
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ catalyst exsolution on perovskite oxides for the production of CO and synthesis gas in ceramic membrane reactors†
Georgios
Dimitrakopoulos
 abc,
Ahmed F.
Ghoniem
*c and
Bilge
Yildiz
*ab
abc,
Ahmed F.
Ghoniem
*c and
Bilge
Yildiz
*ab
aDepartment of Materials Science & Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: byildiz@mit.edu
bDepartment of Nuclear Science & Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
cDepartment of Mechanical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: ghoniem@mit.edu
First published on 25th June 2019
Abstract
This work demonstrates and assesses the concept of in situ catalyst exsolution in ceramic membrane reactors for the co-production of carbon monoxide (CO) and syngas through carbon dioxide (CO2) splitting and methane (CH4) partial oxidation, respectively. We use dense pellets of La0.85Ca0.10Fe0.95Ni0.05O3−δ (LCFN) as a model membrane reactor system. By combining in situ reaction rate measurements under finite oxygen (O2) flux and ex situ material characterization, we show that exsolution of nickel (Ni0) metal nanoparticles takes place in situ on both sides of the LCFN membrane under sufficiently reducing conditions controlled by the presence of CH4. To understand the impact of temperature and inlet fuel concentration on the nucleation, performance and long-term stability of the catalytic particles, exsolution of Ni0 from LCFN was investigated at 900 °C, 950 °C and 1000 °C and with different concentrations of CH4. Nickel particles with sizes ranging between 100 nm and 300 nm formed on LCFN pellets at 950 °C and 1000 °C using CH4 as the in situ reducing agent at a threshold inlet CH4 mole fraction of 16% and 4%, respectively. Nickel exsolution was not observed at 900 °C. No pre-reduction of the sample in a hydrogen (H2) environment was required. As a result, significant performance increase was obtained. Exsolution on LCFN samples at different temperatures results in almost identical performance when compared under similar conditions. Reaction rates obtained after nickel exsolution were stable over long durations, suggesting that the grown particles are resistant to agglomeration in the hydrocarbon environment. Results shown in this work demonstrate that in situ, on demand catalyst exsolution by using the feedstock fuel can be an effective way of improving the surface kinetics of perovskite oxides for ceramic membranes.
1. Introduction
Perovskite oxides are important enablers of a number of technological applications, including oxygen ion1–5 and proton6 conducting membranes, in chemical looping as oxygen carriers,1,7 in solid oxide fuel or electrolysis cells as electrodes2 and as catalysts for several oxidation and reduction reactions.8 In addition to these high temperature applications, perovskite oxides can also serve under ambient conditions as piezoelectric materials,9 as electrolytes for lithium-ion batteries,10 as supercapacitors,11 as gas-sensing materials12 and in resistive switching.13To improve the catalytic activity in energy and fuel conversion processes, a concept termed catalyst exsolution has emerged as a novel approach.14,15 Exsolution produces highly active and dispersed nanometer-sized catalytic particles grown from the bulk of a perovskite backbone. This process relies on the phase precipitation of the transition metal catalyst dopant on the B-site out of the perovskite oxide under sufficiently reducing conditions. The exsolved particles exhibit enhanced high-temperature resistance to agglomeration and deactivation by carbon formation when compared to wet impregnation or atomic layer deposited particles. This is because of the socketed nature of the exsolved particles, restricting their mobility.14,15
Growing catalytic particles from the bulk of a perovskite host has been reported in several studies. For example, Zhou et al. investigated exsolution on La0.8Sr1.2Fe0.9Co0.1O4−δ layered perovskite electrodes for symmetric SOFCs.16 Tsekouras et al. investigated exsolution of nickel (Ni) and iron (Fe) doped La0.4Sr0.4TiO3−δ cathodes for SOFC applications; they showed that the metallic nanoparticles Ni0 or Fe0 can exsolve from the parent perovskite leading to significant performance enhancement.14 Neagu et al. showed that a cobalt (Co)–Ni spinel can exsolve from a La0.7Ce0.1Co0.3Ni0.1Ti0.6O3−δ perovskite and these nanoparticles exhibit high activity towards CO oxidation at temperatures as low as 500 °C.17 Furthermore, Liu et al. investigated CO2 electrolysis showing that a Fe–Ni alloy can exsolve from a La0.6Sr0.4Fe0.8Ni0.2O3−δ porous electrode and the presence of this alloy results in low electrode polarization resistance at temperatures around 850 °C.18 The role of A-site deficiency on Ni0 exsolution from Ni doped La0.7Sr0.3CrO3−δ oxides has also been reported in;19 this study showed that the introduction of 10% A-site deficiency results in more Ni0 on the surface and hence higher performance can be achieved in H2 and H2/H2S environments.19 To the best of our knowledge, the only work that investigated exsolution in ceramic membranes was performed by Paparguriou and Irvine who studied Ni0 exsolution on La0.75Sr0.25Cr0.475Mn0.475Ni0.05O3−δ and La0.75Sr0.25Cr0.475Fe0.475Ni0.05O3−δ.20 In this work, however, only the oxygen loss (reduction) as a function of temperature in 5% H2 (argon balance) and the electronic conductivity of the materials in the same gas environment at equilibrium were reported together with the Ni0 particle morphology; no O2 permeation kinetics measurements were conducted to evaluate the performance and stability of the aforementioned materials under conditions of interest to different chemical reactions. In addition, in all these prior studies, a reducing gas such as H2 was used for the reduction of the perovskite to exsolve the active particles at the surface prior to the measurements of reaction kinetics and performance.
The work reported herein demonstrates a novel concept – in situ catalyst exsolution in ceramic membrane reactors, in this case for CO2 splitting to CO on one side, and CH4 partial oxidation to syngas on the other side, under conditions of finite O2 permeation flux across the membrane. The investigated membrane technology is not new but is certainly challenging. One challenge is that attempting to increase the oxygen permeation flux significantly will result in full oxidation of CH4 to H2O and CO2 instead of CH4 partial oxidation to syngas. Another challenge is that high operating temperatures are required for the CO2 splitting to CO due to enhanced membrane bulk diffusion and surface kinetics. Hence, new methodologies that can enhance the performance of the technology by avoiding deep oxidation of CH4 while decreasing the operating temperature are required, and exsolution of catalysts is one of them.
Conversion of CO2 to CO has been recognized as a means to reduce greenhouse-gas emissions21,22 and produce pure CO that can be used as a fuel, in the chemical industry, or as a reducing agent in metallurgy.1 Syngas, on the other hand, serves as an intermediate mixture for the production of ammonia, methanol and liquid hydrocarbons via Fischer–Tropsch synthesis. The aforementioned processes are described using the following global reactions written in Kröger–Vink notation:
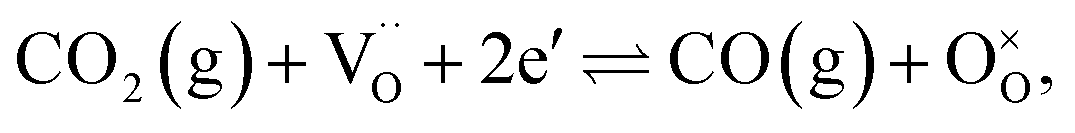 | (1) |
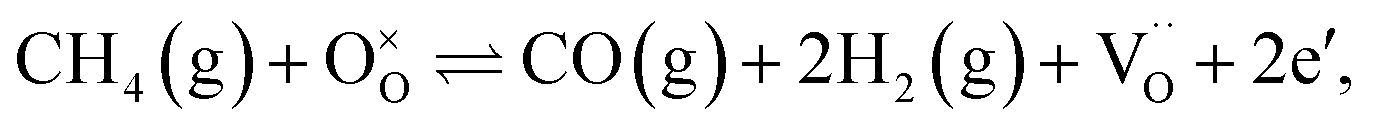 | (2) |
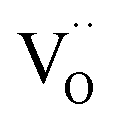 is an oxygen vacancy in an oxygen lattice site,
is an oxygen vacancy in an oxygen lattice site, 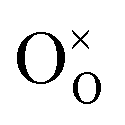 is an oxygen ion incorporated into an oxygen lattice site and e′ denotes electrons required for the ionization of O2.
is an oxygen ion incorporated into an oxygen lattice site and e′ denotes electrons required for the ionization of O2.
