface passivation (Fig. S5, ESI†), which also results in a slight decrease in the specific surface area and pore width measured by N2 physisorption (Fig. S2 and Table S1, ESI†). After surface passivation, the M2-TMDS became highly hydrophobic, while M-DGA-TMDS was partially hydrophobic (Fig. S10, ESI†). Both M2-TMDS and M-DGA-TMDS were used as stationary phases for automated extraction under the above-mentioned experimental conditions, and the extraction chromatogram of M2 showed significant decrease in Th(IV) retention (Fig. S11, ESI†). The signal for thorium appeared almost at the same time as Al(III), Fe(III), Sc(III) and U(VI), and reached rapidly to >90% after 40 min. Since a very limited number of silanols remain accessible on the surface, the amount of Th(IV) recovered by (NH4)2C2O4 decreased substantially from 1820% (compared to the original concentration) for M2 to only 280% for M2-TMDS. In comparison, the enrichment factor dropped to 950% for M-DGA-TMDS, compared to 3500% for M-DGA. The –SiH(Me)2 groups on the surface of M-DGA-TMDS not only significantly reduce the number of accessible silanol groups, but also enhance the hydrophobicity of the material, thus prohibiting the contact between Th(IV) ions and the DGA ligand. Therefore, the presence of free silanol groups is essential for the efficient extraction of Th(IV), as they provide active binding sites and maintain the hydrophilicity of the material. Indeed, the effect of hydrophilic groups in enhancing the ion uptake of sorbents has been discussed previously. For example, Shi et al. showed that the introduction of hydrophilic amino groups (–NH2) on phosphonate-functionalized mesoporous silica surface enhances the accessibility of Th(IV) ions into the inner cavity of the sorbent, although amino groups do not contribute to the complexation with Th(IV).24
Mechanistic studies and comparison with mesoporous silica
To further explore the sorption behavior of M2 and M-DGA, batch solid-phase extraction of Th(IV) from aqueous solutions under various conditions (e.g., contact time, initial Th(IV) concentration, and temperature) were conducted. Compared to continuous flow conditions, in which the precise control of the experiment parameters is difficult, the batch extraction is more practical and could provide some insights into the mechanism of ion uptake. In this work, the pH of all solutions was adjusted to 3.0 (±0.1), and the mass/volume ratio was fixed at 1 mg mL−1. The Th(IV) sorption kinetic studies were carried out for M2 and M-DGA over a time range of 0.5–120 min at room temperature, with an initial Th(IV) concentration of 90 mg L−1. As shown in Fig. 4, the adsorption of Th(IV) increases rapidly within the first 5 min of contact, and the equilibrium was reached within 30 min. Judging from the initial ion uptake (between 1–5 min), the M-DGA shows slightly faster kinetics compared to M2. The kinetic curves of M2 and M-DGA both exhibit excellent fit with a pseudo-second-order kinetic model, with a correlation coefficient of > 0.99 (for the detailed description of the kinetic model fitting, see ESI†). The Qe values calculated from the pseudo-second-order model for M2 (34.7 mg g−1) and M-DGA (55.6 mg g−1) sorbents are also in good agreement with the experimental data Qe,exp (35.0 and 56.1 mg g−1 for M2 and M-DGA, respectively; see Fig. S12 and Table S2†). The equilibrium time of M-DGA (30 min) is close to that of some highly porous and functionalized metal–organic framework (MOF) (less than 1 h),19 and is much faster than the reported values for mesoporous silica-based sorbents (typically 1–2 h),24,27 oxidized multiwalled carbon nanotubes (1 h),58 TiO2 nanoparticles (12 h),59 and hematite (24 h).60 The fast kinetics of the system stem from the strong affinity between Th(IV) ions as Lewis acids and the DGA ligands as Lewis bases, as well as the presence of macropores in the structure. In the bimodal hierarchically porous monolith, the interconnected mesopores and macropores can enhance the mass transport and diffusion to increase the ion uptake by the functional groups, which would be particularly favorable for the application under dynamic conditions.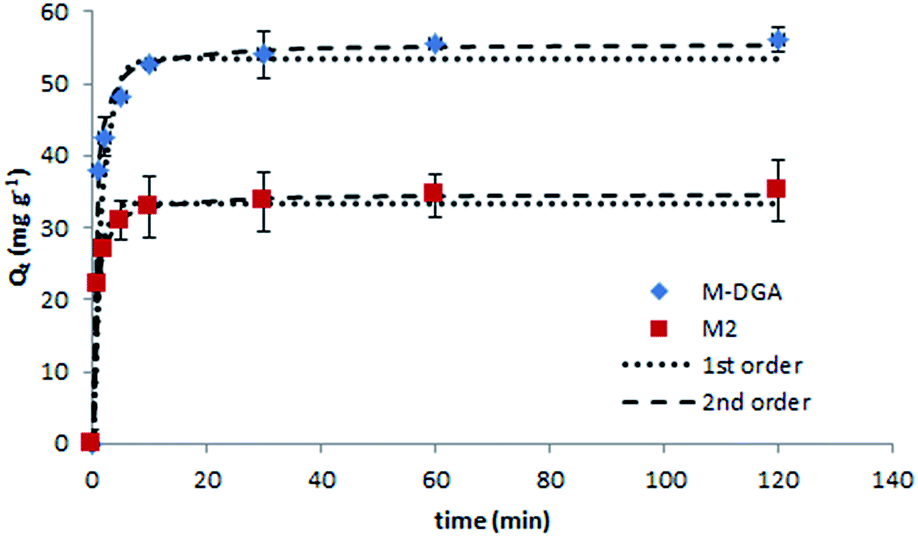 | ||
| Fig. 4 Effect of the contact time on the Th(IV) sorption with kinetic model fittings of pseudo-first- and pseudo-second-order models on M2 and M-DGA at room temperature. | ||
To determine the adsorption process and the sorbent uptake capacity for Th(IV), adsorption experiments were performed for M2 and M-DGA with initial thorium concentrations ranging from 15 to 200 mg L−1 at pH 3.0 (Fig. 5). Linear Langmuir and Freundlich adsorption models were used to fit the experimental data of Th(IV) adsorption. The adsorption isotherm was found to fit better with the Langmuir model with a correlation coefficient of 0.99 for both M2 and M-DGA (Fig. S13, ESI†). The maximum sorption capacity (Qm,cal) was calculated to be 54.3 for M2 and 84.5 mg g−1 for M-DGA, which is in good agreement with experimental values (55.1 and 83.6 mg g−1 for M2 and M-DGA, respectively; Table S3, ESI†). The Langmuir isotherm model suggests a mono-layered and uniform sorption, in which Th(IV) ions are adsorbed by the complexation with the hydroxyl/carbonyl groups on the sorbent surface.
