
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interfacial properties of lipid sponge-like nanoparticles and the role of stabilizer on particle structure and surface interactions†
Maria
Valldeperas
 ab,
Aleksandra P.
Dabkowska
c,
Gunnar K.
Pálsson
d,
Sarah
Rogers
e,
Najet
Mahmoudi
e,
Anna
Carnerup
a,
Justas
Barauskas
f and
Tommy
Nylander
*ab
ab,
Aleksandra P.
Dabkowska
c,
Gunnar K.
Pálsson
d,
Sarah
Rogers
e,
Najet
Mahmoudi
e,
Anna
Carnerup
a,
Justas
Barauskas
f and
Tommy
Nylander
*ab
aPhysical Chemistry, Department Chemistry, Lund University, Lund, Sweden. E-mail: tommy.nylander@fkem1.lu.se
bNanoLund, Lund University, Lund, Sweden
cPharmaceutical Sciences, IMED Biotech Unit, AstraZeneca, Gothenburg, Sweden
dInstitut Laue Langevin, Grenoble, France and Department of Physics, Uppsala University, Uppsala, Sweden
eISIS, STFC, Rutherford Appleton Laboratory, Didcot, UK
fCamurus AB, Ideon Science Park, Lund, Sweden
First published on 1st February 2019
Abstract
The advantage of using nonlamellar lipid liquid crystalline phases has been demonstrated in many applications, such as drug delivery, protein encapsulation and crystallisation. We have recently reported that a mixture of mono- and diglycerides is able to form sponge-like nanoparticles (L3-NPs) with large enough aqueous pores to encapsulate macromolecules such as proteins. Here we use small angle neutron scattering (SANS) to reveal morphology, structural and chemical composition of these polysorbate 80 (P80) stabilized sponge phase nanoparticles, not previously known. Our results suggest that L3-NPs have a core–shell sphere structure, with a shell rich in P80. It was also found that even if P80 is mostly located on the surface, it also contributes to the formation of the inner sponge phase structure. An important aspect for the application and colloidal stability of these particles is their interfacial properties. Therefore, the interfacial behaviour of the nanoparticles on hydrophilic silica was revealed by Quartz crystal microbalance with dissipation (QCM-D) and neutron reflectivity (NR). Adsorption experiments reveal the formation of a thin lipid layer, with the dimension corresponding to a lipid bilayer after L3-NPs are in contact with hydrophilic silica. This suggests that the diglycerol monoleate/Capmul GMO-50/P80 particles reorganize themselves on this surface, probably due to interactions between P80 head group and SiO2.
1. Introduction
Nonlamellar lipid liquid crystalline nanoparticles (LCNP) are widely considered in many applications due to their well-defined internal structure, stability and easily tuneable properties. Varying the lipid composition allows the formation of stable colloidal particle dispersions with inverse cubic, sponge-phase or hexagonal structure (cubosomes, spongosomes and hexosomes, respectively).1–5 In addition, they are advantageous compared to micelles or their lamellar analogues (vesicles), as their surface area per volume ratio is larger and they have a larger capability to solubilize both hydrophilic and hydrophobic molecules.3,6,7 The wide range of applications of these highly versatile particles includes drug delivery, protein encapsulation or crystallisation.3,7–12Most of the LCNP investigated so far had inverse cubic or hexagonal inner structures. More recently, the lipid sponge phase (L3) has attracted more attention due to its flexibility and capacity for forming large aqueous pores able to encapsulate large bioactive molecules.5,13,14 This is more challenging to obtain with other reverse mesophases (cubic or hexagonal), where the cell dimensions are usually smaller or require the use of organic solvents or specific environmental conditions (temperature or pressure).15–17
The interfacial properties of cubosomes and hexosomes have been examined to understand the nature of the interactions between these LCNP and different interfaces that mimic storage or administration vessels, such as glass vials, catheters or syringes.18–22 The adsorption behaviour was observed to depend on the environmental conditions (pH, ionic strength), the surface properties (such as hydrophilicity/phobicity or charge) and the lipid system used to form the LCNP. Vandoolaeghe et al.19,23 studied the adsorption of glycerol monolein (GMO)/pluronic F127 cubosomes on hydrophilic and hydrophobic silica at different conditions. They found that intact cubosomes were adsorbed on hydrophilic silica in the presence of electrolytes. In contrast, a lipid monolayer structure was formed on hydrophobic silica independently of the salt concentration. Soy phosphatidylcholine (SPC)/glycerol dioletate (GDO)/polysorbate 80 (P80) LCNP were found to form lipid multilayers on anionic silica that increase in thickness with P80 concentration.22,24 However on cationic silica, intact LCNP were found to deposit without being dependent on the P80 concentration.
In order to obtain colloidally stable LCNP with a narrow size distribution, a stabilizer that reduces the LCNP aqueous phase interfacial tension, like Pluronic F127 and P80 is required. It has been proposed earlier that P80 not only acts as a dispersing agent found on the outside of the particles, but also contributes to the inner structure organization of the nonlamellar LCNP. This effect is rather pronounced at high P80 concentrations, where even the loss of internal particle structure can occur.22,25 In our earlier work13 with the same DGMO/GMO-50/P80/water system as studied here, we were also able to form nanoparticles with highly swollen sponge phase structures over a wider range than in the pure lipid–aqueous system. Cryo-TEM imaging showed that such phases could be dispersed into sponge-like nanoparticles (L3-NPs) that featured a dense sponge phase core with swollen bilayer structures at the interface. We suggested that P80 was helping in the formation of these structures at the nanoparticle surface and thus, stabilizing the L3-NPs. However, the internal structural and the compositional organisation are still not known. We have here therefore used small angle neutron scattering (SANS) and partial deuteration of the stabilizer to reveal these important properties of the particles. This allowed us to reveal the location of deuterated P80 (dP80), as well as better understand the morphology of these particles as the L3 phase is not as well studied as other lipid liquid crystal (LLC) phases. Furthermore, for the first time interfacial behaviour of L3-NPs on hydrophilic silica were studied by Quartz crystal microbalance with dissipation (QCM-D) and neutron reflectometry (NR). The results suggest that the L3-NPs, that were initially 180 nm in diameter, spread and rearrange into a thin lipid layer onto hydrophilic silica.
2. Experimental
2.1. Materials
A mixture of acylglycerides denoted as Capmul® GMO-50 (Lot No. 100616-8) was provided by Abitec (Janesville, WI). Its composition was 54.7% monoglycerides, 15–35% diglycerides and 2–10% triglycerides with the following fatty acid composition: 84.6% oleic (C18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 6.8% linoleic, 0.8% linolenic and 6.2% saturated acids. Diglycerol monooleate (DGMO) with 88% of diglycerol monoester and 4.9% free glycerol and polyglycerols was received from Danisco A/S (Brabrand, Denmark). The main fatty acid component was oleic acid constituting 90.7%, followed by linoleic (4.2%), saturated (2.9%), eicosenoic (1.2%) and linolenic (0.8%). Polyoxyethylene (20) sorbitan monooleate (polysorbate 80, P80) was supplied by Croda (Chocques, France) for the neutron reflectometry measurements. P80 with hydrogenated and deuterated polyoxyethylene head group (hP80 and dP80), but with hydrogenated oleate chain, used in SANS experiments were synthesised by Oxford Deuteration Laboratory (Dr Robert K. Thomas) as described by Tucker et al.26 Milli-Q purified water (18 MΩ cm) was used for all experiments and heavy water (Sigma-Aldrich) was employed for neutron reflectometry and scattering measurements. All other solvent and reagents were of analytical grade and were used as received.