As a model perovskite oxide base, we used La0.9Ca0.1FeO3−δ (LCF), which is stable under reducing conditions, but its overall performance is limited by the surface reactions at the gas–membrane interfaces. This has been demonstrated using both experimental and numerical investigations of LCF under conditions such as air/inert gas,23–25 air–CH4,26 H2O–CH4,27 H2O–H2,28 CO2–CO/H2/CH4,29,30 air–CO/H231,32 and air/CO.33 To increase the surface kinetics, porous supports on either or both gas–membrane interfaces with/without a catalyst have been used. In this study, we doped the B-site of LCF with 5% Ni while introducing a 5% A-site deficiency that has been shown to facilitate catalyst exsolution19 in order to speed-up the surface kinetics; this model has the composition La0.85Ca0.10Fe0.95Ni0.05O3−δ (LCFN). The feed side of the LCFN in the reactor serves for converting CO2 to CO (eqn (1)), and the fuel side serves for CH4 partial oxidation to syngas (eqn (2)). Our goal is to improve the performance by exsolving active particles on either or both sides of LCFN, and to understand how exsolution at different temperatures affects the performance and stability of the material and catalyst.
In our work, we measured oxygen permeation flux through dense LCFN pellets and CH4 conversion/syngas production at 900 °C, 950 °C and 1000 °C. We demonstrated that exsolution is achieved using CH4 as the fuel in eqn (1) and (2), and this occurs under finite O2 permeation conditions. This finding is important because it shows that exsolution can be triggered using the feedstock fuel of the proposed application, removing any necessity for a pre-reduction step using additional fuels such as hydrogen. Furthermore, exsolution at higher temperatures requires less fuel for catalyst nucleation, consistent with the oxygen effective chemical potential dependency on temperature and fuel concentration. Exsolution on LCFN samples at different temperatures results in almost identical performances when compared under similar conditions. With an inlet CH4 concentration of 18%, the oxygen permeation flux (JO2) is approximately 0.4 μmol cm−2 s−1 at 950 °C and 0.25 μmol cm−2 s−1 at 900 °C. Ex situ characterization of the elemental distribution and morphology of the samples reveals the presence of Ni metal particles on both sides of the membrane, acting as a catalyst for both the CO2 reduction to CO and for the CH4 partial oxidation to syngas. These particles have a distribution of sizes, ranging between 100 nm and 300 nm. Long-term measurements show no performance degradation of the catalysts after exsolution, suggesting that the grown particles are resistant to the hydrocarbon environment and to agglomeration. Finally, the produced syngas at the outlet has a H2 to CO ratio equal to two, and hence can be used directly in Fischer–Tropsch synthesis of liquid hydrocarbons without additional treatment.
2. Experimental
Polycrystalline LCFN powder was prepared using a glycine–nitrate process. First, a stoichiometric amount of CaCO3 (99.999%, MilliporeSigma) was added in deionized water and was fully dissolved by adding nitric acid (99.999%, MilliporeSigma). Subsequently, stoichiometric amounts of La(NO3)3·6H2O (99.999%, MilliporeSigma), Fe(NO3)3·9H2O (99.95%, MilliporeSigma), Ni(NO3)2·6H2O (99.999%, MilliporeSigma) and glycine (99%, MilliporeSigma) were added to the solution one by one in the aforementioned order. The ratio of total metal cations to glycine was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The pH of the solution was adjusted to 2. The mixture was stirred vigorously until a homogeneous solution without precipitation of the nitrates was obtained. The resulting solution was heated on a hot plate at 540 °C until auto-ignition occurred. The raw perovskite ash was ground using an agate mortar and pestle. The powder was calcined in ambient air at 900 °C for 1 hour using a 5 °C min−1 heating and cooling rate.
:1. The pH of the solution was adjusted to 2. The mixture was stirred vigorously until a homogeneous solution without precipitation of the nitrates was obtained. The resulting solution was heated on a hot plate at 540 °C until auto-ignition occurred. The raw perovskite ash was ground using an agate mortar and pestle. The powder was calcined in ambient air at 900 °C for 1 hour using a 5 °C min−1 heating and cooling rate.
To prepare LCFN pellets for permeation measurements, the calcined powder was mixed with 5 wt% polyvinyl butyral and stirred in ethanol to homogenize the mixture. After the evaporation of ethanol, the powder was pressed into disk-shaped pellets using a pressure of 23 MPa and the resulting green bodies were sintered in ambient air at 1250 °C for 8 hours using a heating and cooling rate of 3 °C min−1. After the sintering process, the pellets were polished on both sides using diamond sandpapers of different grit sizes until a smooth, mirror-like surface was obtained. Nickel exsolution from LCFN pellets was investigated at 900 °C, 950 °C and 1000 °C; the samples will be denoted as LCFN-1, LCFN-2 and LCFN-3, respectively. The shrinkage of the pellets, the final thickness after polishing and the density and relative density of the samples are reported in ESI Table S1.†
Permeation measurements across the membrane were conducted using the apparatus shown in ESI Fig. S1.† At the surface of the LCFN facing the feed side of the reactor, the CO2 splitting (eqn (1)) takes place. At the opposite surface, facing the fuel side of the reactor, CH4 partial oxidation (eqn (2)) takes place. During the permeation measurements, pure CO2 flows at the feed side of the reactor at a volumetric flow rate of 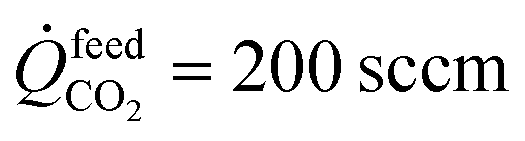 , while at the fuel side, a mixture of CH4 and argon (Ar) flows at different ratios with a fixed total volumetric flow rate of
, while at the fuel side, a mixture of CH4 and argon (Ar) flows at different ratios with a fixed total volumetric flow rate of ![[Q with combining dot above]](https://www.rsc.org/images/entities/i_char_0051_0307.gif) fueltotal = 100 sccm. To estimate the oxygen permeation flux through the material, measurements of the gas-phase composition at the inlet and outlet of the fuel side (CH4 partial oxidation side) are used in a mole balance system of equations similar to the one reported in ref. 31. The nitrogen (N2) mole fraction at the outlet of each side was around 0.1% showing that a gas-tight system was obtained and that the air leaks have a negligible effect on the experimental measurements.
fueltotal = 100 sccm. To estimate the oxygen permeation flux through the material, measurements of the gas-phase composition at the inlet and outlet of the fuel side (CH4 partial oxidation side) are used in a mole balance system of equations similar to the one reported in ref. 31. The nitrogen (N2) mole fraction at the outlet of each side was around 0.1% showing that a gas-tight system was obtained and that the air leaks have a negligible effect on the experimental measurements.
To evaluate the crystal structure of the as-prepared and used samples, we performed X-ray diffraction (XRD) with a PANalytical X'Pert Pro diffractometer using Cu Kα radiation at 45 kV and 40 mA. The obtained XRD patterns were analyzed using the software HighScore Plus. A Zeiss Merlin High-Resolution Scanning Electron Microscope (SEM) equipped with a Backscatter Detector (BSD) operating at 20 kV was used for imaging the morphology of the catalyst particles. To obtain qualitative information about the elemental distribution on the LCFN interfaces, Energy-Dispersive X-ray Spectroscopy (EDS) was conducted using an EDS detector and the software APEX.
ESI Fig. S2† shows the XRD patterns of the as-prepared LCFN powder and dense pellets, confirming that a phase pure material was synthesized. All peaks matched well with those of La0.9Ca0.1FeO3−δ (ICDD no. 01-082-9272). LCFN exhibits an orthorhombic structure in the Pbnm space group; more structural information about the material is reported in ESI Table S2.† ESI Fig. S3† presents a SEM image of the surface of an as-prepared LCFN pellet showing no porosity and a distribution of grain boundary sizes.
3. Results
3.1 Oxygen permeation and CO–syngas production on LCFN dense pellets
Fig. 1 shows the performance of LCFN dense pellets as a function of the inlet CH4 mole fraction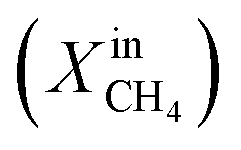 and time at three different exsolution temperatures, 900 °C, 950 °C, and 1000 °C, named LCFN-1, LCFN-2, and LCFN-3, respectively. For LCFN-1, the reactor was heated to 900 °C and was maintained at this temperature with pure CO2 flowing at the feed side and a CH4–Ar mixture flowing at the fuel (sweep) side with a varying
and time at three different exsolution temperatures, 900 °C, 950 °C, and 1000 °C, named LCFN-1, LCFN-2, and LCFN-3, respectively. For LCFN-1, the reactor was heated to 900 °C and was maintained at this temperature with pure CO2 flowing at the feed side and a CH4–Ar mixture flowing at the fuel (sweep) side with a varying 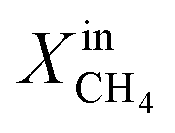 . The experiment is performed in a similar manner for the other two samples at 950 °C and 1000 °C. For LCFN-1, the performance and syngas production are low even at
. The experiment is performed in a similar manner for the other two samples at 950 °C and 1000 °C. For LCFN-1, the performance and syngas production are low even at  . However, for LCFN-2 and LCFN-3, a significant increase in oxygen flux and syngas production is observed as the fuel concentration increases.
. However, for LCFN-2 and LCFN-3, a significant increase in oxygen flux and syngas production is observed as the fuel concentration increases.
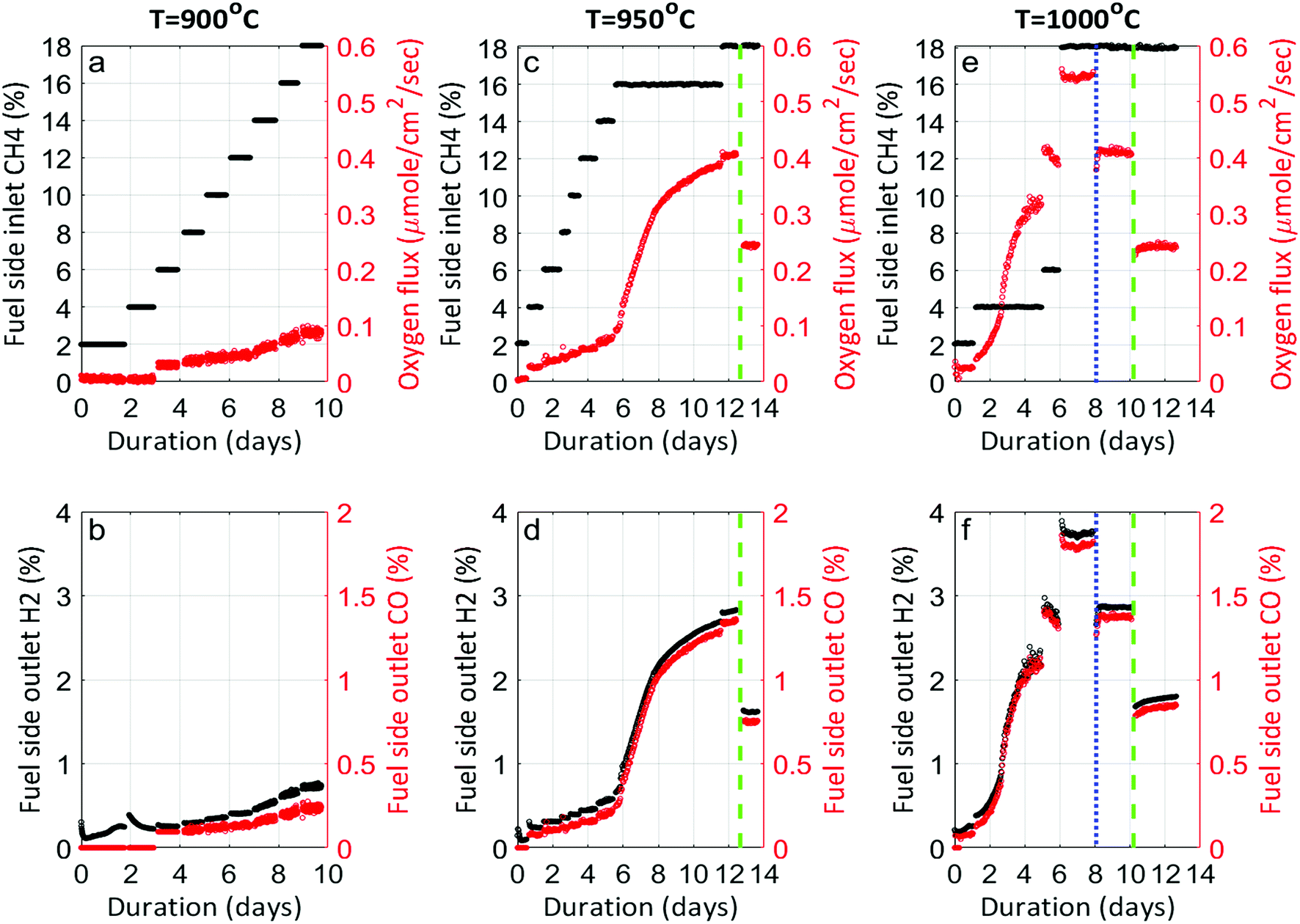 | ||
| Fig. 1 Performance of LCFN-1 (900 °C), LCFN-2 (950 °C) and LCFN-3 (1000 °C) as a function of inlet CH4 mole fraction and time. Top (a, c and e): inlet CH4 mole fraction (black) and oxygen permeation flux (red). Bottom (b, d and f): H2 (black) and CO (red) mole fractions at the outlet of the fuel side. The blue and green vertical dotted lines denote the points where the temperature is reduced from 1000 °C to 950 °C, and from 950 °C to 900 °C, respectively. | ||
More specifically, JO2 and syngas production for LCFN-2 at 950 °C are low until the inlet CH4 mole fraction becomes 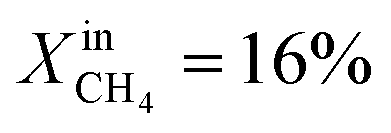 . At this value, the performance increases with a significant increase in JO2 and syngas production. The performance enhancement lasts for approximately 5 days and is linear for the first 2 days followed by an equilibration duration until JO2 levels off at a value of 0.39 μmol cm−2 s−1; at the same time, the fuel side outlet H2 and CO mole fractions become
. At this value, the performance increases with a significant increase in JO2 and syngas production. The performance enhancement lasts for approximately 5 days and is linear for the first 2 days followed by an equilibration duration until JO2 levels off at a value of 0.39 μmol cm−2 s−1; at the same time, the fuel side outlet H2 and CO mole fractions become 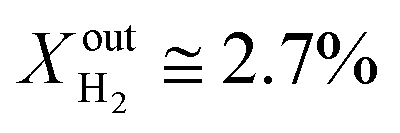 and XoutCO ≅ 1.3%. After the performance has equilibrated, introduction of additional fuel
and XoutCO ≅ 1.3%. After the performance has equilibrated, introduction of additional fuel 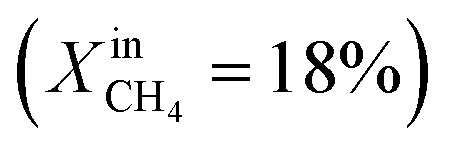 results in an insignificant increase with stable operation for one day.
results in an insignificant increase with stable operation for one day.
To evaluate the performance of LCFN-2 at lower temperatures, the reactor was cooled down from T = 950 °C to T = 900 °C (green dashed line) while 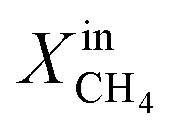 was kept constant at 18%. The performance decreases due to the slower surface kinetics as the operating temperature decreases. In particular, JO2 drops to approximately 0.25 μmol cm−2 s−1, while syngas production decreases to
was kept constant at 18%. The performance decreases due to the slower surface kinetics as the operating temperature decreases. In particular, JO2 drops to approximately 0.25 μmol cm−2 s−1, while syngas production decreases to 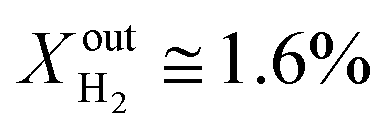 and XoutCO ≅ 0.7%. Despite the temperature-induced initial decrease in the kinetics, the material shows stable operation for 1 day of measurements.
and XoutCO ≅ 0.7%. Despite the temperature-induced initial decrease in the kinetics, the material shows stable operation for 1 day of measurements.
Results for LCFN-3 (for which exsolution is triggered at T = 1000 °C) show trends similar to those for LCFN-2; this time, however, an event of performance increase takes place at  and lasts for 4 days. At the end of the event, JO2 is around 0.3 μmol cm−2 s−1 and the fuel side outlet H2 and CO mole fractions are
and lasts for 4 days. At the end of the event, JO2 is around 0.3 μmol cm−2 s−1 and the fuel side outlet H2 and CO mole fractions are 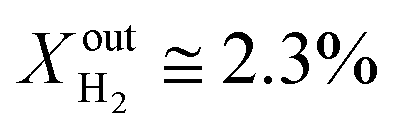 and XoutCO ≅ 1.1%. Increasing the inlet fuel concentration to
and XoutCO ≅ 1.1%. Increasing the inlet fuel concentration to 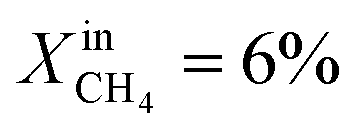 increases the performance but evidence of some degradation is observed with time. Since our findings on LCFN-2 suggest that events of further performance improvement do not take place after the major one (associated with a high performance increase) has happened, this time the inlet fuel concentration was raised to
increases the performance but evidence of some degradation is observed with time. Since our findings on LCFN-2 suggest that events of further performance improvement do not take place after the major one (associated with a high performance increase) has happened, this time the inlet fuel concentration was raised to 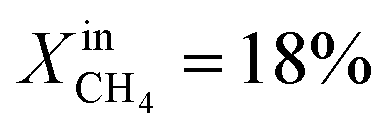 and the performance was measured for a duration of 2 days. At T = 1000 °C and
and the performance was measured for a duration of 2 days. At T = 1000 °C and 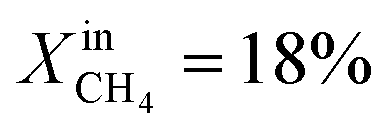 , JO2 approaches 0.55 μmol cm−2 s−1 and the syngas production becomes
, JO2 approaches 0.55 μmol cm−2 s−1 and the syngas production becomes 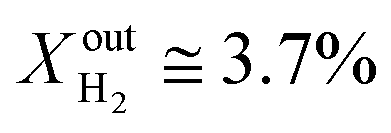 and XoutCO ≅ 1.8%.
and XoutCO ≅ 1.8%.
During the rest of the measurements, the reactor temperature was decreased to evaluate the performance and stability of the sample at lower temperatures. At T = 950 °C and 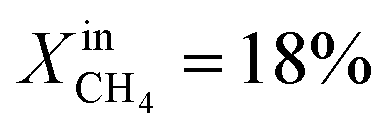 (blue dotted line), JO2 drops to approximately 0.4 μmol cm−2 s−1, which is almost identical to that of the LCFN-2 sample operating under the same conditions; the same is true for the syngas production. Reducing the temperature further to T = 900 °C while maintaining the fuel side inlet mole fraction at
(blue dotted line), JO2 drops to approximately 0.4 μmol cm−2 s−1, which is almost identical to that of the LCFN-2 sample operating under the same conditions; the same is true for the syngas production. Reducing the temperature further to T = 900 °C while maintaining the fuel side inlet mole fraction at 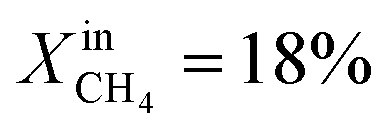 slows down the reaction kinetics. Interestingly, LCFN-2 and LCFN-3 are also found to have approximately the same JO2 and syngas production at T = 900 °C and
slows down the reaction kinetics. Interestingly, LCFN-2 and LCFN-3 are also found to have approximately the same JO2 and syngas production at T = 900 °C and 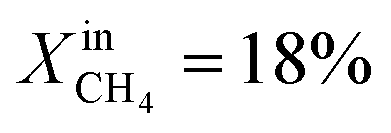 . Stable operation was observed at T = 900 °C and T = 950 °C for a duration of 1–2 days for each temperature, although a slight increase in the performance is observed at T = 900 °C for LCFN-3.
. Stable operation was observed at T = 900 °C and T = 950 °C for a duration of 1–2 days for each temperature, although a slight increase in the performance is observed at T = 900 °C for LCFN-3.
3.2 Structural and chemical characterization of LCFN pellets upon nickel exsolution
To examine the reasons leading to the improved performance of the LCFN-2 and LCFN-3 samples as well as the negligible performance enhancement of LCFN-1, XRD and SEM/EDS with a BSD were used. Fig. 2 shows the XRD patterns of an as-prepared LCFN as a reference, and the LCFN-3 sample exsolved and tested as shown in Fig. 1. XRD patterns of the used LCFN-2 pellet on both sides were identical to those of LCFN-3 and are shown in ESI Fig. S7† for completeness. The XRD patterns of the two sides of the LCFN-1 pellet were identical to those of the as-prepared sample and are also shown in ESI Fig. S6.† Based on the XRD results, both the fuel and feed side of LCFN-3 show two additional peaks that are ascribed to phases other than LCFN. A peak at 2θ = 44.4° is observed on both LCFN sides; based on the phase identification, this peak corresponds to metallic Ni0 in the cubic structure (ICDD no. 01-078-7533). At the fuel side, a peak at 2θ = 35.7° is ascribed to cubic Ca (ICDD no. 01-071-41074) while at the CO2 splitting side, the peak at 2θ = 37.3° corresponds to cubic CaO (ICDD no. 01-080-7710).
 | ||
| Fig. 2 XRD patterns of an as-prepared LCFN pellet, and of the fuel (CH4 oxidation) and feed (CO2 splitting) sides of the LCFN-3 pellet after exsolution and oxygen permeation measurements as described in Fig. 1. The left column includes the XRD patterns in the range 2θ = 20–80°, while the column on the right enlarges the range 2θ = 43.75–45° to show the peaks ascribed to Ni0. | ||
Regarding the peak at 2θ = 44.4°, we note that it cannot be ascribed to nickel oxide (NiO) because if NiO was present, then a peak at 2θ = 43.4° would be observed instead, which corresponds to the 100% intensity peak of the pure cubic NiO phase (ICDD no. 04-005-4791); this peak is absent from the XRD patterns on either LCFN surface. Our results are further confirmed by the work of Neagu et al. who also reported that both metallic Fe and Ni can be detected easily through XRD34 and the work of Tsekouras et al. demonstrating that metallic Ni after exsolution from La0.4Sr0.4Ti0.94Ni0.06O3−δ is present in the XRD pattern at a peak position of 2θ = 44.4°.14
We have also confirmed the presence of the aforementioned phases through EDS mapping. According to Fig. 3, both sides of the material show areas enriched with nickel and this confirms the XRD results shown in Fig. 2. At the same time, this also demonstrates that particles were successfully grown in situ on both sides of LCFN resulting in higher performance. On the fuel side, a distribution of particle sizes is observed ranging between 100 nm and 300 nm. ESI Fig. S4† shows another SEM image and EDS map of a zoomed-out view of the fuel side which also confirms the different Ni0 particle sizes and the presence of areas enriched with Ca. Based on Fig. 3, Ni0 particles on the CO2 splitting side have an average size around 250 nm. The same figure also shows some smaller features that may correspond to smaller Ni0 particles, but resolving these particles with EDS mapping is outside the resolution limit of the instrument used. We note here that areas enriched with CaO were not identified on the sample in the investigated areas. A similar microstructure is observed for the LCFN-2 sample, while no Ni0 particles were observed on the LCFN-1 sample, confirming that, within the range of our operating conditions, Ni0 was exsolved on the surfaces of LCFN-2 and LCFN-3 but not on that of LCFN-1.
 | ||
| Fig. 3 SEM image with a BSD (top row) and nickel EDS maps (bottom row) on the CO2 splitting side (left) and CH4 oxidation side (right) for the LCFN-3 sample after the end of the permeation measurements shown in Fig. 1. | ||
We note here that the particles exsolved on both gas–membrane interfaces have sizes ranging between 100 and 300 nm, which are larger compared to the reported size of exsolved catalysts in other studies.34 We believe that this is related to the material stoichiometry investigated, the operating conditions and the gaseous environment. As demonstrated by Neagu et al.,34 the perovskite stoichiometry and especially the A-site deficiency can promote particle nucleation vs. particle growth. Tuning the LCFN stoichiometry to allow for smaller exsolved particles is part of our future work.
4. Discussion
In the previous section we compared the performance of LCFN dense pellets for the co-production of CO and syngas through CO2 splitting and CH4 partial oxidation, respectively. Results show that reaction rates and the oxygen flux increase for LCFN under selected conditions for several days. Based on structural and elemental characterization of the samples at the end of the experiment, the performance improvement is attributed to exsolution of Ni0 particles on both sides of the investigated pellets.
Table 1 compares the oxygen permeation flux, CH4 conversion (CCH4), H2 yield (YH2) and CO yield (YCO) of the LCFN-2 and LCFN-3 pellets at different temperatures with a pure CO2 flow at the feed side and 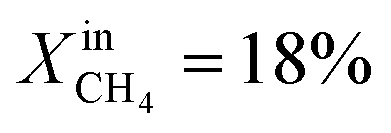 (balance Ar) at the fuel side. Results of LCFN-1 are not reported in the table since exsolution did not occur.
(balance Ar) at the fuel side. Results of LCFN-1 are not reported in the table since exsolution did not occur.
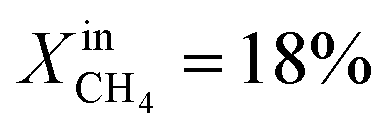 (balance Ar) at the fuel side
(balance Ar) at the fuel side
| Samples | Temperature | Ref. | ||
|---|---|---|---|---|
| 900 °C | 950 °C | 1000 °C | ||
| LCFN-2 | J O2 = 0.24 μmol cm−2 s−1 | J O2 = 0.41 μmol cm−2 s−1 | — | This work |
| C CH4 = 4.6% | C CH4 = 8.0% | |||
| Y H2 = 4.6% | Y H2 = 8.1% | |||
| Y CO = 4.2% | Y CO = 7.7% | |||
| LCFN-3 | J O2 = 0.24 μmol cm−2 s−1 | J O2 = 0.40 μmol cm−2 s−1 | J O2 = 0.55 μmol cm−2 s−1 | This work |
| C CH4 = 5.1% | C CH4 = 8.2% | C CH4 = 10.9% | ||
| Y H2 = 5.1% | Y H2 = 8.2% | Y H2 = 10.9% | ||
| Y CO = 4.8% | Y CO = 7.9% | Y CO = 10.5% | ||
Dense LCF 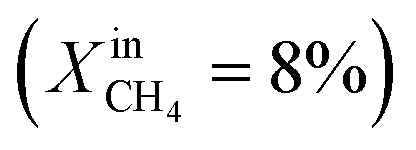 |
— | — | J O2 = 0.13 μmol cm−2 s−1 (at T = 990 °C) | 30 |
Dense LCF 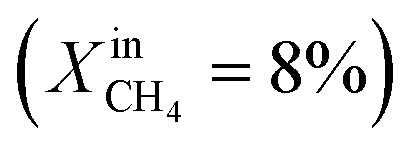 , porous LCF, on both sides , porous LCF, on both sides |
— | — | J O2 = 0.15 μmol cm−2 s−1 (at T = 990 °C) | 30 |
Dense LCF 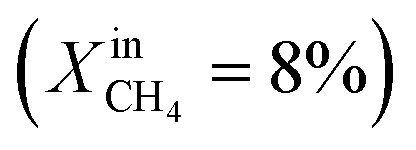 , feed side porous support: 80% LCF–20% CZO, sweep side porous support: 80% LCF–20% LSCF , feed side porous support: 80% LCF–20% CZO, sweep side porous support: 80% LCF–20% LSCF |
— | — | J O2 = 0.17 μmol cm−2 s−1 (at T = 990 °C) | 30 |
To demonstrate the effectiveness of the Ni0 particles exsolved on LCFN, we compare the performance of LCFN with that of the parent material (LCF) in the dense form without and with porous supports on both gas–membrane interfaces.30 According to ref. 30, a membrane assembly that consists of dense LCF with LCF porous supports on both sides exhibits an oxygen permeation flux of 0.15 μmol cm−2 s−1 when operating at 990 °C with 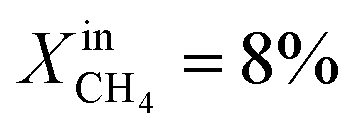 on the fuel side and pure CO2 on the feed side, which is slightly higher than that of dense LCF operating under the same conditions, resulting in an oxygen permeation flux of 0.13 μmol cm−2 s−1. When using dense LCF with porous supports consisting of 80% LCF–20% Ce0.5Zr0.5O2−δ (CZO) on the feed side and 80% LCF–20% (La0.6Sr0.4)0.95Fe0.8Co0.2O3−δ (LSCF) on the fuel side, JO2, taken as a performance measure, improves slightly to 0.17 μmol cm−2 s−1. In this work, we show that the same performance can be achieved at a lower temperature and higher fuel concentration, 900 °C and
on the fuel side and pure CO2 on the feed side, which is slightly higher than that of dense LCF operating under the same conditions, resulting in an oxygen permeation flux of 0.13 μmol cm−2 s−1. When using dense LCF with porous supports consisting of 80% LCF–20% Ce0.5Zr0.5O2−δ (CZO) on the feed side and 80% LCF–20% (La0.6Sr0.4)0.95Fe0.8Co0.2O3−δ (LSCF) on the fuel side, JO2, taken as a performance measure, improves slightly to 0.17 μmol cm−2 s−1. In this work, we show that the same performance can be achieved at a lower temperature and higher fuel concentration, 900 °C and 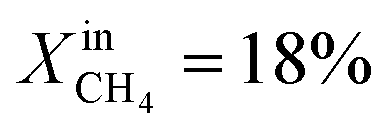 , respectively, by growing Ni0 on both sides of dense LCFN, i.e. without the use of porous supports. Future work will address the performance enhancement when exsolution is triggered on porous supports.
, respectively, by growing Ni0 on both sides of dense LCFN, i.e. without the use of porous supports. Future work will address the performance enhancement when exsolution is triggered on porous supports.
Moreover, during the LCFN measurements, the inlet mole fraction of CH4 on the fuel side was raised for deeper reduction of the membrane material, leading to precipitation of Ni on the surface. Exsolving the catalyst using CH4 has been demonstrated for the first time in our study, and is an important finding because it shows that the feedstock fuel can also serve as a reducing agent in situ, on demand.
Based on Table 1 and within the uncertainties of our measurements, we also observe that the performance of LCFN-2 and LCFN-3 is approximately the same at 900 °C and 950 °C despite the fact that exsolution on LCFN-3 was triggered at 1000 °C and 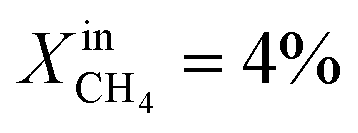 while exsolution on LCFN-2 occurred at 950 °C using
while exsolution on LCFN-2 occurred at 950 °C using 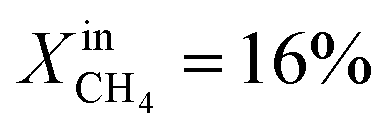 . Because of the different conditions, it is reasonable to conclude that the size and distribution of nickel particles on the interfaces of LCFN-2 and LCFN-3 are not exactly the same. Therefore, the similar performance of these two samples at low temperatures cannot be attributed to an identical microstructure. We suspect that the observed similarity in performance has to do with the rate-limiting step of the overall oxygen permeation process. It is well known that in ceramic membrane reactors, the rate-limiting step of oxygen permeation can be either the incorporation reactions on the gas–membrane interfaces or the diffusion of charged mobile species within the material. Computational work on LCF has shown that, under the conditions investigated in this study, the rate-limiting step is the surface chemistry on the fuel side, i.e. the CH4 oxidation.1,29–31 Given that LCFN does not deviate significantly from LCF in terms of the stoichiometry and crystal structure, it is reasonable to hypothesize that LCFN exhibits the same rate-limiting step, i.e. the CH4 oxidation, prior to exsolution. However, as demonstrated by our results in Fig. 1, upon exsolution of nickel on LCFN, the nickel nanoparticles speed up the CH4 oxidation reaction and the performance is improved. While CH4 oxidation is accelerated, this may not be the case for the bulk diffusion of charged mobile species within the material or for the CO2 splitting reaction. Hence, it is possible that the rate-limiting step is changed after the nickel exsolution, leading to the same overall rate observed in this study. Future work will address this in order to identify the resulting rate limiting step and further accelerate it. In any case, the stability of LCFN is not compromised: the performance of the exsolved particles is steady for a period of 1–2 days, confirming that the exsolved nanoparticles are resistant to agglomeration at high temperatures.
. Because of the different conditions, it is reasonable to conclude that the size and distribution of nickel particles on the interfaces of LCFN-2 and LCFN-3 are not exactly the same. Therefore, the similar performance of these two samples at low temperatures cannot be attributed to an identical microstructure. We suspect that the observed similarity in performance has to do with the rate-limiting step of the overall oxygen permeation process. It is well known that in ceramic membrane reactors, the rate-limiting step of oxygen permeation can be either the incorporation reactions on the gas–membrane interfaces or the diffusion of charged mobile species within the material. Computational work on LCF has shown that, under the conditions investigated in this study, the rate-limiting step is the surface chemistry on the fuel side, i.e. the CH4 oxidation.1,29–31 Given that LCFN does not deviate significantly from LCF in terms of the stoichiometry and crystal structure, it is reasonable to hypothesize that LCFN exhibits the same rate-limiting step, i.e. the CH4 oxidation, prior to exsolution. However, as demonstrated by our results in Fig. 1, upon exsolution of nickel on LCFN, the nickel nanoparticles speed up the CH4 oxidation reaction and the performance is improved. While CH4 oxidation is accelerated, this may not be the case for the bulk diffusion of charged mobile species within the material or for the CO2 splitting reaction. Hence, it is possible that the rate-limiting step is changed after the nickel exsolution, leading to the same overall rate observed in this study. Future work will address this in order to identify the resulting rate limiting step and further accelerate it. In any case, the stability of LCFN is not compromised: the performance of the exsolved particles is steady for a period of 1–2 days, confirming that the exsolved nanoparticles are resistant to agglomeration at high temperatures.
We note here that we have also investigated the possibility of first reducing a LCFN pellet to exsolve the particles and then conducting permeation measurements in a different experiment that involves H2O splitting to H2 on the feed side and H2 oxidation on the fuel side. The LCFN sample in this case was subjected to a sequence of three reduction steps. It was first reduced on both sides at 1000 °C using 5% H2 (balance Ar) for 15 minutes. Then, the gas mixture at the feed side was replaced with 50% H2O (balance N2), while 5% H2 (balance Ar) was still flowing on the fuel side. The measured JO2 was 0.62 μmol cm−2 s−1. A second reduction step under the same conditions was performed for 30 minutes. After switching back to 50% H2O (balance N2) at the feed side, JO2 was reduced to 0.57 μmol cm−2 s−1. A third reduction step took place for 45 minutes, where JO2 dropped further to 0.50 μmol cm−2 s−1. ESI Fig. S5† shows the microstructure of the two sides. Large Ni particles on the order of 500 nm to 1 μm are observed together with large chunks corresponding to Fe2O3. We conclude that the large particles formed after the pre-reduction involving H2 degrades the performance of LCFN due to particle agglomeration and phase decomposition. As shown previously, when exsolving nickel using CH4 on the fuel side, Ni0 particles are smaller in size, and decomposition to form Fe2O3 was not observed.
Although CH4 is introduced in the reactor, products form near the vicinity of the membrane due to gas-phase and heterogeneous reactions that may play a role in the exsolution of Ni. At the fuel side, CH4 can pyrolyze in the gas phase to form carbon (C) and H2. These products may form CO and H2O through oxidation in the gas phase or by lattice oxygen ions; CO can be oxidized further to CO2. Desorption of CO2 and H2O into the gas phase can reform CH4 to syngas through dry- and steam-reforming and hence make the environment near the membrane more reducing; the aforementioned reactions can also take place on the surface of the material.26 Similar gas-phase and surface chemistry interactions have been reported for other fuels and applications, such as ethane oxidative dehydrogenation to ethylene.3,4 At the same time, CO2 splitting to CO on the feed side of the membrane also makes the environment more reducing. The higher the O2 permeation flux through the membrane, the higher the syngas production on the fuel side and CO production on the feed side, and this changes the balance of the equivalent PO2 on the two sides of the material. Note that the focus of this work is to demonstrate how exsolution can be used to enhance the performance of ceramic membrane reactors as well as to investigate under what conditions (temperature and inlet CH4 mole fraction) the exsolution of nickel nanoparticles is triggered on both gas–membrane interfaces. Future work will address the impact of the self-grown nickel particles on the CO2 and CH4 surface chemistry on a fundamental basis.
Another important observation is related to the syngas yield at the outlet of the membrane reactor. In all cases, the  ratio is approximately equal to two. In all our measurements, traces of ethane (C2H6), ethylene (C2H4) and acetylene (C2H2) were measured in the order of 0.01%. O2 at the outlet of each side was measured to be zero. In addition, small concentrations of CO2 around 0.1% are measured at the outlet of the fuel side of the reactor; the same is true for the estimated H2O concentration at the outlet of the fuel side of the reactor (H2O cannot be measured using a gas chromatograph but can be estimated through mass balance between the inlet and outlet of the reactor). All the above results confirm that, overall, the dominant (global) reaction on the fuel side of the reactor is the CH4 partial oxidation to syngas. Compared to syngas production through CH4 steam reforming and dry reforming that yield
ratio is approximately equal to two. In all our measurements, traces of ethane (C2H6), ethylene (C2H4) and acetylene (C2H2) were measured in the order of 0.01%. O2 at the outlet of each side was measured to be zero. In addition, small concentrations of CO2 around 0.1% are measured at the outlet of the fuel side of the reactor; the same is true for the estimated H2O concentration at the outlet of the fuel side of the reactor (H2O cannot be measured using a gas chromatograph but can be estimated through mass balance between the inlet and outlet of the reactor). All the above results confirm that, overall, the dominant (global) reaction on the fuel side of the reactor is the CH4 partial oxidation to syngas. Compared to syngas production through CH4 steam reforming and dry reforming that yield 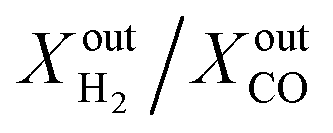 ratios of 3 and 1, respectively, producing syngas at a ratio of
ratios of 3 and 1, respectively, producing syngas at a ratio of 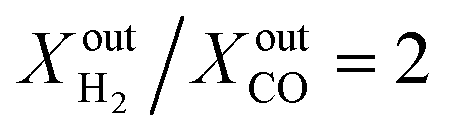 is beneficial in case the syngas is to be used for the production of liquid hydrocarbons via Fischer–Tropsch synthesis because no additional treatment is required. At the same time, the presence of by-products in small concentrations reduces the energy penalty associated with additional downstream separations and this is an advantage for the economic viability of the process.
is beneficial in case the syngas is to be used for the production of liquid hydrocarbons via Fischer–Tropsch synthesis because no additional treatment is required. At the same time, the presence of by-products in small concentrations reduces the energy penalty associated with additional downstream separations and this is an advantage for the economic viability of the process.
5. Conclusions
This work demonstrates that catalyst exsolution can be utilized to enhance the performance and stability of ceramic membranes used for, e.g., CO2 reduction to CO, H2 production by H2O splitting, etc. We applied this concept to the simultaneous splitting of CO2 to CO and CH4 partial oxidation to syngas using dense LCFN pellets. Our results show that significant performance improvement is achieved owing to the in situ growth of Ni0 particles on both sides of LCFN. This was achieved by adjusting the inlet CH4 mole fraction over a threshold value. Exsolution with CH4 acting as both the feedstock and the reducing agent is a novel approach for improving the performance of membrane reactors and is demonstrated here for the first time. Exsolution has been investigated systematically to understand the impact of operating conditions on the growth, performance and stability of the catalytic particles. Our work demonstrates that the resulting Ni0 particles are well dispersed on both the feed and the fuel gas interfaces of the membrane, and exhibit a distribution of sizes ranging between 100 nm and 300 nm. The particles are highly active and stable in the presence of hydrocarbons. The successful in situ growth of catalysts at the surface of the perovskite oxide membrane shown in this work provides insights for enhancing the performance and stability of ceramic membranes towards reactions of interest.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank the Exelon Corporation and the MIT Energy Initiative Seed Fund Program for supporting the work presented in this paper. Also, this work made use of the Materials Research Science and Engineering Center (MRSEC) facilities at MIT’s Materials Research Lab (MRL), supported by the National Science Foundation (NSF) award no. DMR-14-19807.References
- A. F. Ghoniem, Z. Zhao and G. Dimitrakopoulos, Gas oxy combustion and conversion technologies for low carbon energy: Fundamentals, modeling and reactors, Proc. Combust. Inst., 2019, 37, 33–56 CrossRef CAS.
- J. Sunarso, S. S. Hashim, N. Zhu and W. Zhou, Perovskite oxides applications in high temperature oxygen separation, solid oxide fuel cell and membrane reactor: A review, Prog. Energy Combust. Sci., 2017, 61, 57–77 CrossRef.
- G. Dimitrakopoulos, R. C. Schucker, K. Derrickson, J. R. Johnson, K. K. Kopeć, L. Shao, F. Al-Ahmadi and A. F. Ghoniem, Hydrogen and Ethylene Production through Water-Splitting and Ethane Dehydrogenation using BaFe0.9Zr0.1O3−δ Mixed-Conductors, ECS Trans., 2017, 80(9), 181–190 CrossRef CAS.
- R. Schucker, G. Dimitrakopoulos, K. Derrickson, K. K. Kopeć, F. Al-Ahmadi, J. R. Johnson, L. Shao and A. F. Ghoniem, Oxidative Dehydrogenation of Ethane to Ethylene in an Oxygen Ion Transport Membrane Reactor – A Proposed Design for Process Intensification, Ind. Eng. Chem. Res., 2019, 58(19), 7989–7997 CrossRef CAS.
- J. Sunarso, S. Baumann, J. M. Serra, W. A. Meulenberg, S. Liu, Y. S. Lin and J. C. Diniz da Costa, Mixed ionic–electronic conducting (MIEC) ceramic-based membranes for oxygen separation, J. Membr. Sci., 2008, 320, 13–41 CrossRef CAS.
- S. S. Hashim, M. R. Somalu, K. S. Loh, S. Liu, W. Zhou and J. Sunarso, Perovskite-based proton conducting membranes for hydrogen separation: A review, Int. J. Hydrogen Energy, 2018, 43, 15281–15305 CrossRef CAS.
- A. Thursfield, A. Murugan, R. Franca and I. S. Metcalfe, Chemical looping and oxygen permeable ceramic membranes for hydrogen production – a review, Energy Environ. Sci., 2012, 5, 7421–7459 RSC.
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Perovskites as Substitutes of Noble Metals for Heterogeneous Catalysis: Dream or Reality, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS PubMed.
- T. Zheng, J. Wu, D. Xiao and J. Zhu, Recent development in lead-free perovskite piezoelectric bulk materials, Prog. Mater. Sci., 2018, 98, 552–624 CrossRef CAS.
- Y. Li, H. Xu, P. H. Chien, N. Wu, S. Xin, L. Xue, K. Park, Y. Y. Hu and J. B. Goodenough, A Perovskite Electrolyte that is Stable in Moist Air for Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2018, 57, 8587–8591 CrossRef CAS PubMed.
- Y. Maeno, H. Hashimoto, K. Yoshida, S. Nishizaki, T. Fujita, J. G. Bednorz and F. Lichtenberg, Superconductivity in a layered perovskite without copper, Nature, 1994, 372, 532–534 CrossRef CAS.
- X. Liu, B. Cheng, H. Qin, P. Song, S. Huang, R. Zhang, J. Hu and M. Jiang, Preparation, electrical and gas-sensing properties of perovskite-type La1−xMgxFeO3 semiconductor materials, J. Phys. Chem. Solids, 2007, 68, 511–515 CrossRef CAS.
- Y. Wang, Z. Lv, L. Zhou, X. Chen, Y. Zhou, V. A. L. Roy and S. T. Han, Emerging perovskite materials for high density data storage and artificial synapses, J. Mater. Chem. C, 2018, 6, 1600–1617 RSC.
- G. Tsekouras, D. Neagu and J. T. S. Irvine, Step-change in high temperature steam electrolysis performance of perovskite oxide cathodes with exsolution of B-site dopants, Energy Environ. Sci., 2013, 6, 256–266 RSC.
- D. Neagu, T. S. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- J. Zhou, T. H. Shin, C. Ni, G. Chen, K. Wu, Y. Cheng and J. T. S. Irvine, In Situ Growth of Nanoparticles in Layered Perovskite La0.8Sr1.2Fe0.9Co0.1O4−δ as an Active and Stable Electrode for Symmetrical Solid Oxide Fuel Cells, Chem. Mater., 2016, 28, 2981–2993 CrossRef CAS.
- D. Neagu, E. I. Papaioannou, W. K. W. Ramli, D. N. Miller, B. J. Murdoch, H. Ménard, A. Umar, A. J. Barlow, P. J. Cumpson, J. T. S. Irvine and I. S. Metcalfe, Demonstration of chemistry at a point through restructuring and catalytic activation at anchored nanoparticles, Nat. Commun., 2017, 8, 1855 CrossRef PubMed.
- S. Liu, Q. Liu and J. Luo, Highly Stable and Efficient Catalyst with In Situ Exsolved Fe–Ni Alloy Nanospheres Socketed on an Oxygen Deficient Perovskite for Direct CO2 Electrolysis, ACS Catal., 2016, 6, 6219–6228 CrossRef CAS.
- Y. Sun, J. Li, Y. Zeng, B. S. Amirkhiz, M. Wang, Y. Behnamian and J. Luo, A-site deficient perovskite: the parent for in situ exsolution of highly active, regenerable nanoparticles as SOFC anodes, J. Mater. Chem. A, 2015, 3, 11048–11056 RSC.
- D. Paparguriou and J. T. S. Irvine, Nickel nanocatalyst exsolution from (La, Sr) (Cr, M, Ni)O3 (M = Mn, Fe) perovskites for the fuel oxidation layer of Oxygen transport Membranes, Solid State Ionics, 2016, 288, 120–123 CrossRef.
- M. D. Aminu, S. A. Nabavi, C. A. Rochelle and V. Manovic, A review of developments in carbon dioxide storage, Appl. Energy, 2017, 208, 1389–1419 CrossRef CAS.
- X. Wu and A. F. Ghoniem, Mixed ionic-electronic conducting (MIEC) membranes for thermochemical reduction of CO2: A review, Prog. Energy Combust. Sci., 2019, 74, 1–30 CrossRef.
- A. Hunt, G. Dimitrakopoulos, P. Kirchen and A. F. Ghoniem, Measuring the oxygen profile and permeation flux across an ion transport La0.9Ca0.1FeO3−δ membrane and the development and validation of a multistep surface exchange model, J. Membr. Sci., 2014, 468, 62–72 CrossRef CAS.
- A. Hunt, G. Dimitrakopoulos and A. F. Ghoniem, Surface oxygen vacancy and oxygen permeation flux limits of perovskite ion transport membranes, J. Membr. Sci., 2015, 489, 248–257 CrossRef CAS.
- G. Dimitrakopoulos and A. F. Ghoniem, A two-step surface exchange mechanism and detailed defect transport to model oxygen permeation through the La0.9Ca0.1FeO3−δ mixed-conductor, J. Membr. Sci., 2016, 510, 209–219 CrossRef CAS.
- G. Dimitrakopoulos and A. F. Ghoniem, Role of gas-phase and surface chemistry in methane reforming using a La0.9Ca0.1FeO3−δ oxygen transport membrane, Proc. Combust. Inst., 2017, 36, 4347–4354 CrossRef CAS.
- X. Wu, A. F. Ghoniem and M. Uddi, Enhancing co-production of H2 and syngas via water splitting and POM on surface-modified oxygen permeable membranes, AIChE J., 2016, 62, 4427–4435 CrossRef CAS.
- X. Wu, L. Chang, M. Uddi, P. Kirchen and A. F. Ghoniem, Toward enhanced hydrogen generation from water using oxygen permeating LCF membranes, Phys. Chem. Chem. Phys., 2015, 17, 10093–10107 RSC.
- X. Wu and A. F. Ghoniem, Hydrogen-assisted Carbon Dioxide Thermochemical Reduction on La0.9Ca0.1FeO3−δ Membranes: A Kinetics Study, ChemSusChem, 2018, 11, 483–493 CrossRef CAS PubMed.
- X. Wu and A. F. Ghoniem, CO2 reduction and methane partial oxidation on surface catalyzed La0.9Ca0.1FeO3−δ oxygen transport membranes, Proc. Combust. Inst., 2019, 37, 5517–5524 CrossRef CAS.
- G. Dimitrakopoulos and A. F. Ghoniem, Developing a multistep surface reaction mechanism to model the impact of H2 and CO on the performance and defect chemistry of La0.9Ca0.1FeO3−δ mixed-conductors, J. Membr. Sci., 2017, 529, 114–132 CrossRef CAS.
- C. F. Miller, J. Chen, M. F. Carolan and E. P. Foster, Advances in ion transport membrane technology for syngas production, Catal. Today, 2014, 228, 152–157 CrossRef CAS.
- A. S. Yu, J. Kim, T. S. Oh, G. Kim, R. J. Gorte and J. M. Vohs, Decreasing interfacial losses with catalysts in La0.9Ca0.1FeO3−δ membranes for syngas production, Appl. Catal., A, 2014, 486, 259–265 CrossRef CAS.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, In situ growth of nanoparticles through control of non-stoichiometry, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00249a |
| This journal is © The Royal Society of Chemistry 2019 |