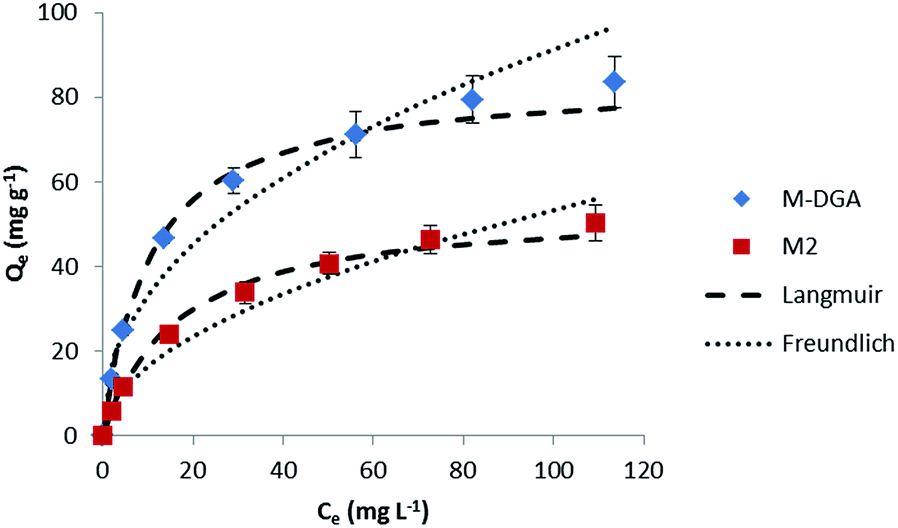 | ||
| Fig. 5 Sorption isotherms of Th(IV) at 298 K for the M2 and M-DGA sorbents, and the corresponding Langmuir and Freundlich models fitting curves. | ||
As mentioned previously, the high adsorption capacity of M2 and M-DGA could be attributed to the abundant amount of accessible silanol groups and DGA ligands. To identify active sorption sites, FTIR spectra of M2 and M-DGA before and after sorption of Th(IV) were compared, as shown in Fig. 6. For M2, the absorption band at 978 cm−1, which is assigned to free O–H of silanols, shifted to 960 cm−1 when the material is saturated with Th(IV), while the similar blue shift was also observed for M-DGA in the same region (from 976 to 961 cm−1), indicating the interaction between silanol groups and Th(IV). Moreover, in the region around 1672 cm−1, where the carbonyl group in DGA is identified (Fig. 6c), the peak shifted to 1648 cm−1 after adsorption of Th(IV). However, no change was observed for the peak at 1550 cm−1 corresponding to amide II (NH stretching). These observations suggest that both free silanol groups and carbonyl groups contribute to the uptake of Th(IV), whereas amino groups do not seem to complex with Th(IV).
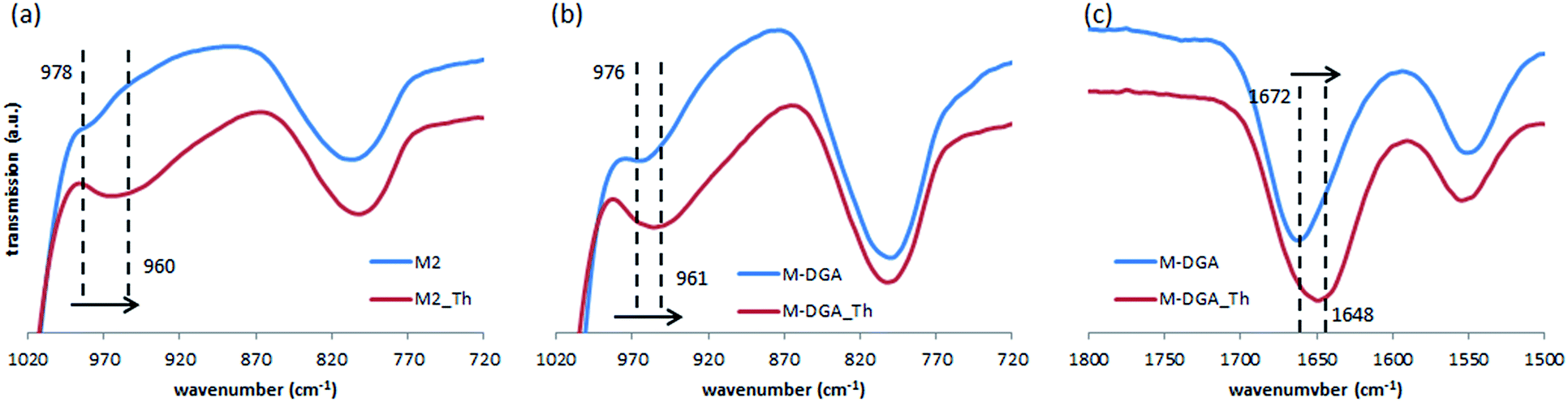 | ||
| Fig. 6 FTIR spectra of M2 (a) and M-DGA (b and c) before and after binding with Th(IV). | ||
To further understand the nature of the sorption process and investigate the influence of temperature, the extraction of Th(IV) by M2 and M-DGA was carried out at temperatures ranging from room temperature (298 K) to 348 K. As shown in Fig. 7(a), the adsorption capacity of the sorbents was largely affected by temperature in the chosen range, with higher temperature favoring the Th(IV) uptake. Thermodynamic parameters such as the standard free energy change (ΔG°), standard enthalpy change (ΔH°) and standard entropy change (ΔS°) were calculated by following eqn (1) and (2):
| ΔG° = ΔH° − TΔS° | (1) |
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kd = −ΔH°/RT + ΔS°/T Kd = −ΔH°/RT + ΔS°/T | (2) |
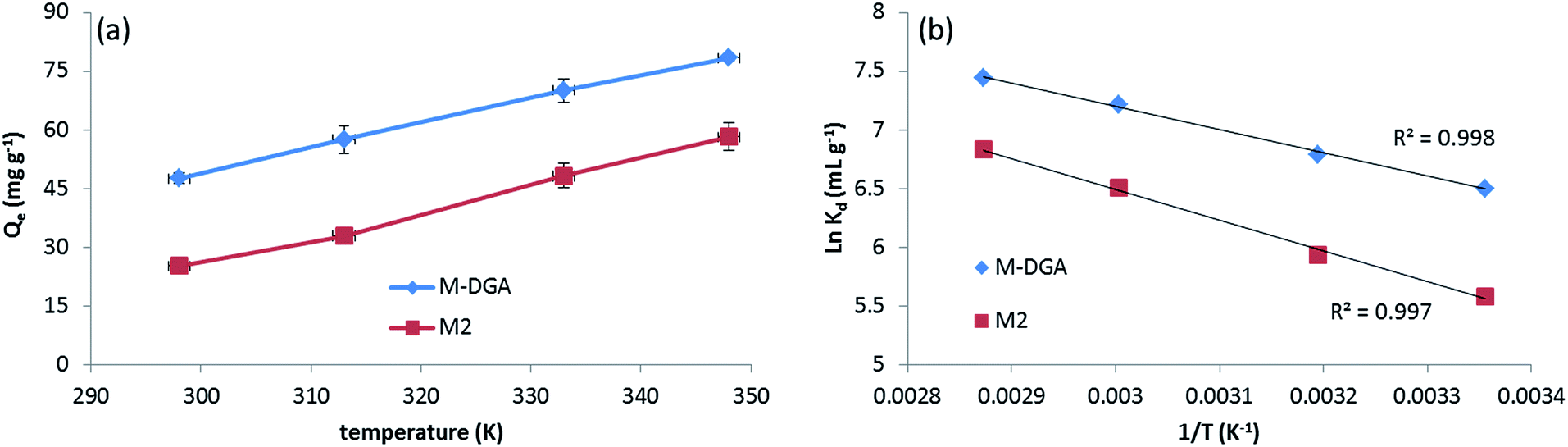 | ||
| Fig. 7 Th(IV) adsorption by M2 and M-DGA at different temperatures (a), and the linear regression of lnKdvs. 1/T (b). | ||
| Materials | ΔH° (kJ mol−1) | ΔS° (J mol−1 K−1) | ΔG° (kJ mol−1) | |||
|---|---|---|---|---|---|---|
| 298 K | 313 K | 333 K | 348 K | |||
| M2 | 21.9 | 119.3 | −13.8 | −15.6 | −17.9 | −19.8 |
| M-DGA | 16.5 | 109.7 | −16.1 | −17.7 | −19.9 | −21.6 |
The positive value of ΔS° suggests an increase in randomness at the solid/liquid interface during the adsorption process. Indeed, for most of Th(IV) adsorbents depending on the completion of the chemisorption, the adsorption is an endothermic process with an increase of randomness,27,58,61 with only few examples of being exothermic process with negative entropy value.20 Upon complexation with active sites, the Th(IV) species undergo dehydration and release water molecules in the aqueous phase, increasing the overall randomness. In the Th 4f XPS spectra of the Th(IV) loaded adsorbents (denoted as M2_Th and M-DGA_Th, Fig. S14, ESI†), the peaks at the binding energy (BE) of 335.26 eV (M-DGA_Th)/336.20 eV (M2_Th) and 344.46 eV (M-DGA_Th)/345.41 eV (M2_Th) were assigned to Th 4f7/2 and Th 4f5/2, respectively, providing evidence of Th(IV) being successfully adsorbed. The spectra are very similar for both M2_Th and M-DGA_Th, which are very close to those of ThO2 (334.6 eV and 343.9 eV for Th 4f7/2 and Th 4f5/2, respectively).62