:1), 6.8% linoleic, 0.8% linolenic and 6.2% saturated acids. Diglycerol monooleate (DGMO) with 88% of diglycerol monoester and 4.9% free glycerol and polyglycerols was received from Danisco A/S (Brabrand, Denmark). The main fatty acid component was oleic acid constituting 90.7%, followed by linoleic (4.2%), saturated (2.9%), eicosenoic (1.2%) and linolenic (0.8%). Polyoxyethylene (20) sorbitan monooleate (polysorbate 80, P80) was supplied by Croda (Chocques, France) for the neutron reflectometry measurements. P80 with hydrogenated and deuterated polyoxyethylene head group (hP80 and dP80), but with hydrogenated oleate chain, used in SANS experiments were synthesised by Oxford Deuteration Laboratory (Dr Robert K. Thomas) as described by Tucker et al.26 Milli-Q purified water (18 MΩ cm) was used for all experiments and heavy water (Sigma-Aldrich) was employed for neutron reflectometry and scattering measurements. All other solvent and reagents were of analytical grade and were used as received.
2.2. Sample preparation
A stock solution composed of DGMO, GMO-50 and P80 was first prepared by co-melting GMO-50 and P80 at 40 °C followed by further weighting of each component into a glass vial. The DGMO/GMO-50 ratio was kept at 40/60 while the lipid/P80 ratio was 70/30. The stock solution was left mixing for 24 h in a roller mixer. Then, sponge-like nanoparticle dispersions were prepared by adding the stock solution into water or heavy water at a concentration of 5 wt% (lipids + P80). The samples were immediately sealed, vigorously hand-shaken and left for 24 h on an orbital shaking table at 300 rpm and room temperature. Samples for QCM-D and neutron reflectometry were heated up at 121 °C and 1.4 bars for 36 min (Prestige Medical Classic 2100 Autoclave, TG-Instrument AB, Helsingborg, Sweden) to remove vesicular aggregates. Samples for SANS measurement were mixed and diluted in appropriate amounts to obtain different contrast and concentrations as specified below. In order to determine the effect of the solvent medium, i.e. D2O versus H2O, on the particle we prepared a sample in the 1:4 H2O:D2O contrast using two different protocols. These two equivalent samples of 1% dilution with identical lipid and P80 composition were prepared by (a) mixing L3-NPs initially prepared in H2O with D2O (1/4 NPs/D2O volume) or (b) mixing L3-NPs initially in D2O with D2O + H2O (1/3/1 NPs/D2O/H2O volume).
2.3. Small angle neutron scattering (SANS)
:4 H2O:D2O as bulk solvent contrast. The reason for the 1:4 H2O/D2O contrast is that it matches the SLD of enzymes, which will be used in future studies. First, 0.5% dP80 micelles were analysed to reveal the degree of deuteration of P80 head group, followed by the evaluation of 5 wt% and 1 wt% L3-NPs in different contrast. Samples were filtered through a 0.45 μm syringe filter (PALL® Acrodisc® 32 mm 0.45 μm Supor® membrane) and measured in 1 mm (H2O and 1:4 H2O:D2O) or 2 mm (D2O) path length quartz cuvettes (Optiglass, UK). The experiment was carried out on the time-of-flight SANS instrument, SANS2D, at the ISIS Neutron and Muon source of the STFC Rutherford Appleton Laboratory. The measurements were performed at two sample-to-detector distances of 4 m and 12 m. The wavelength range was λ = 2–14 Å, which means a total covered q-range of 0.00168–0.98 Å−1, where q = 4π/λsin(θ/2) and θ is the scattering angle. The data reduction and solvent subtraction was performed as described by Wadsäter et al.22
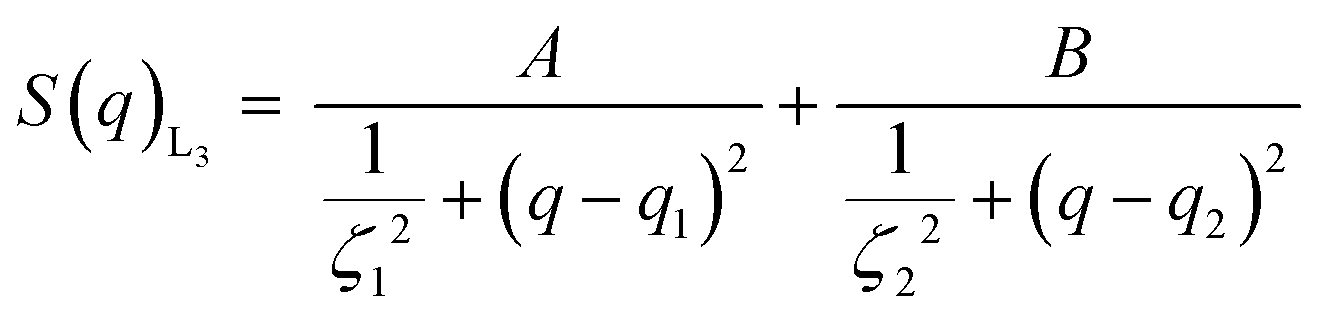 | (1) |
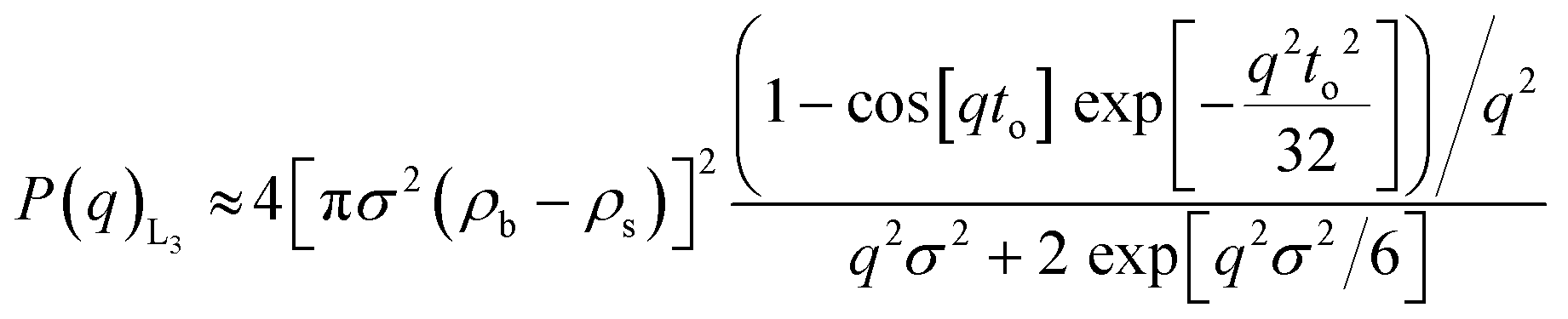 | (2) |
| Compound | Densitya (g cm−3) | Volume (Å3) | SLD (10−6 Å−2) |
|---|---|---|---|
| a All densities were obtained from Sigma-Aldrich.27 b Estimated from DOPC tails divided by 228 plus 2 glycerol groups with density of 1.26 g cm−3. c Calculated from individual components indicated below. d Estimated from known density and molecular weight. e Volume head = volume total − volume tail. f Same DOPC tail volume28 divided by 2, since P80 only have one hydrocarbon chain. g Estimated from individual components and considering a DGMO/GMO-50/P80 ratio of 28/42/30 wt%. | |||
| DGMO | — | 735b | 0.30 |
| GMO-50 | — | — | 0.19c |
| 55% GMO | 0.942 | 628 | 0.21 |
| 35% GDO | 0.934 | 1104 | 0.17 |
| 10% GTO | 0.910 | 1616 | 0.15 |
| P80 | 1.064 | 2041d | 0.55 |
| Fully protonated head | — | 1549e | 0.79 |
| Fully deuterated head | — | 1549 | 6.90 |
| Tail | — | 492f | −0.20 |
| dP80 (estimated) | — | — | 4.87 ± 0.10 |
| Lipid + hP80g | — | — | 0.33 |
| Lipid + dP80g | — | — | 1.63 |
Therefore, the total scattering intensity was described as:
| I(q) = P(q) + S(q)L3·P(q)L3 + background | (3) |
(I) Constrained and simultaneous fit of the same P80 contrast for the two dilutions studied (5 and 1%) were performed in order to obtain more reliable results.
