
Remarkably long-lived excited states of copper photosensitizers containing an extended π-system based on an anthracene moiety†
Robin
Giereth‡
 a,
Immanuel
Reim‡
b,
Wolfgang
Frey
b,
Henrik
Junge
c,
Stefanie
Tschierlei
*a and
Michael
Karnahl
*b
a,
Immanuel
Reim‡
b,
Wolfgang
Frey
b,
Henrik
Junge
c,
Stefanie
Tschierlei
*a and
Michael
Karnahl
*b
aUlm University, Institute of Inorganic Chemistry I, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: stefanie.tschierlei@uni-ulm.de; Web: http://www.tschierlei-group.de
bUniversity of Stuttgart, Institute of Organic Chemistry, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: michael.karnahl@oc.uni-stuttgart.de; Web: http://www.karnahl-group.de
cLeibniz Institute for Catalysis at the University of Rostock (LIKAT), Albert-Einstein-Straße 29a, 18059 Rostock, Germany
First published on 6th November 2018
Abstract
An ideal photosensitizer should strongly absorb visible light over a broad range and exhibit long-lived excited states. Therefore, an anthracene moiety attached to a bipyridine sphere was chosen to study the impact of a carbon extended π-system in the back on the structural, electrochemical and photophysical properties of the resulting copper photosensitizers. Consequently, four novel homo- and heteroleptic Cu(I) complexes of the type [Cu(N^N)2]+ and [(P^P)Cu(N^N)]+ were prepared and fully characterized. The solid state structures possess a strong twist of the anthracene system due to sterical constraints. At the same time the absorption maxima are red-shifted and the excited states display unprecedented lifetimes up to 4 μs in acetonitrile stemming from different excitation processes unique for the anthracene moiety. The excited state properties were then further analyzed by transient absorption spectroscopy and DFT calculations. Moreover, it was found that the anthracene extension has a slight positive effect on the photocatalytic reduction of protons, which was selected as a model reaction to test the capability of these photosensitizers.
Introduction
The adequate and environmentally friendly supply of energy represents a major challenge of this century.1–3 A promising option is given by the increased use of solar energy, which requires the design of efficient and inexpensive photosensitizers for light harvesting.4–8 Furthermore, a suitable photosensitizer should possess a strong absorption in the visible region, long-lived excited states, a reversible electrochemical behavior including an appropriate redox power and a sufficient stability.4,9–12 So far the majority of molecular photosensitizers are based on rare and precious metals, like ruthenium, iridium, platinum, or rhenium, limiting their large-scale application.5,9,10,13–15 In this respect, photoactive copper(I) complexes are considered as a very promising alternative, as evidenced by a strongly increasing number of publications in recent years.16–35The great potential of Cu(I) photosensitizers (CuPS) has already been shown by their successful application in the fields of photo(redox)catalysis,36–39 light-emitting electrochemical cells (LECs)40,41 or dye-sensitized solar cells (DSSCs).42–44 However, there is a great need to further improve these Cu(I) compounds to bring such non-noble systems into practice. In particular, a limited absorption of visible light is a major bottleneck of most of the heteroleptic Cu(I) complexes of the type [(P^P)Cu(N^N)]+,6,12 where N^N indicates a diimine and P^P a diphosphine ligand.
To overcome this issue heteroleptic CuPSs with an extended π-system in the backbone of the diimine ligand have been prepared in a previous study.45 At that time, the selected 3,6-dimethyl-dipyrido[3,2-a:2′,3′-c]phenazine ligand (L3, Me2dppz, Fig. 1) caused increased extinction coefficients, but the visible light absorption was hardly redshifted.45 Furthermore, the bichromophoric character of L3 created a low-lying excited state at the phenazine sphere, resulting in a shortened excited state lifetime and a low photocatalytic performance of the corresponding Cu(I) complex 3 (for comparison included in Fig. 1).45
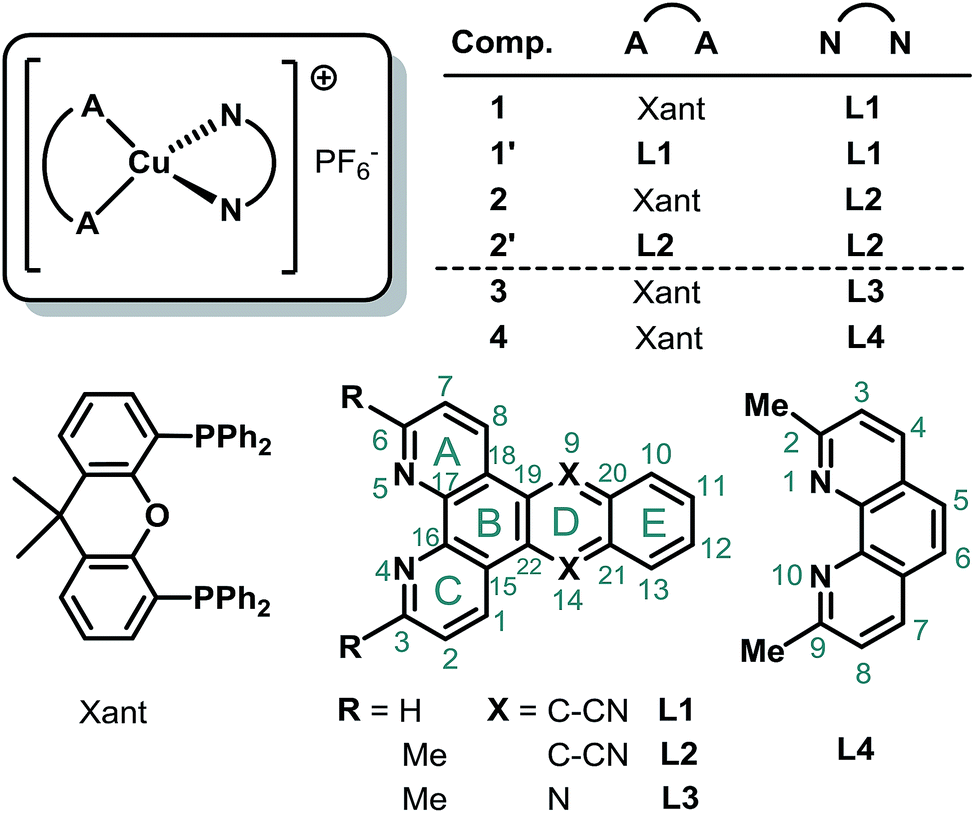 | ||
| Fig. 1 General structure of the ligands L1 to L3 and the resulting heteroleptic Cu(I) complexes 1 and 2 as well as their homoleptic analogues 1′ and 2′. Furthermore, the structures of the reference complexes 3 to 4 and the position numbering as well as the ring labeling scheme (grey color) of the ligands L1 to L4 is given. | ||
Therefore, the present study investigates CuPS with an extended π-system and the impact of replacing the phenazine by an anthracene moiety. In consequence, a systematic series of four novel homo- and heteroleptic Cu(I) complexes using dipyrido[3,2-a:2′,3′-c]anthracene-9,10-dinitrile (L1, dpan, Fig. 1) and 3,6-dimethyl-dipyrido[3,2-a:2′,3′-c]anthracene-9,10-dinitrile (L2) was prepared. Please note, that according to the Hantzsch–Widman system dipyrido[3,2-a:2′,3′-c]anthracene-9,10-dinitrile (dpan) can also be named as naphtho[2,3-f][4,5]phenanthroline-9,14-dicarbonitrile.
