
FeS2 microspheres wrapped by N-doped rGO from an Fe-based ionic liquid precursor for rechargeable lithium ion batteries†
 *a
and
Xiaoying
Huang
a
*a
and
Xiaoying
Huang
a
Abstract
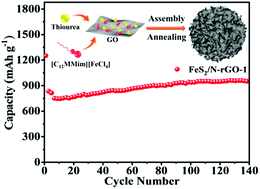
Earth-abundant pyrite (FeS2) is a promising anode material for lithium ion batteries (LIBs) because of its high theoretical specific capacity (894 mA h g−1). However, LIBs using pristine FeS2 usually suffer from volume expansion, dissolution of polysulfides, and low conductivity of Li2S. Herein, FeS2/N-doped reduced graphene oxide microspheres (FeS2/N-rGO) are first synthesized from an Fe-based ionic liquid, [C12MMim]FeCl4 (C12MMim = 1-dodecyl-2,3-dimethylimidazolium), which can not only be used as the metal and nitrogen source but also as an assembly medium and surfactant. As the anode material for rechargeable LIBs, the as-obtained FeS2/N-rGO composites display a specific capacity of 950 mA h g−1 after 140 cycles at a current density of 150 mA g−1 and deliver an average reversible discharge capacity of 973, 867, 778, and 671 mA h g−1 at 0.2, 0.5, 1.0, and 2.0 A g−1, respectively. Even at high current density, the specific capacity can still reach 510 mA h g−1. More importantly, after deep cycling, a high reversible capacity of 973 mA h g−1 can still be recovered when the current density reduced to 0.2 A g−1. This excellent stability and outstanding rate performance are mainly attributed to the suppression of dissolution of polysulfide intermediates and volume expansion by the conductive N-doped rGO matrix.
- This article is part of the themed collection: 2018 Sustainable Energy and Fuels HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

