
Hybridizing amorphous NiOx nanoflakes and Mn-doped Ni2P nanosheet arrays for enhanced overall water electrocatalysis†
Wen-Zhuo
Zhang‡
 a,
Guang-Yi
Chen‡
*a,
Jian
Zhao
*a,
Ji-Cai
Liang
ab,
Guang-Fei
Liu
a,
Bao-Wei
Ji
a and
Li-Feng
Sun
a
a,
Guang-Yi
Chen‡
*a,
Jian
Zhao
*a,
Ji-Cai
Liang
ab,
Guang-Fei
Liu
a,
Bao-Wei
Ji
a and
Li-Feng
Sun
a
aSchool of Automotive Engineering, State Key Laboratory of Structural Analysis for Industrial Equipment, Dalian University of Technology, Dalian, 116024, China. E-mail: chengy@dlut.edu.cn
bKey Laboratory of Automobile Materials, Ministry of Education, and College of Materials Science and Engineering, Jilin University, Changchun, 130025, China. E-mail: jzhao@dlut.edu.cn
First published on 26th August 2019
Abstract
Rational development and facile fabrication of efficient and low-cost bifunctional electrocatalysts for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) play a vital role in hydrogen and oxygen generation by overall water electrocatalysis. Herein, a series of hierarchical core–shell hybrid nanostructured bifunctional electrocatalysts have been successfully synthesized by electrodepositing amorphous nickel oxide nanoflakes (A-NiOx) on the surface of crystalline Mn-doped Ni2P nanosheet arrays (Mn5-Ni2P). Benefitting from the synergistic effect generated from the outer amorphous NiOx and inner highly crystalline Mn5-Ni2P nanosheets, the optimal A-NiOx-20/Mn5-Ni2P sample exhibits quite low overpotentials for the HER (55 mV) and OER (255 mV) to afford a current density of 10 mA cm−2 in alkaline electrolyte. Moreover, the two-electrode water electrolysis device only requires a small cell voltage of 1.54 V to deliver 10 mA cm−2 and shows no obvious attenuation for 20 hours. Our work will provide a valuable method to design and synthesize efficient electrocatalysts for water splitting and other applications of energy conversion and storage.
1. Introduction
The extensive use of fossil fuels has caused serious environmental problems and energy shortage which have promoted the re-emergence of research on renewable hydrogen generation.1–4 Water electrolysis, which is driven by the electrocatalytic HER and OER, has long been regarded as one of the most promising methods to produce clean hydrogen energy.1,5–7 However, the sluggish HER and OER processes still require efficient catalysts to enhance the low electrocatalytic efficiency and reduce the high overpotential.4,8,9 For now, although noble-metal-based electrocatalysts (Pt, RuO2 or IrO2) exhibit excellent electrocatalytic activity, their scarcity and high price considerably impede their wide-spread utilization.6,10–13 Therefore, the exploration of non-noble-metal based effective bifunctional electrocatalysts with high stability for water electrolysis devices is highly desirable.Among all the non-noble-metal based bifunctional electrocatalysts, Ni-based compounds14,15 have received wide research attention. Especially for NiO16,17 and Ni2P,18,19 many prominent researchers are committed to enhance their catalytic performance through various methods, such as doping with heteroatoms,20,21 constructing novel structures,22–24 creating vacancy defects25–27 and so on. However, their electrocatalytic activities still fail to compete with those of noble-metal-based catalysts.28,29 Nowadays, fabrication of amorphous nanomaterials with abundant active sites has been considered as a promising method to enhance the activity of electrocatalysts due to their unique characteristics of short-range order structure.30,31 But the short-range order structure also leads to an unsatisfactory conductivity, which is the fatal weakness of amorphous materials compared with crystal materials.32,33 Therefore, it is of great interest to rationally design a novel structure which can combine the virtues of amorphous and crystalline materials to improve the activity of catalysts.
According to previous research,34–37 construction of hybrid catalysts with heterostructures has been a promising way to fabricate effective electrocatalysts. For example, Wu et al.38 revealed that the coupling interface between MoS2 and Fe5Ni4S8 could facilitate the adsorption of H+ and OH−, according to density functional theory calculations. Zhu et al.39 devised a new approach of developing a Co–Ni3N heterostructure array and found that the heterostructure could facilitate electron transfer between the two different domains. Kim et al.40 found that MoS2 basal planes were activated by decorating with Ni2P nanoparticles, so the Ni2P/MoS2/NRGO catalyst displayed Pt-like HER performance. Han et al.41 found that the Mn–Co–P@MnCo2O4 nanowire array only required 269 mV overpotential to drive a current density of 10 mA cm−2 in 1 M KOH during the OER, which was 93 mV smaller than that of the MnCo2O4 nanoarray. Based on the above research, it can be safely concluded that hybrid catalysts with heterostructures can effectively combine two single-component catalysts and fully exert a synergistic effect, leading to improved physical and chemical properties compared to monolithic catalysts.
Herein, a series of novel hybrid catalysts with 3D heterostructures on Ni foam (NF) have been synthesized by electrodepositing amorphous NiOx nanoflakes (A-NiOx) on the surface of Mn doped-Ni2P nanosheet arrays and acted as bifunctional catalysts for water electrolysis. The inner crystal Mn doped-Ni2P nanosheet arrays which straightly grew on the NF substrate can effectively accelerate the transfer rates of electrons and ions, while the amorphous ultrathin NiOx nanoflakes are beneficial to expose more active surface and reduce the blockage of the active sites of Mn doped-Ni2P. Consequently, the optimal A-NiOx-20/Mn5-Ni2P catalyst exhibits superior bifunctional electrocatalytic performance in alkaline medium, and only needs small overpotentials of 55 mV and 255 mV to afford 10 mA cm−2 for the HER and OER, respectively. More importantly, a home-made two-electrode water electrolysis device by employing the A-NiOx-20/Mn5-Ni2P catalyst as the anode as well as cathode electrode can drive 10 mA cm−2 current density only at a small voltage of 1.54 V, demonstrating its greatly promising prospects in the water electrolysis industry.