All compounds were fully analyzed by means of elemental analysis, mass spectrometry (MS), NMR, UV/vis and emission spectroscopy as well as electrochemical measurements. The resulting structural, photophysical and electrochemical properties were discussed with respect to the reference complexes 3 and 4 (Fig. 1), which possess a Me2phen or Me2dppz ligand, respectively. In addition, the solid-state structures of the two complexes 2 and 2′ (Fig. 1) containing L2 were determined by X-ray crystallography and are compared with the findings of density functional theory (DFT) calculations. Further, the respective light-induced charge transfer processes were calculated by time-dependent DFT (TD-DFT) methods to assist the assignment of the electronic transitions in the absorption spectra. Nanosecond transient absorption spectroscopy has been applied to understand the nature of the triplet excited states of this class of photosensitizers. Finally, the photocatalytic reduction of protons to hydrogen within a fully noble-metal-free system was used to assess the capability of these novel CuPSs. As a result, the combination of structural and spectroscopic characterization as well as application allowed for the identification of structure–activity relationships. Based on these findings future design strategies towards improved CuPSs are proposed, which will contribute to an increased utilization and conversion of sunlight as a promising and clean energy source.
Results and discussions
Synthesis and structural characterization
Starting either from 1,10-phenanthroline-5,6-dione or 2,9-dimethyl-1,10-phenanthroline-5,6-dione the two ligands L1 and L2 were prepared by a condensation reaction with 1,2-bis(cyanomethyl)benzene in the presence of 1,8-diaza-bicyclo[5.4.0]undec-7-ene (DBU).46 Only the addition of this non-nucleophilic and strong base (pKa = 24.34 in MeCN)47 allowed for good yields of L1 (70%) and L2 (76%). Subsequently, the two heteroleptic Cu(I) complexes 1 and 2 (Fig. 1) were synthesized by an one-pot two-step procedure using the [Cu(MeCN)4]PF6 precursor following a slightly modified literature protocol (see ESI†).45,48 Xantphos (Xant) was chosen as a suitable diphosphine ligand owing to its bulkiness and rigid core structure, which were identified as beneficial properties in previous studies.12,36,48 In order to further increase the steric demand and to reduce exciplex quenching, which is induced by fast flattening distortions,49–52 two methyl groups were additionally introduced in 3,6-position of L2.4,12 After purification both heteroleptic CuPSs were obtained as yellow to orange solids in excellent yields (1: 93% and 2: 90%). Likewise the homoleptic counterparts i.e.1′ and 2′ were prepared for comparison prepared using [Cu(MeCN)4]PF6 and two equivalents of the respective diimine ligand. As a result, deeply dark colored and sparingly soluble solids (a slight solubility is given in acetonitrile or dimethylformamide) were received (1′: 73% and 2′: 76%). NMR measurements (1H, 13C, and 31P NMR) and mass spectra (see ESI, Chapter 3†) are all in agreement with the proposed structure. The 31P{1H} chemical shifts of 1 (−12.37 ppm) and 2 (−12.44 ppm) are almost identical to those of the structurally related reference complexes 3 (−12.63 ppm) and 4 (−13.44 ppm, Fig. 1). This implies that the structure around the copper center and the coordination ability of the xantphos ligand is almost unaffected by the variation of the diimine ligand backbone.Single crystals of 2 and 2′ suitable for X-ray crystallography were obtained by slow diffusion of n-pentane into a saturated dichloromethane solution. The subsequent structural analysis of both complexes (Fig. 2 and S6†) revealed a distorted tetrahedral geometry around the copper center, which is commonly observed for these compounds.12,16,29,45,48 Comparison of selected structural parameters of 2 and 2′ (Table 1) with the reference complexes 3 and 4 does not show major differences regarding to the basic coordination geometry (e.g. N1–Cu–N2 for 2: 79.12(7)°, 2′: 80.47(9)°, 3: 80.53(13)° and 4: 80.02(8)°),45 indicating again the small impact of the different diimine ligand backbones on the principal structure around the Cu(I) center. With 87.39° (2) and 87.35° (2′) the angle between the two ligand planes, which are spanned through the chelating heteroatoms and the copper center, differs from the ideal 90° (Table 1). More importantly, the solid state structures exhibit a pronounced torsion of the ligand L2 in the respective complexes 2 and 2′ (Fig. 3). Interestingly, the X-ray analysis of 2 revealed a twisting motion of L2 along the axis through ring B, D and E (cf. ring labelling scheme in Fig. 1). Contrary a butterfly configuration with the rings A and C bended downwards and the two dinitrile groups upwards is present in 2′ (Fig. 3). These findings are also corroborated by our DFT calculations, as well as by calculations carried out by Soulis et al. for structurally related homoleptic Cu(I) systems.34