(II) In the case of the 1:4 H2O:D2O contrast, constrained and simultaneous fit of the two equivalent samples, prepared as described in the sample preparation, was done for only 1% dilution.
The data was normalized by the volume fraction of the scattering entities prior to the fitting. All the parameters were set to be equal except scale factors, polydispersity and background. The solvent volume fraction and the scattering length density (SLD) of the lipid/P80 mixture in the core and the shell were estimated by:
(a) Using the SLD values obtained for the core and shell in different solvent contrast.
(b) Assuming that the solvent volume fraction and SLD of the lipid/P80 mixture were the same independently of the solvent. Volume fractions and SLD values for the core and the shell were considered separately.
These calculations were done comparing two isotopic solvent contrasts simultaneously, and then the results were averaged to obtain the final values. The equations used are shown in the ESI.† It should be noted that not all of the fits gave parameters that were physically possible. For the calculation of lipid/P80 composition and hydration it was not possible to use co-fitting for all solvent contrast used. We therefore compared calculated values for core and shell composition for a pair of two contrasts and the results are shown in Table S1 (ESI†). Although most of values are consistent there are a few pairs of contrast that are not consistent with other values. In such a case the fits that gave the most physically reasonable parameters and gave the most consistent values of two pairs of contrasts were chosen as the best to describe the L3-NPs. Here we also benefitted from the results from the interfacial characterization as well as previous SAXS data. It is possible that a more complex model would have described the system better, but then we would have ended up with even more fitting parameters whose physical validity would be even harder to judge.
2.4. Quartz crystal microbalance with dissipation (QCM-D)
2.5. Neutron reflectometry (NR)
Polished silicon substrates, with spontaneously formed SiO2 layer (Siltronix, Archamps-France) of 8 × 5 × 1.5 cm were used and carefully cleaned before the experiment as follows: (1) at 80 °C for 5 min in a basic solution composed by 25% NH4OH, 30% H2O2 and MilliQ water (1/1/5 volume); (2) rinsed thoroughly and several times with MilliQ water; (3) at 80 °C for 5 min in an acid solution composed by 32% HCl, 30% H2O2 and MilliQ water (1/1/5 volume); and (4) rinsed thoroughly with MilliQ water and kept in water until used. Just before mounting the NR cells, the silicon surfaces were rinsed with MilliQ water and 99.7% ethanol, followed by drying with N2 steam and plasma cleaning for 5 min.
In a specular neutron reflectometry measurement, a neutron beam is reflected against an interface and the reflected neutrons are detected at the same angle as the incoming beam. The reflectivity (R) is then, the intensity ratio between the incident and reflected intensity of neutrons which is measured as a function of the scattering vector (Q) perpendicular to the surface:
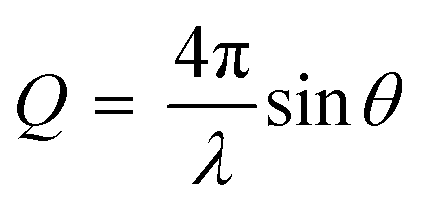 | (4) |
The measurements were performed at the Institute Laue-Langevin (ILL, Grenoble, France) on the Super ADAM reflectometer.34 This instrument operates in a monochromatic mode at the fixed wavelength of 5.183 Å and we used a q-range of 0 Å−1 < q < 0.2 Å−1. The raw data was treated with the SuperADAM Sared program. The absolute reflectivity scale was obtained by normalizing to the direct beam at the same slit setting and then dividing by the monitor reading. This allowed us to account for fluctuations in the neutron flux and to correct for points measured for different lengths of time. Slit openings varied with the incident angle in such a way as to provide a constant illumination of the sample. Since SuperADAM is equipped with a position sensitive 2D detector, the background was measured simultaneously. For this purpose, the average value from the regions of interest on both sides of the specular reflected beam was used.
2.6 Cryogenic transmission electron microscopy (cryo-TEM)
The structure and morphology of the dP80 L3-NPs dispersion in water was determined by using cryo-TEM. The sample was prepared in an automatic plunge freezer (Leica EM GP) to ensure a stable temperature and to prevent the loss of water during sample preparation. The climate chamber temperature was kept at 25 °C, and the relative humidity was kept close to saturation to prevent sample evaporation. The samples were prepared by placing 4 μL of 5 wt% L3-NPs dispersion on glow discharged lacey formvar carbon-coated copper grids (Ted Pella and gently blotted with filter paper to obtain a thin liquid film). Immediately after being blotted, the grids were rapidly plunged into liquid ethane at −183 °C to vitrify the water-rich samples to prevent the formation of ice crystals and to preserve the internal crystalline structure. The vitrified specimens were stored in liquid nitrogen (−196 °C) until measurements were taken. A Fischione Model 2550 cryo transfer tomography holder was used to transfer the samples into the transmission electron microscope, a JEM-2200FS (JEOL), equipped with an in-column energy filter (Omega filter), which allows zero-loss imaging. The accelerating voltage was 200 kV and zero-loss images were recorded with a TVIPS TemCam-F416 digital camera using Serial EM under low-dose conditions with a 10 eV slit. The working temperature was kept below −175 °C.3. Results and discussion
3.1. Modelling lipid sponge-like nanoparticles (L3-NPs)
Small angle neutron scattering (SANS) was used to characterise the L3-NPs structure and morphology as well as to localise the polysorbate 80 (P80) on the L3-NPs. This allowed us to extract information about the role of P80 in the formation and stabilisation of these lipid sponge-like nanoparticles. Deuterated P80 (dP80) was used to highlight its location, as the scattering length density (SLD) of the deuterated head group is higher than that of the lipids and also that of hP80.The presentation, interpretation and discussion of the data are divided in three main sections:
• The structure of the dP80 micelles: simultaneous fit of dP80 micelles in two solvent contrasts was performed using a core–shell sphere model. This allowed us also to extract the SLD of dP80.
• Revealing the inner structure of the nanoparticles: characterised by the sponge phase structure factor, L3-S(q), and form factor, L3-P(q).
• The overall shape and structure of the nanoparticles: described as core–shell spheres.
In order to obtain the results in Sections 3.1.2 and 3.1.3, a constrained and simultaneous fit of the model to SANS data from two samples with the same P80 contrast (either 2 dilutions or 2 sample preparations) were performed. This procedure allowed us to obtain more consistent fits as some contrast are more sensitive than others.
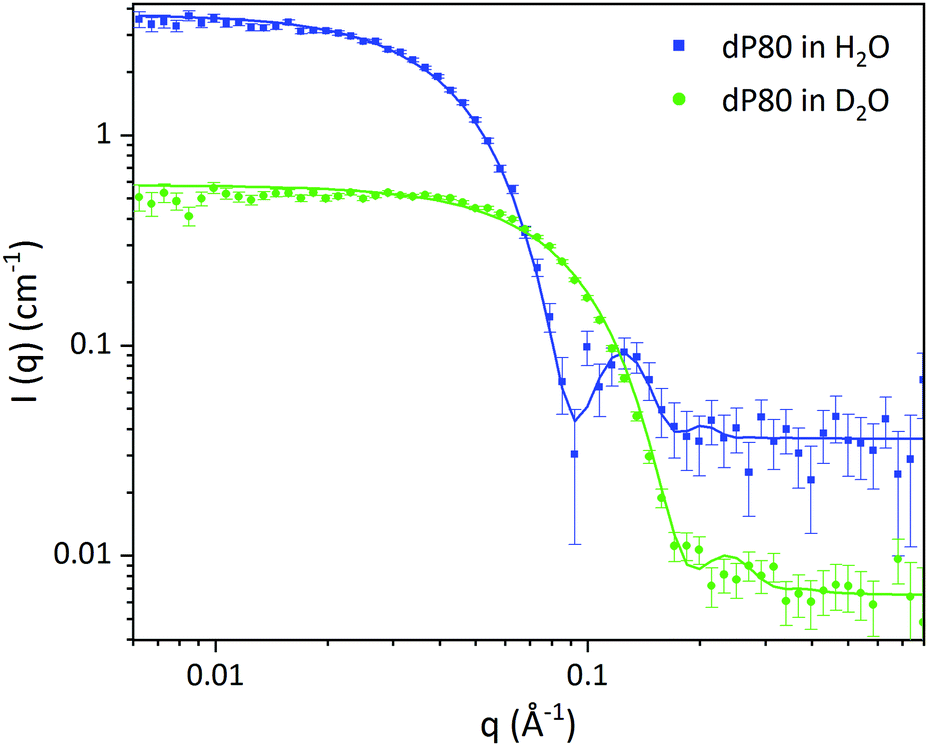 | ||
| Fig. 1 SANS data of dP80 micelles in H2O (blue) and D2O (green). Solid lines represent the core–shell sphere fitted model from Table 2. | ||
| dP80 micelles | D2O | H2O |
|---|---|---|
| Volume fraction of micelles (%) | 4.00 ± 0.03 | |
| Average core diameter (Å) | 46.0 ± 0.3 | |
| Core polydispersity | 0.100 ± 0.004 | |
| Shell thickness (Å) | 21.6 ± 0.2 | |
| SLD core (Å−2) 10−6 | 2.00 ± 0.01 | 0.10 ± 0.02 |
| SLD shell (Å−2) 10−6 | 6.38 ± 0.01 | 1.14 ± 0.02 |
| SLD solvent (Å−2) 10−6 | 6.35 | −0.56 |
| Solvent in core (vol%) | 27.5 ± 0.3 | |
| Solvent in the shell (vol%) | 75.8 ± 0.3 | |
| Background (cm−1) | 0.0065 | 0.036 |
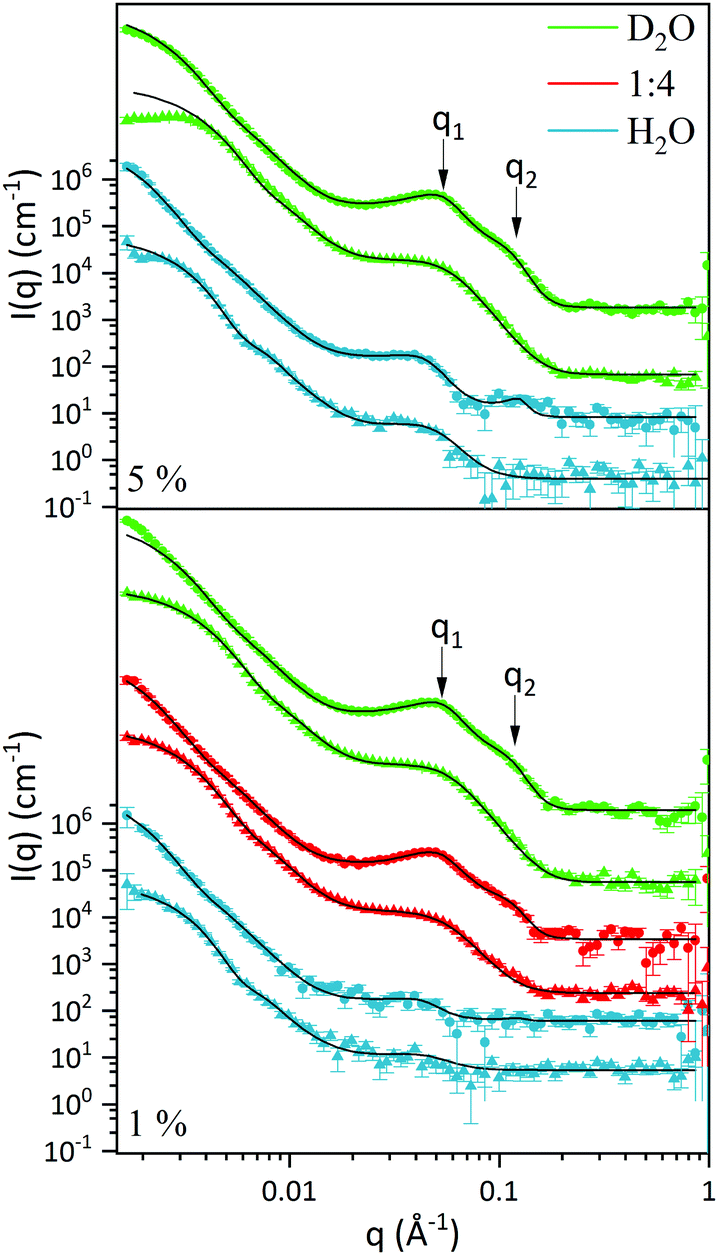 | ||
| Fig. 2 SANS curves of L3-NPs with their corresponding fitted curve (solid line), as a function of P80 and solvent contrast. hP80 and dP80 L3-NPs are depicted as triangles and circles, respectively, while H2O, 1:4 H2O:D2O and D2O solvent contrast are in blue, red and green, respectively. Top plot shows non-diluted samples (5%), while bottom are diluted to 1%. The y-axis scale, indicated for the lowest scattering curve, are the same for all the curves. For the sake of clarity the other scattering curves are shifted upwards without rescaling. | ||
In order to find a realistic model to fit the data recorded in this q-range, we used a combination of sponge phase structure and form factors (S(q)L3 and P(q)L3, respectively) as described in the experimental section and previous work.30
3.1.2.1. L3 correlation peak, S(q)L3. Table 3 shows the results obtained from the fitting, where the peaks’ maxima were fixed. The q1 value was found to be 0.05 Å−1, which agrees quite well with the SAXS results where q1 was 0.047 Å−1. It should be noted that, in spite of the fact that the dP80 and hP80 used here were from a different batch than the hP80 used for the SAXS and cryo-TEM experiments,13 we still found similar sponge phases in terms of dimensions and location in the phase diagram. Comparing different solvent contrast, it can be seen that in H2O the repeat distances are larger than in D2O, as q1 shift towards lower q-values, both for dP80 and hP80. The effect is larger for dP80 than for hP80. This could possibly be due to the sample in H2O being more hydrated than the ones with D2O. However, it should be noted that the shift of q2 is in the opposite direction.
:4 H2O:D2O and H2O
| Solvent contrast | dP80 L3-NPs | hP80 L3-NPs | ||||
|---|---|---|---|---|---|---|
| D2O | 1:4 H2O:D2O |
H2O | D2O | 1:4 H2O:D2O |
H2O | |
| t 0 (Å) | 24.8 ± 0.4 | 26.0 ± 0.3 | 22.6 ± 2.8 | 18.0 ± 0.1 | 18.1 ± 0.6 | 20 ± 3 |
| σ (Å) | 36.9 ± 0.3 | 27.8 ± 0.6 | 93.4 ± 7.5 | 44.9 ± 0.7 | 53 ± 1 | 42 ± 11 |
| q 1 (Å−1) | 0.0524 | 0.05 | 0.0432 | 0.05 | 0.0497 | 0.045 |
| ζ 1 (Å) | 61.4 ± 0.5 | 58.3 ± 1.1 | 67.7 ± 4.2 | 33.1 ± 0.6 | 30.7 ± 0.6 | 44 ± 13 |
| q 2 (Å−1) | 0.109 | 0.115 | 0.126 | |||
| ζ2 (Å) | 26.9 ± 0.6 | 27 ± 2 | 59 ± 19 | |||
3.1.2.2. Discoids form factor, P(q)L3. The bilayer form factor in the bicontinuous L3 phase was described using the one of discoids averaged over random orientations, as used previously by Porcar et al.30 The hydrocarbon core thickness was found to be around 24.5 and 18.8 Å for dP80 and hP80 samples, respectively. Since the dP80 tail extension was found to be 23 Å in the micelles and the hydrocarbon region is the same for DGMO and GMO-50, these results suggest that the lipid and P80 tails are somehow interdigitated or tilted in the L3 bilayer. The lower value for the hP80 sample might be due to the smaller difference in SLD between head group and tail.
3.1.3.1. Differences and similarities between hP80 and dP80 L3-NPs. A core–shell sphere model was used to fit the SANS data for the low q-range, i.e. q < 0.02 Å−1 (Fig. 2). The model was chosen taking into account the sponge-like nanoparticle morphology previously observed by cryo-TEM,13 where the core is characterised by a denser L3 phase while the surface presented swollen bilayer structures. Inter-particle interactions were assumed to be negligible as the concentration of particle was considered to be sufficiently low. Discrepancies could, however, be seen when comparing fitted curves at very low q-values of diluted and non-diluted samples.
Tables 4 and 5 show the results from the fitting, as well as estimated solvent hydration and SLD-solvent corrected values for the core and the shell of L3-NPs. The first aspect that can be noticed is the difference in size between the samples that contain hydrogenated and deuterated P80. In fact, L3-NPs with hP80 are smaller and present lower polydispersity. This size-dependence can also be appreciated from Fig. 2 where samples with hP80 reach a plateau at low q-values that is not present in dP80 samples. These discrepancies could be related to the degree of polymerization of P80 which makes the dP80 and hP80 chemically different. Here it should be noted that the P80 synthesis is very challenging and often give non-negligible differences between batches and there is also the issue of chemical stability. These aspects are discussed in detail by Kishore et al.38 As a result, dP80 samples seem to be more prone to aggregation than hP80 nanoparticles, which could also explain why they are more polydisperse (as also shown in Fig. 3). Therefore, nanoparticles with dP80 and hP80 were considered separately. However, we note that when it comes to phase behaviour, the different P80 samples investigated do not give any large variation.
:4 H2O:D2O and H2O reported in Fig. 2
| Solvent contrast | dP80 L3-NPs | hP80 L3-NPs | ||||
|---|---|---|---|---|---|---|
| D2O | 1:4 H2O:D2O |
H2O | D2O | 1:4 H2O:D2O |
H2O | |
| Average core diameter (Å) | 706 ± 2 | 950 ± 2 | 967 ± 12 | 492 ± 1 | 586 ± 1 | 662 ± 8 |
| Core polydispersity | 0.3 | 0.3 | 0.3 | 0.3 | 0.22/0.28 | 0.19 |
| Shell thickness (Å) | 173 ± 4 | 248 ± 6 | 126 ± 19 | 82 ± 2 | 116 ± 1 | 50 ± 8 |
| SLD core (Å−2) 10−6 | 4.00 ± 0.03 | 3.22 ± 0.05 | 0.15 ± 0.02 | 3.85 ± 0.05 | 3.03 ± 0.02 | −0.197 ± 0.003 |
| SLD shell (Å−2) 10−6 | 6.07 ± 0.01 | 4.83 ± 0.06 | −0.10 ± 0.04 | 5.74 ± 0.04 | 4.510 ± 0.003 | −0.44 ± 0.06 |
| SLD solvent (Å−2) 10−6 | 6.35 | 4.968 | −0.56 | 6.35 | 4.968 | −0.56 |
| Background (cm−1) | 0.897 | 1.11 | 0.667 | 0.695 | 1.78 | 0.35 |
| hP80 L3-NPs | dP80 L3-NPs | |
|---|---|---|
| Average solvent hydration in the core (vol%) | 58.8 ± 0.6 | 55.8 ± 0.4 |
| Average solvent hydration in the shell (vol%) | 89.50 ± 0.01 | 90.6 ± 0.3 |
| Average SLD core (Å−2) 10−6 – solvent corrected | 0.27 ± 0.08 | 1.01 ± 0.04 |
| Average SLD shell (Å−2) 10−6 – solvent corrected | 0.57 ± 0.01 | 3.45 ± 0.07 |
 | ||
| Fig. 3 Cryo-TEM image of L3-NPs with dP80 in H2O solvent. Note that the sample shown here is from a different batch than the SANS sample. | ||
As a general trend, the core dimension of L3-NPs increases with the H2O content while the shell thickness was largest for the 1:4 H2O:D2O solvent contrast and the decreases with D2O followed by H2O. This could be related to the dilutions used for each simultaneous fitting. While for D2O and H2O contrast two dilutions where used (5% and 1%), only one dilution (1%) was used for 1:4 H2O:D2O solvent contrast. The results suggest that the dilution process has an effect on the shell thickness, while the core diameter is less affected by it. This also shows that the inner sponge phase structure can only uptake a certain amount of solvent. Despite this size difference likely associated with dilution, we were able to obtain good fits for 5% and 1% L3-NPs in D2O and H2O.
The results from fitting the core shell model to the experimental data shows that the solvent contrast has a huge impact on the SLD of the core and the shell. This is expected as the sponge phase is an open and flexible system composed of a curved lipid bilayer with interconnected solvent channels that are connected to the bulk solvent. Therefore, when the bulk solvent's contrast changes, the contrast of the solvent inside the sponge mesh changes accordingly. It was found that the solvent present in the core was 59 and 56% for hP80 and dP80 L3-NPs, respectively. This agrees quite well with previous findings that showed that at this DGMO/GMO-50/P80 composition the lipid system could uptake around 60% of water.13 In addition, the solvent content in the shell was found to be around 90%, which means that the contrast between the shell and the surrounding medium is low. This can be explained by the swollen structures observed in the L3-NPs surface (Fig. 3 and Valldeperas et al.13) These structures appear to be composed of curved layers that enhance the contact with the bulk solvent as the surface area to volume ratio is increased. Therefore, it is expected that they are highly hydrated. Interestingly, the L3-NPs in water that contain dP80 show a less swollen surface structure and a denser core than the one we reported previously for hP80 L3-NPs.13 These highly swollen surfaces in the hP80 nanoparticles implies a large amount of solvent in that area, which makes it harder to see in SANS due to the lack of contrast between shell and medium.
3.1.3.2. dP80 stabilizer mostly found on the L3-NPs surface. We previously noted that dP80 was not affecting the sponge correlation peak, but we also saw that the second q2 peak was only visible for particles with dP80. This suggested that P80 somehow participates in the formation of the structure.
The data for hP80 L3-NPs does not allow us to draw any firm conclusions regarding the location of P80 due to the lack of contrast between lipids and hP80. However, it is interesting to notice that the SLD values for the lipid/surfactant mixture in the core and the shell are rather close, i.e. 0.27 and 0.57 × 10−6 Å−2, for the non-deuterated nanoparticles (Table 5). While the core SLD is slightly below the expected value for a homogeneous particle (0.33 × 10−6 Å−2), the shell SLD is very close to the hP80 SLD value of 0.55 × 10−6 Å−2 (Table 1). This suggests that L3-NPs shell is mostly formed by hP80 at the expense of an enriched lipid core. However, the contrast is not large enough to confirm that there is less than 30 wt% of P80 in the core.
In the case of dP80 L3-NPs, it was observed that the SLD of the lipid/surfactant components in the core (1.01 × 10−6 Å−2) is much lower compared to that of the shell (3.45 × 10−6 Å−2). Since the lipids are not deuterated, these high SLD values can only be attributed to dP80. This suggests that deuterated P80 is mostly found on the L3-NPs surface, i.e. stabilizing the lipid nanoparticles and allowing the formation of more swollen bilayers. Moreover, the core SLD value indicates that there is also some dP80 that contributes to the core sponge phase structure, as the SLD is higher than in the non-deuterated system. It is worth noting that the core SLD is lower than the expected SLD of the lipid + P80 mixture (1.63 × 10−6 Å−2), indicating that not all the P80 is in the core and thus, supporting the observations made for the hP80 nanoparticles. Therefore, it can be confirmed that the sponge-like nanoparticles present phase separation with a rich P80 shell. This is in line with what Wadsäter et al.22 previously reported for cubic micellar type of liquid crystalline nanoparticles. Their study focused on the effect of P80 concentration on soy phosphatidylcholine (SPC)/glycerol dioletate (GDO) lipid liquid crystalline nanoparticles. They observed that by increasing the dP80, the shell thickness was larger and richer in P80 at the expense of a GDO enriched core. In addition, the internal structure of the nanoparticles was affected at larger amounts (10 and 15 wt%) of P80, indicating that it was not only on the surface but also on the core of the particles. Yepuri et al. compared the cubic phase nanoparticle structures of phytantriol with two non-ionic stabilizers Tween® 80 (or P80) and Pluronic® F127.39 The location of the dispersing agent was revealed by SANS using deuterated phytantriol. They found that for P80, the surfactant molecules in the phytantriol-d/Tween® 80 followed a self-avoiding walk organization, whereas F127 in phytantriol-d/Pluronic® F127 particles appeared in a more rod-like packing. Here we note that in our case the more even distribution of P80 is favored by the fact that the lipids used and P80 has the same acyl chain.
3.2. Adsorption of L3-NPs on hydrophilic silica
Adsorption experiments were carried out to determine the interfacial properties of L3-NPs with hydrogenated P80. The aim was to understand their colloidal stability and how they interact with a silica interface. This will allow us to understand the driving force for the interaction as well as what happens with L3-NPs when they attach to a surface. Apart from the fundamental aspects, this is directly relevant when these types of dispersions are kept in glass vials for storage purposes. Here we used a negatively charged SiO2 (hydrophilic) as model substrate.The first experiment carried out was to study the adsorption of L3-NPs from pure water with no addition of any salt (pH ≈ 6.5–7), as they are prepared and stored under these conditions. Fig. S3a (ESI†) shows that no adsorption was observed which could be the result of electrostatic repulsion between the negatively charged L3-NPs and silica. Similar results were reported by Vandoolaeghe et al.19 who studied the affinity of GMO/Pluronic F127 cubosomes to hydrophilic SiO2 surfaces in water and at different pH values and ionic strengths. They found that by reducing the pH or adding electrolytes, adsorption of cubosomes took place. Therefore, we followed the same approach by studying the adsorption under low pH conditions (pH = 4 and 5.5) with and without 150 mM NaCl.
The results shown in Fig. 4 and Fig. S2b–d (ESI†), confirm that under these conditions L3-NPs adsorb, although no major differences in frequency and dissipation changes were observed when comparing the different solution conditions. We here focus on the results for pH = 4 with 150 mM, where we expect that the adsorption should not be hampered by repulsion between particles and the surface.
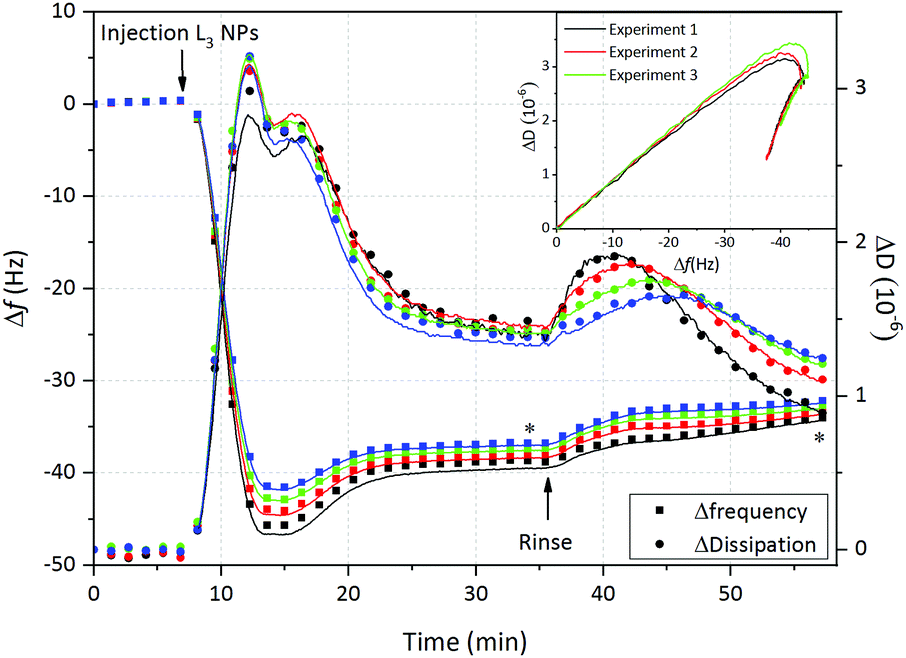 | ||
| Fig. 4 Change in frequency (Δf, squares) and dissipation (ΔD, circles) as a function of time after addition of 0.1 mg ml−1 L3-NPs on hydrophilic silica. Measurements were performed at 10 mM citrate buffer at pH = 4 and 150 mM NaCl. Experimental (dots) and fitted data (solid line) are depicted for four different overtones (5, black; 7, red; 9, green; and 11, blue). Embedded plot shows the ΔD vs. Δf for three equivalent experiments. | ||
Fig. 4 displays the change in frequency (Δf) and dissipation (ΔD) as a function of time after addition of sponge-like nanoparticles on hydrophilic SiO2. After injection there is an immediate decrease of Δf followed by a minimum and then a plateau at slightly higher frequency, which is reached after 30 min. This is accompanied by initially high ΔD values which are reduced once the steady state is achieved. Taking into account that the L3-NPs were initially 180 nm, we could suggest that, as soon as they are in contact with the hydrophilic SiO2, the lipid nanoparticles spread (at least partially) and reorganize themselves forming a lipid + P80 film characterised by the decrease in dissipation and mass (increase in Δf). This is supported by the kink observed in the ΔD vs. Δf plot (embedded in Fig. 4). Similar observations and conclusion regarding change in the soft particle structure on the surface has been previously reported for vesicles.40 It is not surprising that the sponge-like nanoparticles show similarities to the vesicles, as the sponge phase is formed by curved bilayers and it is close to the lamellar phase as showed in the bulk phase behaviour.13 Based on the experimental observation we suggest that the adsorption process is divided in two steps: (1) rapid adsorption of L3-NPs material and (2) rearrangement of material previously adsorbed. Furthermore, it was possible to rule out the adsorption of intact nanoparticles. However, it is difficult to confirm it with QCM-D and neutron reflectometry experiments were therefore conducted to give more insight into this matter.
Once the steady state was reached, the surface was rinsed with the same buffer at a higher flow rate (200 μl min−1). It could be observed on Fig. 4 that the frequency slowly increases, while the dissipation presents an initial rise that is reduced to values slightly below the plateau value. This dissipation bump was not observed when the rinsing flow rate was slower, although dissipation was somewhat reduced (data not shown). These results could suggest that: (a) the hydration of the layer decreases and a more rigid layer is formed and/or (b) excess of L3-NPs, lipid or P80 not attached directly to the surface are washed off. What is clear is that part of the lipid + P80 layer remains on the surface and is hardly disrupted when rinsing, especially when the rinsing flow rate is lower.
Finally, the Voigt model was used to determine the interfacial wet mass (and thickness) after L3-NPs adsorption, as the dissipation values were slightly too high to apply the Sauerbrey equation, indicating that the film formed on hydrophilic silica presented viscoelastic properties. Four overtones were used in order to obtain a good fit of the data that includes the adsorption and rinsing steps. Table 6 shows the thickness and wet mass values at the steady state and after rinsing (values averaged from three repeat experiments). These values also reflect the loss in mass after rinsing, with final values of 655 ng cm−2 and 67 Å for adsorbed mass and thickness, respectively. QCM-D does not give information about the morphology of the film and thus, neutron reflectometry is needed. However, we can speculate that the lipid + P80 film on hydrophilic SiO2 could form a bilayer, as we previously found that for bulk inverse bicontinuous cubic phases (≈50 wt% H2O) the bilayer thickness was 46 Å.13 SAXS results shows that the L3-NPs have a more swollen bicontinuous structure.13 Therefore, they can be expected to give a larger thickness value of surface layer formed from the L3-NPs, which is consistent with QCM-D data (Table 6). Furthermore, QCM-D thickness/adsorbed mass data also include the water coupled to the film.
| Period | Thickness (Å) | Wet mass (ng cm−2) |
|---|---|---|
| After injection of L3-NPs (plateau) | 80 ± 4 | 789 ± 42 |
| After rinsing with buffer | 67 ± 3 | 655 ± 30 |
As NR is less sensitive than QCM-D and the NR surface area was larger, a higher concentration of L3-NPs was used in order to ensure full coverage of the surface. A L3-NPs concentration of 1 mg ml−1 was enough to observe a fringe in NR, as the sample was left to adsorb for 30 min, while in QCM-D the sample was constantly flowed passed the substrate.
Fig. 5 shows the NR data after adsorption of L3-NPs. The data clearly show a strong adsorption of particles as a fringe appears, which is not present for bare silica in D2O buffer. The corresponding reflectivity curves in the H2O solvent contrast is also significantly different. Interestingly, the curves before and after rinsing in D2O contrast do not show much difference, which suggests that the interfacial lipid film structure is not altered by rinsing and that 1 mg ml−1 of L3-NPs was enough to obtain full coverage. This supports what was proposed in the analysis of the QCM-D data: that excess material is not in direct contact with the surface and could be easily removed without affecting the interfacial layer. However, we could not exclude the adsorption of intact nanoparticles onto the lipid film. The minor differences observed were mostly attributed to a change in the SLD of the solvent, as the injected L3-NPs contain some H2O inside the structure that is eliminated by rinsing.
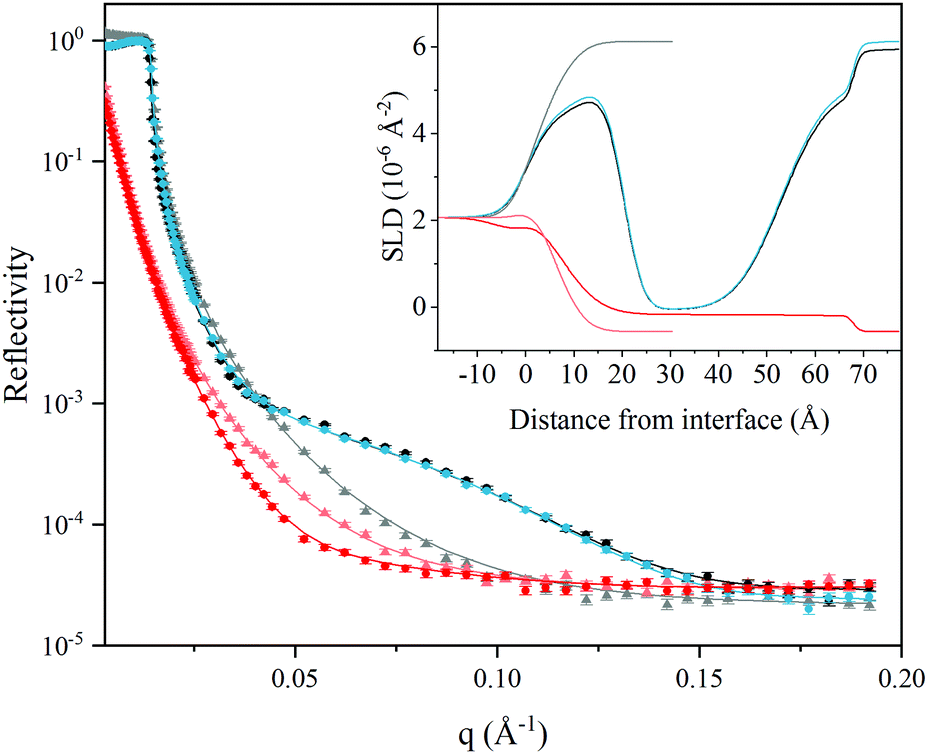 | ||
| Fig. 5 Neutron reflectivity curves as a function of momentum transfer (q) before addition of L3-NPs (grey for D2O and light red for H2O), after adsorption of L3-NPs in deuterated buffer (black) and after rinsing with deuterated (blue) and protonated water (red). Experimental and fitted data are depicted as dots and solid line respectively. Embedded plot show the scattering length density (SLD) values as a function of distance from the interface obtained from the fitting. | ||
Simultaneous fitting of a four-layer model to the data was performed. The results are presented as the SLD profile inserted in Fig. 5 and the fitting parameters are summarised in Table 7. The four-layer model consist of: (1) SiO2; (2) lipid head groups; (3) lipid hydrocarbon tails and; (4) lipid head groups, where the inner and outer head groups were considered equivalent in terms of thickness and SLD. The total thickness of the lipid layers formed from the L3-NPs dispersion was found to be 63 Å bilayer structure on hydrophilic silica, which agrees quite well with QCM-D values after rinsing. Table 7 shows the results obtained from the fit. It is noteworthy that the head group is highly hydrated (≈80%) and it is smaller than the dP80 head group observed by SANS (23 Å). Taking into account that the head group of DGMO and GMO-50 are smaller and that the thickness values are averaged over the whole surface, it makes sense that the observed thickness is reduced. In the case of the hydrocarbon tails, there is almost no solvent, as expected because of its hydrophobicity, and present a thickness of 33 Å. This value is slightly larger than the ones found for the L3-NPs studied by SANS. This suggests that the interfacial bilayer is less compact than the bilayers in L3-NPs. The tail layer thickness is lower than twice the dP80 tail size and thus supports the fact that the tails might be interdigitated or tilted. SLD values for lipid heads and tails agree quite well with the values expected (see Table 1 for more detailed information on head and tail SLD values of each component).
| Thickness (Å) | SLD (10−6 Å−2) | Solvent (vol%) | Roughness (Å) | |
|---|---|---|---|---|
| SiO2 | 5 ± 1.0 | 3.41 | 7 ± 2 | 3 ± 2 |
| Lipid head | 15 ± 1 | 1.24 ± 0.1 | 80 ± 5 | 7.5 ± 0.2 |
| Lipid tail | 33 ± 3 | −0.16 ± 0.1 | 1.78 ± 4 | 3.1 ± 5 |
| Lipid head | 15 ± 1 | 1.24 ± 0.1 | 80 ± 5 | 7.5 ± 0.2 |
The surface excess (Γ) of each layer was estimated in order to compare the neutron reflectivity with the QCM-D values. To do so, the following equation was used:
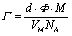 | (5) |
With the experiments performed here it is not possible to distinguish the location of P80 on the interfacial film, as dP80 was not used for these experiments. However, we could speculate that P80 might play an important role in the adsorption process of L3-NPs. Earlier it was suggested24,41,42 that attractive interactions exist between the polyethylene oxide segments of the P80 head group and SiO2. Since P80 is mostly found on the sponge-like nanoparticles surface, the adsorption driving force could be the same here, as no significant changes were obtained between different conditions studied by QCM-D.
4. Conclusion
In this work the role of P80 in the formation of sponge-like nanoparticles was investigated by SANS. It was found that the L3-NPs features local phase separation, as the more swollen bilayer type of structure on the particle surface is rich in P80 at the expense of a more DGMO/GMO-50 rich core. The sponge phase inner structure was however retained and was found to be very similar to the one reported earlier.13 This confirms our hypothesis that the polymer polysorbate 80 not only contributes to the formation of highly swollen inverse bicontinuous phases, but also acts as a stabilizer when these LLC phases are in excess water. Therefore, polysorbate 80 plays an important role here and confers long term stability to these nanoparticles.The interfacial behaviour of L3-NPs was studied by QCM-D and neutron reflectometry. The results reveal that particles spread to form a relatively hydrated 65 Å thick film on hydrophilic silica. Thus, the layer thickness is much lower than the particle size confirming the flexibility of the particles.
These results are of great interest since highly swollen sponge phase nanoparticles can be used as delivery systems, e.g. of bioactive macromolecules. The adsorption data suggests that the particles collapse once they are adsorbed and that is likely to lead to release of the cargo. We envisage that by tuning the stabiliser surface interactions we can potentially achieve targeted release.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the BIBAFOODs project from the European Union's Seventh Framework Programme FP7/2007-2013. We thank the ISIS Neutron and Muon source (UK) and the Super ADAM project at Institut Laue-Langevin (ILL, France), supported by Swedish Research council, for allocating beamtime at SANS2D and SuperADAM beamline, respectively. The deuterated and hydrogenated Tween 80 samples were kindly provided by Dr Robert K. Thomas, University of Oxford, UK. The authors are grateful for the SASView support from Paul Butler and valued discussion provided by Paul Butler and Marc Obiols-Rabasa. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547.References
- K. Larsson, J. Phys. Chem., 1989, 93, 7304–7314 CrossRef CAS.
- K. Larsson, Curr. Opin. Colloid Interface Sci., 2000, 5, 64–69 CrossRef CAS.
- K. Larsson, P. Quinn, K. Sato and F. Tiberg, Lipids. Structure, physical properties and functionality, The Oily Press, Bridgwater, England, 2006 Search PubMed.
- J. Gustafsson, H. Ljusberg-Wahren, M. Almgren and K. Larsson, Langmuir, 1996, 12, 4611–4613 CrossRef CAS.
- J. Barauskas, A. Misiunas, T. Gunnarsson, F. Tiberg and M. Johnsson, Langmuir, 2006, 22, 6328–6334 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, R. Mutafchieva, S. Lesieur and P. Couvreur, Acc. Chem. Res., 2011, 44, 147–156 CrossRef CAS PubMed.
- L. Sagalowicz and M. E. Leser, Curr. Opin. Colloid Interface Sci., 2010, 15, 61–72 CrossRef CAS.
- M. Malmsten, J. Dispersion Sci. Technol., 2007, 28, 63–72 CrossRef CAS.
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4, 449–456 CrossRef CAS.
- M. Caffrey, Curr. Opin. Struct. Biol., 2000, 10, 486–497 CrossRef CAS PubMed.
- C. E. Conn and C. J. Drummond, Soft Matter, 2013, 9, 3449 RSC.
- J. Barauskas and T. Nylander, Delivery and Controlled Release of Bioactives in Foods and Nutraceuticals, Woodhead Publishing Limited, Abington, England, 2008, pp. 107–131 Search PubMed.
- M. Valldeperas, M. Wiśniewska, M. Ram-On, E. Kesselman, D. Danino, T. Nylander and J. Barauskas, Langmuir, 2016, 32, 8650–8659 CrossRef CAS PubMed.
- B. Angelov, A. Angelova, R. Mutafchieva, S. Lesieur, U. Vainio, V. M. Garamus, G. V. Jensen and J. S. Pedersen, Phys. Chem. Chem. Phys., 2011, 13, 3073–3081 RSC.
- H. M. G. Barriga, A. I. I. Tyler, N. L. C. McCarthy, E. S. Parsons, O. Ces, R. V. Law, J. M. Seddon and N. J. Brooks, Soft Matter, 2015, 11, 600–607 RSC.
- A. I. I. Tyler, H. M. G. Barriga, E. S. Parsons, N. L. C. McCarthy, O. Ces, R. V. Law, J. M. Seddon and N. J. Brooks, Soft Matter, 2015, 11, 3279–3286 RSC.
- P. Vandoolaeghe, A. R. Rennie, R. A. Campbell and T. Nylander, Langmuir, 2009, 25, 4009–4020 CrossRef CAS PubMed.
- D. P. Chang, J. Barauskas, A. P. Dabkowska, M. Wadsäter, F. Tiberg and T. Nylander, Adv. Colloid Interface Sci., 2015, 222, 135–147 CrossRef CAS PubMed.
- P. Vandoolaeghe, F. Tiberg and T. Nylander, Langmuir, 2006, 22, 9169–9174 CrossRef CAS PubMed.
- A. J. Tilley, C. J. Drummond and B. J. Boyd, J. Colloid Interface Sci., 2013, 392, 288–296 CrossRef CAS PubMed.
- Y.-D. Dong, I. Larson, T. J. Barnes, C. A. Prestidge, S. Allen, X. Chen, C. J. Roberts and B. J. Boyd, Langmuir, 2012, 28, 13485–13495 CrossRef PubMed.
- M. Wadsäter, J. Barauskas, S. Rogers, M. W. A. Skoda, R. K. Thomas, F. Tiberg and T. Nylander, Soft Matter, 2015, 11, 1140–1150 RSC.
- P. Vandoolaeghe, R. A. Campbell, A. R. Rennie and T. Nylander, J. Phys. Chem. C, 2009, 113, 4483–4494 CrossRef CAS.
- D. P. Chang, M. Jankunec, J. Barauskas, F. Tiberg and T. Nylander, ACS Appl. Mater. Interfaces, 2012, 4, 2643–2651 CrossRef CAS PubMed.
- M. Younus, A. Hawley, B. J. Boyd and S. B. Rizwan, Colloids Surf., B, 2018, 169, 135–142 CrossRef CAS PubMed.
- I. M. Tucker, J. T. Petkov, J. Penfold, R. K. Thomas, P. Li, A. R. Cox, N. Hedges and J. R. P. Webster, J. Phys. Chem. B, 2014, 118, 4867–4875 CrossRef CAS PubMed.
- Sigma-Aldrich, https://www.sigmaaldrich.com, accessed July 2018.
- J. F. Nagle and S. Tristram-Nagle, Biochim. Biophys. Acta, Rev. Biomembr., 2000, 1469, 159–195 CrossRef CAS.
- SasView, https://www.sasview.org/, accessed July 2018.
- L. Porcar, W. A. Hamilton, P. D. Butler and G. G. Warr, Langmuir, 2003, 19, 10779–10794 CrossRef CAS.
- N. Lei, C. R. Safinya, D. Roux and K. S. Liang, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 608–613 CrossRef CAS.
- D. Roux, M. E. Cates, U. Olsson, R. C. Ball, F. Nallet and A. M. Bellocq, Europhys. Lett., 1990, 11, 229–234 CrossRef CAS.
- F. Höök, B. Kasemo, T. Nylander, C. Fant, K. Sott and H. Elwing, Anal. Chem., 2001, 73, 5796–5804 CrossRef.
- A. Vorobiev, A. Devishvilli, G. Palsson, H. Rundlöf, N. Johansson, A. Olsson, A. Dennison, M. Wollf, B. Giroud, O. Aguettaz and B. Hjörvarsson, Neutron News, 2015, 26, 25–26 CrossRef.
- A. Nelson, J. Phys.: Conf. Ser., 2010, 251, 012094 CrossRef.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- A. Amani, P. York, H. de Waard and J. Anwar, Soft Matter, 2011, 7, 2900–2908 RSC.
- R. S. K. Kishore, S. Kiese, S. Fischer, A. Pappenberger, U. Grauschopf and H.-C. Mahler, Pharm. Res., 2011, 28, 1194–1210 CrossRef CAS PubMed.
- N. R. Yepuri, A. J. Clulow, R. N. Prentice, E. P. Gilbert, A. Hawley, S. B. Rizwan, B. J. Boyd and T. A. Darwish, J. Colloid Interface Sci., 2019, 534, 399–407 CrossRef CAS PubMed.
- E. Reimhult, F. Höök and B. Kasemo, J. Chem. Phys., 2002, 117, 7401–7404 CrossRef CAS.
- O. Svensson, K. Thuresson and T. Arnebrant, Langmuir, 2008, 24, 2573–2579 CrossRef CAS PubMed.
- D. P. Chang, M. Jankunec, J. Barauskas, F. Tiberg and T. Nylander, Langmuir, 2012, 28, 10688–10696 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Core–shell sphere model; estimated solvent volume fraction and SLD of the lipid/P80 of the core and shell and equations used for the calculation; Voigt viscoelastic model; shear and viscosity values obtained using the Voigt model; QCM-D results at different conditions; and neutron reflectometry fits for the 5-layer model. See DOI: 10.1039/c8sm02634c |
| This journal is © The Royal Society of Chemistry 2019 |