2. Experimental section
2.1. Chemicals and materials
50% manganese nitrate solution (Mn(NO3)2), ammonium fluoride (NH4F), potassium hydroxide (KOH), urea (CO(NH2)2), nickel acetate (Ni(CH3COO)2), ammonium acetate (CH3COONH4) and sodium hypophosphite (NaH2PO2) were all of analytical grade. The Pt/C catalyst (20 wt%) was bought from Alfa Aesar and deionized water was used throughout. Before use, NFs (thickness: 1 mm) were first treated with acetone and 1 M HCl solution to remove impurities and oxides on the surface, and then they were washed with deionized water and ethanol in turn for each three times.2.2. Synthesis of the Mnx-Ni2P nanosheet arrays
7.5 mmol CO(NH2)2, 4 mmol ammonium fluoride and different amounts of Mn(NO3)2 (3, 5 and 7 mmol) were first dissolved in 35 ml distilled water. Then the aqueous solution and treated NFs (2 cm × 2 cm) were transferred to a 50 ml Teflon-lined stainless steel autoclave, heated to 120 °C and maintained at this temperature for 5 h. After the hydrothermal reaction, the NFs coated with the precursor of Mnx-Ni2P were taken out, rinsed with deionized water and dried at 60 °C. Afterwards, the Mnx-Ni2P (x denotes the mole of Mn(NO3)2 used during the hydrothermal process) was obtained by direct phosphorization of the above precursor at 400 °C for 1 h with a ramp rate of 2 °C min−1 in a N2 atmosphere using NaH2PO2 as the phosphorus source. Typically, the precursor of Mnx-Ni2P was put into a porcelain boat with 20 mmol NaH2PO2 placed 4 cm away from the precursor of Mnx-Ni2P on the upstream side. For comparison, the pure Ni2P sample was also prepared under similar conditions of the Mnx-Ni2P sample except that 5 mmol Ni(NO3)2 was used to substitute Mn(NO3)2 during the hydrothermal process. The mass loadings of Mn3-Ni2P, Mn5-Ni2P, Mn7-Ni2P and pure Ni2P were about 2.2, 2.3, 2.3 and 2.8 mg cm−2.2.3. Synthesis of the A-NiOx/Mn5-Ni2P catalysts
A series of A-NiOx/Mn5-Ni2P catalysts were fabricated by the constant current electrodeposition process in 100 ml of 0.01 M nickel acetate and 0.002 M ammonium acetate mixed aqueous solution with the above obtained Mn5-Ni2P as the working electrode, graphite rod as the counter electrode and standard Ag/AgCl as the reference electrode. After being electrodeposited at a cathodic current of 7.5 mA cm−2 for different times (10 min, 20 min, and 30 min, respectively), the resulting samples were dried at 80 °C, and accordingly named as A-NiOx-10/Mn5-Ni2P, A-NiOx-20/Mn5-Ni2P, and A-NiOx-30/Mn5-Ni2P, respectively. Furthermore, single A-NiOx was also synthesized by direct electrodeposition on the bare NF under the same conditions for 20 min. The mass loadings of A-NiOx-10/Mn5-Ni2P, A-NiOx-20/Mn5-Ni2P, A-NiOx-30/Mn5-Ni2P, and A-NiOx were about 2.8, 3.6, 4.5 and 1.9 mg cm−2, respectively.2.4. Characterization
The microstructure and morphology of the samples were investigated by scanning electron microscopy (SEM, JEOL JSM-6330F) and transmission electron microscopy (TEM, JEOL 2010, 200 kV). Energy dispersive X-ray (EDX) spectroscopy was conducted to analyze the elemental distribution using a JEOL JSM-6330F SEM. X-ray diffraction (XRD) data were recorded on a Rigaku/Max-2550 with Co Kα radiation (λ = 1.7890 Å). X-ray photoelectron spectroscopy (XPS) scans were obtained on a multifunctional imaging electron spectrometer (Thermo ESCALAB 250XI).2.5. Electrochemical tests
The HER and OER measurements were conducted on a CHI660E workstation (Shanghai, Chenhua) with a three-electrode system, by using the prepared catalytic electrode as the working electrode, a graphite rod as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode. The HER measurements were performed in 1 M KOH and the OER measurements were performed in O2-saturated 1 M KOH. Linear sweep voltammetry (LSV) was conducted at a scan rate of 2 mV s−1 for both the HER and OER. Before the test, 50 cyclic voltammetric (CV) cycles were conducted between 0.1 and −0.4 V vs. RHE for the HER and 1.2–1.6 V vs. RHE for the OER with a scan rate of 50 mV s−1. The measured potentials were converted to RHE using eqn (1):| ERHE = Emeasured + 0.059 × pH + 0.242 | (1) |
Electrochemical impedance spectroscopy (EIS) measurements were performed with the same configuration at a bias of −100 mV in a frequency range from 105 to 0.01 Hz. All of the potentials and voltages were iR compensated unless noted. The electrochemical double-layer capacitance (Cdl) of the catalysts was estimated by CV at different scan rates (10, 20, 40, 60, 80, and 100 mV s−1) from 0.1–0.2 V vs. RHE. The Cdl value could be estimated from the slope of the resulting (ja − jc)/2 vs. v plots (ja and jc represent the anodic and cathodic current densities at 0.15 V vs. RHE). The long-term durability tests for the HER and OER were all performed using chronopotentiometric (CP) measurements at a constant current density of 10 mA cm−2 for 20 h.
The overall water electrolysis measurement tests were conducted in a two-electrode system employing the prepared catalytic electrodes both as the cathode and anode. The LSV curves were recorded at a scan rate of 2 mV s−1 without iR correction and the CP measurement was also performed at a current density of 10 mA cm−2 for 20 h. Faradaic efficiency was calculated according to the reported methods.42,43 The amounts of H2 and O2 during the overall water electrolysis at 10 mA cm−2 (Ni foam: 1 × 1 cm2) were measured on a gas chromatograph (Trace 1300). Turnover frequency (TOF) for the HER was calculated using eqn (2):
| TOF = jS/2Fn | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), 2 is the number of electrons involved during the reaction, and n is the number of active sites.
485 C mol−1), 2 is the number of electrons involved during the reaction, and n is the number of active sites.