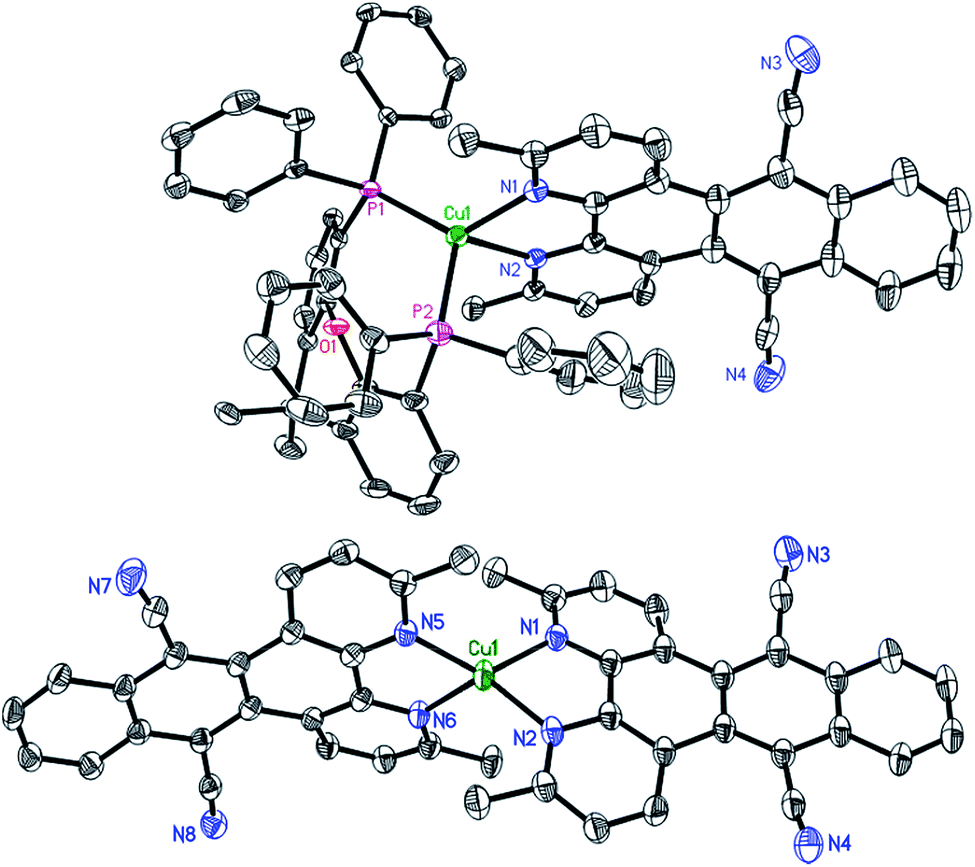 | ||
| Fig. 2 Solid state structures (ORTEP representation) of complex 2 (top) and 2′ (bottom) with thermal ellipsoids at a probability level of 50%. The hydrogen atoms, counter anions and solvent molecules are omitted for clarity. | ||
| 2 X-ray | 2 DFTa | 2′ X-ray | 2′ DFTa | ||
|---|---|---|---|---|---|
| a BP86-D3(BJ)/def2-TZVP. b Ring A/E. c Ring D/E (according to Fig. 1). | |||||
| Cu–N1 | 210.11(18) | 211.88 | Cu–N1 | 202.5(2) | 202.54 |
| Cu–N2 | 211.90(16) | 212.69 | Cu–N2 | 202.5(2) | 202.56 |
| Cu–P1 | 226.08(6) | 232.76 | Cu–N3 | 204.4(2) | 204.21 |
| Cu–P2 | 228.87(6) | 233.49 | Cu–N4 | 205.7(2) | 204.21 |
| N1–Cu–N2 | 79.12(7) | 78.82 | N1–Cu–N2 | 80.47(9) | 81.61 |
| P1–Cu–P2 | 116.69(2) | 118.25 | N5–Cu–N6 | 80.63(8) | 81.60 |
| lpia | 87.39 | 88.54 | lpia | 87.35 | 89.64 |
| C19–C22 | 142.7(3) | 143.83 | 144.7(4)/144.8(3) | 144.50 | |
| C20–C21 | 141.1(4) | 143.24 | 142.2(4)/141.2(4) | 143.05 | |
| Twistb | 28.10 | 28.31 | Twistc | 5.30 | 6.18 |
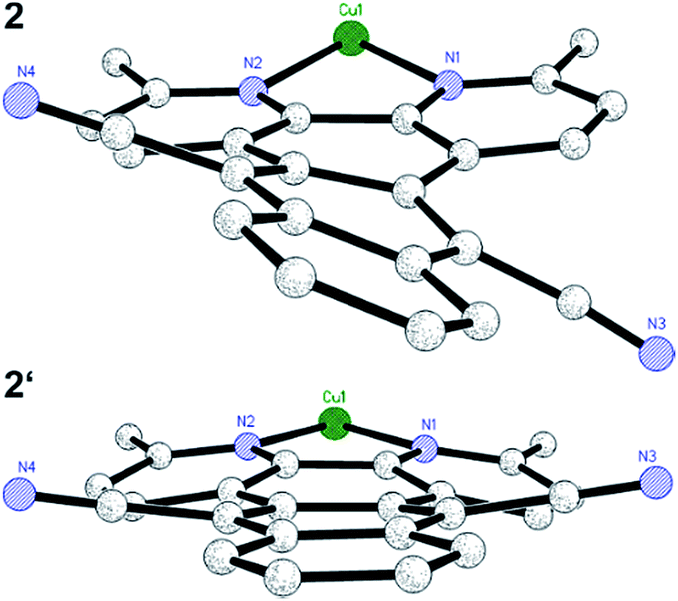 | ||
| Fig. 3 Reduced presentation of the solid state structures of 2 (top) and 2′ (bottom) illustrating the different types of twisting of L2. For the sake of clarity only the Cu–L2 moiety is depicted. | ||
Moreover, there are stacked dimers between neighboring complexes found in the solid state of 2 (Fig. S7†). This observation is in contrast to 3, where no stacking effects are present although being typical for coordination compounds containing a dppz ligand.45,53–56 This stacking interaction could be reasoned by the twist in the ligand backbone, leading to an offset of the copper centres of the dimers along the a-axis, which minimizes the steric repulsion between the bulky xantphos fragments. However, there is no evidence for π–π interactions in the solid state of the homoleptic complex 2′.
Subsequently, DFT calculations were carried out in order to investigate the twisting possibilities of L2 (ESI, Chapter 5†). These DFT calculations indicate that both conformational isomers are very close in energy.
The basic scaffold of L3, which is almost identical to L1 and L2, i.e. the only difference is the central ring D (pyrazine vs. 1,4-benzenedicarbonitrile, Fig. 1), is perfectly planar and symmetric in complex 3.45 This also means, that in L3 the bond lengths at the two joint bonds of ring B/D (C19–C22) and D/E (C20–C21) are similar (142.8(4) and 142.6(4) pm, respectively). In contrast, the bond length C19–C22 in 2 (142.7(3) pm) and 2′ (144.7(4) pm) between the rings B/D is longer compared to C20–C21 (2: 141.1(4) and 2′: 142.2(4) pm and 141.2(4) pm) between D/E due to the exchange of the phenazine nitrogen by a C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group. Especially, in complex 2′ the observed length difference of about 3.6 pm is relatively strong.
N group. Especially, in complex 2′ the observed length difference of about 3.6 pm is relatively strong.
Further, from the crystal structure it becomes obvious, that the bending of the carbonitrile groups (Fig. 3) is predominantly caused by the repulsion with the adjacent hydrogen atoms in 1,8-position (Fig. S8†). These findings are also in accordance with DFT calculations performed by Soulis et al.34 and with calculations for a tetracyano-substituted tetrabenzoheptacene, which also display these conformations caused by steric hindrance between the cyano-group and the adjacent hydrogen atoms.57
In addition to the X-ray structures of 2 and 2′, the structures of all complexes have been optimized on DFT level of theory (ESI, Chapter 5†). The increased steric demand in close vicinity to the copper center leads to a slight elongation of the Cu–N bond lengths by about 10 pm in 2′ compared to 1′, while in the heteroleptic complex 2 an increase of 30 pm for Cu–N as well as for the Cu–P bond lengths compared to 1 are calculated.
Electrochemical and photophysical properties
The electrochemical properties of all four novel Cu(I) compounds were studied by cyclic voltammetry (Fig. 4, Table 2). As a common feature the cyclic voltammograms (CV) of both heteroleptic Cu(I) complexes (Fig. 4) exhibit two reversible reduction processes at −1.14/−1.76 V (1) and −1.19/−1.81 V (2) vs. Fc/Fc+ in acetonitrile solution. Hence, the two additional methyl groups in L2 have only a small impact on the reduction behavior. Most interestingly, the reduction potentials of the homoleptic complex 2′ (−1.18/−1.87 V) perfectly matches the one of the heteroleptic complex 2 (Fig. S28†), implying that both reduction events are related to the ligand L2. Furthermore, the first reduction wave in 1 and 2 occurs at potentials very close to that of the uncoordinated ligand (Ered,L1 = −1.17 V in DMSO)58 and to the structurally related [Ru(bpy)2(L1)]2+ (bpy = 2,2′-bipyridine) complex (Ered = −1.13 V),58 respectively. The uncoordinated ligand L2 itself shows two fully reversible reduction processes at −1.32 and −1.88 V in acetonitrile (Fig. S29†). Compared to the reference complex 3 the reduction events of all complexes containing a dpan ligand are shifted by approx. 300 mV to more positive values. This might be due to the stronger π-accepting ability of the anthracene-9,10-dinitrile subunit of the dpan ligand compared to the phenazine unit in L3. An even larger shift in the first reduction potential (>900 mV) can be observed in comparison to the reference complex 4 with the much smaller Me2phen ligand. Hence, the novel dpan based photosensitizers are easily reduced, but also have a lower reducing power for potential subsequent reactions.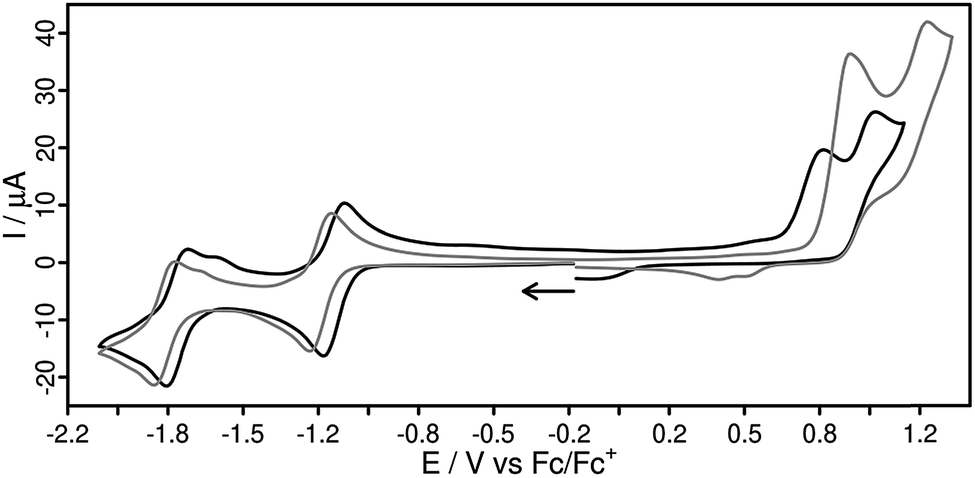 | ||
| Fig. 4 Cyclic voltammograms of 1 (black solid) and 2 (grey) in acetonitrile solution referenced vs. the ferrocene/ferrocenium (Fc/Fc+) couple. Scan rate of 100 mV s−1, with 0.1 M n-Bu4NPF6 as supporting electrolyte. | ||
| λ abs [nm] | ε 400 nm [103 M−1 cm−1] | τ [ns] | E ox [V] | E red [V] | |
|---|---|---|---|---|---|
| a In acetonitrile vs. Fc/Fc+. b In DMF vs. Fc/Fc+. c Irreversible peak. d Extinction coefficients are higher owing to ligand-centered transitions at the Me2dppz. e The emission intensity was below the detection limit. The time constant refers to a non-emissive decay. f Not determined due to insufficient solubility. g Taken from ref. 45 and 71 for comparison. | |||||
| 1 | 376 | 8.3 | 4380 | 0.815c | −1.762 |
| 394 | 7.9 | 1.026c | −1.138 | ||
| 2 | 383 | 11.1 | 1250, 3785 | 0.926c | −1.813 |
| 400 | 10.5 | 1.228c | −1.193 | ||
| 1′ | 377 | 16.4 | n.d. | n.d.f | n.d.f |
| 395 | 15.5 | ||||
| 449 | 4.18 | ||||
| 2′ | 380 | 19.5 | n.d. | 0.191b | −1.873b |
| 398 | 19.2 | −1.179b | |||
| 459 | 9.89 | ||||
| 3 | 364 | 13.5d | 14.5e | n.d. | −1.49 |
| 382 | 11.7d | ||||
| 4 | 378 | 3.1 | 64 | 0.82c | −2.10 |
The second one-electron reduction step can be assigned to the reduction of the bipyridine subunit of L2 (cf. inset of Fig. 6), which is known to be electronically separated from the anthracene moiety.59 Such electronically separated subunits within diimine ligands containing an extended π-system (e.g. dppz) are not uncommon and can be also observed for their Ru(II) complexes.54,58,60
Furthermore, in both heteroleptic complexes 1 and 2 the oxidation is an irreversible process (Fig. 4), whereas it is pseudo-reversible for 2′ (Fig. S35†). This finding is consistent with analogous heteroleptic [(P^P)Cu(N^N)]+ complexes and is most likely caused by a dissociation of a Cu–P bond, resulting in a decomposition of the Cu(II) species.18,45,48,61,62
The UV/vis absorption properties of all Cu(I) complexes have been investigated in acetonitrile solution. The lowest energy absorption bands of the heteroleptic complexes 1 and 2 occur at approx. 376 and 394 nm (Fig. 5). This region can generally be assigned to a metal-to-ligand charge transfer (MLCT) from the d-orbitals of the copper ion to the π* orbital of the diimine ligand and is characteristic for this kind of CuPSs.4,12 Compared to reference 4 all heteroleptic CuPS with an extended π-system (1, 2 and 3) do have increased extinction coefficients (ε, Table 1). In case of 3 the phenazine sphere of L3 is additionally strongly involved in the MLCT transitions.45 This impact of the extended π-system also holds true for 1 and 2, where the anthracene moiety is strongly involved in the MLCT processes (Tables S3 and S4 in the ESI† for detailed transitions). Here, in contrast to 3, the lowest lying excitation is now located on the distant part of the dpan ligand, i.e. ring B, D and E (LUMO, Fig. S19†). This is in accordance with the stronger (electronically) stabilizing impact of the two carbonitrile substituents on the frontier orbitals located on this scaffold. In consequence, these LC transitions superimpose the MLCT transitions of the heteroleptic complexes 1 and 2, and thus, the absorption spectrum also shows the usual band characteristics of anthracene derivatives such as benzo[b]triphenylene-9,14-dicarbonitrile, the phenanthrene-derivative of L1.63 The TD-DFT calculations of the singlet transitions approve this experimental result and reveal that this LC transition starts from ring E and ends up in ring D (Fig. 6), where the electron density is lowered by the dicarbonitrile substituents. Nevertheless, in related Ru(II) and Cu(I) complexes containing L3 also LC transitions occur,45,60 albeit their intensity compared to the MLCT absorption is not so dominant. Further, these LC transitions are not exclusively localized on the backside of the π-extended diimine ligand.45,60
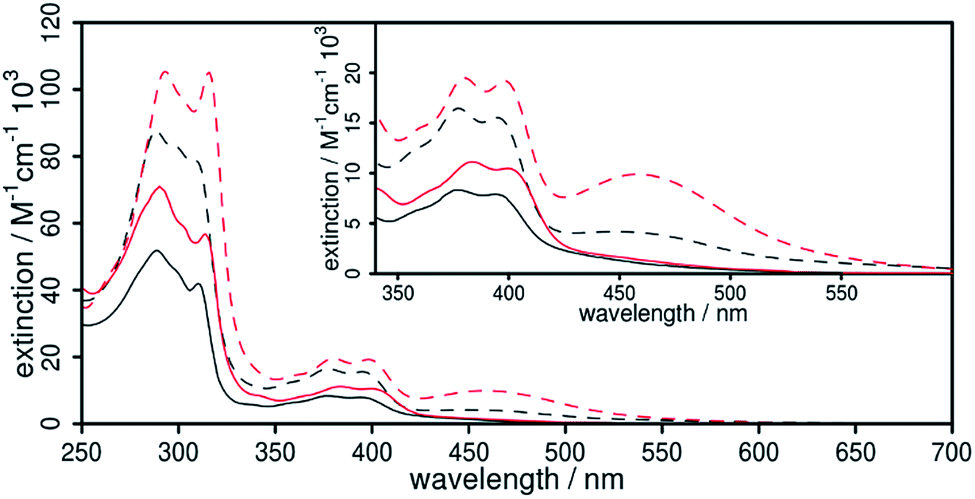 | ||
| Fig. 5 Absorption spectra of 1 (black, solid) and 2 (red, solid) in acetonitrile solution. The corresponding homoleptic complexes 1′ (black) and 2′ (red) are displayed as dashed lines. The inset represents an enlargement of the wavelength range of the metal-to-ligand charge transfer (MLCT) transitions showing a significant increase in the extinction of 2/2′ compared to 1/1′. | ||
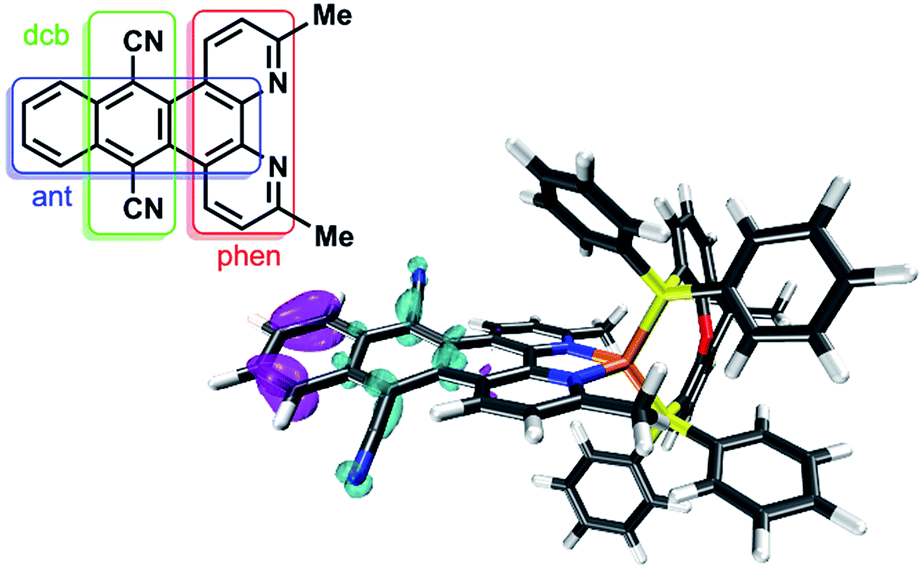 | ||
| Fig. 6 Differential density plot of 2 between the ground state and the second singlet excited state, which is assigned to a ligand-centered (LC) π → π* transition. Cyan corresponds to an increased and purple to a declined electron density. CAM-B3LYP-D3(BJ)/def2-TZVP with CPCM (acetonitrile) solvation model was used for TD-DFT calculations. More details are given in the ESI.† Inset: Labeling scheme and abbreviations of the subunits of the dpan ligand L1. | ||
In the case of the homoleptic Cu(I) complexes 1′ and 2′ the MLCT absorption is shifted more towards lower energy (Fig. 5), and thus, the respective absorption bands are more pronounced as in the related [Ru(bpy)2(L1)]2+ complex (ε439 nm = 14.6 × 103 M−1 cm−1).58
Upon photo-excitation of the complexes 1/1′ and 2/2′ at 355 nm only high energy emission with maxima centered at 425 and 444 nm (Fig. S13†) were observed, respectively. This emission could be assigned to fluorescence of the uncoordinated ligand, while no phosphorescence, originated from the 3MLCT states, was detected under these conditions. Only after exciting 2 with energy below the LC band of L2 a very weak emission signal at 605 nm could be measured, which is in the intensity of the Raman scattering (Fig. S15†). This is in contrast to the related Ru(II) complex [Ru(bpy)2L1]2+, which has a broad and strong emission at 700 nm even in aerated acetonitrile.58 However, our results are in accordance with [Cu(3,6-n-butyl-dipyrido[3,2-a:2′,3′-c]anthracen-9,10-dinitrile)2]+ that also only possess a very weak emission at 815 nm (in degassed dichloromethane with an intensity in the dimension of the Raman scattering).34 The reason for the observed fluorescence at low wavelengths is most likely given by a slight photo-dissociation of the dpan ligand or a dynamic ligand exchange reaction in solution.18,23,61,64 This ligand exchange reaction and the insufficient stability is still a major drawback of most of the heteroleptic CuPS.12,18,23,65,66 However, there is no indication of leftover impurities in the analytical data originating from the work-up procedure (see ESI†).
Transient absorption spectroscopy
To investigate the underlying excited state processes of the complexes 1 and 2 in more detail nanosecond transient absorption measurements were performed. For instance, 3 does not emit light due to radiationless deactivation processes of the excited states. There, the deactivation is clearly accelerated by the extended π-system, and the excited state lifetime of 3 is only 14.5 ns in acetonitrile.45 The situation is totally different for complex 2, where an excited state absorption with a maximum around 460 nm and a broad band up to 720 nm were detected (Fig. 7, top). Interestingly, the spectra have the similar shape as detected for benzo[b]triphenylene-9,14-dicarbonitrile.67 The very weak phosphorescence around 605 nm, which was detected by a steady-state emission spectrometer, was not observed in the transient absorption setup. This underlines that the weak emission entirely occurs in the intensity of the Raman scattering. Furthermore, it takes up to 10 μs to fully deactivate the excited states of 2. | ||
Fig. 7 Transient absorption spectra (top) of 2 in acetonitrile excited at 355 nm after 80 (red), 500 (yellow), 1000 (green), 2000 (cyan), 4000 (blue), 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (violet) and 15000 ns (magenta), kinetics (middle) at 460 (red), 480 (green) and 600 nm (blue). The grey lines represent a bi-exponential fit with τ1 = 1250 ns, τ2 = 3785 ns. The kinetics are plotted on a logarithmic scale for better clarity. The decay times were obtained by a global analysis of the measured data ΔOD with the function 000 (violet) and 15000 ns (magenta), kinetics (middle) at 460 (red), 480 (green) and 600 nm (blue). The grey lines represent a bi-exponential fit with τ1 = 1250 ns, τ2 = 3785 ns. The kinetics are plotted on a logarithmic scale for better clarity. The decay times were obtained by a global analysis of the measured data ΔOD with the function 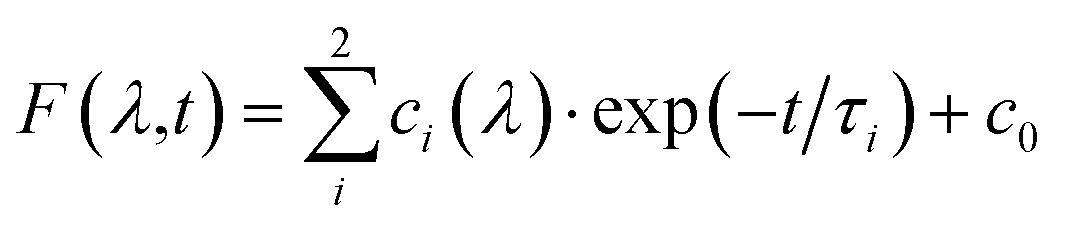 , with the probe wavelength λ, the delay time t and c0 as long-living component. , with the probe wavelength λ, the delay time t and c0 as long-living component. | ||
A detailed global analysis of the time-resolved measurements of 2 in the range from 420 to 720 nm reveals that a bi-exponential fit is required in order to resolve the kinetics. Consequently, two processes with the time constants τ1 = 1.3 μs and τ2 = 3.8 μs are determined for complex 2. From the respective decay associated spectra, where the amplitudes of the time constants are depicted, it is obvious that two depopulation processes occur (Fig S25†). Interestingly, the amplitude spectra of both time constants are comparable in their shape and intensity.
The transient absorption spectra of 1 are similar to 2, where again an excited state absorption with a maximum at about 460 nm is observed (Fig. S24†). The time constant of this deactivation is 4.4 μs. Due to weaker transient signals of 1 compared to the spectra of 2 a second time constant could not be determined. Altogether, 1 and 2 are deactivated via radiationless pathways and with about 4 μs the respective excited states are remarkably long-lived in both complexes. In acetonitrile solution at room temperature these lifetimes are so far, to the best of our knowledge, the longest lifetimes obtained for such heteroleptic CuPSs containing an extended π-system. This is in strong contrast to 3, where the deactivation occurs very fast via the two 3MLCT states located at the phenanthroline and the phenazine moiety of L3.45
However, it seems that these excited states in the Cu(I) complexes are not pure 3MLCT states, because the obtained time constants are unusually long. For the related complex [Ru(L1)(bpy)2]2+ Albano et al. determined at 77 K an excited state lifetime of 464 μs, which they assigned to a triplet intraligand excited state centered on the dicarbonitrile-anthracene moiety of the L1 ligand.58 This assignment is in accordance to the calculated absorption of 1 and 2, where one dominant absorption band belongs to the ligand centered π → π* transition of the anthracene moiety as explained above (Fig. 6, ESI†).
A further indication that the lowest-lying excited states are ligand centered is that for L2 the time constants τ1 = 5.2 μs and τ2 = 22.4 μs were determined in acetonitrile solution. This bi-exponential decay behavior on the microsecond timescale is also in accordance with dicyanoanthracene.68 In about the same time of approx. 7.1 μs the excited state deactivation of benzo[b]triphenylene-9,14-dicarbonitrile occurs.67
Having such long-lived excited states even at room temperature renders 1 and 2 as an attractive class of photosensitizers that can act as light-harvesting units in solar-energy conversion schemes.
Photocatalytic hydrogen production
The photocatalytic reduction of protons to hydrogen was selected as a potential model reaction to test the capability of the novel CuPS. Based on previous results a fully noble metal-free system composed of the respective Cu(I) complex, [Fe3(CO)12] acting as a water reduction catalyst (WRC) and triethylamine as sacrificial reductant (SR) was used for H2 production (Table 3).36,45,48,62 To ensure comparability standard conditions, i.e. 3.5 μmol CuPS and 5.0 μmol [Fe3(CO)12] in 10 mL of THF/TEA/H2O with a volumetric composition of 4:3:1, were applied. Please note, that under these conditions the two homoleptic CuPS 1′ and 2′ could not be tested due to solubility issues (Fig. 8).
| Complex | V 24 h [mL] | V corr [mL] | TONH,Cuc |
|---|---|---|---|
|
a Conditions: CuPS (ca. 3.5 μmol), [Fe3(CO)12] (ca. 5.0 μmol), in THF/TEA/H2O (4:3:1, 10 mL), at 25 °C, Xe light irradiation (output 1.5 W, without light filter), 24 h.
b
V
24 h corrected by a blank volume of 2.3 mL (entries 1 and 2) and 2.8 mL (entries 3 and 4), respectively.
c Besides H2 also up to 60 μmol CO were detected in the gas phase due to the decomposition of the Fe carbonyl catalysts;70 TONH,Cu = n(H)/n(CuPS) with Vm,H2,25°C = 24.48 mL mmol−1.
d Taken from ref. 45 for comparison.
|
|||
| 1 | 5.2 | 2.9 | 39 |
| 2 | 6.0 | 3.7 | 51 |
| 3 | 4.0 | 1.2 | 29 |
| 4 | 16.0 | 13.2 | 305 |
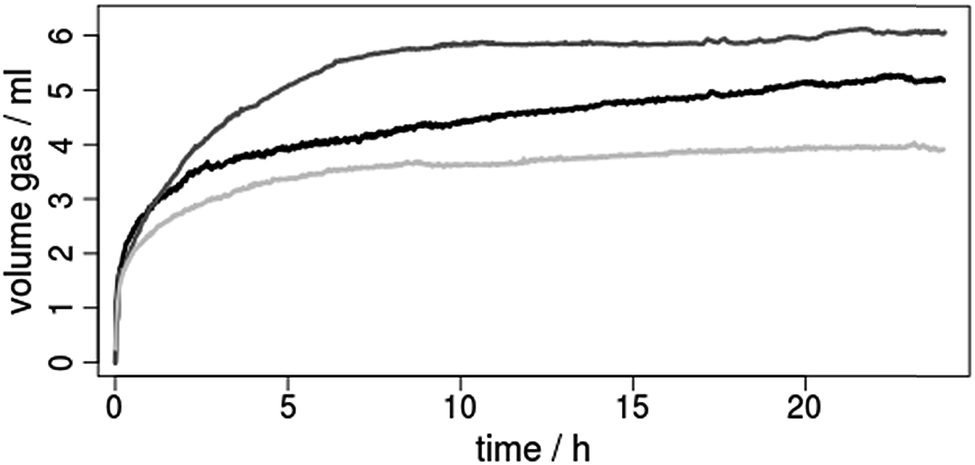 | ||
| Fig. 8 Hydrogen evolution curves for the photocatalytic reduction of protons by using the copper photosensitizers (ca. 3.5 μmol) 1 (black), 2 (dark grey) or 3 (grey) and [Fe3(CO)12] (ca. 5.0 μmol) as water reduction catalyst in a solvent mixture of THF/TEA/H2O (4/3/1). For further reaction details see footnote of Table 3. | ||
The resulting hydrogen evolution curves (Fig. 8) show a low, but significant H2 production within 24 h. The development of the respective turnover numbers over time is presented in the ESI (Fig. S36).† It should be mentioned, that under these conditions also CO is produced (up to 60 μmol), which mainly results from the stepwise decomposition of the [Fe3(CO)12] precursor.69,70 This CO was taken into account for the calculation of the final turnover numbers (Table 3).
Comparison of the catalytic results reveal a clear influence of the diimine ligand on the maximum turnover number (TON) with an order 3 (29) < 1 (39) < 2 (51, Table 3). This means that both the introduction of the anthracene moiety (2vs.3) and of the two methyl substituents in 3,6-position (1vs.2) have a positive effect. On the one hand the improved TON of 2 compared to 1 can be rationalized by a better shielding of the Cu(I) center, which prevents flattening distortion and undesired exciplex quenching in the catalytic environment. On the other hand the long excited state lifetimes induced by the anthracene moiety seem to be superior to the phenazine. However, the maximum TONs of all heteroleptic CuPS 1, 2 and 3 possessing an extended π-system are significantly lower compared to the reference 4 with Me2phen (Table 3).45,71 This is most likely caused by the unfavorable redox potentials of these CuPS. The much less negative first reduction potentials of 1–3 compared to 4 might be not sufficient to reduce the main active species of the iron catalyst during catalysis.64,69 Hence, the improved light harvesting and excited state properties are buried by the electrochemical behavior.
Conclusions
Motivated by the great potential and the large variety of applications of copper photosensitizers (CuPSs) this study deals with novel Cu(I) complexes containing an anthracene moiety at the backbone of the diimine ligand. A couple of two heteroleptic Cu(I) complexes with the general formula [(P^P)Cu(N^N)]+, with N^N = diimine ligand and P^P = xantphos, were synthesized. These CuPSs have then been fully analyzed concerning their structural, electrochemical and photophysical properties. In addition, the light-driven reduction of protons to hydrogen has been performed to demonstrate their general applicability in solar-energy conversion schemes. Most importantly, the heteroleptic CuPSs exhibit remarkably long-lived excited states in the microsecond time range (up to 4.4 μs in acetonitrile solution), which are deactivated without radiation.The complexes have a rather unusual electrochemical behavior, as the first reduction process is located on the very distant part of the dpan ligand. This finding is also reflected in the UV/vis absorption spectra and supported by (TD)-DFT calculations. The maximum of the metal-to-ligand charge transfer band in the visible region is shifted bathochromically of up to 20 nm compared to a CuPS with a phenazine-based diimine ligand. Hence, the intended broadening and red-shift of the visible light absorption was successfully accomplished by extending the π-system with an anthracene moiety.
Notably, with excited state lifetimes of up to 4.4 μs at room temperature these CuPSs offer one of the longest lifetimes measured in acetonitrile solution. This is of particular interest as structurally related Cu(I) complexes with an extended π-system (such as dppz) only possess lifetimes in the nanosecond time regime.
In principle, this feature should promote the ability of these complexes to act as suitable photosensitizers in the field of photo(redox) catalysis. As a result, these CuPSs were successfully applied for light-driven H2 production. Unfortunately, the amount of hydrogen was only slightly increased compared to related heteroleptic Cu(I) complexes with an extended π-system.12 This means that also other parameters than the excited state lifetime and the absorption behavior have to be considered. In this context, particular attention should be given to matching redox potentials.
In summary, the extension of the aromatic system by an anthracene moiety changes the excited state characteristics dramatically compared to a phenazine moiety (dpan vs. dppz ligand). In Cu(I) dpan complexes the excited states are largely dominated by the characteristics of aromatic carbonitrile unit. This finally offers completely new opportunities and design strategies for the modification of the energy level of the triplet metal-to-ligand charge transfer and the ligand centered excited states. For instance, substituents at ring E in the back of the anthracene moiety will be one prominent option to increase the excited state energy.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank A. Kammer (LIKAT Rostock) for performing the hydrogen evolution experiments. M. K. is thankful to the University of Stuttgart and the Fonds der Chemischen Industrie (FCI) for financial support. S. T. grateful acknowledges the German Science Foundation (DFG, TS 330/3-1) and the Fonds der Chemischen Industrie (FCI) for funding. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and by the DFG (INST 40/467-1 FUGG).Notes and references
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- S. Styring, Faraday Discuss., 2012, 357–376 RSC.
- L. Hammarström, Chem, 2016, 1, 515–518 Search PubMed.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- W. T. Eckenhoff and R. Eisenberg, Dalton Trans., 2012, 41, 13004–13021 RSC.
- M. S. Lazorski and F. N. Castellano, Polyhedron, 2014, 82, 57–70 CrossRef CAS.
- N. Armaroli and V. Balzani, Chem.–Eur. J., 2015, 22, 32–57 CrossRef PubMed.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- Y.-J. Yuan, Z.-T. Yu, D.-Q. Chen and Z.-G. Zou, Chem. Soc. Rev., 2017, 46, 603–631 RSC.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268–9274 RSC.
- Y. Halpin, M. T. Pryce, S. Rau, D. Dini and J. G. Vos, Dalton Trans., 2013, 42, 16243–16254 RSC.
- J.-F. Lefebvre, J. Schindler, P. Traber, Y. Zhang, S. Kupfer, S. Gräfe, I. Baussanne, M. Demeunynck, J.-M. Mouesca, S. Gambarelli, V. Artero, B. Dietzek and M. Chavarot-Kerlidou, Chem. Sci., 2018, 9, 4152–4159 RSC.
- S.-M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS PubMed.
- K. Saito, T. Arai, N. Takahashi, T. Tsukuda and T. Tsubomura, Dalton Trans., 2006, 4444–4448 RSC.
- A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J.-J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J.-F. Nierengarten, Inorg. Chem., 2013, 52, 12140–12151 CrossRef CAS PubMed.
- C. E. McCusker and F. N. Castellano, Inorg. Chem., 2013, 52, 8114–8120 CrossRef CAS PubMed.
- M. Sandroni, M. Kayanuma, M. Rebarz, H. Akdas-Kilig, Y. Pellegrin, E. Blart, H. Le Bozec, C. Daniel and F. Odobel, Dalton Trans., 2013, 42, 14628–14638 RSC.
- C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin and N. Robertson, Inorg. Chem., 2014, 53, 10854–10861 CrossRef CAS PubMed.
- R. Czerwieniec, M. J. Leitl, H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS.
- A. J. J. Lennox, S. Fischer, M. Jurrat, S.-P. Luo, N. Rockstroh, H. Junge, R. Ludwig and M. Beller, Chem.–Eur. J., 2015, 22, 1233–1238 CrossRef PubMed.
- M. Schulz, F. Dröge, F. Herrmann-Westendorf, J. Schindler, H. Görls and M. Presselt, Dalton Trans., 2016, 45, 4835–4842 RSC.
- J. Windisch, M. Orazietti, P. Hamm, R. Alberto and B. Probst, ChemSusChem, 2016, 9, 1719–1726 CrossRef CAS PubMed.
- M. Elie, M. D. Weber, F. Di Meo, F. Sguerra, J. Lohier, R. B. Pansu, J. Renaud, H. Matthieu, M. Linares, R. D. Costa and S. Gaillard, Chem.–Eur. J., 2017, 23, 16328–16337 CrossRef CAS PubMed.
- B. Hupp, C. Schiller, C. Lenczyk, M. Stanoppi, K. Edkins, A. Lorbach and A. Steffen, Inorg. Chem., 2017, 56, 8996–9008 CrossRef CAS PubMed.
- J. Kim, D. R. Whang and S. Y. Park, ChemSusChem, 2017, 10, 1883–1886 CrossRef CAS PubMed.
- K. Kubicek, S. Thekku Veedu, D. Storozhuk, R. Kia and S. Techert, Polyhedron, 2017, 124, 166–176 CrossRef CAS.
- M. D. Weber, M. Viciano-Chumillas, D. Armentano, J. Cano and R. D. Costa, Dalton Trans., 2017, 46, 6312–6323 RSC.
- S. Garakyaraghi, C. E. McCusker, S. Khan, P. Koutnik, A. T. Bui and F. N. Castellano, Inorg. Chem., 2018, 57, 2296–2307 CrossRef CAS PubMed.
- D. Hayes, L. Kohler, R. G. Hadt, X. Zhang, C. Liu, K. L. Mulfort and L. X. Chen, Chem. Sci., 2018, 9, 860–875 RSC.
- S. Keller, F. Brunner, J. M. Junquera-Hernández, A. Pertegás, M. La-Placa, A. Prescimone, E. C. Constable, H. J. Bolink, E. Ortí and C. E. Housecroft, ChemPlusChem, 2018, 83, 217–229 CrossRef CAS.
- K. Soulis, C. Gourlaouen, C. Daniel, A. Quatela, F. Odobel, E. Blart and Y. Pellegrin, Polyhedron, 2018, 140, 42–50 CrossRef CAS.
- A. Stoïanov, C. Gourlaouen, S. Vela and C. Daniel, J. Phys. Chem. A, 2018, 122, 1413–1421 CrossRef PubMed.
- S.-P. Luo, E. Mejía, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2012, 52, 419–423 CrossRef PubMed.
- A. C. Hernandez-Perez and S. K. Collins, Acc. Chem. Res., 2016, 49, 1557–1565 CrossRef CAS PubMed.
- O. Reiser, Acc. Chem. Res., 2016, 49, 1990–1996 CrossRef CAS PubMed.
- C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2017, 24, 2039–2058 CrossRef PubMed.
- R. D. Costa, D. Tordera, E. Ortí, H. J. Bolink, J. Schönle, S. Graber, C. E. Housecroft, E. C. Constable and J. A. Zampese, J. Mater. Chem., 2011, 21, 16108–16118 RSC.
- M. D. Weber, E. Fresta, E. M. E. Margaux ElieMargaux, J. Renaud, K. Meyer, S. Gaillard and D. C. Rubén, Adv. Funct. Mater., 2018, 28, 1707423 CrossRef.
- L. N. Ashbrook and C. M. Elliott, J. Phys. Chem. C, 2013, 117, 3853–3864 CrossRef CAS.
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386–8398 RSC.
- S. Wei, Y. Shao, X. Shi, X. Lu, K. Li, Z. Zhao, C. Guo, H. Zhu and W. Guo, Org. Electron., 2016, 29, 142–150 CrossRef CAS.
- M. Heberle, S. Tschierlei, N. Rockstroh, M. Ringenberg, W. Frey, H. Junge, M. Beller, S. Lochbrunner and M. Karnahl, Chem.–Eur. J., 2017, 23, 312–319 CrossRef CAS PubMed.
- W. Lu, D. A. Vicic and J. K. Barton, Inorg. Chem., 2005, 44, 7970–7980 CrossRef CAS PubMed.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- E. Mejía, S.-P. Luo, M. Karnahl, A. Friedrich, S. Tschierlei, A.-E. Surkus, H. Junge, S. Gladiali, S. Lochbrunner and M. Beller, Chem.–Eur. J., 2013, 19, 15972–15978 CrossRef PubMed.
- D. Moonshiram, P. Garrido-Barros, C. Gimbert-Suriñach, A. Picón, C. Liu, X. Zhang, M. Karnahl and A. Llobet, Chem.–Eur. J., 2018, 24, 6464–6472 CrossRef CAS PubMed.
- S. Garakyaraghi, P. Koutnik and F. N. Castellano, Phys. Chem. Chem. Phys., 2017, 19, 16662–16668 RSC.
- M. Iwamura, H. Watanabe, K. Ishii, S. Takeuchi and T. Tahara, J. Am. Chem. Soc., 2011, 133, 7728–7736 CrossRef CAS PubMed.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS PubMed.
- B. Schäfer, H. Görls, M. Presselt, M. Schmitt, J. Popp, W. Henry, J. G. Vos and S. Rau, Dalton Trans., 2006, 2225–2231 RSC.
- C. Kuhnt, M. Karnahl, S. Tschierlei, K. Griebenow, M. Schmitt, B. Schäfer, S. Krieck, H. Görls, S. Rau, B. Dietzek and J. Popp, Phys. Chem. Chem. Phys., 2010, 12, 1357–1368 RSC.
- R. Liu, M.-M. Huang, X.-X. Yao, H.-H. Li, F.-L. Yang and X.-L. Li, Inorg. Chim. Acta, 2015, 434, 172–180 CrossRef CAS.
- W. Villarreal, L. Colina-Vegas, G. Visbal, O. Corona, R. S. Corrêa, J. Ellena, M. R. Cominetti, A. A. Batista and M. Navarro, Inorg. Chem., 2017, 56, 3781–3793 CrossRef CAS PubMed.
- M. Martínez-Abadía, G. Antonicelli, E. Zuccatti, A. Atxabal, M. Melle-Franco, L. E. Hueso and A. Mateo-Alonso, Org. Lett., 2017, 19, 1718–1721 CrossRef PubMed.
- G. Albano, P. Belser, L. De Cola and M. T. Gandolfi, Chem. Commun., 1999, 1171–1172 RSC.
- G. Albano, P. Belser and C. Daul, Inorg. Chem., 2001, 40, 1408–1413 CrossRef CAS PubMed.
- N. Lundin, P. Walsh, S. Howell, A. Blackman and K. Gordon, Chem.–Eur. J., 2008, 14, 11573–11583 CrossRef CAS PubMed.
- Y. Zhang, M. Heberle, M. Wächtler, M. Karnahl and B. Dietzek, RSC Adv., 2016, 6, 105801–105805 RSC.
- R. Giereth, W. Frey, H. Junge, S. Tschierlei and M. Karnahl, Chem.–Eur. J., 2017, 23, 17432–17437 CrossRef CAS PubMed.
- S. Blanc, T. Pigot, C. Cugnet, R. Brown and S. Lacombe, Phys. Chem. Chem. Phys., 2010, 12, 11280–11290 RSC.
- S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P. Schwarzbach, S.-P. Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig and A. Brückner, ACS Catal., 2014, 4, 1845–1849 CrossRef CAS.
- Y. Pellegrin, M. Sandroni, E. Blart, A. Planchat, M. Evain, N. C. Bera, M. Kayanuma, M. Sliwa, M. Rebarz, O. Poizat, C. Daniel and F. Odobel, Inorg. Chem., 2011, 50, 11309–11322 CrossRef CAS PubMed.
- M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann and S. Bräse, Chem.–Eur. J., 2014, 20, 6578–6590 CrossRef CAS PubMed.
- F. Ronzani, E. Arzoumanian, S. Blanc, P. Bordat, T. Pigot, C. Cugnet, E. Oliveros, M. Sarakha, C. Richard and S. Lacombe, Phys. Chem. Chem. Phys., 2013, 15, 17219–17232 RSC.
- L. E. Manring, C. L. Gu and C. S. Foote, J. Phys. Chem., 1983, 87, 40–44 CrossRef CAS.
- H. Junge, N. Rockstroh, S. Fischer, A. Brückner, R. Ludwig, S. Lochbrunner, O. Kühn and M. Beller, Inorganics, 2017, 5, 14 CrossRef.
- I. Reim, B. Wriedt, Ü. Tastan, D. Ziegenbalg and M. Karnahl, ChemistrySelect, 2018, 3, 2905–2911 CrossRef CAS.
- Y. Zhang, P. Traber, L. Zedler, S. Kupfer, S. Gräfe, M. Schulz, W. Frey, M. Karnahl and B. Dietzek, Phys. Chem. Chem. Phys., 2018, 20, 24843–24857 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General information on the experimental and synthetic details, crystallographic data, NMR and UV-vis spectra, cyclic voltammograms as well as results of the (TD-)DFT calculations. CCDC 1864076 and 1864077. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8se00521d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |