
Spin-echo small-angle neutron scattering (SESANS) studies of diblock copolymer nanoparticles†
Gregory N.
Smith‡
 *a,
Victoria J.
Cunningham§
a,
Sarah L.
Canning¶
a,
Matthew J.
Derry
a,
J. F. K.
Cooper
b,
A. L.
Washington
b and
Steven P.
Armes
a
*a,
Victoria J.
Cunningham§
a,
Sarah L.
Canning¶
a,
Matthew J.
Derry
a,
J. F. K.
Cooper
b,
A. L.
Washington
b and
Steven P.
Armes
a
aDepartment of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK
bISIS-STFC, Rutherford Appleton Laboratory, Chilton, Oxon OX11 0QX, UK
First published on 16th November 2018
Abstract
Poly(glycerol monomethacrylate)–poly(benzyl methacrylate) (PGMA–PBzMA) diblock copolymer nanoparticles were synthesized via polymerization-induced self-assembly (PISA) using reversible addition–fragmentation chain-transfer (RAFT) aqueous emulsion polymerization in D2O. Such PISA syntheses produce sterically-stabilized nanoparticles in situ and can be performed at relatively high copolymer concentrations (up to 50 wt%). This PGMA–PBzMA formulation is known to form only spherical nanoparticles in water using aqueous emulsion polymerization (Macromolecules, 2014, 47, 5613–5623), which makes it an ideal model system for exploring new characterization methods. The polymer micelles were characterized using small-angle X-ray scattering (SAXS) and a recently developed form of neutron scattering, spin-echo small-angle neutron scattering (SESANS). As far as we are aware, this is the first report of a study of polymer micelles by SESANS, and the data agree well with reciprocal-space scattering. Using this technique enables characterization of the concentrated, as synthesized dispersions directly without dilution, and this will provide a method to study self-assembled polymer systems that have concentration dependent morphologies, while still maintaining the advantages of scattering techniques.
Polymerization-induced self-assembly (PISA) enables the convenient synthesis of a wide range of diblock copolymer nano-objects directly in water using either RAFT aqueous dispersion polymerization or RAFT aqueous emulsion polymerization.1 Dispersion polymerization requires a monomer that is miscible with the solvent but produces a polymer that is insoluble in the solvent. In water, there are relatively few monomers that meet these requirements. One such vinyl monomer is 2-hydroxypropyl methacrylate (HPMA), which has been well studied in the context of PISA.2 In contrast, most vinyl monomers exhibit relatively low aqueous solubility (less than 20 g L−1) and hence are well suited for aqueous emulsion polymerization. In this case, the monomer only requires sufficient solubility to facilitate mass transport between large monomer droplets and the growing polymer chains.1 Monomers such as benzyl methacrylate meet this requirement,3 and this monomer is used as the core-forming block in the present study. Poly(glycerol monomethacrylate) (PGMA) is used as the steric stabilizer block.
The PISA protocol used to prepare the PGMA–PBzMA diblock copolymers is shown in Scheme 1. 2-Cyano-2-propyl benzodithioate (CPDB) was used as the RAFT chain-transfer agent (CTA), and 4,4′-azobis(4-cyanovaleric acid) (ACVA) was selected as the radical initiator. The reaction conditions indicated in Scheme 1 are the same as those previously reported for the RAFT aqueous emulsion polymerization of BzMA.3 For the present study, this protocol was modified by preparing the nanoparticles in D2O to obtain maximum contrast for neutron scattering experiments. To ensure that the nanoparticles were identical for both X-ray and neutron scattering experiments, D2O was used as the solvent for both.
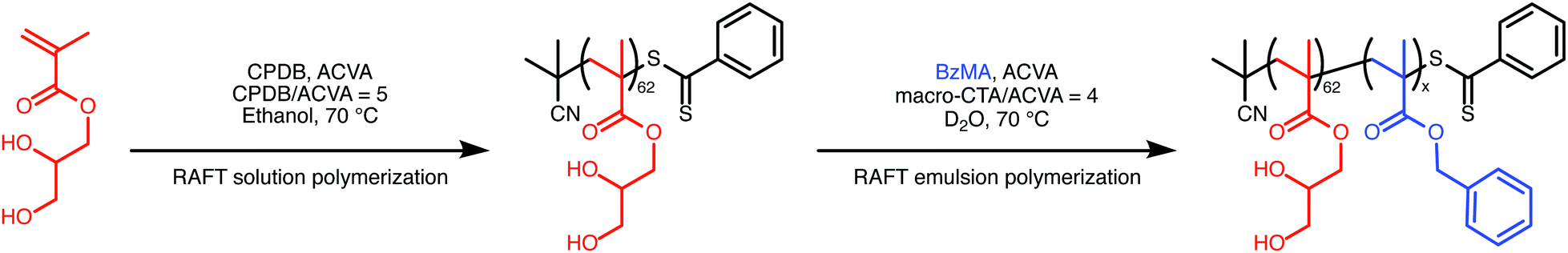 | ||
| Scheme 1 Synthesis of PGMA62–PBzMAx diblock copolymer nanoparticles by (i) RAFT solution polymerization of GMA in ethanol to afford a PGMA62 macromolecular CTA (macro-CTA) and (ii) RAFT aqueous emulsion polymerization of BzMA at 70 °C using the synthesized macro-CTA. | ||
The diblock copolymers were characterized both in solution (using 1H NMR spectroscopy to determine the final BzMA conversion and calculate the mean diblock composition as well as gel permeation chromatography to determine the molar mass and dispersity, Table S2, ESI†) and also as sterically-stabilized nanoparticles (using dynamic light scattering to determine the hydrodynamic radius and transmission electron microscopy to determine the morphology, Table S1 and Fig. S3, ESI†). Monomer conversions, determined by comparing the integrated oxymethylene signals assigned to the BzMA monomer and PBzMA polymer in 1H NMR spectra recorded in dimethylformamide-d7, were extremely high (>99%) for all diblock copolymers. Molar masses (Mn) from gel permeation chromatography (GPC) were comparable to those determined from 1H NMR, and the dispersities (ĐM = Mw/Mn) were relatively low, as expected for a well-controlled RAFT polymerization. The nanoparticles also appear to be spherical, with monotonically increasing Z-average diameters (dZ) and relatively low polydispersity indexes determined from dynamic light scattering (DLS). However, the size distributions are broader and the mean diameters are somewhat lower than data reported for comparable PGMA51–PBzMAx copolymer nanoparticles prepared by RAFT aqueous emulsion polymerization previously (Fig. S4, ESI†).3 These differences may be due to using D2O compared to H2O. However, the discrepancies are relatively small. Crucially for scattering analysis, these PGMA62–PBzMAx nanoparticles maintain the same spherical morphology as that observed when synthesized in H2O.
The structural properties of these sterically-stabilized PGMA62–PBzMAx nanoparticles were studied using small-angle X-ray scattering (SAXS) and spin-echo small-angle neutron scattering (SESANS). (Data for both techniques have been fit to models as described in the text using the SasView small-angle scattering software package.4) The two give complementary details about the polymer spheres, as previously shown for sterically-stabilized polystyrene colloids.5 The ability of each technique to resolve structures depends on the contrast between the blocks and the solvent as well as the length scale that can be accessed. X-ray scattering arises from the interaction of photons with electrons; hence, SAXS is sensitive to differences in electron density between the dispersed phase and the solvent.6 Given the scattering angles accessible by conventional SAXS instruments, this technique is optimal for the determination of both the overall size and internal structure of the nanoparticles and can access interparticle interactions in concentrated dispersions.6 On the other hand, neutron scattering arises from the interaction of neutrons with atomic nuclei; hence, high contrast can be achieved in SANS experiments by dispersing nanoparticles in isotopically-labeled solvent, as done in this study.7 In particular, very high contrasts can be achieved using SESANS, as multiple scattering can be trivially corrected for. Additionally, longer length scales are accessible, enabling both particle size and interparticle interactions up to 1.5 times the particle size to be determined.8,9
Synchrotron SAXS measurements were performed on 1 wt% dispersions of PGMA62–PBzMAx nanoparticles in D2O at the ID02 beamline at the ESRF (Grenoble, France). Full details of the instrument configuration can be found elsewhere.10 Monochromatic X-ray radiation (with wavelength λ = 0.995 Å) was used with a sample-detector distance of 5.0042 m. This gave an accessible Q-range of 0.0015 ≤ Q ≤ 0.15 Å−1. Q is defined as the magnitude of the momentum transfer vector and depends on both λ and θQ (one half of the scattering angle) (Q = (4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθQ)/(λ)). SAXS has proved to be a very powerful technique for determining the structure of diblock copolymer nano-objects in general,1,11 and, more specifically, the PISA syntheses of sterically-stabilized diblock copolymer nanoparticles directly in water.12–15 Experimental SAXS data and corresponding model fits are shown in Fig. 1; I(Q) is essentially independent of Q at low-Q, which is consistent with the formation of spherical nanoparticles. The data have been fitted to a spherical diblock copolymer micelle model.16 The model depends on the scattering length densities (SLD, ρ) of the blocks and solvent and the volume of each of the polymer blocks, all of which were calculated from the known physical properties of the polymers, which are fixed parameters. Further details on the model and calculation of these parameters are provided in the ESI.† The model also depends on the radius of the PBzMA nanoparticle core, which is directly proportional to the aggregation number, and the radius of gyration of the PGMA stabilizer, Rg (initially fixed to a physically reasonable estimate and then allowed to vary).
sinθQ)/(λ)). SAXS has proved to be a very powerful technique for determining the structure of diblock copolymer nano-objects in general,1,11 and, more specifically, the PISA syntheses of sterically-stabilized diblock copolymer nanoparticles directly in water.12–15 Experimental SAXS data and corresponding model fits are shown in Fig. 1; I(Q) is essentially independent of Q at low-Q, which is consistent with the formation of spherical nanoparticles. The data have been fitted to a spherical diblock copolymer micelle model.16 The model depends on the scattering length densities (SLD, ρ) of the blocks and solvent and the volume of each of the polymer blocks, all of which were calculated from the known physical properties of the polymers, which are fixed parameters. Further details on the model and calculation of these parameters are provided in the ESI.† The model also depends on the radius of the PBzMA nanoparticle core, which is directly proportional to the aggregation number, and the radius of gyration of the PGMA stabilizer, Rg (initially fixed to a physically reasonable estimate and then allowed to vary).
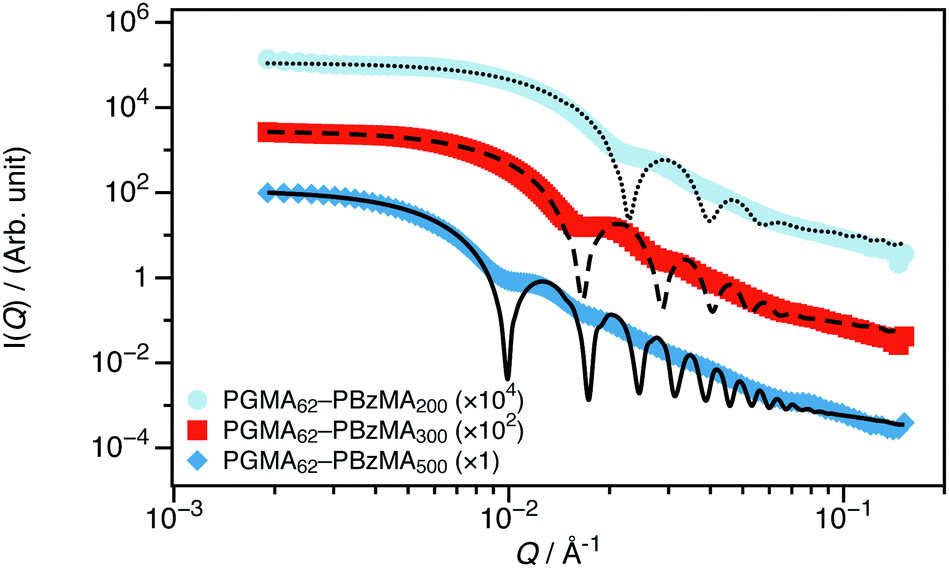 | ||
| Fig. 1 SAXS data recorded for 1 wt% dispersions of PGMA62–PBzMAx nanoparticles in D2O (the core DP, x, is stated in the legend). These data are consistent with the formation of spherical nanoparticles and can be well described using a spherical diblock copolymer micelle model.16 | ||
Both the nanoparticle radius and the mean aggregation number of the micelles increase monotonically as the PBzMA DP is increased, as expected.17 Relatively small spheres with core diameters less than 100 nm were obtained in all cases. A summary of the best fit parameters are given in the ESI† (Table S6). While reciprocal space scattering is well-suited for determining the nanoparticle structure, it is practically more challenging to determine the interparticle interactions. This is because the necessary inclusion of the structure factor (S(Q)) required to model interactions complicates the data analysis, as the scattering intensity becomes the product of S(Q) and the form factor (P(Q)). Additionally, quantification of long-range interactions requires data at extremely low-Q, which is not accessible on all instrumentation or at all facilities. Even when the instrument has the necessary Q-resolution, any multiple scattering from the sample will smear the final signal, lowering the effective resolution even if the instrument is acceptable. Therefore, reciprocal space scattering experiments are often conducted on dilute dispersions, as is the case for this SAXS study.
As a technique, SESANS overcomes this limitation. SESANS is not constrained by the low-Q issues of conventional SANS, as the technique does not require a beam stop and has no lower-Q limit on its detected neutrons. Furthermore, the Fourier relationship between real and reciprocal spaces means that the multiple scattering convolution in reciprocal space is a simple multiplication in real space that can be corrected for analytically. While SESANS lacks the short-range structural resolution offered by reciprocal space SAXS, it enables measurements of objects up to microscopic sizes, quantification of interparticle interactions in dispersions of any concentration, and high contrasts between solute and solvent to be studied from isotopic substitution without concerns over multiple scattering. Recent experiments have shown that SESANS is a powerful method for studying colloidal materials, in particular.5,9,18–22 In this study, we take advantage of these capabilities of SESANS to study PGMA62–PBzMAx nanoparticles directly as synthesized at a concentration of 30 wt%. With the high contrast between the copolymer and solvent attainable, it should be possible to quantify the volume fraction of water (D2O) entrapped by the PGMA-stabilizer as well as the nanoparticle size and interactions.
SESANS measurements were performed directly on all PGMA62–PBzMAx dispersions in D2O without dilution. Measurements were performed on the Offspec beamline at the ISIS Spallation Source (Didcot, UK). The implementation of spin-echo on Offspec is nonstandard, as it encodes spatial, rather than temporal information. For further technical details, see the discussion by Plomp.23 The neutron spin-echo length (Z) depends on the strength (B) of the magnetic field and the angle (θB) of its interface with respect to the neutron beam (Z = (γLmBλ2LcotθB)/(2πh)). In spin-echo mode, Offspec has a usable wavelength (λ) range between 2.2 and 14 Å. The magnetic field is fixed by the spin flippers to 17.1 mT, and angles of 85° and 57° were used. Using values of the gyromagnetic ratio (γL), neutron mass (m), and the Planck constant (h), as well the magnetic field length (L = 1.0 m), the calculated spin-echo lengths for a 10 Å neutron would be 263 Å and 1502 Å respectively, for these angles.
Experimental SESANS data along with model fits are shown in Fig. 2. As the DP of the core-forming PBzMA block is increased from 200 to 500, the first minimum in the normalized spin-echo signal is shifted to longer spin-echo length. This is consistent with the presence of nanoparticles of increasing size. The scattering data from the nanoparticles were modeled as non-interacting hard spheres.24,25 This model was selected for several reasons. The SESANS signal will be dominated by the particle's primary length scale and interparticle structure factor, so additional model parameters would be adding extraneous degrees of freedom while providing little statistically significant detail about the sample. This is the same approach used for core–shell polymer nanoparticles studied by SESANS previously.5 In contrast to this previous study, where concentrated dispersions were prepared to study the interactions between colloids, we perform experiments on colloidal dispersions that were synthesized as a concentrate. SESANS has been shown to be a robust method to study sterically-stabilized colloids5 and is therefore ideally suited to characterizing the nanoparticles that are synthesized by PISA. As can be seen in Fig. 2, the data can be successfully modeled by treating the particles as homogeneous spheres. The mean diameters obtained for these sterically-stabilized nanoparticles using SAXS and SESANS compare reasonably well to the volume-weighted hydrodynamic diameters from DLS (dV), shown in Table 1. The uncertainties shown are the standard deviation of repeated measurements for DLS and are estimated errors from fitting for SAXS and SESANS. The certainty of the SAXS diameters is overstated, as the maximum precision from SAS fitting is on the order of ±1 Å.26 The agreement between DLS and SAXS or SESANS is best for the technique where the particle size best matches the optimal length scale (small particles for SAXS and large particles for SESANS). For the same Q-range in a SAXS measurement, large particles will scatter less within this range than small particles, and therefore, sizes of larger particles will be less certain. The reverse is true for SESANS, as the technique's assumption that all scattered neutrons are incident on the detector grows less appropriate as the particles grow smaller and the associated scattering angles expand beyond the bounds of the detector. For example, the SESANS data for the PGMA62–PBzMA200 spheres can only be well fit if the fitting window is restricted to Z < 1000 Å. In a DLS measurement, the certainty of the sizes of all particles should be similar. This is precisely what we observe when comparing SAXS or SESANS data to DLS data.
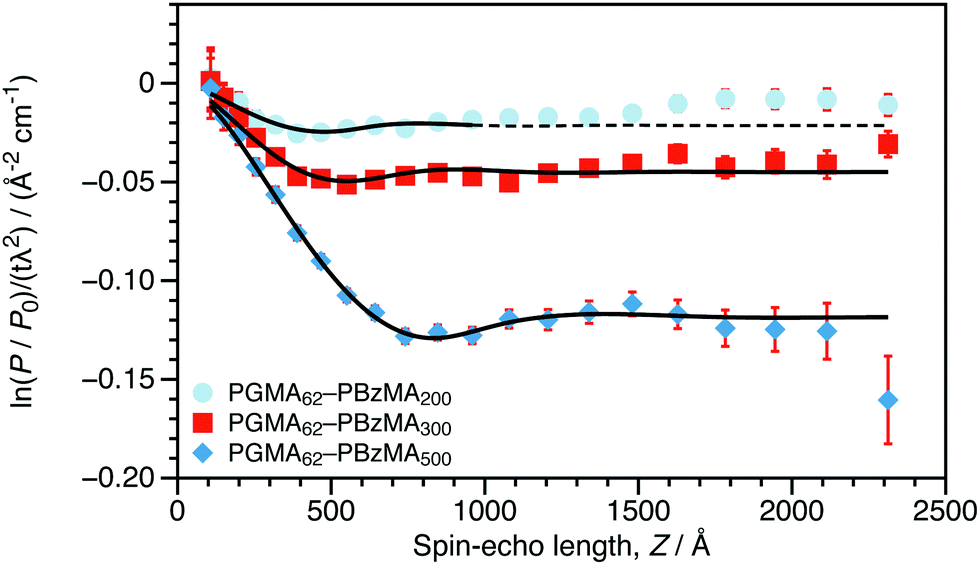 | ||
| Fig. 2 Normalized SESANS signal as a function of spin-echo length (Z) for 30 wt% dispersions of PGMA62–PBzMAx nanoparticles in D2O (the core DP, x, is stated in the legend). These data were fitted to a spherical model with hard sphere interactions24,25 and a variable scattering length density (Δρ) between that of the sphere and the solvent. The data for the PGMA62–PBzMA200 spheres are fit only for Z < 1000 Å. | ||
| DLS dV/nm | SAXS d/nm | SESANS d/nm | |
|---|---|---|---|
| SAXS: d = 2r + 4Rg; SESANS: d = 2r; DLS uncertainties are standard deviations from repeated measurements, and SAXS and SESANS uncertainties are estimated errors from data fitting. | |||
| PGMA62–PBzMA200 | 43 ± 1 | 44.12 ± 0.02 | 62 ± 13 |
| PGMA62–PBzMA300 | 58 ± 1 | 58.49 ± 0.03 | 66 ± 10 |
| PGMA62–PBzMA500 | 98 ± 1 | 94.15 ± 0.04 | 97 ± 5 |
From the fits to the SESANS data (d and Δρ), it is possible to determine the composition of the spheres. The SLD (ρt) of a multicomponent particle is a volume fraction (ϕ) weighted sum of the SLDs of the individual components 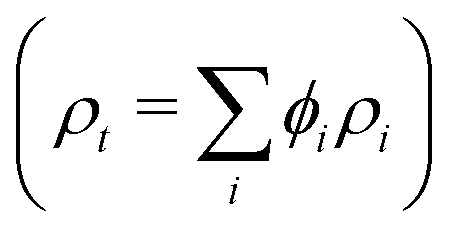 . For these sterically-stabilized nanoparticles, the three components are the PGMA chains in the shell (ρs and ϕs), the solvating D2O in the shell (ρm and ϕm), and the PBzMA core (ρc and ϕc). The volume fractions of the stabilizer and core for a non-solvated nanoparticle are known from the diblock composition, and the SLDs for all three pure components are also known. Thus, it is possible to calculate the ϕm that would be required to give the experimental ρt. These ρm values are shown in Table 2 for the three PGMA62–PBzMAx nanoparticles studied. The corresponding ϕm values calculated from the SAXS fitting (assuming that the stabilizer thickness is 2Rg) are shown for comparison. These two methods compare favorably, although the best agreement is for the largest sphere where SESANS fitting is more reliable, as discussed above. Using a similar approach, the mean aggregation numbers (nagg) for these nanoparticles can be determined. In the case of the SAXS data, this parameter is determined by dividing the nanoparticle core volume by the molecular volume of a single PBzMA chain. For the SESANS measurements, the fraction of the sphere that is occupied by polymer and not D2O is determined by scaling the total sphere volume by (1 − ϕm) and dividing by the volume of the entire diblock chain. As shown in Table 2, the nagg values calculated from these two approaches are gratifyingly similar. Although there are discrepancies, the two techniques require different approaches to calculating structural parameters (from the geometry for SAXS and from the contrast for SESANS), which can explain this. While consistent values can be obtained from both SESANS and SAXS, the fact that the structural information from the SESANS measurements comes largely from the fit value of Δρ and that it agrees well with the SAXS data is particularly satisfying.
. For these sterically-stabilized nanoparticles, the three components are the PGMA chains in the shell (ρs and ϕs), the solvating D2O in the shell (ρm and ϕm), and the PBzMA core (ρc and ϕc). The volume fractions of the stabilizer and core for a non-solvated nanoparticle are known from the diblock composition, and the SLDs for all three pure components are also known. Thus, it is possible to calculate the ϕm that would be required to give the experimental ρt. These ρm values are shown in Table 2 for the three PGMA62–PBzMAx nanoparticles studied. The corresponding ϕm values calculated from the SAXS fitting (assuming that the stabilizer thickness is 2Rg) are shown for comparison. These two methods compare favorably, although the best agreement is for the largest sphere where SESANS fitting is more reliable, as discussed above. Using a similar approach, the mean aggregation numbers (nagg) for these nanoparticles can be determined. In the case of the SAXS data, this parameter is determined by dividing the nanoparticle core volume by the molecular volume of a single PBzMA chain. For the SESANS measurements, the fraction of the sphere that is occupied by polymer and not D2O is determined by scaling the total sphere volume by (1 − ϕm) and dividing by the volume of the entire diblock chain. As shown in Table 2, the nagg values calculated from these two approaches are gratifyingly similar. Although there are discrepancies, the two techniques require different approaches to calculating structural parameters (from the geometry for SAXS and from the contrast for SESANS), which can explain this. While consistent values can be obtained from both SESANS and SAXS, the fact that the structural information from the SESANS measurements comes largely from the fit value of Δρ and that it agrees well with the SAXS data is particularly satisfying.
| ϕ m | n agg | |||
|---|---|---|---|---|
| SESANS | SAXS | SESANS | SAXS | |
| PGMA62–PBzMA200 | 0.58 | 0.33 | 825 | 486 |
| PGMA62–PBzMA300 | 0.36 | 0.21 | 1120 | 947 |
| PGMA62–PBzMA500 | 0.09 | 0.11 | 3179 | 2833 |
The advantage of using SESANS to study these PGMA62–PBzMAx nanoparticles is that no a priori assumptions are required, and all structural information is a consequence of the large isotopic contrast between the copolymer and the solvent. The reasonable agreement found between structural properties calculated from fitting reciprocal space SAXS data and real space SESANS data promises wider application of this new form of neutron scattering to diblock copolymer nano-objects.
Clearly, SESANS is a useful and informative technique for characterizing concentrated dispersions of relatively large nanoparticles. The results in this study show that using SESANS makes it straightforward to determine the properties of model polymer nanoparticles, providing complementary information to other forms of scattering. The ability to study concentrated dispersions is advantageous for PISA, in particular, as it known that the concentration of nano-objects can impact the morphology that are formed even for equivalent diblock copolymers.2,27 The ability to study such dispersions directly, therefore, is appealing, particularly in cases where dilution results in morphological reorganization. As advances continue to be made in both spin-echo scattering instrumentation and data analysis, we expect that SESANS will become an important tool for studying dispersions of diblock copolymer nano-objects in the future.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
GNS and SPA acknowledge the ERC (PISA 320372) and EPSRC (EP/J007846) for funding. The ESRF and ISIS (experiment RB1610048) are acknowledged for allocation of beamtime and the UK Science and Technology Facilities Council for funding travel. The personnel of the ID02 beamline and Mr T. J. Neal, Miss D. Beattie, and Mr E. J. Cornel are acknowledged for assistance with SAXS measurements. This work benefited from the use of the SasView application, originally developed under NSF Award DMR-0520547. SasView also contains code developed with funding from the EU Horizon 2020 programme under the SINE2020 project Grant No. 654000.References
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- M. Doucet, et al., SasView Version 4.1, Zenodo, http://dx.doi.org/10.5281/zenodo.438138 Search PubMed.
- K. van Gruijthuijsen, W. G. Bouwman, P. Schurtenberger and A. Stradner, Europhys. Lett., 2014, 106, 28002 CrossRef.
- B. R. Pauw, J. Phys.: Condens. Matter, 2013, 25, 383201 CrossRef.
- M. J. Hollamby, Phys. Chem. Chem. Phys., 2013, 15, 10566–10579 RSC.
- M. T. Rekveldt, Nucl. Instrum. Methods Phys. Res., Sect. B, 1996, 114, 366–370 CrossRef CAS.
- A. L. Washington, X. Li, A. B. Schofield, K. Hong, M. R. Fitzsimmons, R. Dalgliesh and R. Pynn, Soft Matter, 2014, 10, 3016–3026 RSC.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS.
- J. S. Pedersen and C. Svaneborg, Curr. Opin. Colloid Interface Sci., 2002, 7, 158–166 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- M. K. Kocik, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2014, 10, 3984–3992 RSC.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS PubMed.
- V. J. Cunningham, L. P. D. Ratcliffe, A. Blanazs, N. J. Warren, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Polym. Chem., 2014, 5, 6307–6317 RSC.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- T. Krouglov, W. H. Kraan, J. Plomp, M. T. Rekveldt and W. G. Bouwman, J. Appl. Crystallogr., 2003, 36, 816–819 CrossRef CAS.
- T. Krouglov, W. G. Bouwman, J. Plomp, M. T. Rekveldt, G. J. Vroege, A. V. Petukhov and D. M. E. Thies-Weesie, J. Appl. Crystallogr., 2003, 36, 1417–1423 CrossRef CAS.
- W. G. Bouwman, T. V. Krouglov, J. Plomp, S. V. Grigoriev, W. H. Kraan and M. T. Rekveldt, Phys. B, 2004, 350, 140–146 CrossRef CAS.
- T. V. Kruglov, W. G. Bouwman, I. M. de Schepper and T. M. Rekveldt, Phys. B, 2005, 356, 218–222 CrossRef CAS.
- S. R. Parnell, A. L. Washington, A. J. Parnell, A. Walsh, R. M. Dalgliesh, F. Li, W. A. Hamilton, S. Prevost, J. P. A. Fairclough and R. Pynn, Soft Matter, 2016, 12, 4709–4714 RSC.
- J. Plomp, PhD thesis, TU Delft, 2009.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-Rays, John Wiley & Sons, New York, 1955 Search PubMed.
- J. K. Percus and G. J. Yevick, Phys. Rev., 1958, 110, 1–13 CrossRef CAS.
- S. Nave, J. Eastoe, R. K. Heenan, D. Steytler and I. Grillo, Langmuir, 2000, 16, 8741–8748 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SESANS data available from ISIS Data Journal. See DOI: 10.5286/ISIS.E.83549865. Additional data available from the Zenodo repository. See DOI: 10.5281/zenodo.1308812. See DOI: 10.1039/c8sm01425f |
| ‡ Present address: Niels Bohr Institute, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark. E-mail: gregory.smith@nbi.ku.dk |
| § Present address: Scott Bader Company Ltd., Wollaston, NN29 7RL, UK. |
| ¶ Present address: Fujifilm Speciality Ink Systems Ltd, Pysons Road, Broadstairs, Kent CT10 2LE, UK. |
| This journal is © The Royal Society of Chemistry 2019 |