
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The physico-chemistry of adhesions of protein resistant and weak polyelectrolyte brushes to cells and tissues†
Edward J.
Cozens
ab,
Dexu
Kong
ab,
Nima
Roohpour
c and
Julien E.
Gautrot
 *ab
*ab
aInstitute of Bioengineering and Materials Science, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: j.gautrot@qmul.ac.uk
bSchool of Engineering and Materials Science, Queen Mary University of London, Mile End Road, London, E1 4NS, UK
cConsumer Healthcare R&D, GlaxoSmithKline, St George's Avenue, Weybridge, Surrey, KT13 ODE, UK
First published on 5th December 2019
Abstract
The non-specific adhesion of polymers and soft tissues is of great interest to the field of biomedical engineering, as it will shed light on some of the processes that regulate interactions between scaffolds, implants and nanoparticles with surrounding tissues after implantation or delivery. In order to promote adhesion to soft tissues, a greater understanding of the relationship between polymer chemistry and nanoscale adhesion mechanisms is required. In this work, we grew poly(dimethylaminoethyl methacrylate) (PDMAEMA), poly(acrylic acid) (PAA) and poly(oligoethylene glycol methacrylate) (POEGMA) brushes from the surface of silica beads, and investigated their adhesion to a variety of substrates via colloidal probe-based atomic force microscopy (AFM). We first characterised adhesion to a range of substrates with defined surface chemistry (self-assembled monolayers (SAMs) with a range of hydrophilicities, charge and hydrogen bonding), before studying the adhesion of brushes to epithelial cell monolayers (primary keratinocytes and HaCaT cells) and soft tissues (porcine epicardium and keratinized gingiva). Adhesion assays to SAMs reveal the complex balance of interactions (electrostatic, van der Waals interactions and hydrogen bonding) regulating the adhesion of weak polyelectrolyte brushes. This resulted in particularly strong adhesion of PAA brushes to a wide range of surface chemistries. In turn, colloidal probe microscopy on cell monolayers highlighted the importance of the glycocalyx in regulating non-specific adhesions. This was also reflected by the adhesive properties of soft tissues, in combination with their mechanical properties. Overall, this work clearly demonstrates the complex nature of interactions between polymeric biomaterials and biological samples and highlights the need for relatively elaborate models to predict these interactions.
1. Introduction
The non-specific interactions between polymeric biomaterials and soft tissues play an important role in the performance of implants, tissue engineering scaffolds and drug delivery systems.1–5 Physico-chemical interactions are particularly important in regulating the strength of bioadhesives, such as skin adhesives for surgical applications,6–8 hydrogels for epicardial placement and stem cell delivery,9 soft tissue adhesion for tissue regeneration3,4 or mucoadhesives for dental adhesion.10,11 Although a range of chemical functions and molecules have been introduced in biomaterials to promote covalent coupling and adhesion to soft tissues, the control and regulation of non-specific physico-chemical interactions with surrounding tissues post-implantation and delivery can play an important role and even dominate adhesion performance.11,12 In addition, polymeric coatings introduced around nanomaterials for drug and gene delivery, or for imaging, can significantly impact their biodistribution and systemic persistence/clearance.1,2,13 Soft tissue adhesion is a complex process involving non-specific interactions, interpenetration of polymer networks and soft tissue interfaces, and molecular coupling; these are regulated by the mechanical properties of the bulk biomaterial, the soft tissue and their interface.12,14 Typically, after bringing a polymer into contact with a soft tissue, initial physical entanglement will be followed by secondary bonding to strengthen adhesive interactions.15 As is often the case in Nature, these secondary bonds will be formed by multiple individually weak non-covalent bonds that combine to form a stable, highly specific, reversible and strong intermolecular connection. These intermolecular interactions typically involve a combination of hydrogen bonds, electrostatic interactions and van der Waals interactions.16 The design of interfaces with controlled chemistry and intermolecular interactions is particularly important to the modelling and understanding of biointerface adhesion.In addition, tissue adhesion is further complicated by the secretion of biomacromolecules from cells constituting the corresponding soft tissues, modulating their interactions with contacting polymers. For example, salivary glands in the oral mucosa secrete mucins, a variety of antimicrobial substances, salts, water, and intestinal proteins that modulate mucosal adhesion. Hence mucoadhesives must adhere to the moist surface of the oral mucosa whilst resisting the flushing action of the saliva and competition with associated biomacromolecules.17,18
Polymer brushes are interesting model biointerfaces for the study of physico-chemical parameters affecting bioadhesion. Indeed, polymer brushes offer unique control of the chemistry of interfaces, enabling the formation of both very hydrophobic and hydrophilic polymer coatings, as well as polyelectrolytes. Furthermore, the morphology and mechanics of these interfaces can be regulated via the control of brush grafting density, thickness, swelling and conformation.19,20 The wealth of monomers that can be incorporated in polymer brushes has enabled a wide range of properties for these coatings, including protein resistance,21 thermoresponsiveness,22,23 electrolyte responsiveness24,25 and bacterial resistance.26,27 In addition, brushes can be readily grown from a wide range of substrates, making these coatings attractive for a range of biomedical applications from implant design28 to gene delivery.29,30
The adhesive and anti-fouling properties of polymer brushes and their nanoscale mechanics have been characterised relatively extensively by atomic force microscopy (AFM). Colloidal probe microscopy (CPM) utilises an AFM cantilever with a spherical particle attached at the apex in order to study the nano-scale forces between a particle and substrate with defined physico-chemical properties. The most widely used microspheres for CPM is silica, as they have a low roughness, are commercially available in a variety of sizes, and their surfaces can be readily chemically modified.31,32 The use of CPM has enabled the characterisation of protein adhesion to polymer brushes,33–35 as well as bacterial adhesion.27 In these studies, neutral and zwitterionic brushes that are particularly well solvated were shown to effectively limit adhesion of unwanted biomacromolecules and components of the bacterial membranes such as lipopolysaccharides. In addition, CPM was used to quantify adhesion and friction to hydrophobic and fluorophilic brushes; the brushes displaying increasing degrees of fluorination displayed reduced adhesion and friction (silica colloidal probes).36 The conformation and swelling of polymer brushes also strongly affect adhesion strength and contact mechanics, therefore highlighting the impact of environmental factors on non-specific adhesion.37 Such impact of the environment is also strikingly illustrated by the response of polyelectrolyte brushes to pH, electrolyte chemistry and ionic strength. Hence the strength of adhesion of silica microparticles to poly(2-vinylpyridine) brushes is enhanced by phosphate and sulfate electrolytes, compared to chloride, and is reduced at higher ionic strength.38 Overall, although the non-specific adhesion of solid substrates, bacteria and proteins to polymer brushes has been widely studied, limited studies have focused on non-specific adhesion to cells and tissues.
In this report, we study the adhesion of three types of polymer brushes to cells and tissues. We selected poly(acrylic acid) (PAA), poly(dimethylaminoethyl methacrylate) (PDMAEMA) and poly(oligoethylene glycol methacrylate) (POEGMA) as three important types of polymer brushes with distinct physico-chemical properties (anionic, cationic and neutral hydrophilic polymers, respectively). PAA displays a high density of carboxylic acid groups and negative charges (at high pH), is biocompatible and is widely used as biomaterials39 to promote mucoadhesion,40–42 as drug delivery systems,43 for surface coating and modification44,45 and for microdevice and sensor design.39 PDMAEMA is a weak polybasic polymer with pH-responsive properties46 that has applications in gene delivery.30,47 Finally, POEGMA is known for its excellent protein resistance arising from the high density of ethylene glycol moieties on the polymer backbone, its ability to grow at high grafting densities and its hydrophilicity.48 As such, its applications include surface modifications and coatings that resist protein absorption and thus prevent bacterial adhesion and biofilm formation.48–50 We first examine the adhesion of these three brushes to a range of model monolayers presenting a range of chemistries (surface charge, hydrogen bonding and hydrophilicity). We then explore the impact of brush chemistry on adhesion to model cell monolayers (epithelial cell sheets) and investigate the impact of their glycocalyx on adhesion. Indeed, most epithelial cells are surrounded by a glycoprotein and glycolipid layer associated with their membrane and known as the glycocalyx; this ranges from 7 nm in thickness for red blood cells up to several 100 nm in some cases.51 Consequently, when an implanted material comes into contact with epithelial cell sheets, often lining the surface of tissues, the glycocalyx is the first point of contact and its interaction with the material is of significant importance. Finally, we explore brush adhesion to tissues (gingival epithelium and epicardium) and discuss the impact of brush chemistry on adhesive behaviour.
2. Experimental section
2.1. Chemicals and materials
tert-Butyl acrylate (tBA, Mn = 128.17, 98%), 2-(dimethylamino)ethyl methacrylate (DMAEMA, Mn = 157.21, 98%), oligo(ethylene glycol methyl ether methacrylate) (OEGMA, Mn = 300), poly(L-lysine) (PLL), N,N,N′,N′′,N”-pentamethyldiethylenetriamine (PMDETA, 99%), 2,2′-bipyridyl (bipy, ≥99%), triethylamine (≥99.5%), copper chloride (Cu(I)Cl), copper bromide (Cu(I)Br and Cu(II)Br2), trifluoroacetic acid (TFA, 99%), toluene (anhydrous, 99.8%), iodomethane (99%), (3-aminopropyl)triethoxysilane (APTES, 99%), triethoxy(octyl)silane (97%), trichloro(1H,1H,2H,2H-perfluorooctyl)silane (97%), 11-mercaptoundecanoic acid (MUDA, 95%), 11-mercapto-1-undecanol (97%), Dulbecco's phosphate buffered saline (PBS), neuraminidase (from Clostridium Perfringens), heparinase III (from Flavobacterium Heparinum), wheat germ agglutinin (WGA, lectin from Triticum vulgaris, FITC conjugate), formaldehyde solution (4% in PBS) (PFA) and 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Sigma-Aldrich and used as received. Cu(I)Cl and Cu(I)Br were kept under vacuum until use. The silane initiator (3-trimethoxysilyl)-propyl 2-bromo-2-methylpropionate was purchased from Gelest. Acetone (technical), versene solutions (0.2 g L−1 EDTA in PBS), keratinocyte serum-free medium (KSFM), Dulbecco's modified Eagle's medium (DMEM), trypsin (0.25%), L-glutamine, penicillin–streptomycin, bovine pituitary extract (BPE), human recombinant epidermal growth factor (EGF), and goat anti-mouse IgG (H + L) secondary antibodies (conjugated to AlexaFluor 488) were purchased from Thermo Fisher Scientific. Fibronectin solution (from human plasma) and the anti-heparin/heparan sulfate antibody were purchased from Merck Millipore. Foetal bovine serum (FBS) was purchased from Labtech. Collagen I (type I) was purchased from Corning. Dichloromethane (DCM, ≥99.8%) was purchased from Honeywell. Dimethylformamide (DMF, ≥99.9%) was purchased from VWR Chemicals. AFM probes (ORC8-10) were purchased from Bruker. Silica particles (unfunctionalised) were purchased from microParticles GmbH (mean diameter of 19.59 μm) and Bangs Laboratories (mean diameter of 300 nm). SEMGLU was purchased from Kleindiek Nanotechnik GmbH, Germany. Silicon wafers (100 mm diameter, 〈100〉 orientation, polished on one side/reverse etched) were purchased from Compart Technology Ltd. All Plasma treatment was carried out using a Henniker Plasma Vacuum System HPT-200. Human keratinocyte HaCaT cells and Human primary epidermal keratinocytes were kindly provided by Dr John Connelly, Blizard Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London.2.2. Polymer brush synthesis on planar silicon substrates
The brushes were synthesized by the “grafting from” method using atom transfer radical polymerization (ATRP).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)) at room temperature. The samples were then washed thoroughly in ethanol, followed by deionised water, and then dried under a stream of nitrogen.
:1 (v/v), 15 mL) was used. The monomer was used as purchased without any need for initial purification, and polymerization was carried out at room temperature and was stopped by immersing samples in deionised water, rinsing with ethanol and drying under a stream of nitrogen.
:1 (v/v), 15 mL) was used. For PAA, PDMAEMA and POEGMA brushes polymerization times were adjusted accordingly to give dry ellipsometric thicknesses of 30 nm; these thicknesses equated to polymerization times of 5 h, 20 min and 30 min, respectively.
:1 (v/v)) at room temperature. The samples were then washed thoroughly in ethanol, followed by deionised water, and then dried under a stream of nitrogen.
:1 (v/v), 15 mL) was used. The monomer was used as purchased without any need for initial purification, and polymerization was carried out at room temperature and was stopped by immersing samples in deionised water, rinsing with ethanol and drying under a stream of nitrogen.
:1 (v/v), 15 mL) was used. For PAA, PDMAEMA and POEGMA brushes polymerization times were adjusted accordingly to give dry ellipsometric thicknesses of 30 nm; these thicknesses equated to polymerization times of 5 h, 20 min and 30 min, respectively.
Fourier Transform Infrared-Attenuated Total Reflectance Spectroscopy (ATR-FTIR) was carried out using a Bruker Tensor 27 with an MCT detector (liquid N2 cooled). Spectra were acquired at a resolution of 4 cm−1 with a total of 256 scans per run. Nitrogen was run through the system during measurements in order to limit the effects of environmental fluctuations. Contact angle measurements (using the sessile drop method) were taken using a Kruss DSA100 instrument using 3 μL droplets of deionised water. Measurements were carried out on three independent samples and for each sample an average reading was taken using measurements taken from three separate regions of the sample.
XPS was carried out on a Thermo Scientific K-Alpha X-ray XPS System using a monochromatic AlKα source. This was operated at 150 W under ultrahigh vacuum conditions (10−9 mbar). As the surfaces were not electrically conductive, a built-in spectrometer charge neutralizing system was used which compensates for sample charging during measurements by flooding the surface with low energy electrons (<20 eV). To be able to compare data, the C1s hydrocarbon peak at 285.0 eV was used as an external standard for calibration of the binding energy (BE) scale. In order to identify the main peaks by their binding energies, survey spectra were collected from 1200 to 0 eV. In addition, high-resolution XPS spectra were acquired for O1s, C1s, and Si2p signals. An analyzer pass energy of 1 eV and 0.1 eV was used for the survey spectra and high resolution spectra, respectively.
2.3. Synthesis of polymer brush-coated silica nanoparticles
For grafting of the silane initiator the beads were transferred from water to ethanol and then to toluene using the aforementioned method. 50 mg of the beads were dispersed in a solution of anhydrous toluene (1 mL), triethylamine (50 μL) and silane initiator (10 μL) and left stirring overnight. The beads were then washed in toluene three times using the sonication-centrifugation method and then transferred to acetone and left in 1 mL of the solvent in the fridge ready for the polymerization step.
Polymerization was then carried out at 60 °C whilst stirring. Degassing was continued for a further 30 min and the reaction vessel was then covered with parafilm. To terminate polymerization, the reaction mixture was bubbled with oxygen for a couple of min. The SiO2–polymer suspension was then centrifuged, washed in acetone three times and transferred into DCM/TFA (10:1 (v/v), 1 mL) and left stirring overnight at room temperature. The resulting PAA beads were washed in DCM three times and transferred into acetone for storage in the fridge.
For synthesis of PDMAEMA polymer brushes, 5 mL of the DMAEMA monomer solution (see Section 2.2 for preparation protocol) was added to a 5 mL silica dispersion (50 mg SiO2–silane beads in water/ethanol (4:1 (v/v)), degassed under argon for 30 min). Argon bubbling was continued and polymerization was allowed to proceed at room temperature. The reaction was stopped with oxygen bubbling and the beads were washed in water/ethanol (4:1 (v/v)). Synthesis of POEGMA polymer brushes was identical except that 5 mL of the OEGMA monomer solution (see Section 2.2) was instead added to the silica bead dispersion. The grafting density of polymer brushes that were previously grown from silica nanoparticles using this protocol was 0.5 chains per nm2, with dry thicknesses of 30 nm (and a Mn of 50 kDa).30
2.4. Formation of self-assembled monolayers (SAMs) on silicon substrates
The Table S1 (ESI†) gathers the contact angle data and ellipsometric thicknesses for all SAMs generated on silicon and gold-coated substrates.2.5. Colloidal probe force microscopy
 | ||
| Fig. 1 Quantitative analysis of colloidal probe adhesion profiles. (A) Extraction of detachment work and force from AFM retraction curves. (B) Physical representation of the detachment length, taken from the retraction portion of the AFM curves, and the three different polymers which are used to functionalise the colloidal probes. The detachment length is defined as the distance between the colloidal probe and the substrate at the maximum negative force value. | ||
For measurements on soft tissues, the Young's modulus was additionally characterised from the AFM curves. This parameter was quantified using a custom-built Matlab script based on the Oliver–Pharr method for nanoindentation.59
2.6. Preparation of cell monolayers
000 cells per well (13000 cells per cm2), and left to form a confluent monolayer for 24 h in an incubator (37 °C and 5% CO2). After three aspirations with PBS, confluent monolayer samples were fixed with 4% formaldehyde solution in PBS (PFA) for 10 min, washed with PBS and left in the fridge ready for AFM measurements.
000 cells per well (13000 cells per cm2) and left to form a confluent monolayer for 24 h in an incubator (37 °C and 5% CO2). After three aspirations with PBS, confluent monolayer samples were fixed with 4% PFA for 10 min, washed with PBS and left in the fridge ready for AFM measurements.
:1000) were prepared by dissolving 5 mg 4′,6-diamidino-2-phenylindole dihydrochloride in 1750 μL deionised water. Goat anti-mouse IgG (H + L) secondary antibodies conjugated to AlexaFluor 488 (1 μg mL−1) were used for immunofluorescence staining. Fluorescence microscopy images were acquired with a Leica DMi8 fluorescence microscopy (CTR compact lamp; 63 × 1.30 NA, oil lens; DFC9000 GT camera). For each sample and condition three fluorescence microscopy images were taken. Quantification of the fluorescence intensities and profiles was then performed using ImageJ.
2.7. Preparation of tissue samples
Fresh soft tissue samples were obtained from C Humphreys & Sons Abattoir in Chelmsford, Essex. Both the epicardial and gingival samples were extracted from freshly slaughtered 6 to 7-month-old pigs. The animals were sacrificed and the relevant parts were extracted at the abattoir; these were then delivered by courier to the lab on the same day as the slaughter. The samples were then washed and stored in PBS overnight at 5 °C. All AFM adhesion testing was then carried out within 48 hours from the initial sacrifice of the animals.Gingival samples were extracted using a scalpel from the lingual side of the lower mandible. These samples were taken from the region of the keratinized attached gingiva which is the gingival portion that lies between the free gingival groove and the mucogingival junction (Fig. S2B, ESI†). Epicardial samples were extracted from the wall of the left and right ventricles. These samples were taken from the healthy areas of the myocardium away from any major adipose tissue deposits or prominent blood vessels (Fig. S2C, ESI†). For AFM testing, results were obtained across samples from at least two different animals for each condition tested. Fig. S2A (ESI†) shows an example of loaded epicardial samples in the AFM.
2.8. Statistics
A one-way ANOVA test with Tukey's post hoc analysis was used to determine statistical significance. In the case of the quantification of the glycocalyx, the analysis was carried out pairwise. For box and whisker diagrams the box represents the 1st, 2nd and 3rd quartiles, as standard, and the whiskers represent the standard deviations of data sets. Mean values for the data sets are also shown. In all other figure types (and for in-text referencing) standard errors are reported.3. Results and discussion
3.1. Development of polymer-brush functionalised colloidal probes
To study polymer brush interactions with cell monolayers and tissues, we grew polymer brushes from silica microparticles (19.6 μm), via atom transfer radical polymerization (ATRP). The growth of PDMAEMA and POEGMA brushes from silica particles was directly adapted from protocols previously reported.29,60 To generate PAA brushes, we opted for the controlled growth of PtBA brushes, prior to their conversion upon mild treatment with dilute trifluoroacetic acid.52,61 The growth kinetics of the PtBA brushes (monitored via ellipsometry) was 3–4 fold faster than that previously reported in similar conditions, but appeared relatively linear (Fig. 2A). However, Lego et al. had monitored brush growth via AFM, potentially underestimating thicknesses measured, due to the compression of the brush by the AFM tip.62 In addition, the silane monolayers generated in the present study may display a higher density, resulting in higher brush densities and thickness growth profiles, although direct comparison is not possible.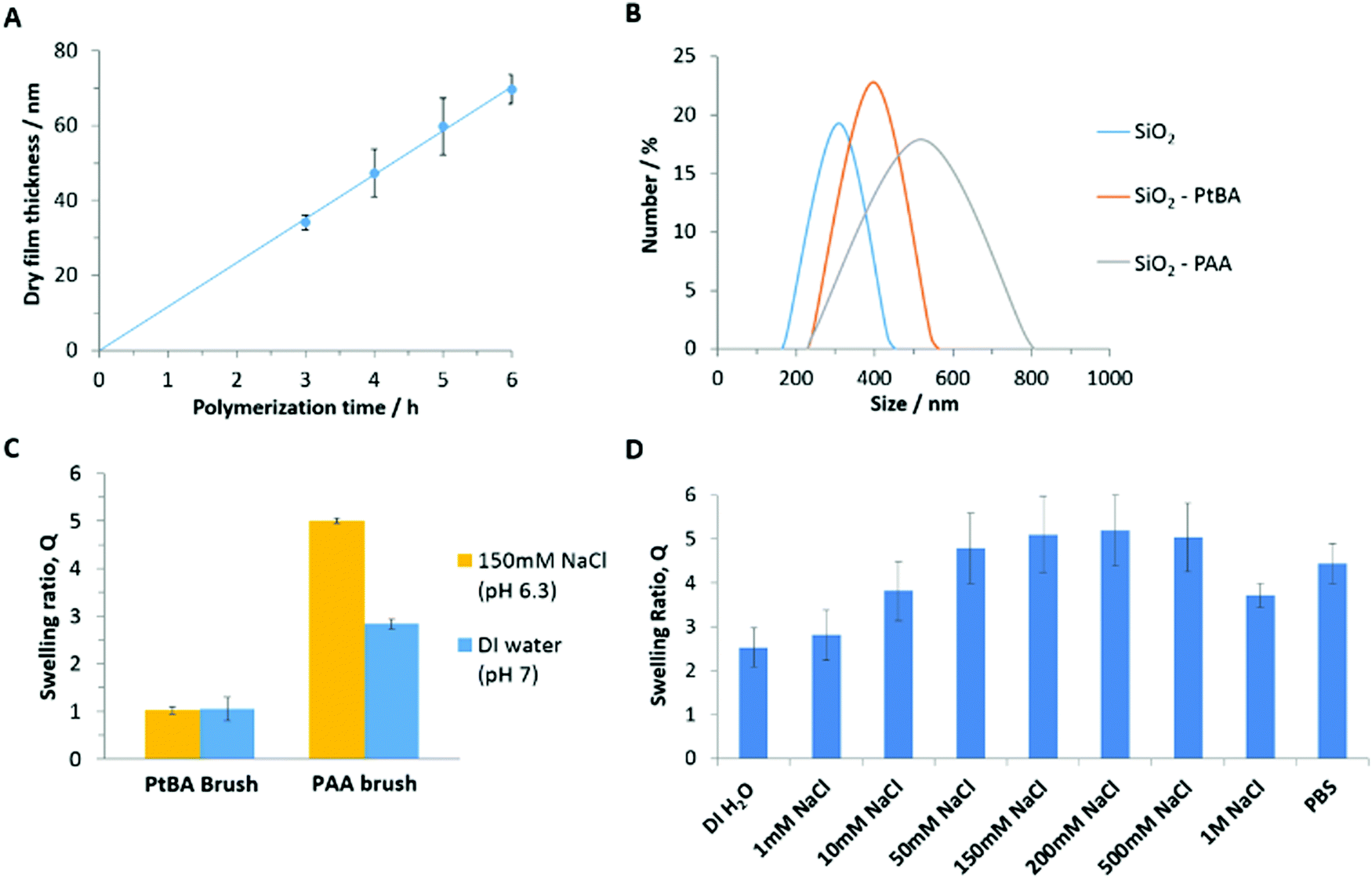 | ||
| Fig. 2 Functionalisation of silicon substrates and silica microparticles with PAA brushes. (A) Kinetics of the growth of PtBA polymer brushes from silicon substrates, monitored by ellipsometry. (B) Dynamic light scattering data of functionalised and non-functionalised 300 nm silica microparticles. SiO2 beads and PtBA functionalised beads were dispersed in ethanol and PAA in deionised water. (C) Ellipsometric swelling ratio measured for PtBA and PAA brushes in deionised water (DI) and 150 mM NaCl aqueous solutions. (D) Ellipsometric swelling ratio of PAA brushes (pH kept at 7.0 ± 0.4, except for PBS which had a pH of 7.4). | ||
The deprotection of PtBA brushes was subsequently monitored by ellipsometry (Fig. S3, ESI†), indicating a rapid decrease in the dry thickness of the brush upon incubation in DCM/TFA solutions, due to the loss of tert-butyl groups and associated chain relaxation. After overnight incubation in the DCM/TFA solution, the polymer thickness dropped to around 50% of its original value. This is in line with previous reports61 and with the predicted reduction in molar mass of repeat units and associated dry film thickness (52%).63 The FTIR spectra of PtBA brushes before and after deprotection confirmed the full conversion to PAA brushes (Fig. S4, ESI†), with the clear disappearance of the typical tert-butyl bending bands at 2976, 1392 and 1367 cm−1, and the shift (and broadening) of the carbonyl band from 1730 to 1712 cm−1.61,64 This conversion is further confirmed by XPS spectra of the PtBA brushes before and after deprotection (Fig. S5, ESI†). The C1s range of the spectra was deconvoluted into three peaks corresponding to carbon atoms associated with C–C, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks. The peak associated with carbon atoms involved in C–O bonds was found to shift and broaden upon deprotection. In addition, the atomic percentages and corresponding binding energies of the C1s and O1s signals were extracted from the XPS survey spectra (Fig. S5C, ESI†), clearly indicating a reduction in the carbon content relative to oxygen following the deprotection of tBA groups. Both observations are in accordance with the expected change in the chemical structure of the brush, and in agreement with previously reported XPS characterisation data for these surfaces.65–67
O peaks. The peak associated with carbon atoms involved in C–O bonds was found to shift and broaden upon deprotection. In addition, the atomic percentages and corresponding binding energies of the C1s and O1s signals were extracted from the XPS survey spectra (Fig. S5C, ESI†), clearly indicating a reduction in the carbon content relative to oxygen following the deprotection of tBA groups. Both observations are in accordance with the expected change in the chemical structure of the brush, and in agreement with previously reported XPS characterisation data for these surfaces.65–67
The hydrodynamic diameter of nanoparticles coated with PtBA and PAA brushes was compared to that of uncoated nanoparticles (see Fig. 2B). The diameter of uncoated nanoparticles was found to be 320 ± 10 nm and increased to 390 ± 10 nm and 530 ± 10 nm for SiO2–PtBA and SiO2–PAA particles, respectively. This is in good agreement with the expected dry thickness of PtBA brushes (not swollen in ethanol), which were found to grow to 60 nm in 5 h, from silicon substrates (Fig. 2A). The significant increase in hydrodynamic diameter observed for PAA-coated nanoparticles is in agreement with their pronounced swelling in deionised water (the diameters measured correspond to a swelling greater than 3) and the decrease in hydrophobicity associated with the deprotection of tBA groups. In addition, the ζ-potentials measured for SiO2–PtBA and SiO2–PAA particles were 23.1 ± 0.1 mV and −29.3 ± 0.1 mV, respectively, although these values are only qualitatively indicating the charge of the associated particles (PtBA particles did not resuspend in aqueous solutions, therefore preventing direct comparison with PAA-coated particles). Hence the charge reversal of the ζ-potential is consistent with a significant change in surface chemistry of these particles and the introduction of negatively charged carboxyl groups following the deprotection step.