The benefit of a bimodal pore structure was further evidenced when compared with standard mesoporous materials. The batch sorption capacity of Th(IV) at the same initial Th(IV) concentration (120 mg L−1, pH = 3.0) by pure MCM-41, KIT-6, and corresponding sorbents functionalized with DGA are shown in Fig. 8. Although MCM-41 has the largest specific surface area (1410 m2 g−1) and provides the highest silanol density on the surface, its 2-D hexagonal structure and relatively small pore size (4.0 nm) are not ideal for the adsorption (Fig. S3, ESI†). In comparison, the interconnected 3-D cubic structure of pure KIT-6 is more favorable, since more silanol groups become accessible to Th(IV). After functionalization with DGA, the MCM-41-DGA and KIT-6-DGA possess a comparable amount of ligands grafted (Fig. S4, ESI†), and the sorbents showed enhanced extraction capacity compared to the pristine materials. For the monolithic materials, the superior sorption capacity can be attributed to their hierarchical porous 3-D structure, which reduces the risk of pore blocking during functionalization, and allows the full use of binding sites to achieve a high ion uptake, meanwhile promoting mass transport to facilitate solution permeation. Furthermore, the macropores in the monolith M-DGA can significantly reduce the back pressure associated with the column compared to mesoporous silica powders such as MCM-41-DGA (Fig. 8b), although the two materials possess similar mesopore size (3.2–3.3 nm). Therefore, the use of silica monoliths exhibiting a bimodal, hierarchical porosity as stationary phase is of particular interest in dynamic separation systems, as it provides fast kinetics and high adsorption capacities, and overcomes the back pressure issue associated with silica particle-based columns.
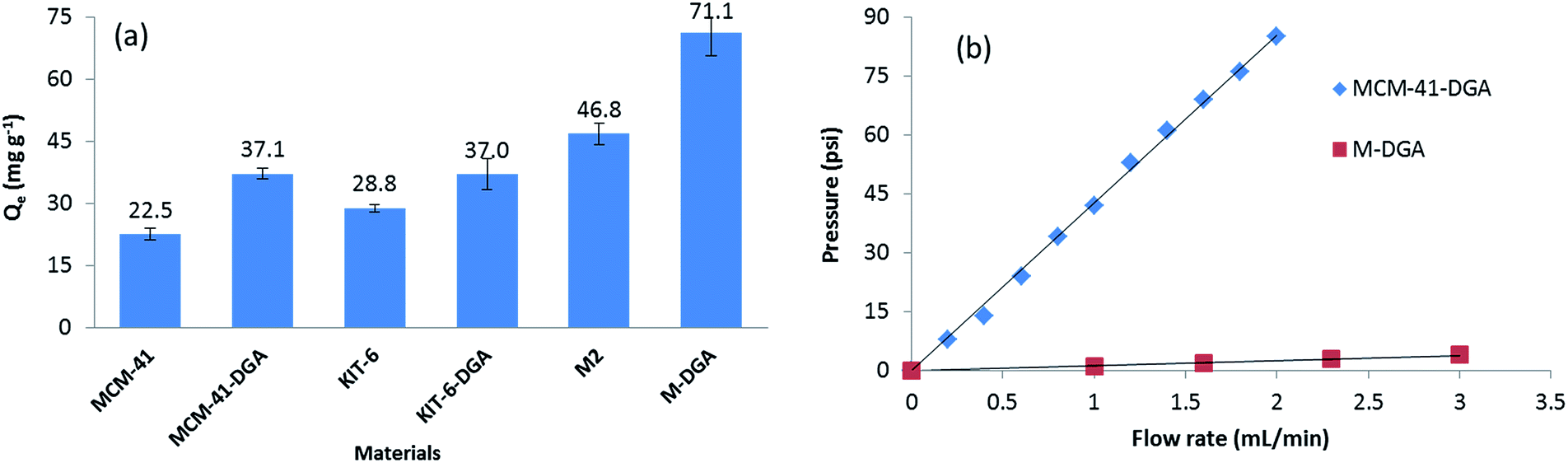 | ||
| Fig. 8 Comparison of Th(IV) sorption capacity by various mesoporous silica materials (a), and comparison of the back pressure associated with columns using MCM-41-DGA powder and hierarchically structured monolithic silica M-DGA as stationary phase (b). | ||
Application to real-world samples and stability tests
To test the applicability of the monolithic sorbents for Th(IV) sequestration in real-world samples, two leachates obtained from the digestion of mineral samples were analyzed: OKA-2 (britholite), a Canadian Certified Reference Materials Project (CCRMP) rare earth mineral reference material, and bauxite residue (red mud), an industrial waste from aluminum production. The initial concentration of the elements after digestion is shown in Table S4, ESI†. Beside relatively high concentration of thorium, both samples are characterized by the high ionic strength and contain a variety of competitive metals (e.g., Al(III), Fe(III), Ca(II), Na(I), and Ti(IV)). Although the interaction between the sorbents and Th(IV) is strongly favored over all other interactions, the high ionic strength of the leachates will likely increase the electrostatic repulsion between the material surface and Th(IV), and reduce the diffusion rate of thorium. To overcome this issue, we increased the contact time of every aliquot (15 mL for OKA-2 and 30 mL for the bauxite residue) through a decrease of substrate flow rate to 0.5 mL min−1. The efficiency of dynamic extraction by M-DGA is represented in Fig. 9, with focus on Th(IV), the competitive ions (Al(III), Fe(III), U(VI), Na(I), Ca(II), and Ti(IV)), and certain REEs with concentrations that are potentially economically exploitable. Detailed results on the extraction efficiency of each element are shown in the ESI (Table S5, ESI†). The initial concentration of Th(IV) in OKA-2 solution is 159 mg L−1 (∼7 × 10−5 mol L−1), which is concentrated enough to result in the precipitation of Th(IV) as amorphous Th(IV) hydroxide or hydrous oxide from the aqueous phase at pH 3.0.24 Therefore, the digested solution of OKA-2 was diluted 4 times with nanopure water, and then used for the following extraction tests (the final pH was adjusted to 3.0). During a running time of 30 min, the M-DGA reduced the Th(IV) concentration by 95% in diluted OKA-2. The extraction performance of the material remains efficient and selective even in the complex ionic matrix, with less than 2% of U(VI) and, in general, less than 0.5% of REEs being extracted. The amount of Al(III), Fe(III) and Sc(III) were reduced by 63%, 54%, and 68%, respectively, mainly due to their low initial concentration. This extraction pattern will potentially facilitate the individual separation of REEs in the following steps, since most of the undesired elements in the matrix are eliminated. Under the similar condition, M-DGA extracted approximately 81% of Th(IV) from a 30 mL bauxite residue solution. The slightly lower extraction efficiency can be attributed to the relative high concentration of Sc(III), Al(III) and Fe(III), which could compete with Th(IV) for the active binding sites (silanols and DGA). Interestingly, the concentration of Ti(IV) in the bauxite residue was reduced by approximately 60% by the column, which could be explained by the high affinity of Ti(IV) with DGA and free silanol groups.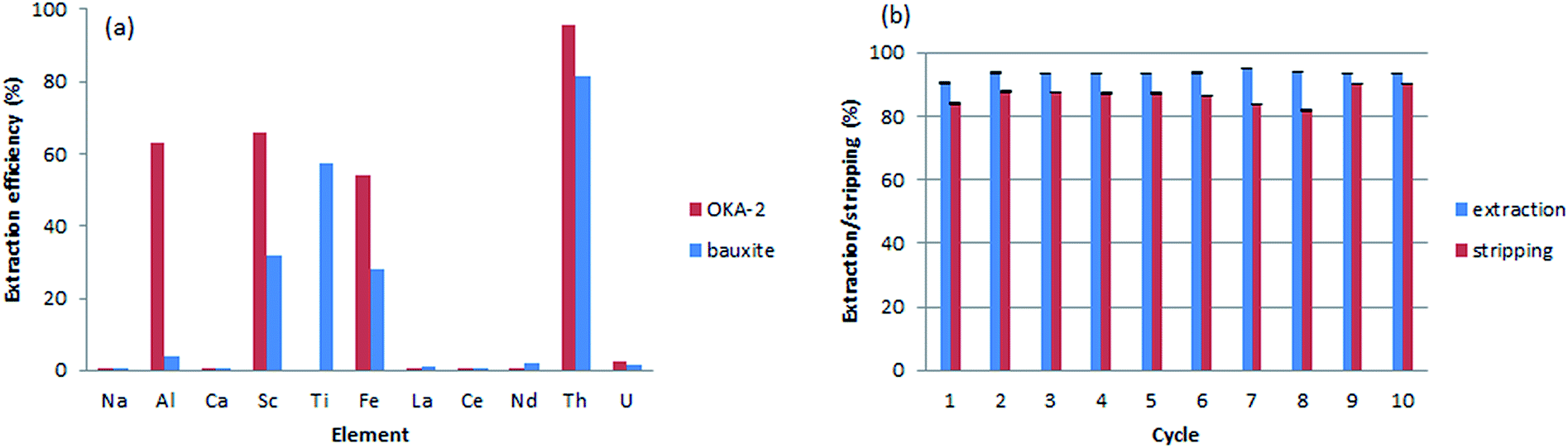 | ||
| Fig. 9 Selective extraction of Th(IV) from real-world mineral samples by M-DGA (a) and reusability performance of the sorbent over 10 extraction–stripping–regeneration cycles in the dynamic system (b). | ||
A crucial aspect for the application of the materials is their reusability under the extraction conditions. Ten cycles of extraction–stripping–regeneration experiments were performed to assess the stability of the functionalized monolith under dynamic conditions. Briefly, in each cycle, 15 mL of a diluted OKA-2 solution was passed through the column during 30 min. The retained elements were then recovered by a solution of (NH4)2C2O4 (0.05 M, 15 mL) at a flow rate of 1 mL min−1, followed by 15 mL diluted HNO3 (pH 3.0) to remove the remaining oxalate salt and regenerate the column. As shown in Fig. 9b, no significant loss in Th(IV) extraction capacity was observed even after 10 extraction–stripping–regeneration cycles. The majority of adsorbed Th(IV) was recovered by the elution of the oxalate salt (>82%). In general, approximately 93% of Th(IV) was extracted from 150 mL diluted OKA-2 solution, in which 86% of Th(IV) was recovered. The unaccounted amount of Th(IV) is likely to be found in the diluted HNO3 used for column regeneration, as a small portion of oxalate was trapped in the dead volume of the material during column extraction. After reusability assessment, the monolith was washed by water and characterized. The results obtained from N2 physisorption analysis showed an increase in both specific surface area (from 485 to 537 m2 g−1) and pore size (from 3.3 to 3.5 nm, Fig. S15, ESI†), which is probably due to loss (leaching) of some organic moieties tethered to the silica surface. The thermogravimetric analysis confirmed this result, as a reduction in the mass loss from 14.3 to 10.1% in the temperature range 100–700 °C (Fig. S16, ESI†). Furthermore, the 13C CP/MAS NMR spectrum showed the decrease in intensity of the peak at 61 ppm and disappearance of the one at 16 ppm, relative to the fresh material, indicating partial hydrolysis of the grafted ligand under the experimental condition, especially the ethoxysilane groups (Fig. S17, ESI†).31 However, the major peaks corresponding to carbonyl group and structural carbon remained well-defined, indicating that the overall structure of the organic ligand is well preserved.