According to the research of Hu et al.,44 the number of active sites was calculated by the CV measurement with a scan rate of 50 mV s−1 in a potential range of −0.2 to 0.6 V (vs. RHE) in 1 M PBS (pH = 7). Assuming a one electron redox process, the upper limit of active sites could be obtained according to eqn (3):
| n = Q/2F | (3) |
3. Results and discussion
3.1. Synthetic protocol and materials characterization
The fabrication processes of A-NiOx-T/Mnx-Ni2P catalysts are illustrated in Scheme 1, including the hydrothermal process, high-temperature phosphorization and electrodeposition. The pretreated NFs with a smooth surface were used as the precursor, substrate and current collector. Firstly, the precursor of Mnx-Ni2P nanosheet arrays was straightly grown on the NFs by a hydrothermal self-growth process. Mn(NO3)2 was purposely chosen as the reactant because it can be decomposed to produce HNO3 during the hydrothermal process, so the surface of NFs would be partly etched by the acid and Ni2+ ions would be released into the reaction solution. With the hydrothermal reaction proceeding, the Mn-doped Ni-carbonate hydroxide precursor nanosheets successfully grew on NFs. After that, the above self-grown nanosheet precursor was then used as the starting material and converted into highly crystalline Mnx-Ni2P nanosheets through a high-temperature phosphorization process at 400 °C. Then, the amorphous NiOx nanoflakes were coated on the surface of Mnx-Ni2P nanosheets by a simple electrodeposition method to form the final A-NiOx/Mnx-Ni2P hierarchical core–shell electrodes.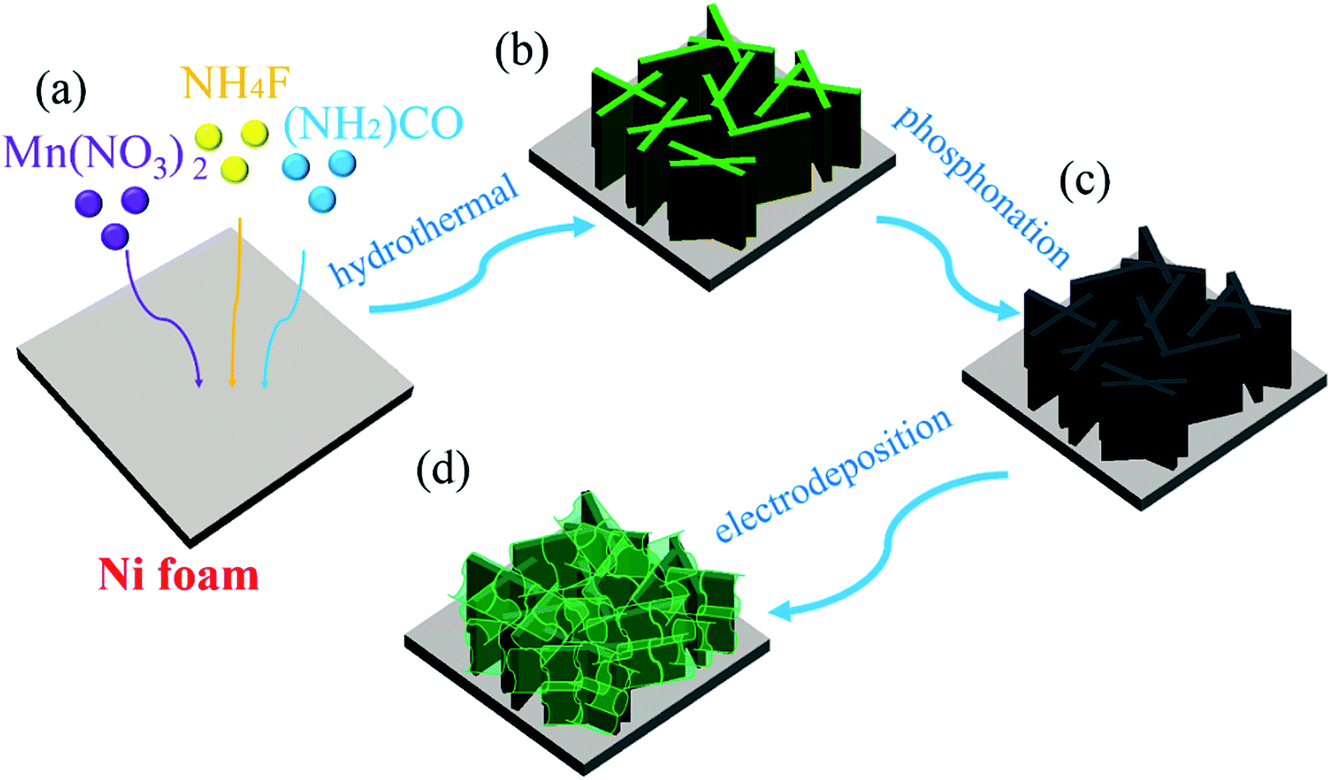 | ||
| Scheme 1 Schematic synthesis procedure of the A-NiOx-T/Mn5-Ni2P catalytic electrodes. | ||
The morphology and crystalline structure of Mnx-Ni2P were first characterized by SEM and XRD. As shown in Fig. 1a–c, all the Mnx-Ni2P (x = 3, 5, and 7) samples present a similar morphology with little difference and the original smooth Ni foams are fully covered with aligned nanosheet arrays. The EDS result presented in Fig. S1† shows that the Ni, P and Mn elements exist in all Mnx-Ni2P samples and the ratio of Mn to P is about 8.1%, 11.2% and 16.0% in Mn3-Ni2P, Mn5-Ni2P and Mn7-Ni2P, respectively. Moreover, the XRD patterns of Mn3-Ni2P, Mn5-Ni2P and Mn7-Ni2P shown in Fig. 1d indicate that except the strong diffraction peaks of the NF substrate, the apparent four peaks at about 47.5°, 52.2° (overlapping with the diffraction peak of Ni), 55.4° and 63.6° can be indexed to the (111), (021), (210) and (003) planes of Ni2P (JCPDS no. 65-3544). It is noteworthy that there are not any XRD diffraction signals belonging to manganese phosphide species in all Mnx-Ni2P materials, demonstrating that Mn is doped into Ni2P. For comparison, pure Ni2P nanosheet arrays on NFs were also synthesized and characterized by SEM, XRD and EDS (Fig. S2†). The in situ self-supported Mnx-Ni2P nanosheet arrays have not only good electrical conductivity but also excellent mechanical properties, making them a suitable skeleton for further electrodeposition of other catalytically active materials. In order to determine which one among all the Mnx-Ni2P samples and pure Ni2P nanosheet arrays is suitable as the skeleton for further electrodeposition of amorphous NiOx, we first evaluated their electrocatalytic performance from LSV curves. As shown in Fig. S3,† the Mn5-Ni2P sample has the best electrocatalytic activity for both the HER and OER at the same time. So the Mn5-Ni2P sample was chosen as the target material for further investigation.
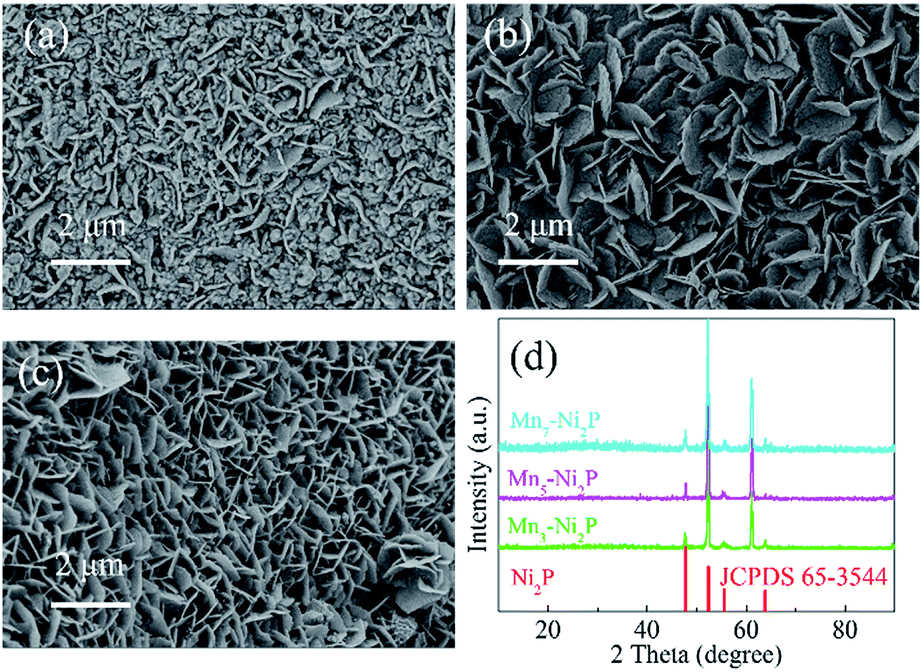 | ||
| Fig. 1 SEM images of (a) Mn3-Ni2P, (b) Mn5-Ni2P and (c) Mn7-Ni2P. (d) XRD patterns of Mnx-Ni2P. | ||
The low-magnification SEM image (Fig. 2a) reveals that after the electrodeposition process for 20 min, the resulting A-NiOx-20/Mn5-Ni2P sample also presented the morphology of nanosheet arrays, which is similar to the Mn5-Ni2P precursor. However, the high-magnification SEM image (Fig. 2b) definitely indicates that the nanosheet arrays are actually coated with continuous chiffon-like nanoflakes. The ultrathin nanoflakes were interconnected with each other to form a higher specific surface structure and complete electron and ion transfer paths. The XRD pattern presented in Fig. 2c shows that no diffraction peaks of NiOx species can be detected for the A-NiOx-20/Mn5-Ni2P sample after the electrodeposition process, indicating that the electrodeposited NiOx is amorphous. However, the EDS spectra and corresponding elemental mapping images (Fig. 2d and S4†) clearly reveal the existence of Ni, Mn, P and O elements in the A-NiOx-20/Mn5-Ni2P sample and the elements are distributed uniformly. The hybrid structure of A-NiOx-20/Mn5-Ni2P was further proved by the TEM image (Fig. 2e) and HRTEM image (Fig. 2f). The distinct lattice fringes with a d-spacing of 2.212 Å can be attributed to the (111) plane of Ni2P. The amorphous nature of outer NiOx nanoflakes was also confirmed by the HRTEM image and the fast Fourier transform (FFT) image, which is in agreement with the above XRD result. The morphologies of A-NiOx-10/Mn5-Ni2P and A-NiOx-30/Mn5-Ni2P were also studied by SEM. As shown in Fig. S5a,† the morphology of A-NiOx-10/Mn5-Ni2P is very similar to that of A-NiOx-20/Mn5-Ni2P but with little difference in which the quantity of amorphous NiOx nanoflakes is less than that of the A-NiOx-20/Mn5-Ni2P sample. However, along with the further increase of the electrodeposition time to 30 minutes, more NiOx was deposited on the surface of Mn5-Ni2P nanosheet arrays to finally form a layered nanocomposite structure which looks very similar to a dry ground (Fig. S5b†).