The swelling of the corresponding brushes was further characterised via in situ ellipsometry. The swelling ratio (Q), defined as the ratio between the wet (L) and dry (D) ellipsometric thicknesses (Q = L/D), was close to unity prior to deprotection of PtBA brushes (Fig. 2C), as expected from the hydrophobicity of tert-butyl acrylate repeat units. In contrast, PAA brushes were characterised by high swellings in deionised water and 150 mM NaCl solutions; this is in agreement with the expected behaviour of weak polyelectrolytes in the osmotic regime, characterised by high proton dissociation at neutral pH, substantial electrostatic repulsion between repeat units and the associated stretching of polymer chains.39,63,68 The swelling ratio of PAA brushes was further characterised in PBS and in aqueous NaCl solutions with a range of ionic strengths (Fig. 2D). The swelling was observed to initially increase with the ionic strength, due to increasing proton dissociation and associated hydrophilicity. As the salt concentration increased further, the brush appeared to enter the neutral brush and salted brush regime and electrostatic interactions became largely screened, resulting in a decrease in brush thickness.63,68 Finally, contact angle measurements were in agreement with the increase in hydrophilicity and brush swelling. Following the deprotection of PtBA brushes, average contact angles shifted from 87.0 ± 0.6° to 37.4 ± 0.7° for PtBA and PAA brushes, respectively (Table S1, ESI†).
3.2. Polymer brush adhesion to SAMs
Prior to studying polymer brush adhesion to cells and tissues, we first investigated their adhesion to model substrates (self-assembled monolayers) presenting chemical moieties with a range of hydrophilicities, hydrogen bonding and charge. Fig. 3 presents the detachment forces observed for the adhesion of PAA, PDMAEMA and POEGMA brushes to hydroxyl-terminated monolayers alongside the corresponding representative lift curves. Overall, similar adhesions were measured to both 11-mercapto-1-undecanol monolayers assembled to gold and silicon oxide interfaces. As the surface packing density for monolayers assembled on silica24,69 and gold70 substrates are reported to be in the region of 5 molecules per nm2, the similarity of these adhesion profiles is justified. POEGMA displayed little adhesion to these interfaces, in good agreement with its neutral structure, lacking strong proton acceptor or donor functions. POEGMA brushes indeed typically display moderate hydrophilicity and excellent anti-fouling properties.71,72 In contrast, PAA and PDMAEMA displayed moderate adhesions to both surfaces. At the near-neutral pH at which these measurements were carried out (the pH of deionised water and NaCl solutions was in the range of 7.0 ± 0.4, whereas that of PBS solutions was 7.4), PAA brushes are globally negatively charged (we found a ζ-potential of −29 mV)73,74 and PDMAEMA brushes are positively charged (ζ-potential of 40 mV),29,75 whereas silica substrates are negatively charged (ζ-potential of −40 mV).74,76 Hence, in the case of PDMAEMA brushes, electrostatic interactions should lead to increased adhesion. However, PAA and PDMAEMA brush adhesion (detachment force) to silanol and 11-mercapto-1-undecanol monolayers are typically comparable and, in some cases, increased in the case of PAA (in deionised water and in PBS, in the case of 11-mercapto-1-undecanol monolayers; Fig. 3A and C). In addition, higher ionic strength (150 mM NaCl) did not lead to substantial reduction in adhesion force, as would be predicted in the case of oppositely charged surfaces. These changes were qualitatively reflected in the corresponding detachment work (see Fig. S6A and B, ESI†). Therefore, weak adhesion of PAA and PDMAEMA brushes to moderately hydrophilic and moderately charged silanol and 11-mercapto-1-undecanol monolayers appeared to be dominated by weak hydrogen bonding. In PBS solutions, adhesion forces measured for PDMAEMA and POEGMA brushes remained comparable to those measured in 150 mM NaCl solutions. Indeed, PBS has a NaCl concentration of 137 mM, similar to that of the NaCl solution used, and the pH of both solutions were relatively close (pH 7.4 and pH 7.0 ± 0.4, for PBS and NaCl, respectively). For PAA brushes, adhesion forces were increased in PBS compared with 150 mM NaCl (forces increased from 0.69 to 1.29 nN and 0.82 to 6.60 nN for silanol and 11-mercapto-1-undecanol SAMs, respectively). This phenomenon is associated with a reduction of the swelling of PAA brushes in PBS, compared to 150 mM NaCl, and may indicate the contribution of phosphate to hydrogen bonding at the surface.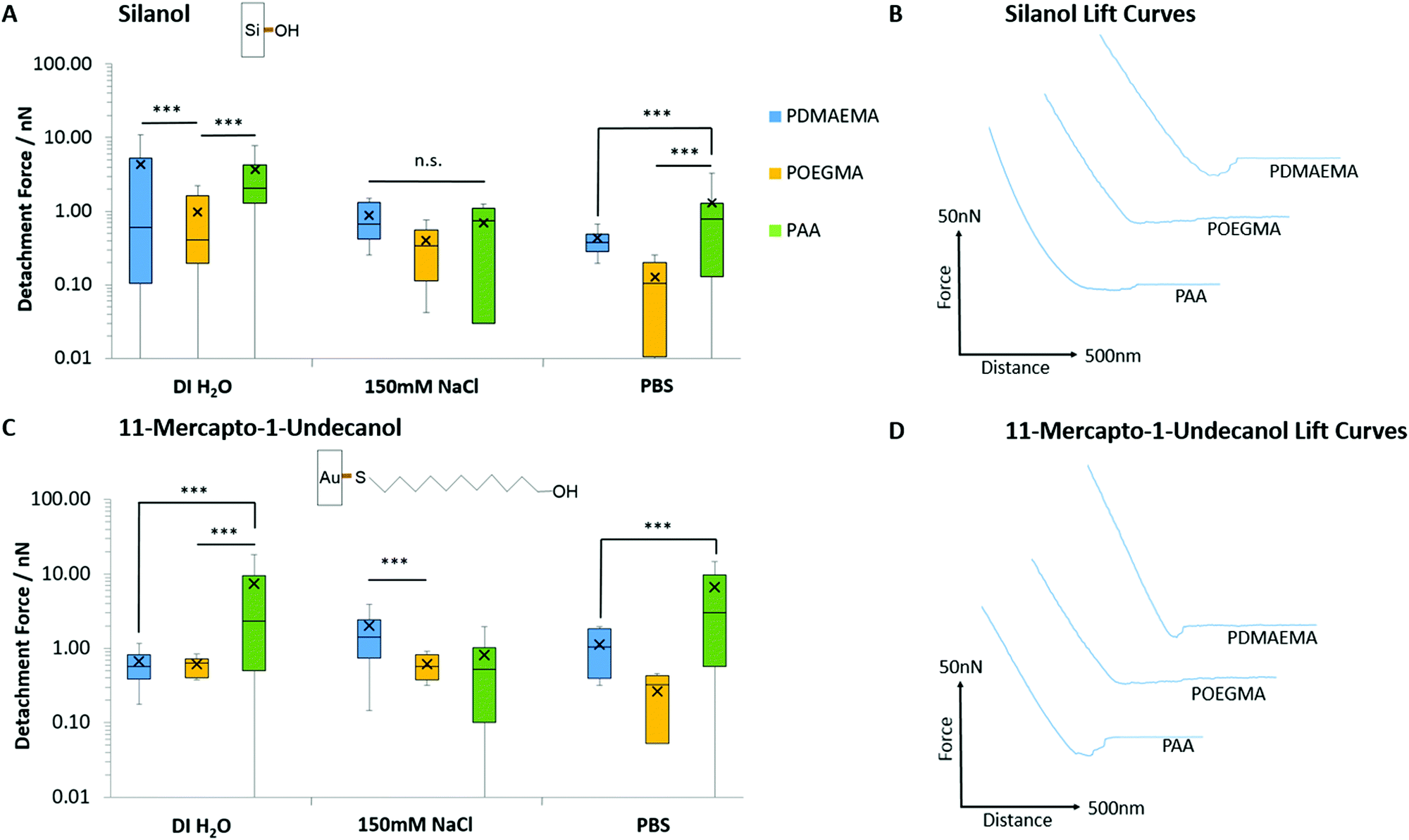 | ||
| Fig. 3 Detachment forces and corresponding representative lift curves for the adhesion of PAA, PDMAEMA and POEGMA brushes to hydroxyl-terminated monolayers. Representative curves are taken from adhesion experiments performed in deionised water. (A) Detachment forces to silicon oxide interfaces. (B) Representative lift curves for adhesion to silanol monolayers. (C) Detachment forces to 11-mercapto-1-undecanol monolayers assembled on gold-coated silicon substrates. (D) Representative lift curves for adhesion to 11-mercapto-1-undecanol monolayers. Data is plotted as means, with box plots. ***p ≤ 0.001. n.s., non-significant. | ||
Fig. 4 presents the adhesion forces measured for the three different polymer brushes to charged model substrates (associated detachment work values are given in Fig. S6, ESI†). As expected, negatively charged PAA brushes strongly adhered to positively charged quaternary ammonium monolayers (QAPTES); this is further evidenced in Fig. 5A where the jump-to-contact is indicative of a strong attractive interaction. In the corresponding representative lift curves (Fig. 5B), it is also clear that interactions between PAA brushes and oppositely charged monolayers (APTES and QAPTES) are strong (adhesion forces >10.16 nN in deionised water) and decrease at higher ionic strength (Fig. 4D), as would be expected from the corresponding screening of coulombic forces. Indeed, at neutral pH we found ζ-potentials of 13 mV77,78 and 37 mV for APTES and QAPTES, respectively, whereas ω-mercaptoundecanoic acid (MUDA) displays a ζ-potential of −40 mV.79 This behaviour contrasts with the adhesion of poly(2-vinylpyridine), which displayed increased adhesion forces at higher ionic strength.80,81 This was attributed to changes in brush conformation from the salted regime to the osmotic regime. We also note that the ionic strength range at which brush adhesion was maximised was above the range of ionic strength presently tested (matching that of physiological buffers such as PBS). The neutral POEGMA, in contrast, displayed interactions below 3.45 nN, comparable to what was measured on neutral hydroxyl functionalised interfaces (detachment forces were reduced for hydroxyl SAMs in comparison to charged monolayers, however, from the detachment work (Fig. S6, ESI†), interactions are of a similar scale), in agreement with its neutral charge. The adhesive response of PDMAEMA to these three charged monolayers was more surprising as, although it adhered weakly to APTES surfaces (<4.05 nN), its adhesion to MUDA was not as significantly increased (<5.93 nN), as was observed in the case of PAA adhering to APTES interfaces. This indicates that other interactions dominate the adhesive behaviour of PDMAEMA to the charged monolayers studied.
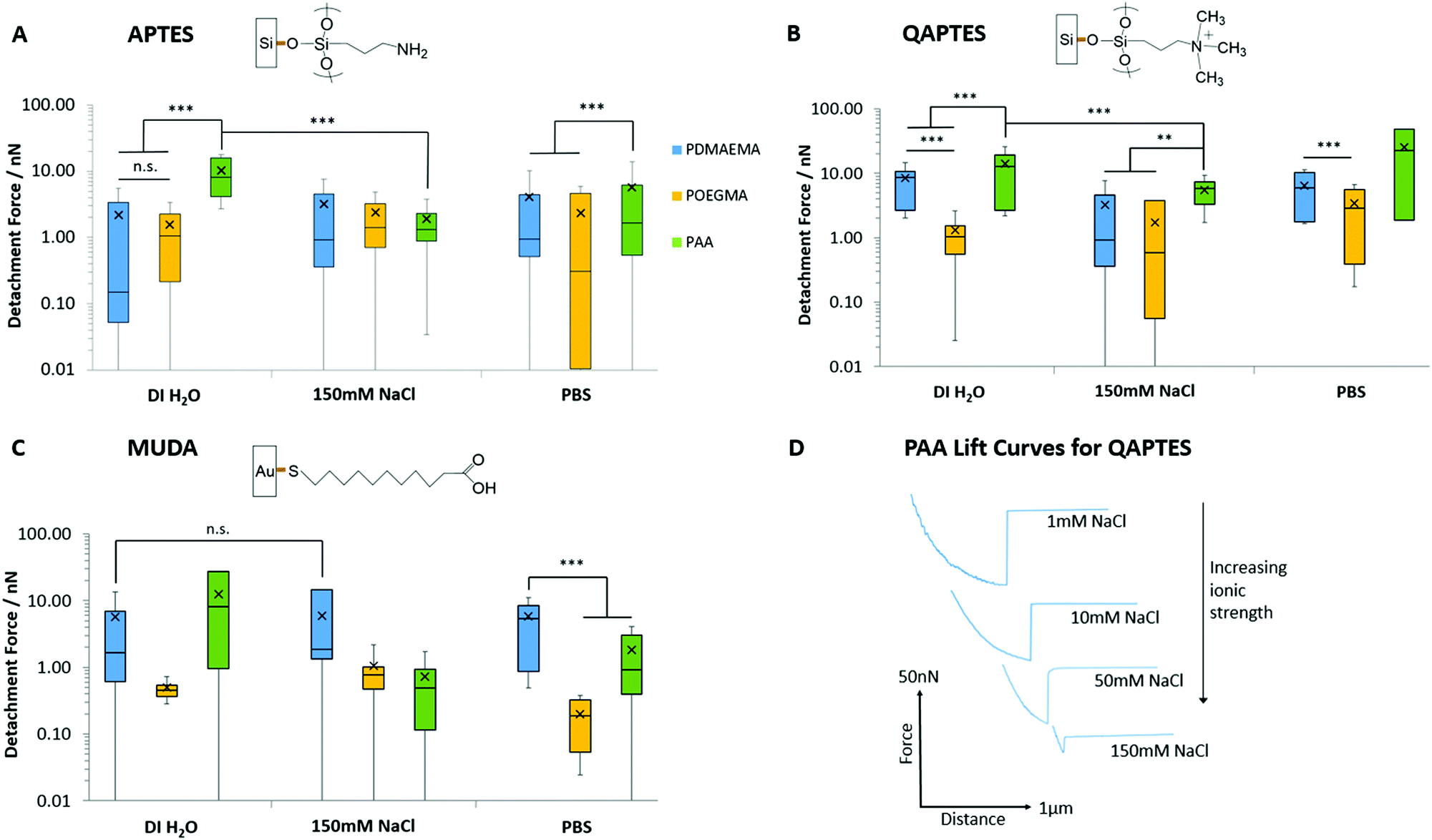 | ||
| Fig. 4 Detachment forces measured for the adhesion of PAA, PDMAEMA and POEGMA brushes to charged monolayers. (A) Detachment forces to ammonium (APTES) monolayers. (B) Detachment forces to quaternary ammonium (QAPTES) monolayers. (C) Detachment forces to undecanoic acid (MUDA) monolayers assembled on gold-coated silicon substrates. (D) Representative lift curves for adhesion of PAA to QAPTES monolayers in solutions of increasing ionic strengths. Data is plotted as means, with box plots. **p ≤ 0.01. ***p ≤ 0.001. n.s., non-significant. | ||
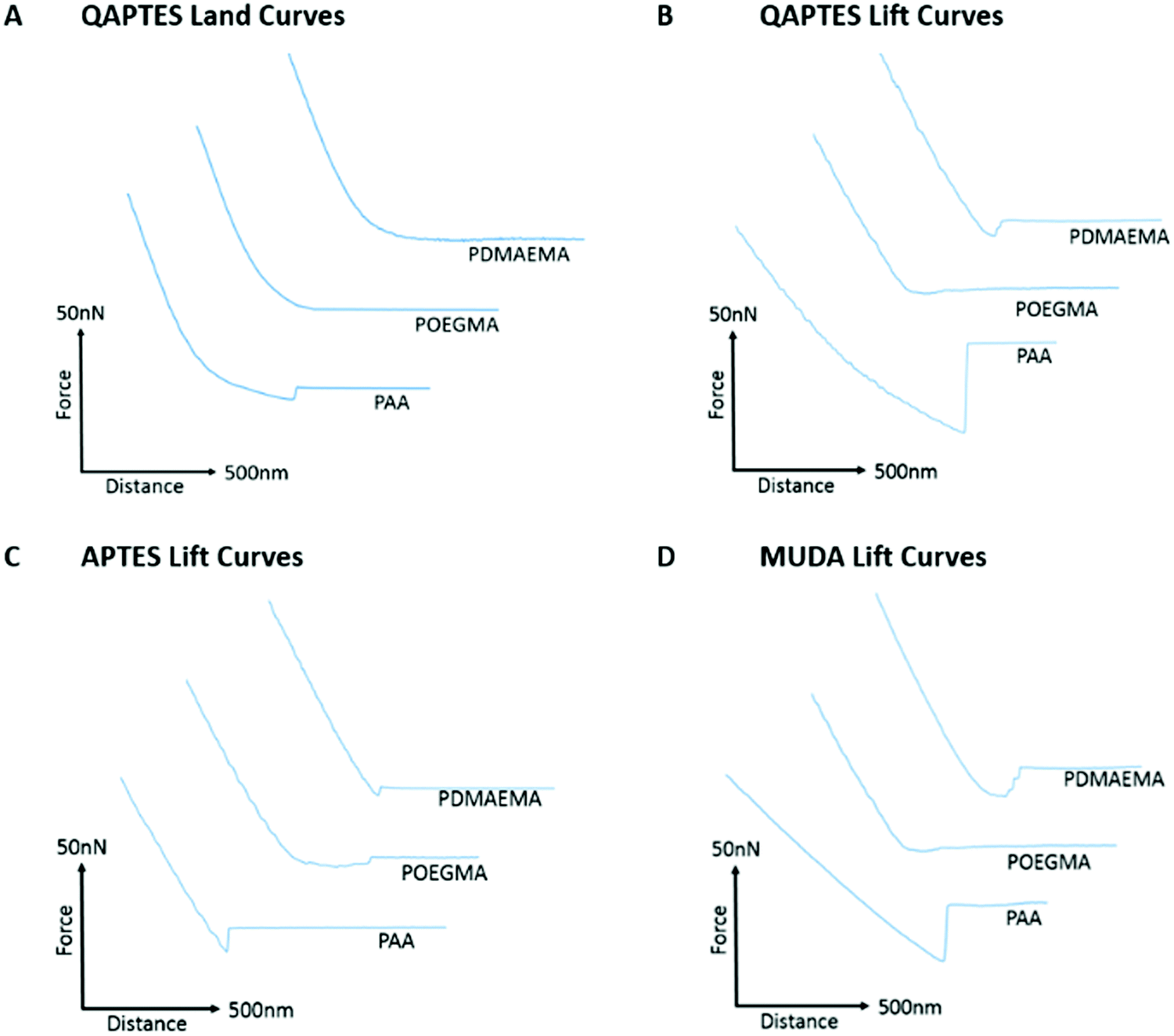 | ||
| Fig. 5 Representative lift and land curves for the adhesion of PAA, PDMAEMA and POEGMA brushes to charged monolayers. All curves are taken from adhesion experiments performed in deionised water. (A) Land curves representative of adhesion of brushes to QAPTES monolayers. (B) Lift curves representative of adhesion to quaternary ammonium (QAPTES) monolayers. (C) Lift curves representative of adhesion to ammonium (APTES) monolayers. (D) Lift curves representative of adhesion to undecanoic acid (MUDA) monolayers assembled on gold. | ||
In addition to electrostatic interactions, hydrogen bonding may significantly impact the adhesion strength of PAA and PDMAEMA brushes to charged monolayers, owing to their acid/base character. Indeed, in contrast to PAA brushes, which showed a marked decrease in interactions at increasing ionic strength, the adhesion of PDMAEMA brushes to MUDA monolayers remained unaffected by increasing ionic strength (150 mM NaCl vs. deionised water; Fig. 4C). At neutral pH (and slightly below), PDMAEMA is only partially charged and a substantial proportion of amine moieties are not protonated.29,82 Similarly, APTES and MUDA monolayers and PAA brushes will only be partially deprotonated at the neutral pH of the solutions used in this study. Therefore, hydrogen bonding between polymer brushes and APTES and MUDA monolayers may significantly contribute to bonding and adhesion profiles. Furthermore, although hydrogen bonding is reported to be influenced by the ionic strength of the medium, predicting the impact of electrolytes on hydrogen bonding remains difficult. Hence, cations are known to alter the melting temperature of double-stranded oligonucleotides, although this is via their combined impact on coulombic repulsion between phosphates and on hydrogen bonding between bases.83 Electrolytes were shown to reinforce hydrogen bonding84 and to perturb networks of intra-molecular hydrogen bonds of water molecules.85 In addition, electrolytes such as phosphates are particularly prone to hydrogen bond and alter interactions with biomacromolecules.86,87 The relatively high interactions of PAA and PDMAEMA to MUDA and APTES, respectively (Fig. 4 and 5), in particular in PBS solutions, may therefore be explained by hydrogen bonding between the corresponding interfaces, perhaps stabilised by phosphate ions.
Further to the evidence for strong hydrogen bonding between polymer brushes and monolayers, the adhesion behaviour of PDMAEMA brushes to QAPTES also implies some impact of hydrophobic interactions on adhesion strength (Fig. 4B and 5B). Indeed, QAPTES is highly positively charged and cannot directly be involved in hydrogen bonding with other molecules. Therefore, the high charge density of cationic PDMAEMA (at neutral pH) should result in repulsive forces. In contrast to this expected behaviour, we observed no evidence of repulsion in the corresponding landing traces (Fig. 5A) and relatively strong adhesions can be observed in the retraction traces (Fig. 5B). Quaternary ammoniums such as those of QAPTES are known to display some level of hydrophobicity, enabling the solubility of salts in some organic solvents. In addition, PDMAEMA also shows significant hydrophobicity and a clear pH-responsive behaviour, as detailed above, especially close to its pKa and above.29 Therefore, hydrophobic interactions between these two interfaces are likely to play an important role in determining adhesion profiles.
In order to probe further into the impact of hydrophobic forces in regulating adhesion of PAA, PDMAEMA and POEGMA brushes, we generated octyl and perfluorooctyl SAMs on silicon substrates (with contact angles of 96.3 ± 1.3° and 103.7 ± 1.4°, respectively). Significant adhesion was measured for PDMAEMA and PAA brushes, whilst POEGMA brushes displayed lower adhesions, but higher than what was reported for hydrophilic SAMs (Fig. 6; detachment works follow similar trends, see Fig. S6, ESI†). In particular, PDMAEMA displayed strong adhesion to octyl SAMs (>5.13 nN) and the perfluorooctyl SAMs (10.30 nN in 150 mM NaCl solutions). PAA displayed overall weaker adhesion to octyl SAMs, especially at high ionic strength, but relatively high adhesion to perfluorooctyl SAMs. The relatively strong adhesion of weak polyelectrolyte brushes to hydrophobic surfaces is likely due to their partial protonation/deprotonation at neutral pH and associated moderate hydrophobicity.29,82 Hence, PDMAEMA brushes were found to adhere relatively strongly to PDMAEMA surfaces at neutral pH,88 whereas little adhesion was observed between two symmetrical hydrophilic polymer brushes.89 Similarly, polymer brushes displaying a Lower Critical Solution Temperature (LCST) were reported to strongly adhere (symmetrical interface bonding) above their LCST, but displayed weak interactions below their LCST.90 Interestingly, the adhesion force of sparse (grafted to) PAA brushes was also reported to be significantly higher to alkyl monolayers than corresponding hydroxyl and carboxylic acid-terminated SAMs,91,92 although this was for single desorption events rather than the cumulative desorption forces associated with full detachment of the tip. To account for such strong interactions between PAA brushes and hydrophobic SAMs, Friedsam et al. proposed that the structure of water close to these interfaces (and the lack of hydrogen bonding of interfacial water molecules, in contrast to the networks formed with hydroxyl and carboxylic acid terminated SAMs) led to easier displacement of water molecules closely associated with the hydrophobic SAMs, compared to hydrophilic SAMs. Indeed, the profiles of desorption of our PAA brushes displayed strong adhesion forces with a sharp detachment step in the case of alkyl SAMs (>6.43 nN detachment force, >2.78 fJ detachment work and <22.3 nm detachment lengths; Fig. S7, ESI†), whereas detachment from hydrophilic silanol SAMs displayed an overall weak adhesion force, but a more gradual detachment profile and increased detachment length (3.69 nN detachment force, 0.48 fJ detachment work and 68.4 nm detachment length; Fig. S7, ESI†). Such difference in the structure of water at hydrophobic and hydrophilic SAMs is also supported by molecular dynamics studies that give evidence for the occurrence of 0.3 nm gaps between vicinal water and hydrophobic SAMs.93 Similarly, such water structuring was found to impact the adhesion of moderately hydrophobic peptides presenting catechol residues (DOPA), which showed increased bonding forces to hydrophobic SAMs, despite increased hydrogen bonding to hydrophilic SAMs.94 Hence, our results also support the occurrence of an aqueous interfacial layer that differentially regulates adhesion of moderately hydrophobic polymer brushes to hydrophobic SAMs (Fig. 7).