Conclusions
Herein, a bimodal, hierarchically structured macroporous–mesoporous monolithic silica functionalized with chelating ligand diglycolamide (DGA) was fabricated for highly selective and efficient uptake of Th(IV) from aqueous solution in a solid-phase extraction system. A post-synthetic treatment with mildly concentrated ammonium solution was necessary to obtain large, centimeter-size monoliths. These monolithic silica materials possess robust mechanical properties, adjustable and interconnected macropores that facilitate rapid mass transport, as well as tunable and well-defined mesopores for functionalization. Upon grafting of DGA, enhanced Th(IV) uptake was achieved with high enrichment factor and selectivity, using the functionalized monolith as a column-type fixed bed sorbent for continuous flow extraction. Mechanistic studies on Th(IV) adsorption by the sorbent under batch conditions demonstrated that the hierarchical pore structure is advantageous for fast kinetics and high extraction capacity.Further work is in progress to prepare carbon-based, hierarchically structured monolithic sorbents with higher acid tolerance for actinide removal at low pH. Furthermore, novel functional groups (extractants) will be designed to further enhance the extraction performance, reusability and radiation stability, and ultimately, the new SPE systems will be assessed for their applicability in separation of actinides from spent fuel and mineral deposits.
Experimental
Chemical and materials
Tetraethoxysilane (TEOS, 98%), poly(ethylene glycol) (PEG, with an average molecular weight of 35000 g mol−1), hexadecyltrimethylammonium bromide (C16TAB, 95%), Pluronic P123 (EO20PO70EO20), (3-aminopropyl)triethoxysilane (APTS, 98%), diglycolyl chloride (DGCl, 95%), 1,1,3,3-tetramethyldisilazane (TMDS, 97%), ammonia (29%), and hydrofluoric acid (HF, 40%) were purchased from Sigma-Aldrich. Dichloromethane was purchased from Fisher Scientific. Nitric acid (65%) was purchased from Merck. These chemicals were used without further purification. Toluene, hexane, and triethylamine (Et3N, 99%) were purchased from Sigma-Aldrich, distilled using Na/benzophenone, dried over molecular sieves, and stored under nitrogen. Solutions of Th, U, rare earth elements (Sc, Y, La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu) and additional ions (Al and Fe) were prepared from the reference standard solutions (Plasma, Cal, SCP Science) using nanopure water. Certified reference material OKA-2 was purchased from CANMET (Ottawa, ON). Raw bauxite residue (red mud) was obtained from Rio Tinto Alcan, QC, Canada.
Synthesis of diglycol-2,4-diamido-propyltriethoxysilane (DGA-APTS)
The DGA-APTS was synthesized and purified as previously reported.54 Typically, a mixture of APTS (7.8 mL, 16.8 mmol) and triethylamine (11.2 mL, 0.80 mol) in 30 mL of toluene was added slowly into a solution of DGCl (2.00 mL, 8 mmol) in 30 mL of toluene at 0 °C. The reaction was allowed to warm to room temperature overnight. The reaction was filtered and the filtrate was evaporated and dried under vacuum to give 7.37 g of thick yellow oil (yield = 85%). 1H NMR (chloroform-d) (Fig. S1, ESI†): δ: 6.77 (br, 2H, NHAPTS), 3.99 (s, 4H, CH2–O), 3.77 (q, 12H, CH2–OSiAPTS), 3.27 (q, 4H, CH2–NHAPTS), 1.62 (m, 4H, CH2–CH2APTS), 1.17 (t, 18H, CH3APTS), 0.60 (t, 4H, CH2–SiAPTS).Synthesis of the silica monoliths
The pristine silica monoliths were prepared following the procedure described by Smått et al.38 TEOS was added to a mixture of PEG (35000) dissolved in concentrated nitric acid solution and nanopure water. The mixture was subsequently stirred at room temperature until a clear solution was obtained. At this stage, C16TAB was added and stirred for additional 10 min until the surfactant was completely dissolved. The initial TEOS/HNO3/H2O/PEG/C16TAB molar ratio was 1.00/0.25/14.69/0.54/0.0899. The sol was then transferred to molds (PVC tubes sealed by laboratory parafilms), followed by aging at 40 °C for 3 days. The wet silica monoliths obtained after the aging step are transferred to a 500 mL polyethylene bottle and immersed in 200 mL of NH4OH solution (concentration varied from 0.5 to 2 mol L−1) at 90 °C for 8 h, or in 200 mL of NH4OH (2 mol L−1) at 75 °C. The monoliths were named as M0.5, M1, and M2 for 0.5, 1, and 2 mol L−1 of NH4OH, M0 for monoliths without solvent exchange, and M2_75 for treatment using 2 mol L−1 of ammonia solution at 75 °C, respectively. The monoliths were then acidified with 100 mL HNO3 (0.1 mol L−1), washed thoroughly with ethanol (25 wt%), and dried on a flat surface under air for 3 days at 50 °C. The surfactant from the as-synthesized monoliths was then removed by calcination at 550 °C in air for 5 h, with the temperature ramping at 1 °C min−1.
Synthesis of mesoporous MCM-41 and KIT-6 silica powders
The MCM-41 material was obtained following the method reported by Kleitz et al.63 C16TAB (9.65 g) was dissolved in distilled water (480 g) at 35 °C under stirring. After complete dissolution, NH4OH (29%, 36.5 mL) was added, and the temperature was reduced to 25 °C. After 15 min, TEOS (40 g) was added at once and the mixture was left for 2 h under stirring, followed by an aging step at 90 °C for 3 days under static conditions. The resulting white solid was filtered, dried at 100 °C for 24 h, and washed with HCl–ethanol mixture. The surfactant was then removed by calcination at 550 °C for 5 h.The KIT-6 material was synthesized following the method reported by Kleitz et al.64 Pluronic P123 (9.0 g) was dissolved in distilled water (325.0 g), followed by addition of HCl (37%, 17.4 g) under vigorous stirring. After complete dissolution, n-butanol (BuOH, 9.0 g) was added. The reaction mixture was left under stirring at 35 °C for 6 h, after which TEOS (19.4 g) was added at once to the homogeneous solution. The molar composition of the starting mixture was TEOS/P123/HCl/H2O/BuOH = 1.0/0.017/1.83/195/1.31. The mixture was stirred at 35 °C for 24 h, followed by static aging step at 100 °C for 24 h. The resulting solid product was filtered, and dried for 24 h at 100 °C. The template was removed by extraction in ethanol–HCl mixture, followed by calcination in air at 550 °C for 5 h.
Functionalization and passivation of the materials
Prior to the grafting procedure, the silica monolith, MCM-41, and KIT-6 powders were activated overnight at 150 °C under vacuum. To functionalize the monolith, the pristine monolithic silica was first immersed in dry toluene (50 mL) under inert condition for 20 min. The DGA-APTS (0.54 g) was then added into the solvent, and the mixture was refluxed at 120 °C for 24 h under stirring. To functionalize MCM-41 and KIT-6 powders, 1.0 g of the support material was dispersed in 100 mL of dry toluene under inert atmosphere. After 20 min, DGA-APTS (0.54 g) and Et3N (1.2 mL) were added at once to the suspension. The resulting mixture was left under reflux conditions for 24 h. After the modification procedure, the silica powder was filtered, washed thoroughly with toluene. For all the functionalized materials, unreacted silane molecules were removed by Soxhlet extraction in dichloromethane for at least 12 h. The functionalized monolithic and powder materials are noted as M-DGA, MCM-41-DGA and KIT-6-DGA, respectively.Surface passivation of monolithic silica was carried out for M2 and M-DGA. The monoliths were immersed in 20 mL dry hexane, followed by addition of TMDS (2 mL) into the solvent. The mixture was stirred at room temperature overnight. The unreacted TMDS molecules were removed by Soxhlet extraction in CH2Cl2 for 12 h. The passivated materials were denoted as M2-TMDS and M-DGA-TMDS.
Characterization techniques
N2 adsorption–desorption isotherms were measured at −196 °C using an Autosorb-1-C sorption analyzer (Quantachrome Instruments, Boynton Beach, FL, USA). Prior to the analysis, the samples were outgassed for at least 12 h at 200 °C (pure silica) or at 80 °C (functionalized materials). The specific surface area (SBET) was determined using the Brunauer–Emmett–Teller equation in the range 0.05 ≤ P/P0 ≤ 0.20 and the total pore volume (Vpore) was measured at P/P0 = 0.95. The pore size distributions were calculated using NLDFT methods considering sorption of nitrogen at −196 °C in cylindrical silica pores from the desorption branch (for powder materials) or from the adsorption branch (for monolithic materials). The apparent density of the monolithic materials was also determined using the N2 physisorption data, as described by Weinberger et al.45 Mercury intrusion measurements were carried out at 25 °C with a Pascal 140/440 porosimeter (Thermo Fisher Scientific, Rodano, Italy), in a pressure range from 0–400 MPa. The surface tension of mercury was assumed to be 0.48 N m−1, and a contact angle of 140° was chosen. Transmission electron microscopy (TEM) images were taken using a Titan G2 ETEM with an accelerating voltage of 300 kV. Before the analysis, the powder sample was dispersed in methanol for 15 min through ultrasonication. The suspension is dropped onto a holey carbon-coated copper grid and dried under air. High resolution scanning electron microscopy images (HRSEM) were taken using a Verios 460 (FEI) at a landing voltage of 0.5 kV in the deceleration mode (stage bias voltage: 5 kV). The samples were mounted without metal coating (KAIST, Daejeon, Republic of Korea). X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Kratos AXIS-ULTRA spectrometer with a monochromatic Al X-ray source operated at 300 W. High energy resolution spectra (Th 4f) were recorded at 20 eV or 40 eV pass energy and step size of 0.05 eV or 0.1 eV. Elemental analysis was performed by the combustion method using a CHNS Analyzer Flash 2000, Thermo Scientific. FTIR spectra were recorded using a Nicolet Magna FTIR spectrometer with a narrow band MCT detector (Specac Ltd., London). Spectra were obtained from 64 scans with a 4 cm−1 resolution. Simultaneous thermogravimetric analysis–differential thermal analysis (TGA–DTA) was performed using a Netzsch STA 449C thermogravimetric analyzer, under air flow of 20 mL min−1, with a heating rate of 10 °C min−1 from 35 °C to 700 °C. Solid state magic angle spinning (MAS) NMR spectra were obtained on a Bruker DRX300 MHz spectrometer. The 29Si MAS and 13C cross-polarization (CP)/MAS NMR spectra were measured at 59.60 MHz using 4 mm rotors spinning at 9 kHz. The chemical shifts are reported in ppm relative to tetramethylsilane (TMS) for 29Si and adamantane for 13C.Automated extraction studies
Prior to analysis, the sorbents were outgassed at 200 °C (pristine silica materials) or 80 °C (functionalized materials) overnight. The monolith was inserted into a metal-free chromatography column (purchased from BioSafe Column System, 10 mm × 5 cm, Biotech USA, LLC) connected to a chromatographic pump (ICS-3000 single pump, Dionex). Prior to the extraction tests, the column was conditioned with a solution of diluted HNO3 (pH = 3.0 ± 0.1). During the automated extraction analysis, a solution containing 300 ppb (μg L−1) of rare earth elements (REEs), Al(III), Fe(III), Th(IV), and U(VI) (pH = 3.0 ± 0.1) was pumped through the column, followed by a solution of ammonium oxalate ((NH4)2C2O4, 0.05 mol L−1) to extract the retained elements. The mixed solutions were prepared by the dilution of calibration standards, and the pH was adjusted by diluted HNO3 or NH4OH. The outlet channel of the column was directly linked to the ICP-MS/MS (model 8800, Agilent Technologies) for on-line analysis to obtain the corresponding chromatogram. The flow rate for extraction was fixed at 1 mL min−1. For the MCM-41-DGA, the silica powder was packed into the column, and the back pressure associated with the column was measured by Chromeleon chromatography data system (CDS) software.Solid-phase batch extraction studies
Typical batch solid-phase extraction (SPE) experiments (kinetics, isotherms, and thermodynamic analysis) were carried out in a 50 mL centrifuge tube, where solutions of Th(IV) and additional ions (Al(III), Fe(III), U(VI) and REEs(III)) in diluted HNO3 were prepared in the desired concentrations depending on the experimental requirements. The values of pH were adjusted by adding negligible volumes of diluted HNO3 or NH4OH. The mass/volume ratio was fixed at 1 mg L−1. The kinetic studies were performed for the pristine monolith M2 and M-DGA, using a solution of Th(IV) (90 mg L−1) at pH 3.0. The contact time was varied from 0.5 min to 2 h. The isotherm studies were performed using solutions of Th(IV) with concentration ranging from 10 to 160 mg L−1 for pristine M2 and from 15 to 200 mg L−1 for M-DGA, and the contact time was 2 h. Each condition was run in triplicate and only the average value is shown. The sorption capacity (Qe, mg g−1) was calculated using the following eqn (3):| Qe = V/m × (C0 − Cf) | (3) |
Thermodynamic data for M2 and M-DGA were obtained by carrying out the sorption of Th(IV) at different temperatures (298, 313, 333, and 353 K). The corresponding thermodynamic parameters were calculated based on the distribution coefficient Kd (L mg−1, eqn (4)):
| Kd = V/m × (C0 − Cf)/Cf | (4) |
Application to mineral leachates and reusability tests
Commercially available certified reference material (CRM) containing REEs, Th(IV), and U(VI), e.g., britholite (OKA-2), was used to assess extraction performances. The microwave digestion (MWD) of three samples (0.1 g each) by concentrated HNO3 (10 mL) and HF (1 mL) was performed in high pressure Teflon vessels using a Mars5 microwave oven (CEM Corporation, Matthews, NC, USA), as reported previously.65 The temperature of the mixed acid was ramped from 20 °C to 180 °C within 25 min and maintained at 180 °C for 15 min, followed by cooling down to room temperature for 90 min. The digestion mixture was then filtered through a 0.22 μm syringe filter. The resulting solution was diluted to 500 mL and the pH was adjusted by KOH and HNO3.The digestion of bauxite residue was performed by a DigiPREP block digestion (SCP Science, Montreal). Three samples of bauxite residue (0.1 g each) were mixed with 10 mL of H2SO4 (pH = 1) in 20 mL beakers covered with watch glass. The mixture was heated to 75 °C for 30 min with the temperature ramp of 2 °C min−1. After cooling to room temperature, the mixture was filtered through a 0.45 μm syringe filter. The resulting solutions were combined and diluted to 500 mL and the pH was adjusted by KOH and HNO3. The original composition in digested OKA-2 and bauxite residue solutions was investigated by ICP-MS/MS and ICP-OES (Optima 3000, Perkin-Elmer), and the corresponding concentrations are listed in the Table S4, ESI†.