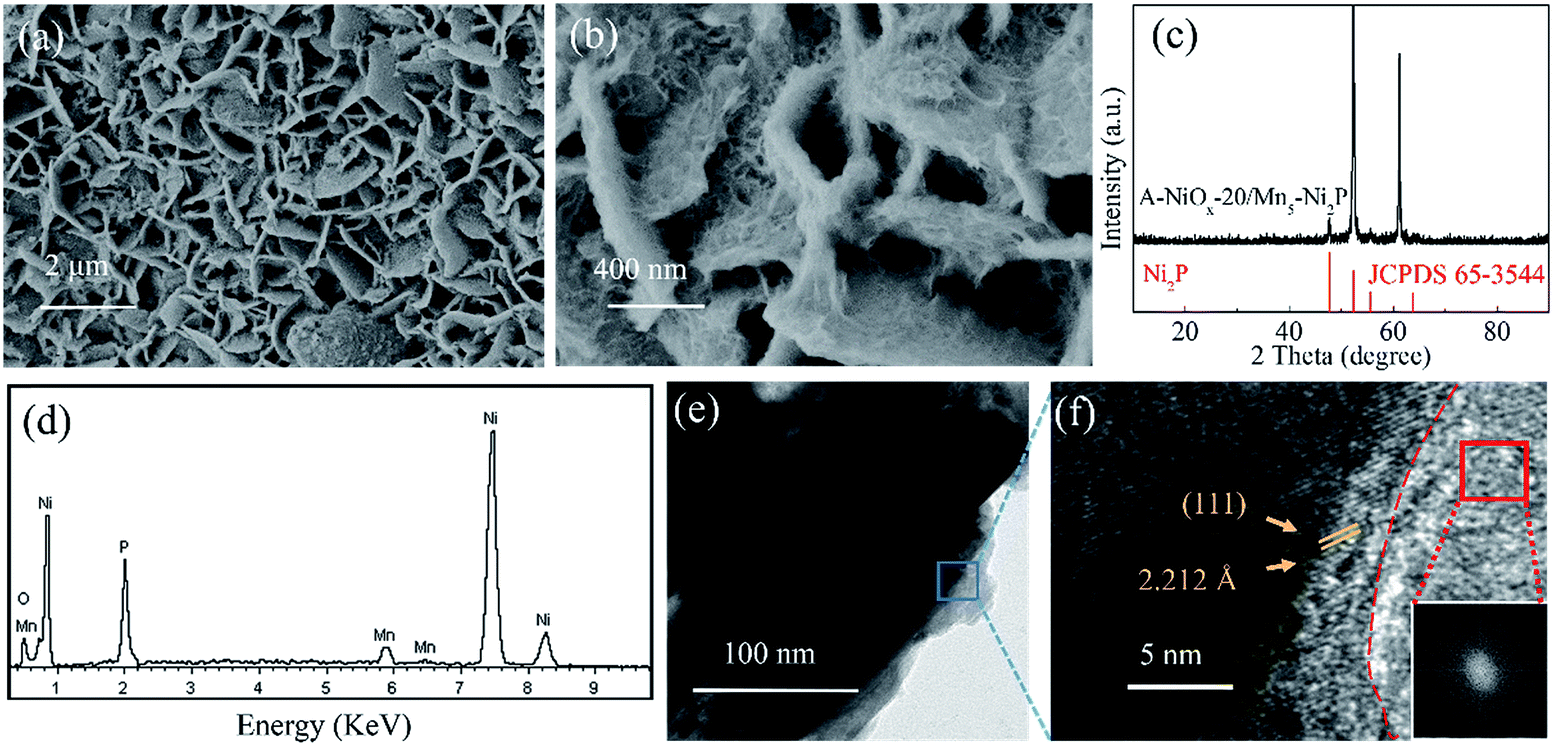 | ||
| Fig. 2 (a and b) SEM images, (c) XRD pattern, (d) EDS spectrum, (e) TEM image and (f) HRTEM image of A-NiOx-20/Mn5-Ni2P (insert of (f) is the FFT for the red framed region). | ||
XPS was used to analyze the chemical surface electronic states of A-NiOx-20/Mn5-Ni2P and Mn5-Ni2P samples (Fig. 3). As shown in Fig. 3a, the Mn 2p spectrum could be fitted to two main peaks around ∼643 eV and ∼654 eV assigned to Mn 2p3/2 and Mn 2p1/2,45 while the peaks around ∼643 eV can be divided into two peaks at ∼641 eV and ∼645 eV, which represent Mn2+ and Mn3+, respectively.45,46 In the Ni 2p region (Fig. 3b), the peaks around ∼853 eV and ∼870 eV can be attributed to the Ni 2p3/2 orbitals in Ni2P.47 Moreover, the peaks around ∼855 eV and ∼873 eV with two satellite peaks around ∼861 eV and ∼879 eV can be attributed to the NiOx species in A-NiOx-20/Mn5-Ni2P. The peaks at the same position are also observed for Mn5-Ni2P but much lower than that of A-NiOx-20/Mn5-Ni2P, which is due to the surface oxidation of Mn5-Ni2P.47,48 This superficial oxidation phenomenon can also be observed in the P 2p region (Fig. 3c). The two peaks at ∼129 and ∼134 eV corresponding to the phosphide49 and phosphate/phosphite are owing to the surface oxidation state of P.50 High-resolution O 1s spectra are shown in Fig. 3d. The peak at ∼531.5 eV can be observed for Mn5-Ni2P, which is assigned to phosphate species,32,51 while the additional strong peak at ∼530 eV corresponds to the oxide species in A-NiOx-20/Mn5-Ni2P, also indicating that the outside surface nanoflakes are NiOx species.51,52
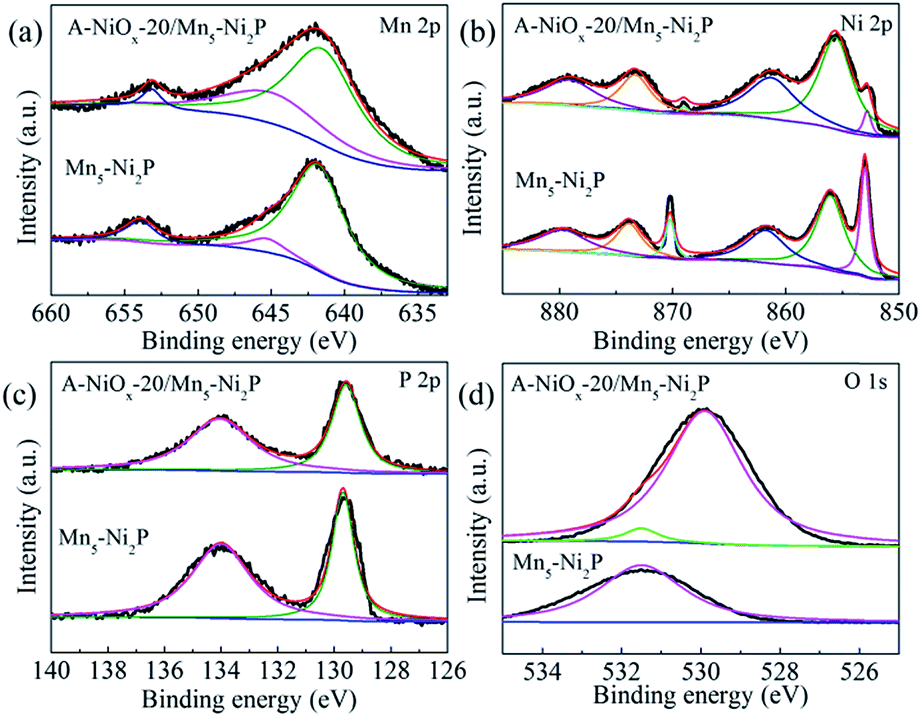 | ||
| Fig. 3 The high resolution XPS spectra of A-NiOx-20/Mn5-Ni2P and Mn5-Ni2P. (a) Mn 2p, (b) Ni 2p, (c) P 2p and (d) O 1s. | ||
3.2. Electrocatalytic performance of the prepared catalysts
The HER performances of the prepared catalytic electrodes were investigated in 1 M KOH electrolyte. For comparison, the bare Mn5-Ni2P and single A-NiOx electrodes were also studied under the same conditions. As presented in Fig. 4a, the iR compensated LSV curves clearly show that the A-NiOx-20/Mn5-Ni2P electrode only requires a small overpotential (η) of 55 mV to afford a current density (j) of 10 mA cm−2, which is obviously more outstanding than that of A-NiOx-10/Mn5-Ni2P (η = 76 mV), A-NiOx-30/Mn5-Ni2P (η = 65 mV), Mn5-Ni2P (η = 96 mV) and A-NiOx (η = 126 mV). In order to avoid the interference of scan rate, controlled-current electrolysis (CCE) at different current densities was conducted for 1 h to determine the accurate overpotential for the HER.53,54 As shown in Fig. S6,† the A-NiOx-20/Mn5-Ni2P electrode can achieve 10, 50, and 100 mA cm−2 at small overpotentials of about 55, 132 and 166 mV respectively, and performs better than many reported catalysts as shown in Table S1.† Moreover, the A-NiOx-20/Mn5-Ni2P sample also displays a superior exchange current density (j0 = 0.62 mA cm−2) and mass activity (20 mA mg−1 at an overpotential of 150 mV) as shown in Fig. S7 and S8.† All the above results indicate the excellent HER activity of the A-NiOx-20/Mn5-Ni2P electrode and prove that the hybridization of amorphous NiOx and crystallized Mn5-Ni2P in a suitable manner can have a great promoting effect on the HER performance. The intrinsic activity of active sites was evaluated by TOF based on the electrochemical method.55,56 As shown in Fig. S9,† the TOF value of A-NiOx-20/Mn5-Ni2P is 0.81 s−1 at an overpotential of 200 mV, which is about 2 and 4.5 times that of Mn5-Ni2P (0.40 s−1) and A-NiOx (0.18 s−1), revealing that A-NiOx-20/Mn5-Ni2P delivers higher intrinsic activity than the single-components of Mn5-Ni2P and A-NiOx. Furthermore, A-NiOx-20/Mn5-Ni2P also possesses a better Tafel slope of 37 mV dec−1 than that of Mn5-Ni2P (59 mV dec−1) and A-NiOx (76 mV dec−1), suggesting admirable HER kinetics (Fig. 4b). The HER kinetics were further verified by the EIS measurement (Fig. 4c). Naturally, the crystalline Mn5-Ni2P shows superior conductivity (Rct = 4.0 Ω) to A-NiOx with amorphous structure (Rct = 6.1 Ω). However, the heterostructured A-NiOx-20/Mn5-Ni2P electrode unexpectedly shows the smallest Rct of 2.3 Ω among all three electrodes, which is attributed to the surface interconnected nanoflakes. They can join the independent Mn5-Ni2P nanosheets to form a complete charge transport network. Moreover, the Cdl was also estimated by the CV cycles (Fig. S10a–c†) to represent the electrochemically active surface area.30,55 As shown in Fig. S10d,† the A-NiOx-20/Mn5-Ni2P sample possesses the highest Cdl value of 66.2 mF cm−1 which is much higher than that of A-NiOx (33.2 mF cm−2) and Mn5-Ni2P (22.3 mF cm−1). The above measurement results clearly prove that the novel heterostructure of A-NiOx-20/Mn5-Ni2P can fully exert the advantages of crystal and amorphous materials, leading to the admirable intrinsic activity, excellent conductivity and abundant active sites. In view of the favorable HER activity of A-NiOx-20/Mn5-Ni2P catalytic electrodes, the stability and durability were evaluated by long-term electrolysis at a current density of 10 mA cm−2 (Fig. 4d). The overpotential of the A-NiOx-20/Mn5-Ni2P electrode shows a negligible increase after the CP determination for 20 h, demonstrating its excellent durability and stability for the HER. The excellent stability also can be verified from another side by SEM, XRD and HRTEM. The morphology and crystal structure of the A-NiOx-20/Mn5-Ni2P catalytic electrode are perfectly maintained after the durability test (Fig. S11†).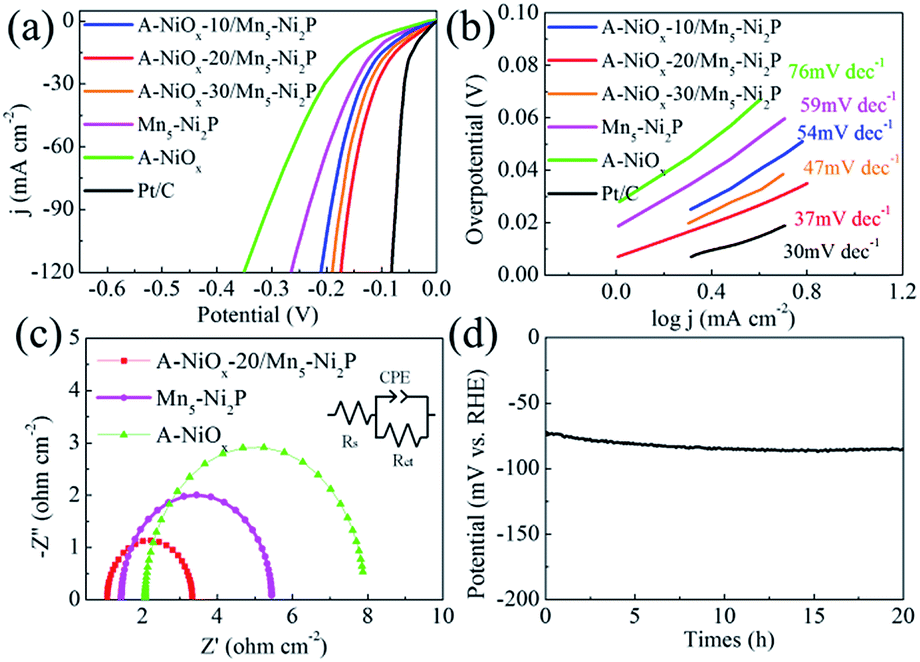 | ||
| Fig. 4 (a) LSV curves of A-NiOx-T/Mn5-Ni2P, Mn5-Ni2P, A-NiOx and Pt/C for the HER. (b) Corresponding Tafel plots of A-NiOx-T/Mn5-Ni2P, Mn5-Ni2P, A-NiOx and Pt/C. (c) Nyquist plots of A-NiOx-20/Mn5-Ni2P, Mn5-Ni2P and A-NiOx at −100 mV vs. RHE (insert is the equivalent circuit). (d) Long-term HER stability test of A-NiOx-20/Mn5-Ni2P at 10 mA cm−2 (without iR correction). | ||
Moreover, the high resolution XPS spectra shown in Fig. S12† demonstrate that the peaks at ∼855, ∼873 eV in the Ni 2p region and ∼530 eV in the O 1s region corresponding to NiOx do not change obviously, implying that the amorphous shell catalyst is still NiOx. However, the intensity of peaks at ∼853 and ∼129 eV belonging to Ni2P obviously decreases and a peak at ∼530.5 eV attributed to the O–H appears, indicating that the surface of the Ni2P core catalyst is partially oxidized into amorphous Ni(OH)x. As displayed in Fig. S13,† the XRD result shows that only the crystal Ni2P phase exists in A-NiOx-20/Mn5-Ni2P after the HER, implying that the produced Ni(OH)x and the NiOx shell are all amorphous. The above results definitely demonstrate that the surface of the core catalyst Ni2P is oxidized to amorphous Ni(OH)x after the HER stability test. According to previous reports,48,54 the thickness of Ni(OH)x on the surface of Ni2P is usually very thin, while as for our A-NiOx-20/Mn5-Ni2P catalyst, the generated amorphous Ni(OH)x was mixed with the amorphous NiOx shell, so it is very hard to determine the thickness of the Ni(OH)x layer.