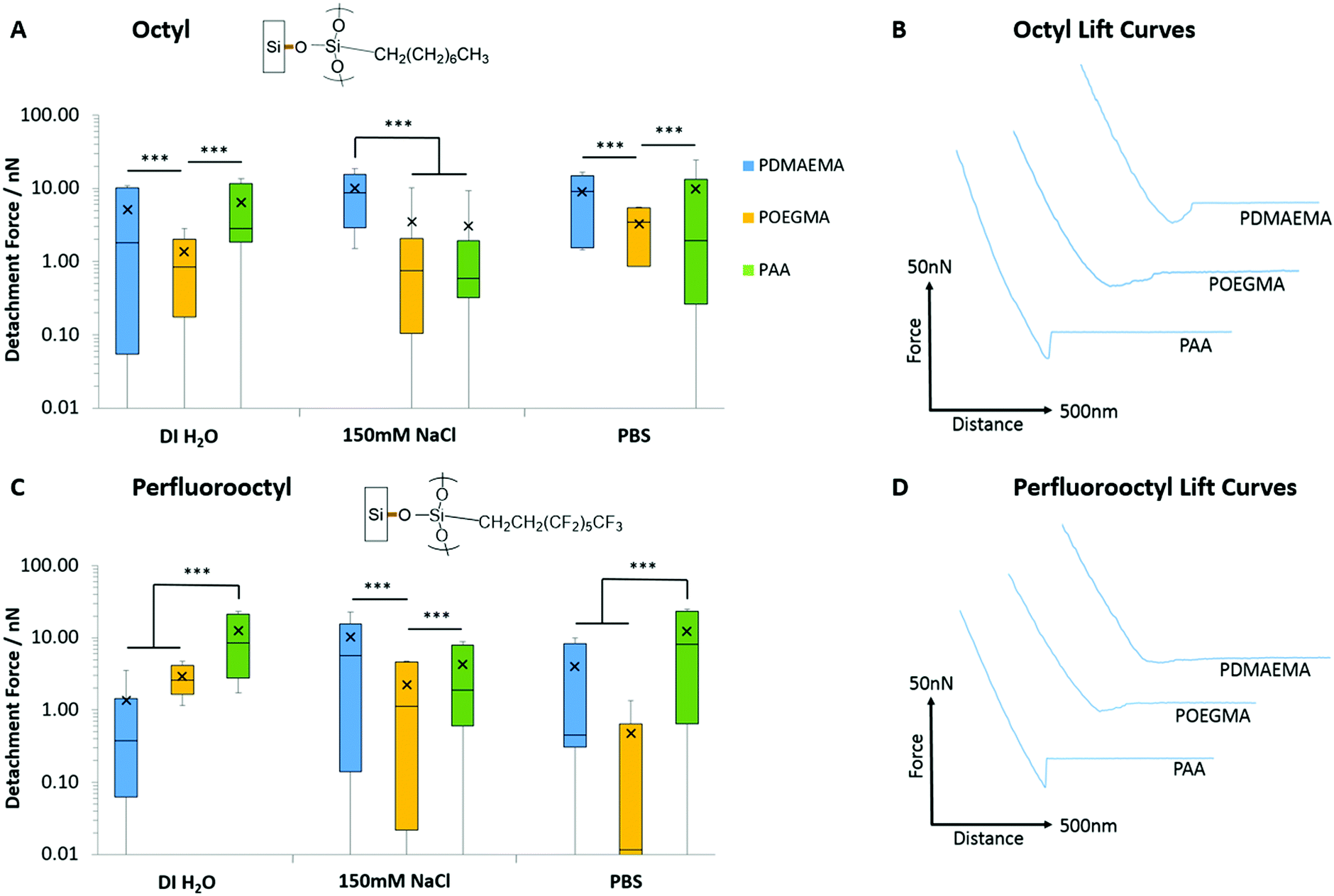 | ||
| Fig. 6 Detachment forces and corresponding representative lift curves for the adhesion of PAA, PDMAEMA and POEGMA brushes to hydrophobic monolayers. Representative curves are taken from adhesion experiments performed in deionised water. (A) Detachment forces to octyl monolayers. (B) Representative lift curves for adhesion to octyl monolayers. (C) Detachment forces to perfluorooctyl monolayers. (D) Representative lift curves for adhesion to perfluorooctyl monolayers. Data is plotted as means, with box plots. ***p ≤ 0.001. | ||
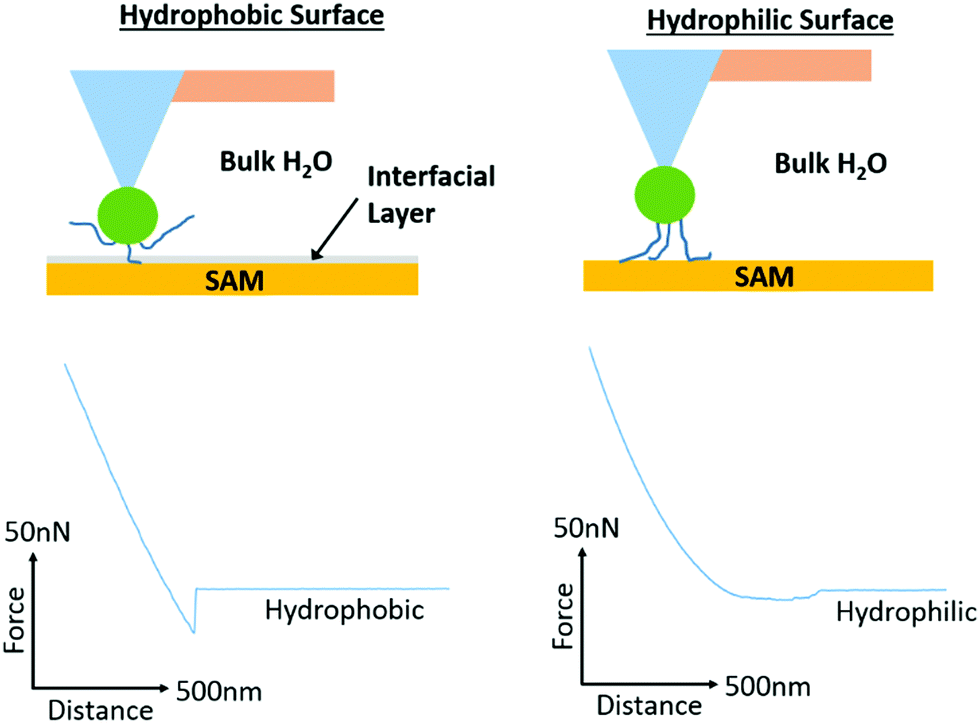 | ||
| Fig. 7 Proposed mechanism of adhesion of PAA brushes to hydrophobic and hydrophilic monolayers. Representative lift curves are taken from adhesion measurements for alkyl (octyl and perfluorooctyl) and hydrophilic (silanol) SAMs submerged in deionised water. The desorption profiles indicate strong adhesion forces, with a sharp detachment step in the case of alkyl SAMs but overall weak adhesion forces with a more gradual detachment profile for hydrophilic SAMs. These profiles support the occurrence of an aqueous interfacial layer that differentially regulates adhesion of moderately hydrophobic polymer brushes to hydrophobic SAMs.91,92 | ||
3.3 Polymer brush adhesion to cell monolayers
Having studied the impact of substrate chemistry in a set of monolayers with a range of hydrophobicity/hydrophilicity, charge and hydrogen bonding potential, we next turned our attention to the adhesion profile of weak polyelectrolyte brushes and POEGMA to epithelial cell sheets. These biointerfaces can be regarded as simplified systems to the understanding of tissue bonding due to the greater cellular homogeneity within cultured cell sheets, their planarity and relative rigidity (at the macroscale, owing to the rigidity of the underlying substrate). We focused on two epithelial models: HaCaT cells (a human epidermal cell line) and primary keratinocytes (stem cells responsible for the homeostasis of the interfollicular epidermis).95Fig. 8A and B present the adhesion profiles and quantification of adhesion forces of POEGMA, PDMAEMA and PAA brushes to cell sheets of keratinocytes and HaCaT cells. Due to the high density and the length of their oligo(ethylene glycol) side chains, POEGMA brushes are known for their excellent protein and bacterial resistance.71,72 The exact detailed mechanism of their protein resistance remains only partially understood, but was proposed to result from their combined hydration shell and steric hindrance (and high chain densities), restricting the infiltration of biomacromolecules.49,50 In agreement with these reports, POEGMA brushes were found to display very low adhesion to both HaCaT and primary keratinocyte monolayers, with adhesion forces below 0.29 nN (Fig. 8A and B). Such adhesions are lower than those measured for POEGMA, in the case of hydrophobic or charged SAMs, indicating relatively hydrophilic and weakly charged cell membranes. This is in contrast with the moderate adhesion forces measured between POEGMA brushes and bacteria,27 which typically display relatively charged and hydrophobic membranes (with lipopolysaccharides).26
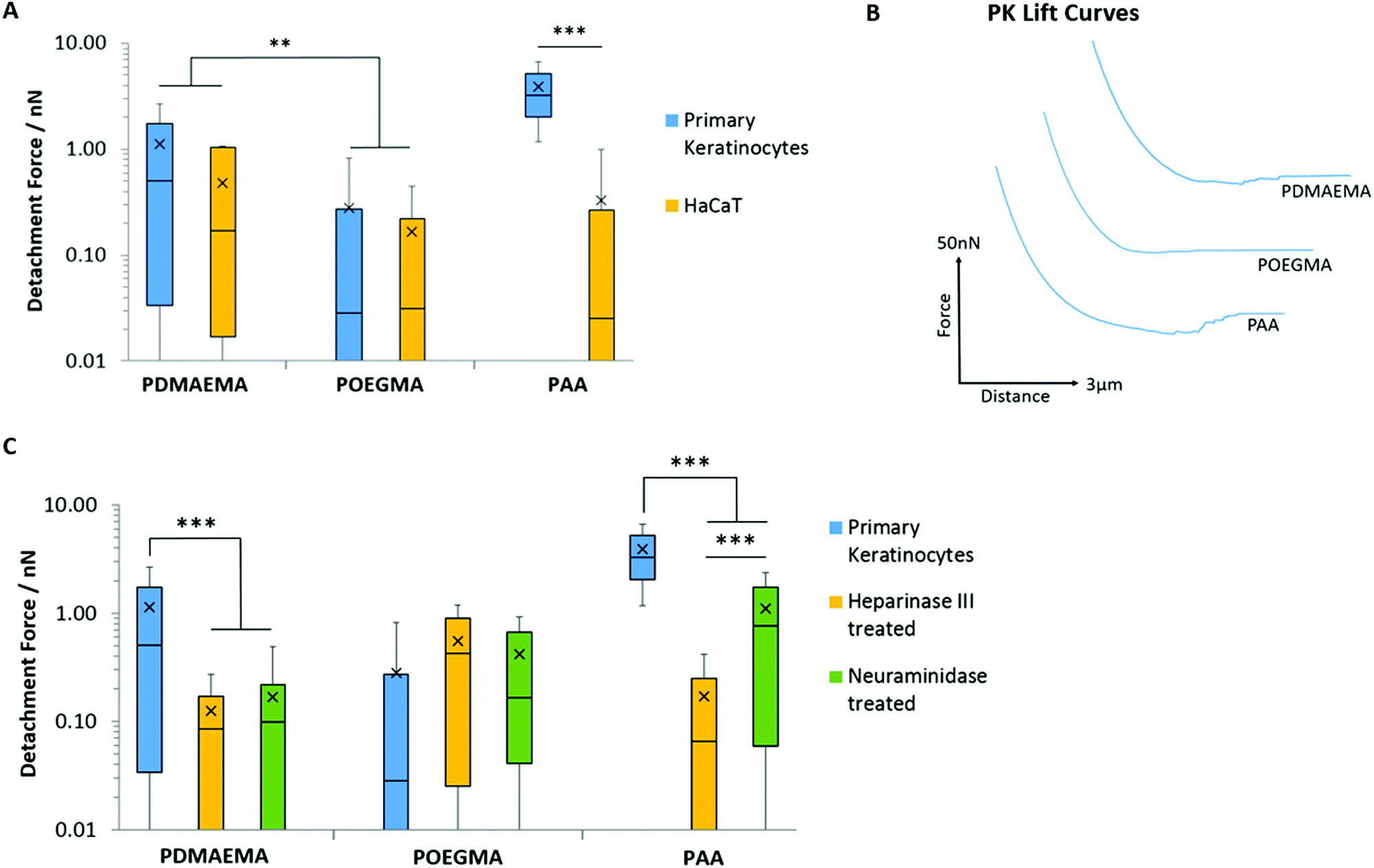 | ||
| Fig. 8 Characterisation of adhesive interactions between polymer brushes and cell monolayers. Testing was carried out on samples submerged in PBS. (A) Detachment forces between polymers and primary keratinocyte (PK) and HaCaT cell monolayers. (B) Representative AFM lift curves displaying the interaction between the polymers and primary keratinocyte cells. (C) Detachment forces between polymers and primary keratinocyte monolayers with and without enzymatic treatment. Data is plotted as means, with box plots. **p ≤ 0.01. ***p ≤ 0.001. | ||
PDMAEMA displayed relatively weak interactions with both cell monolayers; this is associated with low adhesion forces and detachment work (<1.14 nN and <0.87 fJ, respectively; Fig. 8A and B and Fig. S8A, ESI†). Although higher than that measured for POEGMA, such low adhesion is surprising considering the high positive ζ-potential of PDMEAMA brush-based colloids and their rapid uptake by cells, for example for gene delivery applications.30,96,97 Hence, it is possible that the rapid fouling of cationic polymer brushes by proteins and components found in the medium, results in the substantial modification of the PDMAEMA brush surface and associated decrease in ζ-potential,98 leading to a masking of short-range hydrophobic interactions and hydrogen bonding.
In contrast, PAA brushes displayed strong adhesions to primary keratinocytes (3.91 nN and 4.41 fJ, respectively; Fig. 8A and B and Fig. S8A, ESI†). This behaviour was associated with substantially longer detachment lengths than those reported for SAMs (752 ± 17 nm, compared to lengths typically <100 nm for SAMs), suggesting that the retraction of PAA-coated colloids is associated with substantial deformation of the cell membrane, contributing to the overall retraction profile. Strikingly, the adhesion of PAA brushes to HaCaT cells was very low (0.33 nN). To test whether fouling occurred at the brush surface, repeated measurements (600 adhesion and retraction events) were carried out and plotted as a function of cycle number (Fig. S8B, ESI†). The scatter of the data as function of cycle number, compared to the overall average, clearly indicates no significant positive or negative deviations as a function of time. To account for the high adhesion of PAA brushes to primary keratinocytes, we proposed that the glycocalyx (a proteoglycan brush-like layer that coats the cell membrane)99 of the epithelial cell sheets studied differed. Staining of the glycocalyx with wheat germ agglutinin (WGA) and specific immunostaining of the heparin component of the glycocalyx indeed revealed striking differences in the abundance and localisation of the glycocalyx in primary keratinocytes and HaCaT cells (Fig. 9).
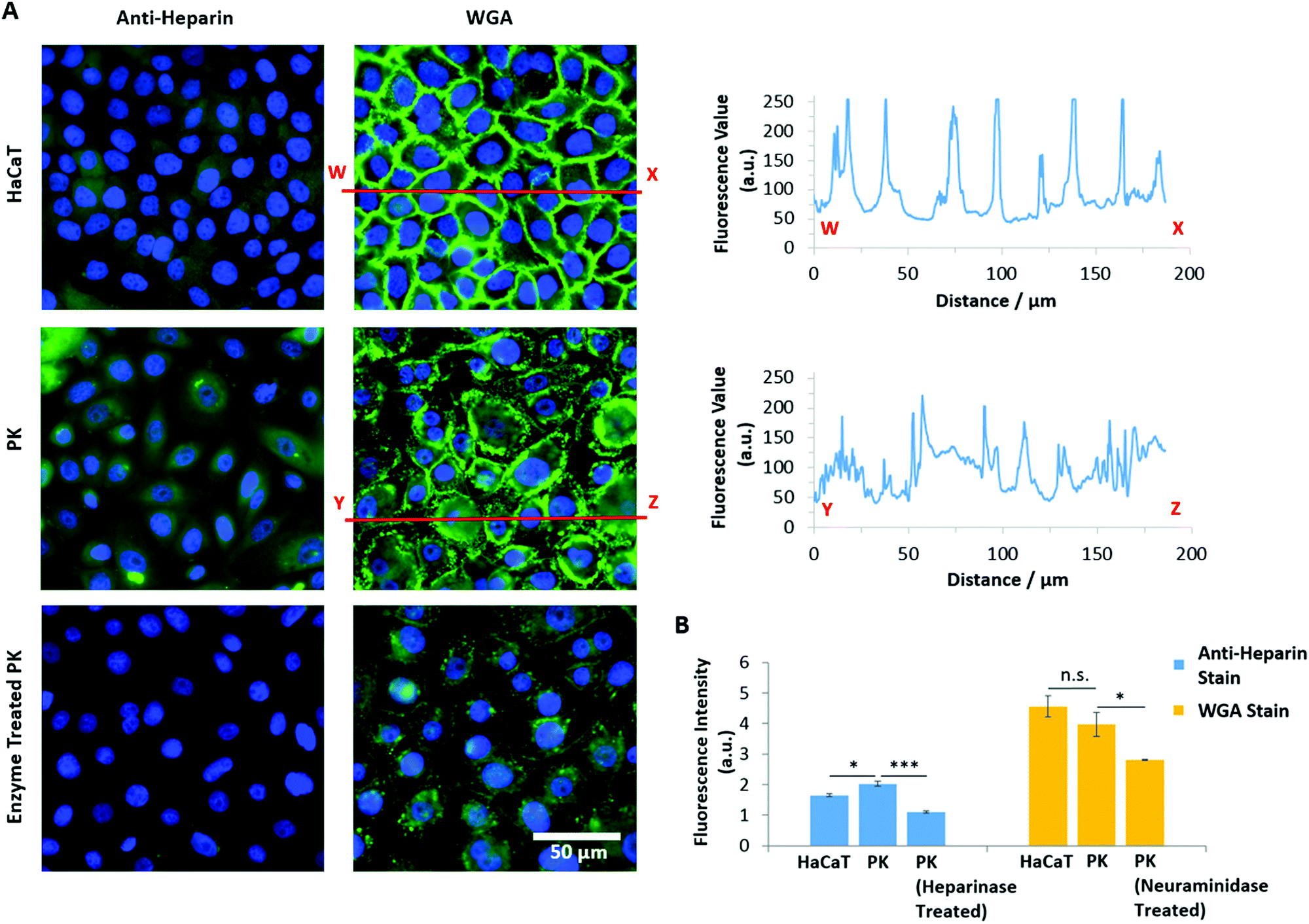 | ||
| Fig. 9 Quantification of the glycocalyx in primary keratinocytes (PK) and HaCaT cells through staining with wheat germ agglutinin (WGA) and specific immunostaining of the heparin component of the glycocalyx using anti-heparin. (A) Representative fluorescence images (blue, DAPI; green, anti-heparin or WGA) for HaCaT cells and primary keratinocytes. Primary keratinocytes were treated with heparinase III and neuraminidase and staining was carried out using anti-heparin and WGA, respectively. Cross-sections in the fluorescence values illustrate the differing localisation of the glycocalyx for HaCaT and primary keratinocyte cells. (B) Fluorescence intensity values for HaCaT cells, primary keratinocytes and enzyme treated primary keratinocytes, stained with either anti-heparin or WGA. Data is plotted as means. *p ≤ 0.05. ***p ≤ 0.001. n.s., non-significant. | ||
Heparin was found to be expressed at lower levels in HaCaT cells, compared to primary keratinocytes (Fig. 9A and B). In contrast, non-specific staining of the glycocalyx using WGA did not show the same trend; instead of a diffuse relatively homogenous staining, the glycocalyx was strongly localised at cell–cell junctions in the case of HaCaT monolayers (Fig. 9A). With primary keratinocytes, the glycocalyx was spread more uniformly on the apical membrane, with little sequestration at cell junctions. This was quantified by plotting the intensity profile of WGA sequestration in HaCaT cells and keratinocytes. This differential regulation of the localisation of the glycocalyx is proposed to result from the culture conditions: HaCaT cells were cultured in normal DMEM medium, which contains high levels of Ca2+, enabling the formation of cell junctions, whereas keratinocytes were cultured in KSFM, a medium containing low levels of Ca2+, in which cadherin-mediated junctions are typically not stabilised.100 Overall, these results indicate that the localisation and abundance of the glycocalyx in HaCaT cells and keratinocytes differs significantly, and this correlates with the changes in adhesion measured for polyelectrolyte brushes to the corresponding cell monolayers.
To further test the impact of the glycocalyx on colloidal probe adhesion to primary keratinocytes, we treated cell sheets with neuraminidase (broad-spectrum enzyme cleaving the glycocalyx) and heparinase (enzyme specifically cleaving heparin components).101 The efficiency of such cleavage was confirmed by staining and fluorescence microscopy (Fig. 9B). Following such treatment, we found that PAA brush adhesion was significantly reduced (Fig. 8C and Fig. S8C, ESI†). In particular, heparinase treatment resulted in a reduction of adhesion forces to levels comparable to POEGMA adhesion on untreated cells. Neuraminidase treatment had a more modest impact, indicating that, although cleavage of heparin accounts for most of the adhesion strength of PAA brushes to cell membranes, full cleavage of the glycocalyx reveals buried domains and potentially directly exposes the phospholipid bilayer, accounting for the moderate adhesion measured. These observations are in good agreement with the work of Servais et al., who reported that the adhesive force of pectin/CMC formulations to the mesothelium of several different tissues (investigated via uniaxial tensile strength tests using a customized apparatus for load/displacement measurements) was reduced after treatment of these samples with neuraminidase; in some cases this was by as much as 50%.102 The dependence of weak polyelectrolyte adhesion to cells, mediated by the glycocalyx, is therefore demonstrated across multiple length scales. Similarly, enzymatic cleavage of the glycocalyx components had a significant impact on the adhesion of PDMAEMA brushes, indicating that steric repulsion may also be responsible for the weak adhesion profile of these brushes to cell monolayers; this further suggests that fouling of the PDMAEMA surface is responsible for such low adhesion, despite the absence of medium or serum in the testing conditions. Surprisingly, the adhesion of POEGMA brushes slightly increased after enzymatic treatment of primary keratinocytes (to 0.55 and 0.42 nN after heparinase and neuraminidase treatment, respectively). This suggests that such enzymatic treatment leads to the exposure of residues, perhaps with higher hydrophobicity, as this was a particular type of interaction that promoted stronger adhesion of POEGMA brushes with SAMs.
3.4. Polymer brush adhesion to soft tissue samples
We next studied the adhesion of polymer brushes to soft epithelial tissues: the gingiva (which structure and homeostasis is regulated by gingival keratinocytes) and the epicardium (a membrane to which adhesion is particularly relevant for epicardial placement strategies).103,104Fig. 10 presents the detachment force and work measured during the adhesion of PDMAEMA, POEGMA and PAA brushes to porcine gingiva and epicardium samples. Overall, interactions of polymer brushes were stronger with the epicardium compared to the gingiva. As expected, adhesion between POEGMA brushes and both tissue types was minimal, consistent with the protein resistance of this polymer brush. Adhesion forces and work of PAA and PDMAEMA brushes to gingival epithelium were increased compared to that of POEGMA but remained overall relatively weak (<2.72 nN). In contrast, the adhesion of PAA and PDMAEMA brushes to the epicardium increased significantly (3.58 and 5.67 nN, respectively). Hence PDMAEMA was found to adhere relatively strongly to the epicardium, perhaps reflecting a higher coulombic attraction or hydrogen bonding with this tissue. The adhesion of PAA brushes to the epicardium was in line, although slightly lower than that measured to primary keratinocyte monolayers. These differences in adhesion to the epicardium and gingiva likely reflect differences in the composition of the cell surface, and in particular that of the glycocalyx in these two tissues.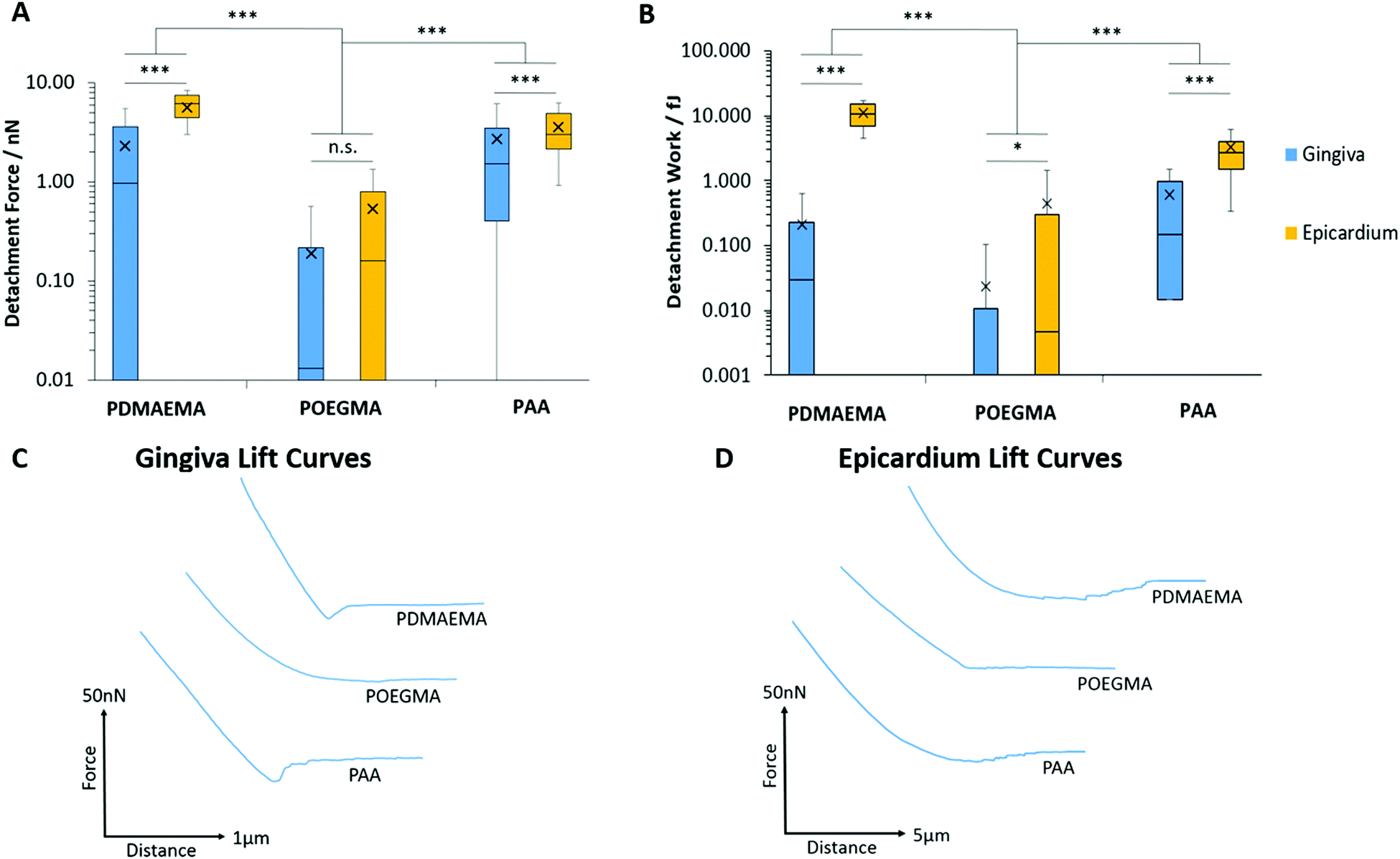 | ||
| Fig. 10 Characterisation of adhesive interactions between polymer brushes and porcine tissue samples. Testing was carried out on samples submerged in PBS. (A) Detachment force of epicardium and keratinized gingiva to polymers. (B) Detachment work between polymers and soft tissue samples. (C) Representative AFM lift curves between polymers and gingival samples. (D) Representative AFM lift curves between polymers and epicardium. Data is plotted as means, with box plots. *p ≤ 0.05. ***p ≤ 0.001. n.s., non-significant. | ||
To the best of our knowledge, the epicardial glycocalyx has not yet been characterised. The parietal and visceral pericardium (epicardium) are known to be similar in structure, both comprising a serosal (mesothelial cell) component adjacent to a fibrous tissue layer.105 The parietal pericardium has been shown to display a rich glycocalyx coating, in particular, rich in sialic acid residues.106 Assuming there is similarity in the glycocalx structure of both of these layers, the higher level of sialic acid content at the surface of the epicardium could qualitatively account for the strong adhesion observed for PDMAEMA brushes to this tissue.
In the oral mucosa, the epithelium forms a keratinized layer in which the glycocalyx interpenetrates with the salivary film.107 The presence of a glycocalyx layer on the surface of corneocytes has been evidenced via electron microscope,108,109 but to the best of our knowledge the exact composition of this layer is yet to be determined. The salivary film, which is estimated to be 70–100 μm thick,110 contains several molecules which include a large concentration of mucins111 and a number of different types of bacterial species. These bacteria have been shown to adhere to each other, as well as to the surface of the oral epithelium via a network of their glycocalyx.112,113 As such, there are a number of potential molecules that could contribute to polymeric adhesion to the keratinized gingiva; additional enzymatic studies will be required to identify the specific molecules present and the role that each plays in adhesion.
However, it should be noted that significant differences in tissue stiffness could account for at least some of the differences in adhesion measured between these two tissues. Indeed, the epicardium was found to be significantly softer than the gingival epithelium. This can be clearly seen in the large retraction lengths measured for the epicardium, especially for PDMAEMA brushes, compared to those measured for the gingiva and even cell monolayers (Fig. 10C and D). To confirm such differences in stiffness at the microscale, we measured the Young's modulus of these tissues, based on the curves obtained from the AFM adhesion data, using the Oliver–Pharr method to quantify corresponding moduli.59 The gingiva was found to be significantly stiffer than the epicardium (1020 ± 130 and 20.7 ± 0.5 kPa, respectively), suggesting that the colloidal probe indentation may result in substantial conformal deformation of the epicardium, and therefore an associated increase in contact area. As such, our data suggests that tissue biochemistry and stiffness combine to regulate the strength of adhesion of soft polymeric interfaces.
4. Conclusions
Overall, our results indicate that the adhesion profile of weak polyelectrolyte brushes is relatively complex, arising from combinations of electrostatic and van der Waals interactions, as well as hydrogen bonding. The response of such adhesive behaviour to electrolytes and changes in the Debye length is in turn equally complex, and is further modulated by the formation of hydrogen bonds with electrolytes (such as phosphates). In this respect, the comparison of adhesion profiles to model substrates with those observed with cells and tissues is particularly insightful. Specifically, based on our data, we propose that: (1) adhesion to cells and tissues is primarily mediated by hydrogen bonding and electrostatic forces, based on the differential adhesion profiles observed for the three brushes tested on biointerfaces and their comparison with the response to SAMs; (2) the glycocalyx has a profound impact on such adhesion and its composition (and abundance) at the surface of tissues will modulate soft matter adhesion; (3) the mechanics of the tissue/substrate (non-deformable in the context of SAMs) has a strong impact on the measured adhesion, in particular at the microscale, since it will dictate contact areas promoting adhesion. Therefore, our results demonstrate that simple considerations of electrostatic and hydrophobic interactions between polymer brushes, and even relatively simple interfaces (self-assembled monolayers), cannot fully account for adhesive profiles. In this context, in studies of brush adhesion to cells and tissues, the prediction of interactions remains particularly difficult to establish; relatively elaborate models, such as molecular dynamics simulations based on atomistic and coarse grain models, will be essential to capture the complex nature of polymer brush-interactions with biological samples, whether bacteria, eukaryotic cells or tissues. Our study also points to the need for an improved understanding and characterisation of the glycocalyx of the epithelium, or the surface chemistry of tissues, to which biomaterials bonding is required. Some studies have explored the structure and morphology of the glycocalyx in epithelial layers, but this is not systematic across all tissues, and often remains incomplete.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Ms Yaqi You for her help with tissue dissection, Dr William Megone for his help with AFM and Dr Danyang Li for her help with polymer brush synthesis. Funding from the Engineering and Physical Sciences Research Council (grant EP/M507532/1) is gratefully acknowledged.References
- J. L. Markman, A. Rekechenetskiy, E. Holler and J. Y. Ljubimova, Adv. Drug Delivery Rev., 2013, 65, 1866–1879 CrossRef CAS PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed.
- V. Bhagat and M. L. Becker, Biomacromolecules, 2017, 18, 3009–3039 CrossRef CAS PubMed.
- S. Tiwari, R. Patil and P. Bahadur, Polymers, 2018, 11, 1 CrossRef PubMed.
- T. N. Dinh, S. Hou, S. Park, B. A. Shalek and K. J. Jeong, ACS Biomater. Sci. Eng., 2018, 4, 3471–3477 CrossRef CAS PubMed.
- C. Ghobril and M. W. Grinstaff, Chem. Soc. Rev., 2015, 44, 1820–1835 RSC.
- N. Annabi, A. Tamayol, S. R. Shin, A. M. Ghaemmaghami, N. A. Peppas and A. Khademhosseini, Nano Today, 2014, 9, 574–589 CrossRef CAS PubMed.
- E. Bressan, V. Favero, C. Gardin, L. Ferroni, L. Iacobellis, L. Favero, V. Vindigni, M. Berengo, S. Sivolella and B. Zavan, Polymers, 2011, 3, 509–526 CrossRef CAS.
- J. R. Venugopal, M. P. Prabhakaran, S. Mukherjee, R. Ravichandran, K. Dan and S. Ramakrishna, J. R. Soc., Interface, 2012, 9, 1–19 CrossRef CAS PubMed.
- W. Samprasit, R. Kaomongkolgit, M. Sukma, T. Rojanarata, T. Ngawhirunpat and P. Opanasopit, Carbohydr. Polym., 2015, 117, 933–940 CrossRef CAS PubMed.
- J. Tsibouklis, A. M. Middleton, N. Patel and J. Pratten, J. Biomed. Mater. Res., Part A, 2013, 101, 3327–3338 Search PubMed.
- M. L. B. Palacio and B. Bhushan, Philos. Trans. R. Soc., A, 2012, 370, 2321–2347 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- M. Mehdizadeh and J. Yang, Macromol. Biosci., 2013, 13, 271–288 CrossRef CAS PubMed.
- Y.-C. Nho, J.-S. Park and Y.-M. Lim, Polymers, 2014, 6, 890–898 CrossRef.
- J. Wong, A. Chilkoti and V. T. Moy, Biomol. Eng., 1999, 16, 45–55 CrossRef CAS PubMed.
- C. a Squier and M. J. Kremer, J. Natl. Cancer Inst. Monogr., 2001, 52242, 7–15 CrossRef PubMed.
- M. Derrien, M. W. van Passel, J. H. van de Bovenkamp, R. Schipper, W. de Vos and J. Dekker, Gut Microbes, 2010, 1, 254–268 CrossRef PubMed.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H. A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, M. Houska, V. Šubr, A. B. Alles, E. Hasan, J. E. Gautrot and W. T. S. Huck, Macromol. Rapid Commun., 2011, 32, 952–957 CrossRef CAS PubMed.
- A. J. Hackett, J. Malmström, P. J. Molino, J. E. Gautrot, H. Zhang, M. J. Higgins, G. G. Wallace, D. E. Williams and J. Travas-Sejdic, J. Mater. Chem. B, 2015, 3, 9285–9294 RSC.
- A. M. Jonas, K. Glinel, R. Oren, B. Nysten and W. T. S. Huck, Macromolecules, 2007, 40, 4403–4405 CrossRef CAS.
- K. Y. Tan, J. E. Gautrot and W. T. S. Huck, Langmuir, 2011, 27, 1251–1259 CrossRef CAS PubMed.
- D. E. S. Santos, D. Li, M. Ramstedt, J. E. Gautrot and T. A. Soares, Langmuir, 2019, 35, 5037–5049 CrossRef CAS PubMed.
- O. Rzhepishevska, S. Hakobyan, R. Ruhal, J. Gautrot, D. Barbero and M. Ramstedt, Biomater. Sci., 2013, 1, 589–602 RSC.
- C. Rodriguez-Emmenegger, S. Janel, A. de los Santos Pereira, M. Bruns and F. Lafont, Polym. Chem., 2015, 6, 5740–5751 RSC.
- T. A. Petrie, J. E. Raynor, D. W. Dumbauld, T. T. Lee, S. Jagtap, K. L. Templeman, D. M. Collard and A. J. Garcia, Sci. Transl. Med., 2010, 2, 45ra60 Search PubMed.
- J. E. Gautrot, J. M. Rosenholm, D. Li, T. Gulin-Sarfraz, A. S. Sharili and M. Krishnamoorthy, Biomacromolecules, 2017, 18, 4121–4132 CrossRef PubMed.
- D. Li, A. S. Sharili, J. Connelly and J. E. Gautrot, Biomacromolecules, 2018, 19, 606–615 CrossRef CAS PubMed.
- D. J. D’Sa, H.-K. Chan and W. Chrzanowski, J. Colloid Interface Sci., 2014, 426, 190–198 CrossRef PubMed.
- H.-J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- P. Schön, E. Kutnyanszky, B. ten Donkelaar, M. G. Santonicola, T. Tecim, N. Aldred, A. S. Clare and J. J. G. Vancso, Colloids Surf., B, 2013, 102, 923–930 CrossRef PubMed.
- Y. Inoue, T. Nakanishi and K. Ishihara, Langmuir, 2013, 29, 10752–10758 CrossRef CAS PubMed.
- Y. Inoue, T. Nakanishi and K. Ishihara, React. Funct. Polym., 2011, 71, 350–355 CrossRef CAS.
- N. S. Bhairamadgi, S. P. Pujari, C. J. M. Van Rijn and H. Zuilhof, Langmuir, 2014, 30, 12532–12540 CrossRef CAS PubMed.
- Y. Yu, B. D. Kieviet, E. Kutnyanszky, G. J. Vancso and S. De Beer, ACS Macro Lett., 2015, 4, 75–79 CrossRef CAS.
- M. M. Elmahdy, A. Drechsler, E. Bittrich, P. Uhlmann and M. Stamm, Colloid Polym. Sci., 2014, 292, 1999–2012 CrossRef CAS.
- M.-J. Yin, M. Yao, S. Gao, A. P. Zhang, H.-Y. Tam and P.-K. A. Wai, Adv. Mater., 2016, 28, 1394–1399 CrossRef CAS PubMed.
- N. Salamat-Miller, M. Chittchang and T. P. Johnston, Adv. Drug Delivery Rev., 2005, 57, 1666–1691 CrossRef CAS PubMed.
- G. P. Andrews, T. P. Laverty and D. S. Jones, Eur. J. Pharm. Biopharm., 2009, 71, 505–518 CrossRef CAS PubMed.
- A. V. Dubolazov, Z. S. Nurkeeva, G. A. Mun and V. V. Khutoryanskiy, Biomacromolecules, 2006, 7, 1637–1643 CrossRef CAS PubMed.
- Y. Huang, H. Yu and C. Xiao, Carbohydr. Polym., 2007, 69, 774–783 CrossRef CAS.
- E. De Giglio, S. Cometa, N. Cioffi, L. Torsi and L. Sabbatini, Anal. Bioanal. Chem., 2007, 389, 2055–2063 CrossRef CAS PubMed.
- W. Lee, T. G. Lee and W. G. Koh, J. Ind. Eng. Chem., 2007, 13, 1195–1200 CAS.
- S. Sanjuan, P. Perrin, N. Pantoustier and Y. Tran, Langmuir, 2007, 23, 5769–5778 CrossRef CAS PubMed.
- C. V. Synatschke, A. Schallon, V. Jérôme, R. Freitag and A. H. E. Müller, Biomacromolecules, 2011, 12, 4247–4255 CrossRef CAS PubMed.
- A. Hucknall, S. Rangarajan and A. Chilkoti, Adv. Mater., 2009, 21, 2441–2446 CrossRef CAS.
- S. Swar, V. Zajícová, J. Müllerová, P. Šubrtová, J. Horáková, B. Dolenský, M. Řezanka and I. Stibor, J. Mater. Sci., 2018, 53, 14104–14120 CrossRef CAS.
- M. Heuberger, T. Drobek and N. D. Spencer, Biophys. J., 2005, 88, 495–504 CrossRef CAS PubMed.
- D. Boettiger and B. Wehrle-Haller, J. Phys.: Condens. Matter, 2010, 22, 194101 CrossRef CAS PubMed.
- B. Lego, M. François, W. G. Skene and S. Giasson, Langmuir, 2009, 25, 5313–5321 CrossRef CAS PubMed.
- F. Jenkins and H. White, Fundamentals of Optics, McGraw-Hill Primis Custom Publishing, 4th edn, 2001, pp. 474–496 Search PubMed.
- F. Qu, D. Li, X. Ma, F. Chen and J. E. Gautrot, Biomacromolecules, 2019, 20, 2218–2229 CrossRef CAS PubMed.
- J. E. Sader, I. Larson, P. Mulvaney and L. R. White, Rev. Sci. Instrum., 1995, 66, 3789–3798 CrossRef CAS.
- L. O. Heim, T. S. Rodrigues and E. Bonaccurso, Colloids Surf., A, 2014, 443, 377–383 CrossRef CAS.
- Y. An, D. Li, N. Roohpour, J. E. Gautrot and A. H. Barber, Dent. Mater., 2016, 1–9 Search PubMed.
- F. Hang, D. Lu, R. J. Bailey, I. Jimenez-Palomar, U. Stachewicz, B. Cortes-Ballesteros, M. Davies, M. Zech, C. Bödefeld and A. H. Barber, Nanotechnology, 2011, 22, 365708 CrossRef PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564–1583 CrossRef CAS.
- K. Y. Tan, H. Lin, M. Ramstedt, F. M. Watt, W. T. S. Huck and J. E. Gautrot, Integr. Biol., 2013, 5, 899–910 CrossRef CAS PubMed.
- B. Lego, W. G. Skene and S. Giasson, Macromolecules, 2010, 43, 4384–4393 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- T. Wu, P. Gong, I. Szleifer, P. Vlček, V. Šubr and J. Genzer, Macromolecules, 2007, 40, 8756–8764 CrossRef CAS.
- K. A. Davis and K. Matyjaszewski, Macromolecules, 2000, 33, 4039–4047 CrossRef CAS.
- R. Dong, S. Krishnan, B. A. Baird, M. Lindau and C. K. Ober, Biomacromolecules, 2007, 8, 3082–3092 CrossRef CAS PubMed.
- P. Louette, F. Bodino and J.-J. Pireaux, Surf. Sci. Spectra, 2005, 12, 22–26 CrossRef CAS.
- N. D. Treat, N. Ayres, S. G. Boyes and W. J. Brittain, Macromolecules, 2006, 39, 26–29 CrossRef CAS.
- R. Israels, F. A. M. Leermakers and G. J. Fleer, Macromolecules, 1994, 27, 3087–3093 CrossRef CAS.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 1999, 121, 7409–7410 CrossRef CAS.
- H. Hinterwirth, S. Kappel, T. Waitz, T. Prohaska, W. Lindner and M. Lämmerhofer, ACS Nano, 2013, 7, 1129–1136 CrossRef CAS PubMed.
- H. Ma, J. Hyun, P. Stiller and A. Chilkoti, Adv. Mater., 2004, 16, 338–341 CrossRef CAS.
- J. E. Gautrot, B. Trappmann, F. Oceguera-Yanez, J. Connelly, X. He, F. M. Watt and W. T. S. Huck, Biomaterials, 2010, 31, 5030–5041 CrossRef CAS PubMed.
- A. Drechsler, A. Synytska, P. Uhlmann, M. M. Elmahdy, M. Stamm and F. Kremer, Langmuir, 2010, 26, 6400–6410 CrossRef CAS PubMed.
- S. E. Burke and C. J. Barrett, Langmuir, 2003, 19, 3297–3303 CrossRef CAS.
- G. Liu, D. Wu, C. Ma, G. Zhang, H. Wang and S. Yang, ChemPhysChem, 2007, 8, 2254–2259 CrossRef CAS PubMed.
- L. N. Nagy, R. Mizsei, T. Kiss, Z. Varga, I. C. Szigyártó, M. Pálmai, A. Bóta, T. Kremmer, G. Tárkányi and J. Mihály, J. Colloid Interface Sci., 2012, 390, 34–40 Search PubMed.
- Z. Wu, H. Xiang, T. Kim, M. S. Chun and K. Lee, J. Colloid Interface Sci., 2006, 304, 119–124 CrossRef CAS PubMed.
- S. Vaidya, P. Thaplyal and A. Ganguli, Nanoscale Res. Lett., 2011, 6, 169 CrossRef PubMed.
- S. Ansar, S. Chakraborty and C. Kitchens, Nanomaterials, 2018, 8, 339 CrossRef PubMed.
- M. M. Elmahdy, A. Drechsler, E. Bittrich, P. Uhlmann and M. Stamm, Colloid Polym. Sci., 2014, 292, 1999–2012 CrossRef CAS.
- M. Stamm, F. Kremer, A. Synytska, P. Uhlmann and A. Drechsler, Langmuir, 2012, 28, 15555–15565 CrossRef PubMed.
- W. M. De Vos, F. A. M. Leermakers, A. De Keizer, M. A. C. Stuart and J. M. Kleijn, Langmuir, 2010, 26, 249–259 CrossRef CAS PubMed.
- M. Schlierf, S. Ramakrishnan, A. Keller, G. Krainer and G. Grundmeier, Small, 2017, 13, 1702100 CrossRef PubMed.
- T. Urbic, Chem. Phys. Lett., 2014, 610–611, 159–162 CrossRef CAS PubMed.
- J. T. OBrien and E. R. Williams, J. Am. Chem. Soc., 2012, 134, 10228–10236 CrossRef CAS PubMed.
- Y. Yang and Q. Cui, J. Phys. Chem. B, 2007, 111, 3999–4002 CrossRef CAS PubMed.
- G. R. J. Thatcher, D. R. Cameron, R. Nagelkerke and J. Schmitke, J. Chem. Soc., Chem. Commun., 1992, 386 RSC.
- M. Raftari, Z. J. Zhang, S. R. Carter, G. J. Leggett and M. Geoghegan, Macromolecules, 2015, 48, 6272–6279 CrossRef CAS.
- S. Sakata, Y. Inoue and K. Ishihara, Langmuir, 2015, 31, 3108–3114 CrossRef CAS PubMed.
- A. Synytska, E. Svetushkina, N. Puretskiy, G. Stoychev, S. Berger, L. Ionov, C. Bellmann, K. J. Eichhorn and M. Stamm, Soft Matter, 2010, 6, 5907–5914 RSC.
- C. Friedsam, A. D. C. Bécares, U. Jonas, M. Seitz and H. E. Gaub, New J. Phys., 2004, 6, 9 CrossRef.
- C. Friedsam, M. Seitz and H. E. Gaub, J. Phys.: Condens. Matter, 2004, 16, S2369–S2382 CrossRef CAS.
- R. Godawat, S. N. Jamadagni and S. Garde, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15119–15124 CrossRef CAS PubMed.
- Z. A. Levine, M. V. Rapp, W. Wei, R. G. Mullen, C. Wu, G. H. Zerze, J. Mittal, J. H. Waite, J. N. Israelachvili and J.-E. Shea, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4332–4337 CrossRef CAS PubMed.
- F. M. Watt and H. Green, Nature, 1982, 295, 434–436 CrossRef CAS PubMed.
- A. P. Majewski, U. Stahlschmidt, V. Jéroîme, R. Freitag, A. H. E. Müller and H. Schmalz, Biomacromolecules, 2013, 14, 3081–3090 CrossRef CAS PubMed.
- S. Agarwal, Y. Zhang, S. Maji and A. Greiner, Mater. Today, 2012, 15, 388–393 CrossRef CAS.
- K. Y. Tan, H. Lin, M. Ramstedt, F. M. Watt, W. T. S. Huck and J. E. Gautrot, Integr. Biol., 2013, 5, 899–910 CrossRef CAS PubMed.
- S. Weinbaum, J. M. Tarbell and E. R. Damiano, Annu. Rev. Biomed. Eng., 2007, 9, 121–167 CrossRef CAS PubMed.
- D. W. Owens, V. G. Brunton, E. K. Parkinson and M. C. Frame, Biochem. Biophys. Res. Commun., 2000, 269, 369–376 CrossRef CAS PubMed.
- A. Singh, S. C. Satchell, C. R. Neal, E. A. McKenzie, J. E. Tooke and P. W. Mathieson, J. Am. Soc. Nephrol., 2007, 18, 2885–2893 CrossRef CAS PubMed.
- A. B. Servais, A. Kienzle, C. D. Valenzuela, A. B. Ysasi, W. L. Wagner, A. Tsuda, M. Ackermann and S. J. Mentzer, Tissue Eng., Part A, 2018, 24, 199–206 CrossRef CAS PubMed.
- M. Araña, J. J. Gavira, E. Peña, A. González, G. Abizanda, M. Cilla, M. M. Pérez, E. Albiasu, N. Aguado, M. Casado, B. López, S. González, M. Soriano, C. Moreno, J. Merino, J. M. García-Verdugo, J. Díez, M. Doblaré, B. Pelacho and F. Prosper, Biomaterials, 2014, 35, 143–151 CrossRef PubMed.
- N. Tano, T. Narita, M. Kaneko, C. Ikebe, S. R. Coppen, N. G. Campbell, M. Shiraishi, Y. Shintani and K. Suzuki, Mol. Ther., 2014, 22, 1864–1871 CrossRef CAS PubMed.
- E. R. Rodriguez and C. D. Tan, Prog. Cardiovasc. Dis., 2017, 59, 327–340 CrossRef PubMed.
- A. Ohtsuka, S. Yamana and T. Murakami, Histochem. Cell Biol., 1997, 107, 441–447 CrossRef CAS PubMed.
- Y. Collan and A.-M. Raeste, Eur. J. Oral Sci., 1978, 86, 374–385 CrossRef CAS PubMed.
- V. Nguyen, A. Ndoye and L. Hall, J. Cell Sci., 2001, 114, 1189–1204 CAS.
- K. Wolff and E. Schreiner, J. Invest. Dermatol., 1968, 51, 418–430 CrossRef CAS PubMed.
- L. M. C. Collins and C. Dawes, J. Dent. Res., 1987, 66, 1300–1302 CrossRef CAS PubMed.
- A. Sengupta, D. Valdramidou, S. Huntley, S. J. Hicks, S. D. Carrington and A. P. Corfield, Arch. Oral Biol., 2001, 46, 529–538 CrossRef CAS PubMed.
- L. Vitkov, W. D. Krautgartner, M. Hannig and K. Fuchs, FEMS Microbiol. Lett., 2001, 202, 25–30 CrossRef CAS PubMed.
- M. Chevalier, C. Sakarovitch, I. Precheur, J. Lamure and V. Pouyssegur-Rougier, Acta Odontol. Scand., 2015, 73, 267–273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm01403a |
| This journal is © The Royal Society of Chemistry 2020 |