The digested bauxite residue and OKA-2 solutions were used to evaluate the capture efficiency of M-DGA for Th(IV) under dynamic conditions, as described above. Briefly, M-DGA (0.89 g) was placed inside the column. Then, 15 mL of mineral sample was passed through the column with a nominal flow rate of 0.5 mL min−1 to ensure sufficient contact time between the column and solution (about 30 min for each aliquot). The adsorbed ions were then stripped by loading 15 mL (NH4)2C2O4 (0.05 mol L−1) at a flow rate of 1 mL min−1. After recovery of retained elements, diluted HNO3 (pH 3.0, 15 mL) was used to remove traces of (NH4)2C2O4 and regenerate the column. The total interval for each cycle is 60 min. The same extraction-recovery-regeneration recycle was repeated 10 times using diluted OKA-2 solution to assess the reusability and stability of the material.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the Natural Sciences and Engineering Research Council of Canada (NSERC) and Fonds de recherche du Québec – Nature et technologies (FRQNT) for financial support (2018-PR-207062). NSERC supported this work through a Strategic Project Grant (Grant no. STPGP 463032-14). This research was undertaken, in part, thanks to the funding from the Canada Research Chairs program. F. K. also acknowledges the funding support of the University of Vienna (Austria). The authors thank Dr Roland Marschall and Prof. Bernd M. Smarsly (both Justus-Liebig-University Giessen, Germany) for their support. The authors thank Prof. Ryong Ryoo (IBS and KAIST, Daejeon, Republic of Korea) for providing access to high-resolution TEM and SEM.Notes and references
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435 CrossRef PubMed.
- C. Lin, H. Wang, Y. Wang, L. Zhou and J. Liang, Int. J. Environ. Anal. Chem., 2011, 91, 1050–1061 CrossRef CAS.
- Z. Zhu, Y. Pranolo and C. Y. Cheng, Miner. Eng., 2015, 77, 185–196 CrossRef CAS.
- A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273 RSC.
- T. J. Tranter, in Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, Woodhead Publishing, 2011, pp. 377–413 Search PubMed.
- J. Florek, S. Giret, E. Juère, D. Larivière and F. Kleitz, Dalton Trans., 2016, 45, 14832–14854 RSC.
- S. A. Ansari and P. K. Mohapatra, J. Chromatogr. A, 2017, 1499, 1–20 CrossRef CAS PubMed.
- W. A. Abbasi and M. Streat, Solvent Extr. Ion Exch., 1998, 16, 1303–1320 CrossRef CAS.
- P. W. Naik, P. S. Dhami, S. K. Misra, U. Jambunathan and J. N. Mathur, J. Radioanal. Nucl. Chem., 2003, 257, 327–332 CrossRef CAS.
- A. Mellah, S. Chegrouche and M. Barkat, J. Colloid Interface Sci., 2006, 296, 434–441 CrossRef CAS PubMed.
- W. Chouyyok, J. W. Pittman, M. G. Warner, K. M. Nell, D. C. Clubb, G. A. Gill and R. S. Addleman, Dalton Trans., 2016, 45, 11312–11325 RSC.
- J. Shen, W. Chai, K. Wang and F. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 22440–22448 CrossRef CAS PubMed.
- X. Han, M. Xu, S. Yang, J. Qian and D. Hua, J. Mater. Chem. A, 2017, 5, 5123–5128 RSC.
- S. Demir, N. K. Brune, J. F. Van Humbeck, J. A. Mason, T. V. Plakhova, S. Wang, G. Tian, S. G. Minasian, T. Tyliszczak, T. Yaita, T. Kobayashi, S. N. Kalmykov, H. Shiwaku, D. K. Shuh and J. R. Long, ACS Cent. Sci., 2016, 2, 253–265 CrossRef CAS PubMed.
- B. Li, Q. Sun, Y. Zhang, C. W. Abney, B. Aguila, W. Lin and S. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 12511–12517 CrossRef CAS PubMed.
- Y. Yuan, Y. Yang, X. Ma, Q. Meng, L. Wang, S. Zhao and G. Zhu, Adv. Mater., 2018, 30, 1706507 CrossRef PubMed.
- C. Xiao, Z. Hassanzadeh Fard, D. Sarma, T.-B. Song, C. Xu and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 16494–16497 CrossRef CAS PubMed.
- D. Sarma, C. D. Malliakas, K. S. Subrahmanyam, S. M. Islam and M. G. Kanatzidis, Chem. Sci., 2016, 7, 1121–1132 RSC.
- N. Zhang, L.-Y. Yuan, W.-L. Guo, S.-Z. Luo, Z.-F. Chai and W.-Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 25216–25224 CrossRef CAS PubMed.
- X.-G. Guo, S. Qiu, X. Chen, Y. Gong and X. Sun, Inorg. Chem., 2017, 56, 12357–12361 CrossRef CAS PubMed.
- X. Wang and N. Ye, Electrophoresis, 2017, 38, 3059–3078 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, D. Earl Lyndsey, W. Abney Carter, W. Lukasz, K. Thallapally Praveen and S. Ma, Adv. Mater., 2018, 30, 1705479 CrossRef PubMed.
- P. J. Lebed, J.-D. Savoie, J. Florek, F. Bilodeau, D. Larivière and F. Kleitz, Chem. Mater., 2012, 24, 4166–4176 CrossRef CAS.
- L.-Y. Yuan, Z.-Q. Bai, R. Zhao, Y.-L. Liu, Z.-J. Li, S.-Q. Chu, L.-R. Zheng, J. Zhang, Y.-L. Zhao, Z.-F. Chai and W.-Q. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 4786–4796 CrossRef CAS PubMed.
- L.-Y. Yuan, L. Zhu, C.-L. Xiao, Q.-Y. Wu, N. Zhang, J.-P. Yu, Z.-F. Chai and W.-Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 3774–3784 CrossRef CAS PubMed.
- P. D. Hopkins, T. Mastren, J. Florek, R. Copping, M. Brugh, K. D. John, M. F. Nortier, E. R. Birnbaum, F. Kleitz and M. E. Fassbender, Dalton Trans., 2018, 47, 5189–5195 RSC.
- Z. Yang, G. Chen, H. Weng, W. Shen, Z. Huang and M. Lin, J. Mater. Sci., 2018, 53, 3398–3416 CrossRef CAS.
- J. Górka, R. T. Mayes, L. Baggetto, G. M. Veith and S. Dai, J. Mater. Chem. A, 2013, 1, 3016–3026 RSC.
- M. Carboni, C. W. Abney, K. M. L. Taylor-Pashow, J. L. Vivero-Escoto and W. Lin, Ind. Eng. Chem. Res., 2013, 52, 15187–15197 CrossRef CAS.
- Y. Wang, Z. Gu, J. Yang, J. Liao, Y. Yang, N. Liu and J. Tang, Appl. Surf. Sci., 2014, 320, 10–20 CrossRef CAS.
- Y. Hu, E. Drouin, D. Larivière, F. Kleitz and F.-G. Fontaine, ACS Appl. Mater. Interfaces, 2017, 9, 38584–38593 CrossRef CAS PubMed.
- L. Lefrançois-Perreault, S. Giret, M. Gagnon, J. Florek, D. Larivière and F. Kleitz, ACS Appl. Mater. Interfaces, 2017, 9, 12003–12012 CrossRef PubMed.
- S. Yang, J. Qian, L. Kuang and D. Hua, ACS Appl. Mater. Interfaces, 2017, 9, 29337–29344 CrossRef CAS PubMed.
- J. Fasihi, N. Bavarsad, S. Shariati and K. Ashtari, Int. J. Environ. Anal. Chem., 2016, 96, 789–800 CrossRef CAS.
- X. Zheng, E. Liu, F. Zhang, Y. Yan and J. Pan, Green Chem., 2016, 18, 5031–5040 RSC.
- K. Nakanishi and N. Soga, J. Am. Ceram. Soc., 1991, 74, 2518–2530 CrossRef CAS.
- S. A. El-Safty, J. Porous Mater., 2010, 18, 259–287 CrossRef.
- J.-H. Smått, S. Schunk and M. Lindén, Chem. Mater., 2003, 15, 2354–2361 CrossRef.
- K. Nakanishi, Y. Sato, Y. Ruyat and K. Hirao, J. Sol-Gel Sci. Technol., 2003, 26, 567–570 CrossRef CAS.
- A. Feinle, M. S. Elsaesser and N. Hüsing, Chem. Soc. Rev., 2016, 45, 3377–3399 RSC.
- A. Galarneau, A. Sachse, B. Said, C.-H. Pelisson, P. Boscaro, N. Brun, L. Courtheoux, N. Olivi-Tran, B. Coasne and F. Fajula, C. R. Chim., 2016, 19, 231–2676 CrossRef CAS.
- H.-C. zur Loye, T. Besmann, J. Amoroso, K. Brinkman, A. Grandjean, C. H. Henager, S. Hu, S. T. Misture, S. R. Phillpot, N. B. Shustova, H. Wang, R. J. Koch, G. Morrison and E. Dolgopolova, Chem. Mater., 2018, 30, 4475–4488 CrossRef CAS.
- J. Florek, F. Chalifour, F. Bilodeau, D. Larivière and F. Kleitz, Adv. Funct. Mater., 2014, 24, 2668–2676 CrossRef CAS.
- W. C. Wilfong, B. W. Kail, T. L. Bank, B. H. Howard and M. L. Gray, ACS Appl. Mater. Interfaces, 2017, 9, 18283–18294 CrossRef CAS PubMed.
- C. Weinberger, S. Vetter, M. Tiemann and T. Wagner, Microporous Mesoporous Mater., 2016, 223, 53–57 CrossRef CAS.
- R. Guillet-Nicolas, R. Ahmad, K. A. Cychosz, F. Kleitz and M. Thommes, New J. Chem., 2016, 40, 4351–4360 RSC.
- K. A. Cychosz, R. Guillet-Nicolas, J. García-Martínez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 RSC.
- A. Galarneau, B. Lefèvre, H. Cambon, B. Coasne, S. Valange, Z. Gabelica, J.-P. Bellat and F. Di Renzo, J. Phys. Chem. C, 2008, 112, 12921–12927 CrossRef CAS.
- E. Juère, J. Florek, D. Larivière, K. Kim and F. Kleitz, New J. Chem., 2016, 40, 4325–4334 RSC.
- T. Yokoyama, A. Makishima and E. Nakamura, Anal. Chem., 1999, 71, 135–141 CrossRef CAS PubMed.
- V. Neck and J. I. Kim, Radiochim. Acta, 2009, 89, 1–16 Search PubMed.
- M. Eskandari Nasab, Fuel, 2014, 116, 595–600 CrossRef CAS.
- Y. Chen, Y. Wei, L. He and F. Tang, J. Chromatogr. A, 2016, 1466, 37–41 CrossRef CAS PubMed.
- J. Florek, A. Mushtaq, D. Larivière, G. Cantin, F.-G. Fontaine and F. Kleitz, RSC Adv., 2015, 5, 103782–103789 RSC.
- L.-Y. Yuan, Y.-L. Liu, W.-Q. Shi, Z. Li, J.-H. Lan, Y.-X. Feng, Y.-L. Zhao, Y.-L. Yuan and Z.-F. Chai, J. Mater. Chem., 2012, 22, 17019 RSC.
- S. Giret, Y. Hu, N. Masoumifard, J.-F. Boulanger, E. Juère, F. Kleitz and D. Larivière, ACS Appl. Mater. Interfaces, 2018, 10, 448–457 CrossRef CAS PubMed.
- L. L. Crowe and L. M. Tolbert, Langmuir, 2008, 24, 8541–8546 CrossRef CAS PubMed.
- M. Wang, X. Tao and X. Song, J. Radioanal. Nucl. Chem., 2011, 288, 859–865 CrossRef CAS.
- G. Zhijun, N. Lijun and T. Zuyi, J. Radioanal. Nucl. Chem., 2005, 266, 333–338 CrossRef.
- P. Reiller, F. Casanova and V. Moulin, Environ. Sci. Technol., 2005, 39, 1641–1648 CrossRef CAS PubMed.
- J. Wang, P. Liu, Z. Li, W. Qi, Y. Lu and W. Wu, Materials, 2013, 6, 4168–4185 CrossRef CAS PubMed.
- B. W. Veal and D. J. Lam, Phys. Rev. B, 1974, 10, 4902–4908 CrossRef CAS.
- F. Kleitz, W. Schmidt and F. Schüth, Microporous Mesoporous Mater., 2003, 65, 1–29 CrossRef CAS.
- F. Kleitz, S. H. Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- L. Whitty-Léveillé, K. Turgeon, C. Bazin and D. Larivière, Anal. Chim. Acta, 2017, 961, 33–41 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, 13C cross polarization (CP) and 29Si magic-angle spinning (MAS) NMR spectra, FT-IR spectra, plots of TGA–DTA, linear regression and the corresponding parameters of the kinetics and adsorption isotherm experiments, N2 sorption isotherms and mercury porosimetry data of the materials, XPS spectra of the monolithic materials saturated with Th(IV), and extraction chromatograms of the functionalized and passivated monolithic materials. See DOI: 10.1039/c8ta07952h |
| This journal is © The Royal Society of Chemistry 2019 |