The electrocatalytic performance of the as-prepared electrodes towards the OER was evaluated using the same three-electrode system in O2-saturated 1 M KOH electrolyte. The three electrodes all have a distinct oxidation peak between 1.3 V and 1.5 V vs. RHE due to the oxidation of Ni species (Fig. 5a).57 The A-NiOx-20/Mn5-Ni2P catalytic electrode possesses the best OER activity, and only needs a small potential of 1.485 V to afford 10 mA cm−2, much better than that of A-NiOx-10/Mn5-Ni2P (1.52 V), A-NiOx-30/Mn5-Ni2P (1.505 V), Mn5-Ni2P (1.53 V) and A-NiOx (1.567 V). The CCE was also used to determine the accurate overpotential for the OER at different current densities. As shown in Fig. S14,† the A-NiOx-20/Mn5-Ni2P electrode only requires small overpotentials of about 255, 295 and 320 mV to achieve 10, 50 and 100 mA cm−2 respectively, and is superior to many reported OER catalysts in alkaline medium (Table S2†). A-NiOx-20/Mn5-Ni2P also has high mass activity (63 mA mg−1 at a small overpotential of 350 mV), outstanding kinetics (Tafel slope is 55 mV dec−1) and remarkable stability (the overpotential negligibly increased after the 20 h CP measurement) during the OER process (Fig. 5b–d). The morphological and structural changes of the A-NiOx-20/Mn5-Ni2P catalyst after the OER process were also investigated by SEM, HRTEM, XRD, XPS and EDS. As shown in Fig. S15a,† although some agglomeration took place and the surface of the nanosheets became rougher compared to the HER process, the hybrid composite nanostructure was still basically preserved after the OER process. However, the HRTEM image (Fig. S15b†) clearly shows a typical triple layer structure after the OER stability test. The outer layer is the amorphous material, and the middle and the inner layer are the crystal material with lattice spacings of 3.908 and 2.212 Å, corresponding well to the (006) plane of Ni(OH)2 and the (111) plane of Ni2P, respectively. The XPS results of the A-NiOx-20/Mn5-Ni2P catalyst after the OER process are shown in Fig. S16.† The peaks at ∼855, ∼873 and ∼530 eV can also be observed obviously in Ni 2p and O 1s regions, indicating that the outer layer amorphous material is NiOx. However, peaks at ∼853 and ∼129 eV corresponding to Ni2P disappear, while a new peak at ∼530.5 eV ascribed to the O–H species appears, implying that the middle layer crystal Ni(OH)2 is transformed from the core catalyst Ni2P. This change can also be verified by the XRD test, and the intensity corresponding to the Ni2P peaks clearly decreased and the other two diffraction peaks belonging to Ni(OH)2 (JCPDS no. 38-0751) appeared (Fig. S17a†). Moreover, the EDS result shown in Fig. S17b† demonstrates that the intensity of the P peak obviously decreased while the intensity of the O peak greatly increased. The above results indicate that after the OER stability test, the surface of the core catalyst Ni2P was oxidized to Ni(OH)2, which is very common in phosphide-based catalysts for the OER and consistent with previous literature reports.58,59
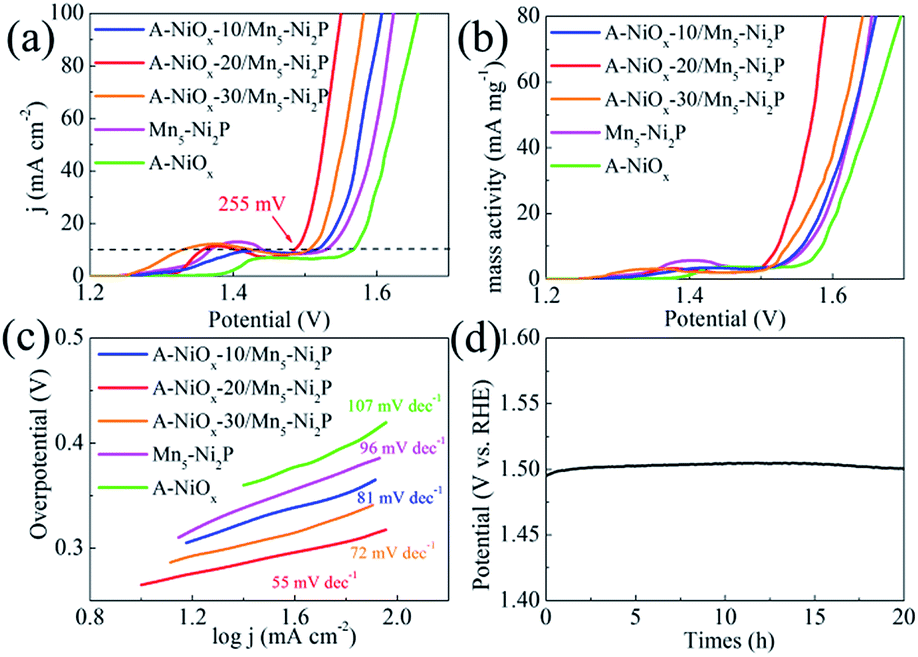 | ||
| Fig. 5 (a) LSV curves of A-NiOx-T/Mn5-Ni2P, Mn5-Ni2P and A-NiOx for the OER. (b) Mass activity of A-NiOx-T/Mn5-Ni2P, Mn5-Ni2P and A-NiOx for the OER. (c) Tafel plots for A-NiOx-T/Mn5-Ni2P, Mn5-Ni2P, A-and NiOx. (d) Long-term OER stability test of A-NiOx-T/Mn5-Ni2P at 10 mA cm−2 (without iR correction). | ||
Considering the outstanding bifunctional catalytic performance of the A-NiOx-20/Mn5-Ni2P electrode, we further used the prepared electrode as the anode as well as the cathode to prepare a two-electrode device for the overall water splitting in alkaline electrolyte (1 M KOH). As displayed in Fig. 6a, only a small voltage of 1.54 V is needed for A-NiOx-20/Mn5-Ni2P||A-NiOx-20/Mn5-Ni2P to achieve a current density of 10 mA cm−2, which is much smaller than that of Mn5-Ni2P||Mn5-Ni2P (1.65 V), A-NiOx||A-NiOx (1.71 V), and even some reported Ir/C||Pt/C couples (1.62 V).60
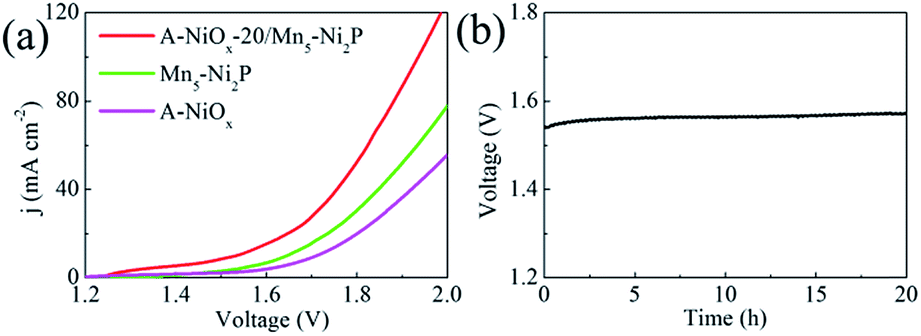 | ||
| Fig. 6 (a) LSV curves of A-NiOx-20/Mn5-Ni2P||A-NiOx-20/Mn5-Ni2P, Mn5-Ni2P||Mn5-Ni2P and A-NiOx||A-NiOx for overall water splitting (without iR correction). (b) Long-term overall water splitting stability test of A-NiOx-20/Mn5-Ni2P||A-NiOx-20/Mn5-Ni2P at 10 mA cm−2(without iR correction). | ||
Furthermore, the faradaic efficiency of the alkaline electrolyzer was measured at a current density of 10 mA cm−2 as shown in Fig. S18.† The measured molecular ratio of H2 to O2 is about 2:1 and the faradaic efficiency of A-NiOx-20/Mn5-Ni2P is determined to be 100% for the HER and 98% for the OER, indicating that any competitive side reaction can be ruled out. Furthermore, the A-NiOx-20/Mn5-Ni2P electrode was very stable during overall water splitting. As shown in Fig. 6b, the voltage of the electrolytic device at 10 mA cm−2 showed no fluctuation for 20 h. The above results indicate that the A-NiOx-20/Mn5-Ni2P electrode is a promising bifunctional electro-catalyst towards overall water splitting.
4. Conclusions
In conclusion, a series of hierarchical core–shell hybrid nanostructured A-NiOx-T/Mn5-Ni2P catalytic electrodes have been prepared by growing amorphous NiOx on highly crystalline and conductive Mn-doped Ni2P nanosheet arrays by a simple and scalable method. Benefitting from the large surface area provided by the outer amorphous NiOx and good conductivity offered by the inner Mn5-Ni2P nanosheets, the optimal A-NiOx-20/Mn5-Ni2P electrode shows excellent bifunctional catalytic performance in alkaline electrolyte, and is better than the single-component counterparts and some reported electrocatalysts. Furthermore, the alkaline electrolytic device fabricated by using A-NiOx-20/Mn5-Ni2P as both the cathode and anode only needs a small voltage to deliver 10 mA cm−2 for at least 20 hours, indicating that it is a promising and practical candidate for overall water splitting and other energy related applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Fundamental Research Funds for the Central Universities of China (no. DUT17LK01) and the National Natural Science Foundation of China (no. 51575088).Notes and references
- B. Raush, M. D. Symes, G. Chisholm and L. Cronin, Science, 2014, 345, 1326–1330 CrossRef.
- X. Li, X. Hao, A. Abudula and G. Guan, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- X. Xia, C. Wang, L. Liu, Y. Du and W. Ye, Sustainable Energy Fuels, 2018, 2, 444 RSC.
- C. Meng, T. Ling, T. Y. Ma, H. Wang, Z. Hu, Y. Zhou, J. Mao, X. W. Du, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2017, 29, 1604607 CrossRef.
- X. Zhang, X. Zhang, H. Xu, Z. Wu, H. Wang and Y. Liang, Adv. Funct. Mater., 2017, 27, 1606635 CrossRef.
- S. Lian, M. P. Browne, C. Dominguez, S. N. Stamatin, H. Nolan, G. S. Duesberg, M. E. G. Lyons, E. Fonda and P. E. Colavita, Sustainable Energy Fuels, 2017, 1, 780–788 RSC.
- S. Hirai, K. Morita, K. Yasuoka, T. Shibuya, Y. Tojo, Y. Kamihara, A. Miura, H. Suzuki, T. Ohno, T. Matsuda and S. Yagi, J. Mater. Chem. A, 2018, 6, 15102–15109 RSC.
- W. Li, X. Gao, D. Xiong, F. Wei, W. G. Song, J. Xu and L. Liu, Adv. Energy Mater., 2017, 7, 1602579 CrossRef.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Xu, J. Li, D. Xiong, B. Zhang, Y. Liu, K. H. Wu, I. Amorim, W. Li and L. Liu, Chem. Sci., 2018, 9, 3470–3476 RSC.
- W. Liu, K. Du, L. Liu, J. Zhang, Z. Zhu, Y. Shao and M. Li, Nano Energy, 2017, 38, 576–584 CrossRef CAS.
- M. Dave, A. Rajagopal, M. D. Ruttensperger, B. Schwarz, F. Nagele, L. Daccache, D. Fantauzzi, T. Jacob and C. Streb, Sustainable Energy Fuels, 2018, 2, 1020–1026 RSC.
- Y. Sun, K. Xu, Z. Wei, H. Li, T. Zhang, X. Li, W. Cai, J. Ma, H. J. Fan and Y. Li, Adv. Mater., 2018, 30, 1802121 CrossRef.
- H. Li, S. Chen, H. Lin, X. Xu, H. Yang, L. Song and X. Wang, Small, 2017, 13, 1701487 CrossRef.
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou, M. T. Soo, M. Hong, X. Yan, G. Qian, J. Zou, A. Du and X. Yao, Adv. Mater., 2017, 29, 1700017 CrossRef.
- J. Zheng, W. Zhou, T. Liu, S. Liu, C. Wang and L. Guo, Nanoscale, 2017, 9, 4409–4418 RSC.
- J. Liu, Y. Zheng, Y. Jiao, Z. Wang, Z. Lu, A. Vasileff and S. Z. Qiao, Small, 2018, 14, 1704073 CrossRef.
- L. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- Y. Pan, N. Yang, Y. Chen, Y. Lin, Y. Li, Y. Liu and C. Liu, J. Power Sources, 2015, 297, 45–52 CrossRef CAS.
- R. Wu, B. Xiao, Q. Gao, Y. R. Zheng, X. S. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., 2018, 130, 15671–15675 CrossRef.
- Z. Du, N. Jannatun, D. Yu, J. Ren, W. Huang and X. Lu, Nanoscale, 2018, 10, 23070–23079 RSC.
- M. Miao, R. Hou, Z. Liang, R. Qi, T. He, Y. Yan, K. Qi, H. Liu, G. Feng and B. Y. Xia, J. Mater. Chem. A, 2018, 6, 24107–24113 RSC.
- X. Liu, X. Wang, X. Yuan, W. Dong and F. Huang, J. Mater. Chem. A, 2016, 4, 167–172 RSC.
- C. Du, M. Shang, J. Mao and W. Song, J. Mater. Chem. A, 2017, 5, 15940–15949 RSC.
- E. A. Duran, Y. Meas, J. J. P. Bueno, J. C. Ballesteros and G. Trejo, Electrochim. Acta, 2018, 268, 49–58 CrossRef.
- T. Zhang, M. Y. Wu, D. Y. Yan, J. Mao, H. Liu, W. B. Hu, X. W. Du, T. Ling and S. Z. Qiao, Nano Energy, 2018, 43, 103–109 CrossRef CAS.
- W. Cheng, H. Zhang, X. Zhao, H. Su, F. Tang, J. Tian and Q. Liu, J. Mater. Chem. A, 2018, 6, 9420–9427 RSC.
- M. Gong, W. Zhou, M. C. Tsai, J. Zhou, M. Guan, M. C. Lin, B. Zhang, Y. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. Dai, Nat. Commun., 2014, 5, 4695 CrossRef CAS.
- Y. Zhang, Y. Liu, M. Ma, X. Ren, Z. Liu, G. Du, A. M. Asiri and X. Sun, Chem. Commun., 2017, 53, 11048–11051 RSC.
- C. J. Chen, C. W. Liu, K. C. Yang, L. C. Yin, D. H. Wei, S. F. Hu and R. S. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 37142–37149 CrossRef CAS.
- B. Chang, S. Hao, Z. Ye and Y. Yang, Chem. Commun., 2018, 54, 2393–2396 RSC.
- L. Yang, Z. Guo, J. Huang, Y. Xi, R. Gao, G. Su, W. Wang, L. Cao and B. Dong, Adv. Mater., 2017, 29, 1704574 CrossRef.
- W. Xu, S. Zhu, Y. Liang, Z. Cui, X. Yang and A. Inoue, J. Mater. Chem. A, 2018, 6, 5574–5579 RSC.
- K. Guo, Z. Zou, J. Du, Y. Zhao, B. Zhou and C. Xu, Chem. Commun., 2018, 54, 11140–11143 RSC.
- C. Hu, L. Zhang, Z. J. Zhao, J. Luo, J. Shi, Z. Huang and J. Gong, Adv. Mater., 2017, 29, 1701820 CrossRef.
- J. Chang, Q. Lv, G. Li, J. Ge, C. Liu and W. Xing, Appl. Catal., B, 2017, 204, 486–496 CrossRef CAS.
- W. Li, S. Zhang, Q. Fan, F. Zhang and S. Xu, Nanoscale, 2017, 9, 5677–5685 RSC.
- Y. Wu, F. Li, W. Chen, Q. Xiang, Y. Ma, H. Zhu, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, Adv. Mater., 2018, 30, 1803151 CrossRef.
- C. Zhu, A. L. Wang, W. Xiao, D. Chao, X. Zhang, N. H. Tiep, S. Chen, J. Kang, X. Wang, J. Ding, J. Wang, H. Zhang and H. J. Fan, Adv. Mater., 2018, 30, 1705516 CrossRef.
- M. Kim, M. A. R. Anjum, M. Lee, B. J. Lee and J. S. Lee, Adv. Funct. Mater., 2019, 29, 1809151 CrossRef.
- J. Han, S. Hao, Z. Liu, A. M. Asiri, X. Sun and Y. Xu, Chem. Commun., 2018, 54, 1077–1080 RSC.
- P. W. Menezes, C. Panda, S. Loos, F. B. Bruns, C. Walter, M. Schwarze, X. Deng, H. Dau and M. Driess, Energy Environ. Sci., 2018, 11, 1287 RSC.
- L. M. Cao, Y. W. Hu, S. F. Tang, A. Iljin, J. W. Wang, Z. M. Zhang and T. B. Lu, Adv. Sci., 2018, 5, 1800949 CrossRef.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- X. Zhang, W. Gu and E. Wang, Nano Res., 2017, 10, 1001–1009 CrossRef CAS.
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, J. Am. Chem. Soc., 2016, 138, 4006–4009 CrossRef CAS.
- W. Li, D. Xiong, X. Gao, W. G. Song, F. Xia and L. Liu, Catal. Today, 2017, 287, 122–129 CrossRef CAS.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4067–4077 CrossRef CAS.
- J. Xu, J. P. S. Sousa, N. E. Mordvinova, J. D. Costa, D. Y. Petrovykh, K. Kovnir, O. I. Lebedev and Y. V. Kolenko, ACS Catal., 2018, 8, 2595–2600 CrossRef CAS.
- Y. Pan, Y. Liu, J. Zhao, K. Yang, J. Liang, D. Liu, W. Hu, D. Liu, Y. Liu and C. Liu, J. Mater. Chem. A, 2015, 3, 1656–1665 RSC.
- W. X. Lu, B. Wang, W. J. Chen, J. L. Xie, Z. Q. Huang, W. Jin and J. L. Song, ACS Sustainable Chem. Eng., 2019, 7, 3083–3091 CrossRef CAS.
- C. Jian, Q. Cai, W. Hong, J. Li and W. Liu, Small, 2018, 14, 1703798 CrossRef.
- D. C. Liu, L. M. Cao, Z. M. Luo, D. C. Zhong, J. B. Tan and T. B. Lu, J. Mater. Chem. A, 2018, 6, 24920 RSC.
- F. S. Zhang, J. W. Wang, J. Luo, R. R. Liu, Z. M. Zhang, C. T. He and T. B. Lu, Chem. Sci., 2018, 9, 1375 RSC.
- A. B. Laursen, K. R. Patraju, M. J. Whitaker, M. Retuerto, T. Sarkar, N. Yao, K. V. Ramanujachary, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2015, 8, 1027–1034 RSC.
- Y. Pan, Y. Liu, Y. Lin and C. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 13890–13901 CrossRef CAS.
- Q. Yan, T. Wei, J. Wu, X. Yang, M. Zhu, K. Cheng, K. Ye, K. Zhu, J. Yan, D. Cao, G. Wang and Y. Pan, ACS Sustainable Chem. Eng., 2018, 6, 9640–9648 CrossRef CAS.
- D. Li, H. Baydoun, B. Kulikowski and S. L. Brock, Chem. Mater., 2017, 29, 3048–3054 CrossRef CAS.
- J. Xu, D. Xiong, I. Amorim and L. Liu, ACS Appl. Nano Mater., 2018, 1, 617–624 CrossRef CAS.
- X. Fan, Y. Liu, S. Chen, J. Shi, J. Wang, A. Fan, W. Zan, S. Li, W. A. Goddard and X. M. Zhang, Nat. Commun., 2018, 9, 1809 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00396g |
| ‡ Wen-Zhuo Zhang and Guang-Yi Chen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |