
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction mechanism, norbornene and ligand effects, and origins of meta-selectivity of Pd/norbornene-catalyzed C–H activation†
Tao
Yang
 *a,
Chuncai
Kong
a,
Shengchun
Yang
a,
Zhimao
Yang
a,
Sen
Yang
a and
Masahiro
Ehara
*bc
*a,
Chuncai
Kong
a,
Shengchun
Yang
a,
Zhimao
Yang
a,
Sen
Yang
a and
Masahiro
Ehara
*bc
aSchool of Science, MOE Key Laboratory for Non-Equilibrium Synthesis and Modulation of Condensed Matter, Xi'an Jiaotong University, Xi'an 710049, Shaanxi, China. E-mail: taoyang1@xjtu.edu.cn
bResearch Center for Computational Science, Institute for Molecular Science, Nishigonaka 38, Myodaiji, Okazaki 444-8585, Japan. E-mail: ehara@ims.ac.jp
cElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Kyoto 615-8520, Japan
First published on 23rd October 2019
Abstract
The reaction mechanism, ligand and norbornene effects, and origins of meta-selectivity in Pd/norbornene-catalyzed alkylation and arylation via C–H activation are theoretically elucidated by DFT computation. The reaction proceeds through six major steps: ortho-C–H activation, norbornene insertion into Pd–C bonds, meta-C–H activation, meta-C–C bond formation, β-carbon elimination, and protodemetallation. Both ortho-C–H and meta-C–H activations undergo a concerted metalation–deprotonation pathway. The meta-C–C bond formation, which is the selectivity-determining step, follows a Pd(IV) pathway via oxidative addition on a Pd(II) five-membered-ring intermediate. The oxidative addition of alkyl iodide adopts an SN2 pathway, whereas aryl iodide prefers concerted oxidative addition rather than the SN2 pathway. The Pd(II) pathway via meta-C–C reductive coupling on dinuclear palladium species is not the dominant pathway because of the low concentration of Pd(0)L2. For methylation, with norbornene and pyridine (or its derivative) as ligands, the C–C reductive coupling and C–C reductive elimination from the Pd(IV) intermediate are found to be the selectivity-determining steps for meta-functionalization and benzocyclobutene formation, respectively, whereas the use of large ligands such as acridine and quinoline-type ligands (L1 and L2) moves the selectivity-determining step back to the oxidative addition and the C–C reductive elimination steps on the Pd(II) intermediate. The geometric and electronic properties of L1 and L2 further suppress the benzocyclobutene formation by increasing the energy difference between meta-functionalization and benzocyclobutene formation. The combination of 2-carbomethoxynorbornene and L2 promotes meta-ethylation and -arylation by disfavoring the C–C reductive elimination steps from the Pd(II) and Pd(IV) intermediates as well as slightly favoring the oxidative addition step.
Introduction
Direct C–H functionalization, which allows the conversion of C–H bonds to C–C or C–X bonds, has attracted wide interest as a powerful synthesis methodology in the past few decades.1 Highly selective C–H functionalization of arenes which occurs via activation of the inert C–H bond can be utilized for various promising applications in many fields including pharmaceutical, agrochemical and materials industries.2 However, achieving site selective C–H functionalization of arenes is still challenging, especially in the development of synthetically useful remote arene C–H activation. In particular, meta- and para-selective functionalizations have emerged as important targets very recently, because ortho-C–H functionalization methods have been extensively exploited.2,3 Up to now, there are a limited number of available transition-metal-catalyzed meta-C–H functionalizations such as steric-controlled formal meta-C–H functionalization,4meta-C–H functionalization via traceless directing groups,5 chelation-assisted meta-C–H arylation,6 and meta-C–H functionalization utilizing nitrile-containing templates.7Inspired by the Catellani reaction,8,9 Yu’s group developed a new approach for meta-selective C–H functionalization of arenes containing an amide-directing group (phenyl acetamides) by employing norbornene (N1) and a Pd(II) catalyst with pyridine-based ligand L1 synthesized by their group (catalytic system A),10 as shown in Scheme 1. The use of this catalyst switches ortho-selectivity to meta-selectivity and achieves the highly selective meta-alkylation and -arylation. More interestingly, modifying the norbornene (N1) to 2-carbomethoxynorbornene (N2) and changing the ligand L1 to a quinoline-based ligand L2 (catalytic system B), Yu and coworkers significantly improved the efficiency and scope of this meta-selective C–H functionalization.11 For example, only functionalization of aryl iodides with either an ortho-coordinating group or multiple electron-withdrawing substituents could be achieved with catalytic system A, but the use of catalytic system B could overcome this limitation and a broad range of aryl iodides can be employed for the meta-arylation protocol. This synthesis strategy has been successfully extended to meta-C–H chlorination, amination, and alkynylation.12 Almost simultaneously, Dong and coworkers reported a similar Pd/norbornene-catalyzed meta-arylation reaction with dimethylamine as the directing group and AsPh3 as the ligand, which is also effective with the aryl iodides with ortho-coordinating groups.13 Very recently, Yu’s group further extended this approach and successfully realized remote enantioselective meta-C–H arylation and alkylation by using Pd and chiral 2-carbomethoxynorbornene.14
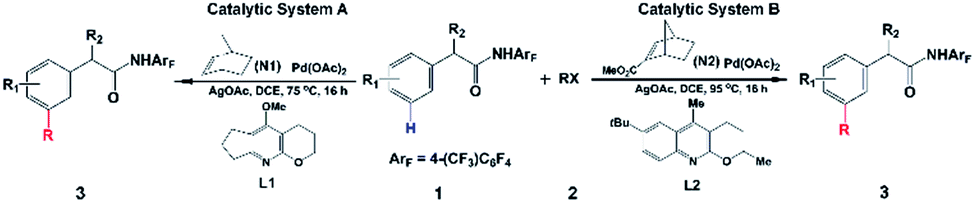 | ||
| Scheme 1 Pd/norbornene-catalyzed meta-selective C–H activation of arenes.9,10 | ||
As shown in Scheme 2, the amide-directed ortho-C–H activation, forming the ortho-palladacycle I, was proposed to be the first step of the meta-C–H functionalization of phenyl acetamides. Then, norbornene inserts into the Pd–C bond of I followed by meta-C–H activation, producing palladacycle II. Subsequent reaction of II with a coupling partner (alkyl or aryl iodides) affords palladacycle IV with a functionalized meta position via intermediate III. This step, which is known as the key pathway of the Catellani process, usually contains oxidative addition, reductive elimination and β-carbon elimination. Final protodemetallation of the aryl–palladium bond delivers the desired product. Because of the presence of intermediates I, II, and IV, the side reactions including ortho-C–H functionalization, ortho,meta-C–H difunctionlization and C–C reductive elimination of palladacycle II to form benzocyclobutene can also take place. However, with the aid of the pyridine-based ligand L1 or L2, ortho-functionalized and ortho,meta-difunctionalized products were not observed experimentally, and the benzocyclobutene formation was minimized.
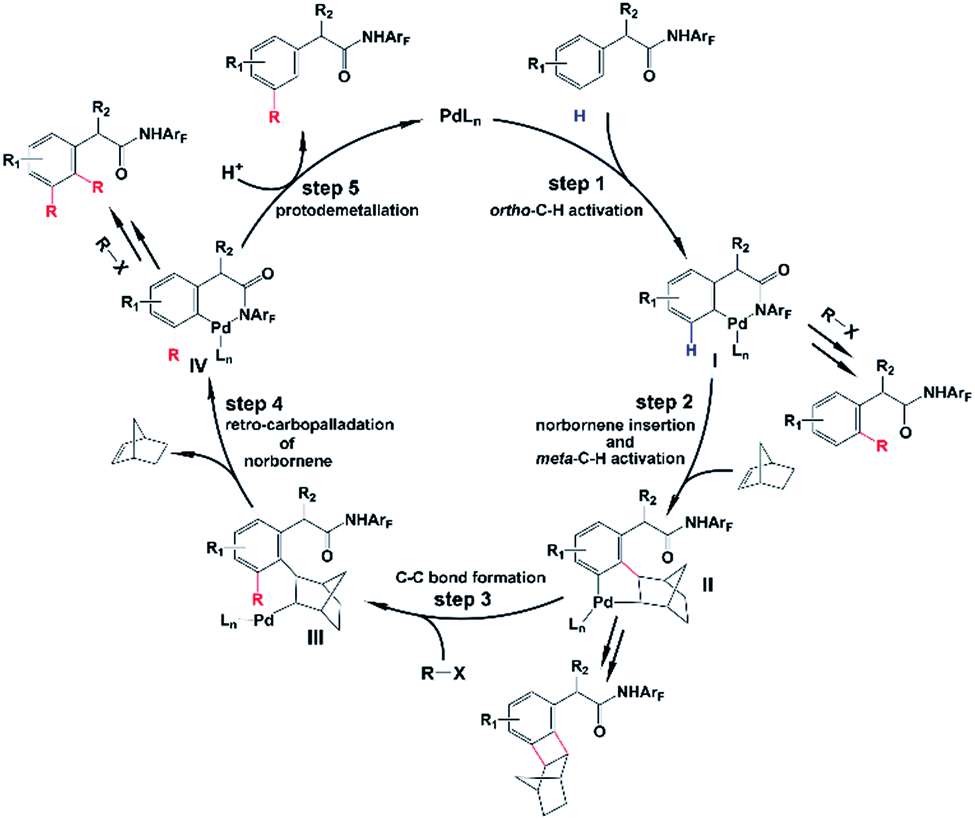 | ||
| Scheme 2 Proposed mechanism for meta-selective C–H activation catalyzed by the Pd/norbornene system. | ||
Even though the Pd/norbornene-catalyzed meta-C–H functionalization of phenyl acetamides has been well utilized experimentally, important questions have been unsolved. For example, the reaction mechanism is not clear, especially the transformation step from palladacycle II to III is completely uncertain. There are two different pathways proposed in the literature, the Pd(IV) pathway and Pd(II) pathway (also known as the transmetalation15 pathway) which was proposed by Cardenas and Echavarren,16 as shown in Scheme 3. The reaction pathway significantly depends on the reactant species. Previous mechanistic studies demonstrated that the Pd/norbornene-catalyzed reaction of metallacycles II with alkyl halides follows the Pd(IV) pathway via oxidative addition.8a,17,18 However, for the aryl–aryl coupling, those two pathways could compete. By using an ethylene bridge to replace the norbornene unit, Cardenas and Echavarren performed theoretical calculations on simplified systems and revealed that the Pd(IV) pathway is favored over the Pd(II) pathway, which was also found by Wu and Lei.16,19 Catellani and Derat theoretically confirmed that the intermediate palladacycle without ortho-substitution preferably undergoes the transmetalation pathway, whereas these palladacycles could probably undergo oxidative addition of an aryl halide when an ortho-substituent is present.20 Moreover, the origins of selectivity of meta-C–H functionalization over other side reactions are not disclosed. Experimentalists need the knowledge about how much the ligand and norbornene influence the selectivity and scope of this reaction. However, the knowledge and answers for all these questions have not been presented at all.
 | ||
| Scheme 3 Proposed pathways of the C–C bond formation step. | ||
The theoretical answers to the above open questions will undoubtedly deepen the understanding of the title reaction, assist in the design of more efficient and reactive norbornene and ligands, and promote further development of the meta-selective C–H functionalization methodology. In the present study, we performed theoretical calculations to elucidate the reaction mechanism, origins of selectivities, and effects of norbornene and ligands on palladium/norbornene-catalyzed amide-directed meta-selective C–H alkylation and arylation of phenyl acetamides.
Computational methods
All the structures were optimized in the gas phase by employing the ωB97XD functional21 using the effective core potentials (Lanl2dz)22 and basis sets for Ag, Pd and I and the 6-31G(d) basis set for all the other atoms. Frequency calculations were performed at the same level of theory to obtain zero-point vibrational energy and thermal corrections at three different temperatures, 348.15, 363.15, and 368.15 K, which were used experimentally. In the test calculations, the M06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and B3LYP-D3 (Becke–Johnson damping, BJ)24 results are very close to the ωB97XD ones.25 Single-point energy calculations were carried out at the M06/SDD26-6-311++G(d,p) level, using the gas-phase optimized geometries, to evaluate electronic energies. This type of theoretical method has been frequently used in previous theoretical studies on Pd-catalyzed C–H activations.27 Solvation free energy was calculated by using the SMD28 model with two experimentally employed solvents, 1,2-dichloroethane (DCE) and trifluorotoluene (PhCF3).29 The partial charges were obtained from natural bond orbital (NBO)30 analysis at the B3LYP/SDD-6-311++G(d,p)-SMD level. All theoretical calculations were conducted by employing the Gaussian 09 program.31 Molecular structures were illustrated with CYLView.32
23 and B3LYP-D3 (Becke–Johnson damping, BJ)24 results are very close to the ωB97XD ones.25 Single-point energy calculations were carried out at the M06/SDD26-6-311++G(d,p) level, using the gas-phase optimized geometries, to evaluate electronic energies. This type of theoretical method has been frequently used in previous theoretical studies on Pd-catalyzed C–H activations.27 Solvation free energy was calculated by using the SMD28 model with two experimentally employed solvents, 1,2-dichloroethane (DCE) and trifluorotoluene (PhCF3).29 The partial charges were obtained from natural bond orbital (NBO)30 analysis at the B3LYP/SDD-6-311++G(d,p)-SMD level. All theoretical calculations were conducted by employing the Gaussian 09 program.31 Molecular structures were illustrated with CYLView.32
Results and discussion
Several pyridine-based ligands have been used experimentally.10,11 When pyridine (Py) was used as the ligand, both the meta-functionalized product and benzocyclobutene were produced with the same yield. The ligands L1 and L2 were found experimentally to be better than pyridine and to significantly improve the yield of the meta-functionalized product. However, to clarify the possible competing pathways of catalytic cycles, pyridine is used as a ligand in calculations here, to save computation time.1. Initiation and substrate binding
In 2014 Yu’s group reported a pyridine-based ligand-promoted C(sp3)–H activation and obtained a five-membered-ring cyclopalladation intermediate as crystals,33 analogous to the six-membered-ring cyclopalladacycle I. Theoretical calculations by Wang et al. also confirmed that the catalytic cycle involving the five-membered-ring cyclopalladation intermediate is formed from square planar Pd(II) bis-ligated complexes.34 On the basis of those results, both trans and cis square planar Pd(II) di-pyridine complexes, which are formed from precatalyst Pd3(OAc)6, have been proposed, as shown in Fig. 1. Though the formation of the cis-form 2 is not exergonic, we will not rule out any of them in this stage.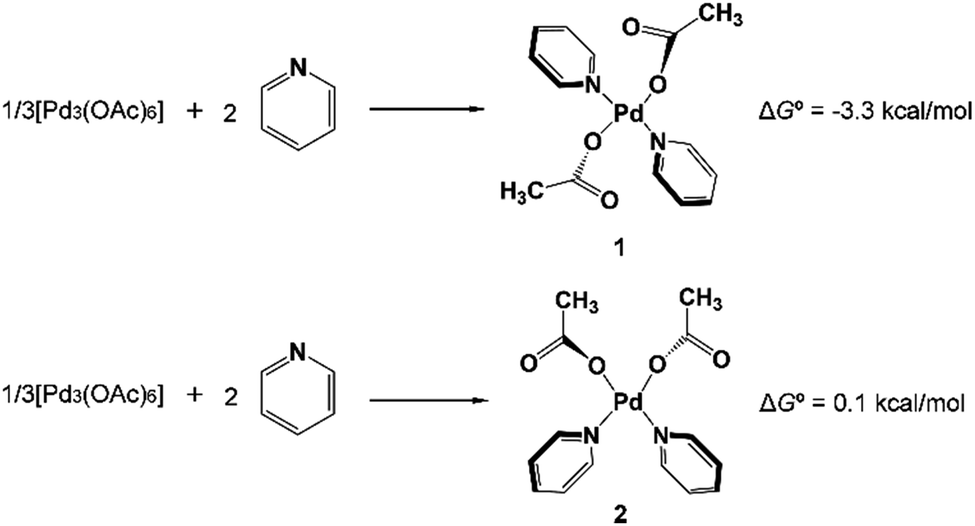 | ||
| Fig. 1 Formation of the most stable trans-PdPy2(OAc)2 (1) and cis-PdPy2(OAc)2 (2). | ||
The N-2,3,5,6-tetrafluoro-4-trifluoromethylphenylamide substrate (1a), which was derived from phenylacetic acid, was applied experimentally to the investigation of the ligand effect. Because the deprotonation reaction takes place with ΔG° = −4.6 kcal mol−1, as shown in Scheme 4, three possible coordination modes such as a charge-neutral N-donor amide (1a), a deprotonated N-donor amidate anion (1a′) and, its resonance form, an O-donor imidate (1a′′) are examined here. In a pre-catalyst 3 that is the isomer of the pre-catalyst 1, the substitution of acetate for 1a′ occurs through a transition state TS4 with a Gibbs activation energy (ΔG°‡) of 25.3 kcal mol−1 and a Gibbs reaction energy (ΔG°) of 3.1 kcal mol−1, as shown in Fig. 2, to afford Pd(OAc)(1a′)(Py)24. However, the substitution reaction of 1a and 1a′′ needs higher activation energy, indicating that 1a′ is more reactive to form an active species than 1a and 1a′′; see Fig. S1 and S2 in the ESI.†
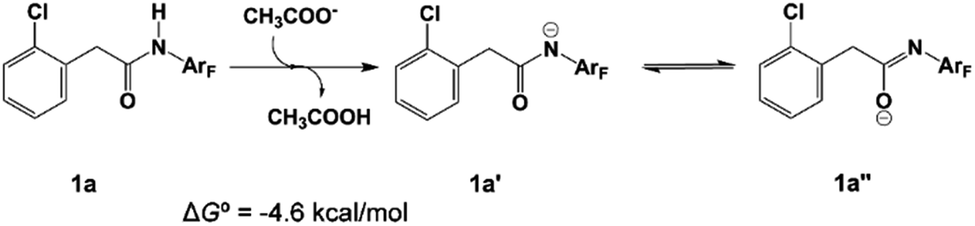 | ||
| Scheme 4 The deprotonation reaction. | ||
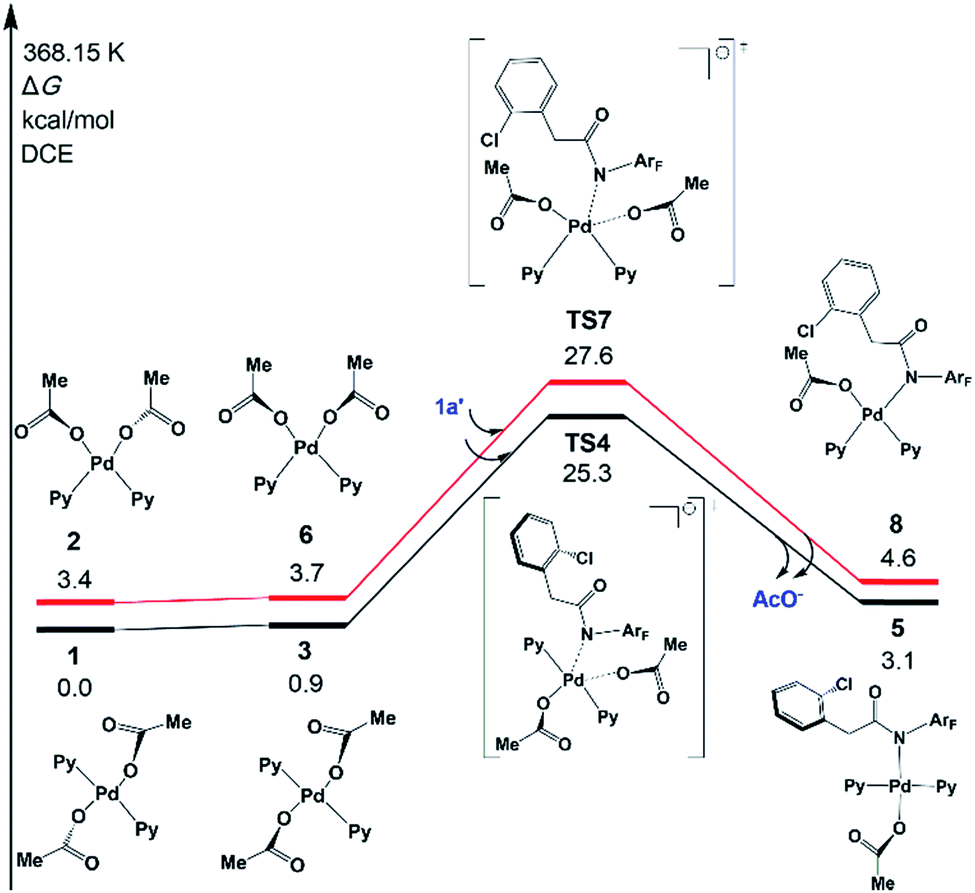 | ||
| Fig. 2 Gibbs free energy profiles for the substitution reactions of 1 and 2 with 1a′. Both trans- and cis-Pd(Py)2(OAc)2 pathways are included. | ||
A similar reaction that occurs from less stable cis complex 2 proceeds via a higher-energy transition state TS7 (ΔG°‡ = 27.6 kcal mol−1) to afford complex 8 (ΔG° = 4.6 kcal mol−1), indicating that 5 is preferably formed over 8. Further examination revealed that through releasing one 2-pyridine ligand, both 5 and 8 could lead to the formation of the same bidentate carboxylate complex, which could undergo the ortho-C–H activation (Fig. S3†). The reaction mechanism that starts from 5 will be discussed in the following section.
2. ortho-C–H activation
Although the concerted metalation–deprotonation (CMD)35 mechanism which is assisted by the acetate has been demonstrated to be a main C–H activation pathway, several other possible pathways were proposed in the previous mechanistic studies, including σ-bond metathesis and oxidative addition of the C–H bond.7c We investigated those three pathways and two new possible pathways, an AcOH-assisted CMD mechanism, in which AcOH plays a role in proton shuttle, and direct ortho-C–H activation via the CMD pathway, in which 1a does not coordinate with Pd of 1 but reacts directly with ortho-C–H of 1 (see Fig. S4 in the ESI†).The original CMD pathway was found to be the most favored mechanism among the five possible pathways, which agrees with previous mechanistic studies on palladium(II) acetate-catalyzed C–H activation reactions.35 It is noteworthy that the direct CMD between complex 1a and trans-Pd(Py)2(OAc)2 occurs at the ortho-C–H bond with a moderately higher activation energy than that of the original CMD pathway, indicating that the auxiliary additive might not be necessary for the reaction.36 Very recently, Yu’s group realized Pd/N2-catalyzed auxiliary-free meta-C–H arylation of phenylacetic acids with a pyridine-type ligand,12f which is consistent with the present calculation results.
Fig. 3 depicts the most favored CMD pathway of ortho-C–H activation with the Gibbs energy profile starting from 5. The pathway starts from the attack of carboxylate oxygen on the Pd center and the dissociation of one pyridine ligand (TS9) occurs, resulting in a bidentate acetate intermediate 10. Then, 10 isomerizes to an intermediate 11, in which the ortho-C–H approaches the Pd center. No transition state has been located between 10 and 11. Starting from 11, the ortho-C–H bond of the phenyl group is activated and cleaved with the assistance of the bound acetate viaTS12 to afford an intermediate 13 with acetic acid being weakly bound to the Pd center. Further ligand exchange yields the six-membered-ring cyclopalladacycle 14. ortho-C–H activation viaTS12 needs the highest ΔG°‡ in the above steps but it is attainable at an energy of 24.8 kcal mol−1. As shown in Fig. 4, the Pd–C distance in TS12 becomes 2.121 Å and the H atom is transferred from the ortho-C to the acetate with C–H and O–H distances of 1.321 and 1.347 Å, respectively.
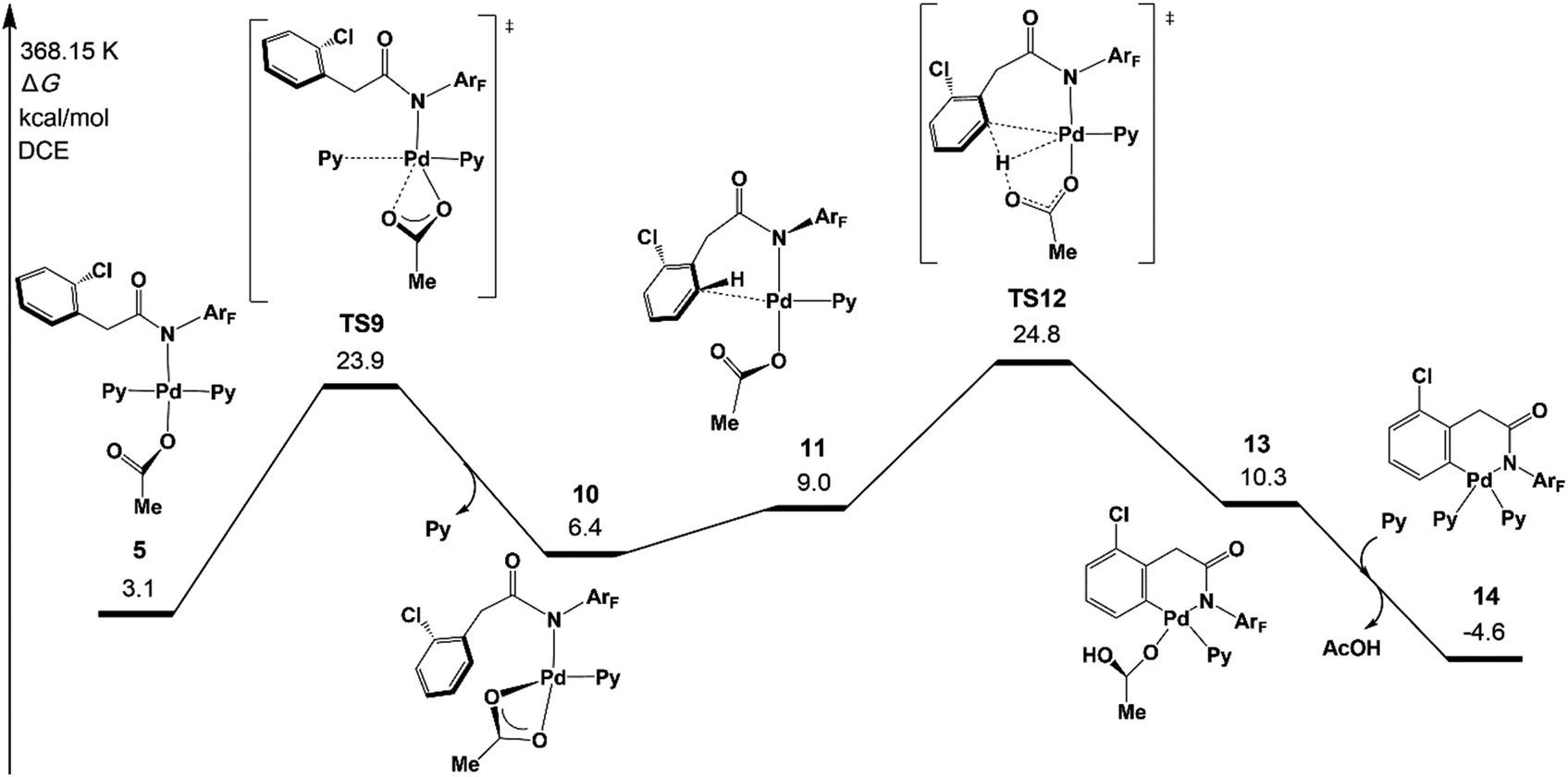 | ||
| Fig. 3 Gibbs free energy profile for the concerted metalation–deprotonation (CMD) pathway of ortho-C–H activation. | ||
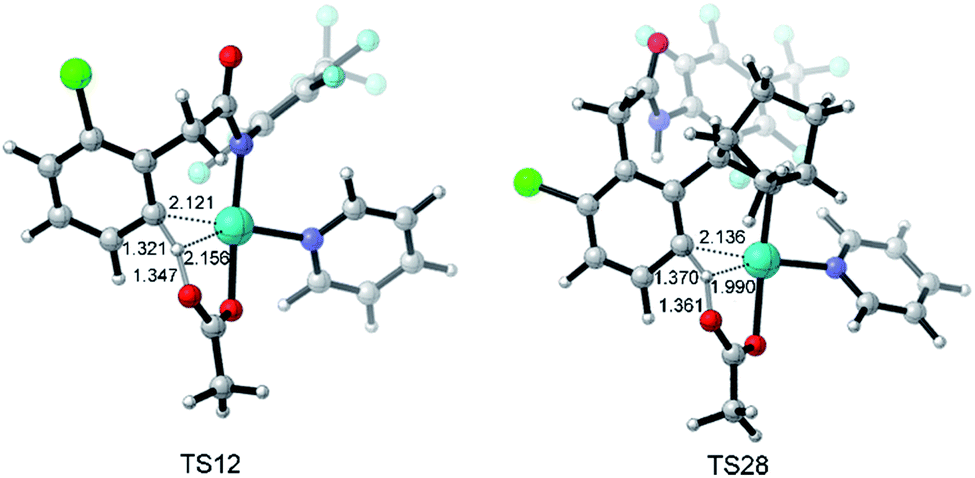 | ||
| Fig. 4 Optimized structures of transition states TS12 and TS28 with selected distances given in angstroms. | ||
3. Norbornene insertion and meta-C–H activation
As shown in Fig. 5, there are two key steps in norbornene insertion. The first key step is to provide an empty coordination site for norbornene. This process occurs via the protonation reaction of 14, in which the weakly bound acetic acid protonates the amide auxiliary with the Pd−N bond scission (TS16). The protonation reaction affords an intermediate 17, in which a weak interaction exists between Pd and N atoms. Because the Pd–N bond in 17 is weaker than in 14, the NHArF group easily dissociates from Pd followed by norbornene coordination to yield an intermediate 18. The second key step is norbornene insertion via transition state TS19, which is the insertion of the norbornene double bond into the Pd–Ph bond to form an intermediate 20. Then, 20 isomerizes to an intermediate 21.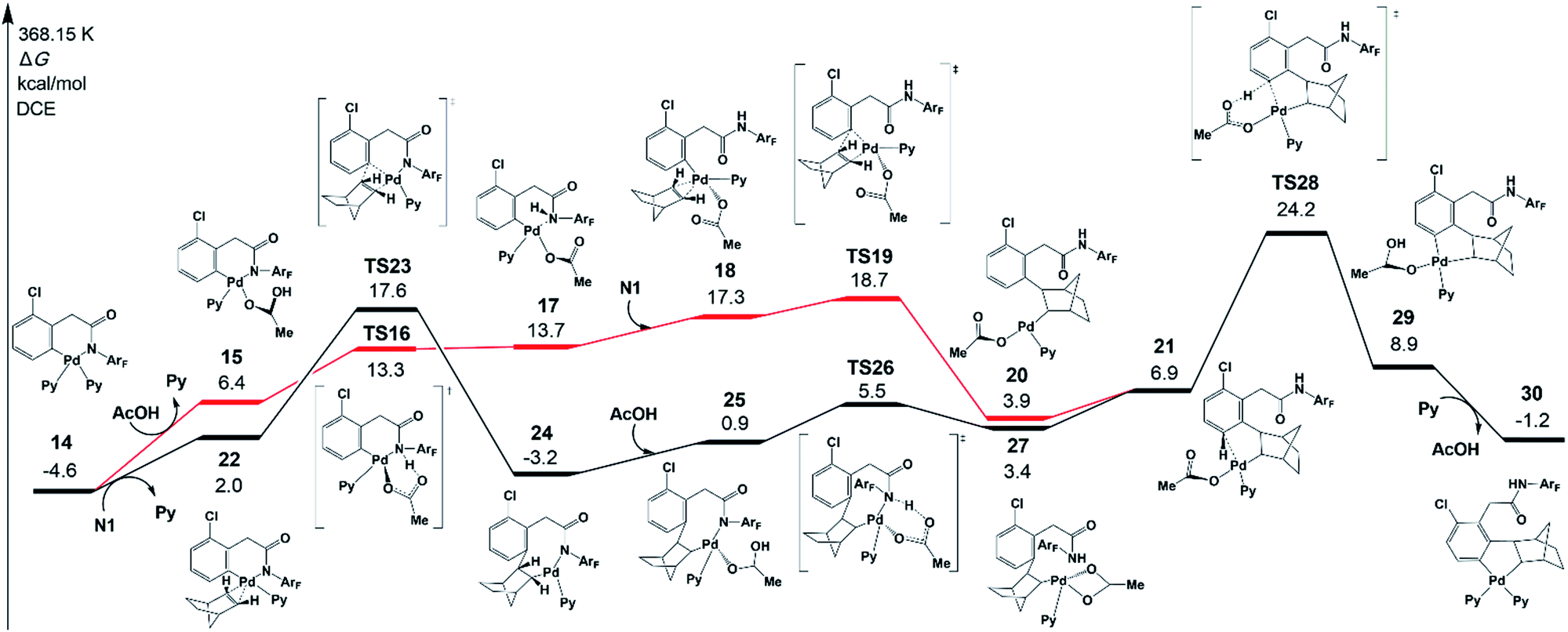 | ||
| Fig. 5 Gibbs free energy profile for the norbornene insertion. | ||
The highest-energy point in this pathway is TS19, which is 18.7 kcal mol−1 in energy. As shown by the black line in Fig. 5, there is another pathway that starts from norbornene insertion into the Pd–Ph bond viaTS23, leading to intermediate 24. After the coordination of AcOH to the Pd center (25), the protonation reaction takes place viaTS26 and the intermediate 27 is formed, followed by the isomerization to 21. The highest-energy point in this pathway is norbornene exo-migratory insertion viaTS23 (17.6 kcal mol−1),38 revealing that this pathway is preferred. The exo-migratory insertion agrees with the experimental observation.11
Fig. 5 also presents the CMD pathway of meta-C–H activation, TS28, in which the meta-C–H bond cleavage occurs with the aid of the acetate, with an activation energy of 24.2 kcal mol−1. Then, a five-membered-ring cyclopalladation intermediate 29 with weakly bound acetic acid is afforded. Further ligand exchange generates intermediate 30.37 Several other possible meta-C–H activation pathways, such as the oxidative addition of the C–H bond and an AcOH-assisted CMD pathway, have also been examined (Fig. S5†). However, they need larger activation energy, compared with the CMD pathway shown in Fig. 5. As shown in Fig. 4, the Pd–Hmeta-C distance in TS28 is 2.136 Å with C–H and O–H distances of 1.370 and 1.361 Å, respectively.
4. Carbon–carbon bond formation
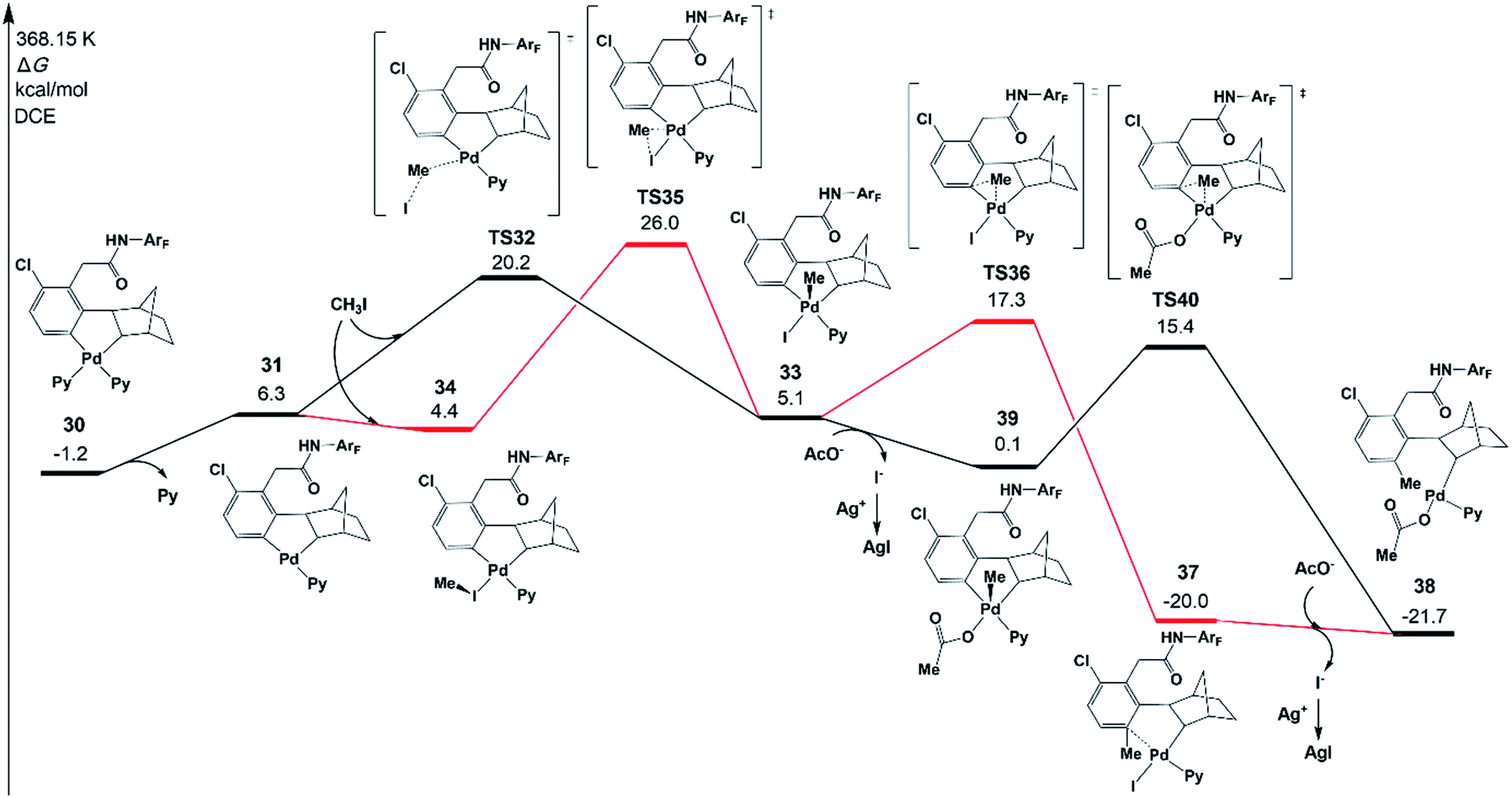
It has been demonstrated experimentally that the Pd/norbornene-catalyzed reaction of alkyl halides follows the Pd(IV) pathway via oxidative addition.8a,c,17 Further mechanistic study by Lautens and coworkers has proved that the oxidative addition prefers an SN2 pathway over concerted oxidative addition.18 A recent experimental/theoretical study on a selective Pd/norbornene-catalyzed trifluoroethylation reaction of indoles by Liu, Lan, and coworkers also supported the SN2 mechanism for the oxidative addition step.39 In the case of Pd/norbornene-catalyzed arylation, two possible pathways have been considered for the Pd/norbornene-catalyzed C–C bond formation step: the Pd(II) and Pd(IV) pathways, as depicted in Scheme 3. The Pd(IV) pathway starts with concerted oxidative addition of an aryl halide to the Pd(II) center, followed by the C–C reductive coupling. The Pd(II) pathway undergoes transmetalation with another Pd(II) complex, leading to the formation of dinuclear palladacycle intermediate VI. Subsequent C–C reductive elimination leads to the formation of an intermediate III with a new C–C bond.Two possible Pd(IV) pathways starting from five-membered-ring cyclopalladation 30 are investigated first in the present study, as shown in Fig. 6. The ligand dissociation from 30 results in an intermediate 31 with an unsaturated coordination site. The SN2 mechanism takes place via transition state TS32 with a ΔG°‡ of 20.2 kcal mol−1, resulting in a Pd(IV) five-membered-ring intermediate 33. In the case of the concerted oxidative addition pathway, the unsaturated coordination site near the meta-carbon is coordinated by CH3I to form an intermediate 34, and then, oxidative addition occurs via a three-membered-ring type oxidative addition transition state TS35 (26.0 kcal mol−1), leading to 33. Therefore, the SN2 mechanism is preferred over concerted oxidative addition.40
 | ||
| Fig. 6 Gibbs free energy profile for the possible Pd(IV) pathways of the C–C bond formation step. | ||
If the intermediate 33 undergoes the C–C reductive coupling first via transition state TS36 (17.3 kcal mol−1) to form an intermediate 37, in which the meta-C is functionalized by the methyl group, the abstraction of iodide from 37 by silver(I) will take place and acetate is bound on the Pd center, generating an intermediate 38. However, the abstraction of iodide could occur first from 33, generating an intermediate 39 with an acetate. The subsequent C–C reductive coupling viaTS40 needs only 15.4 kcal mol−1, indicating that this pathway is favored.41
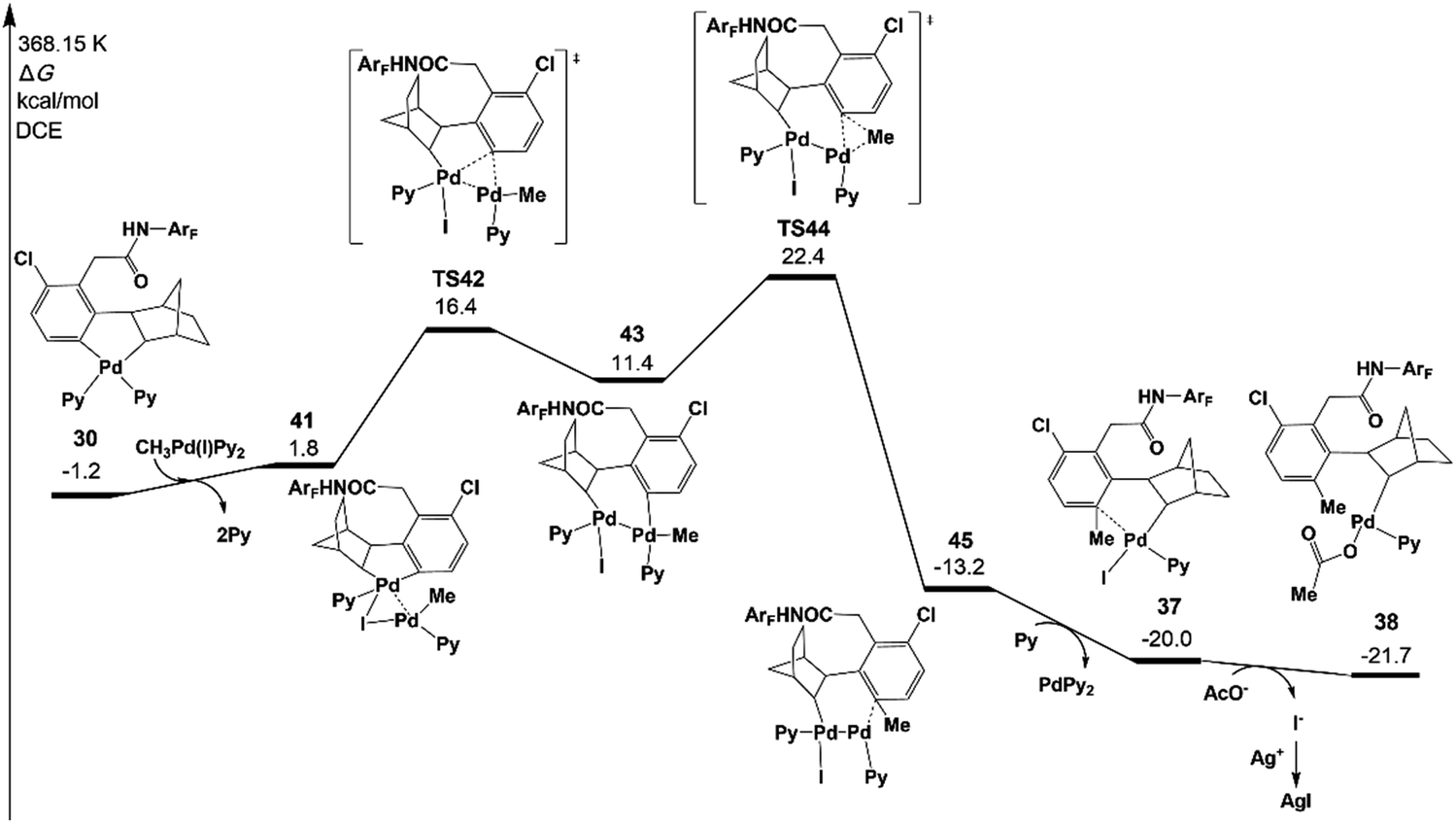
Fig. 7 presents the free energy profile of the Pd(II) pathway (the transmetalation pathway), which starts with an association intermediate 41 from CH3Pd(I)Py2 and intermediate 30. Then, the meta-C transfers from the first palladium atom of 41 to the second palladium atom in 43viaTS42. The reductive elimination occurs viaTS44 on the second palladium atom with an energy of 22.4 kcal mol−1.42,43
 | ||
| Fig. 7 Gibbs free energy profile for the Pd(II) pathway of the C–C bond formation step. | ||
The resulting intermediate 45 could transfer to 36via the release of Pd(0)Py2. The activation energy of the Pd(II) pathway is not so high and CH3Pd(I)Py2 could be generated from the oxidative addition of Pd(0)Py2 (Scheme 3).44 However, no Pd(0) species but only the Pd(II)(OAc)2 catalyst was added at the very beginning in the experiments. Pd(0)Py2 could be formed only if benzocyclobutene is produced (see Section 6), which is usually unfavoured and occurs at a very late stage. Thus, in the following sections, the activation energy of the Pd(II) pathway viaTS43 will be just provided and not be discussed unless the formation of Pd(0) species is preferred.
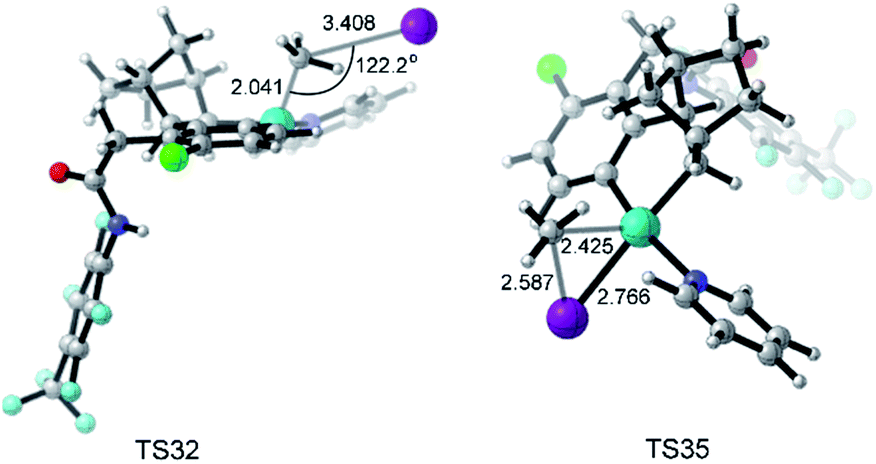
The key transition states TS32 and TS35 of SN2 and concerted oxidative addition pathways are shown in Fig. 8. TS32 is a typical SN2 transition state with a Pd–C–I bond angle of 122.2°. The concerted oxidative addition transition state TS35 has a three-membered ring with a Pd–C distance of 2.425 Å.
 | ||
| Fig. 8 Optimized structures of transition states TS32 and TS35 with selected distances given in angstroms and angles in degrees. | ||
5. β-carbon elimination and protodemetallation
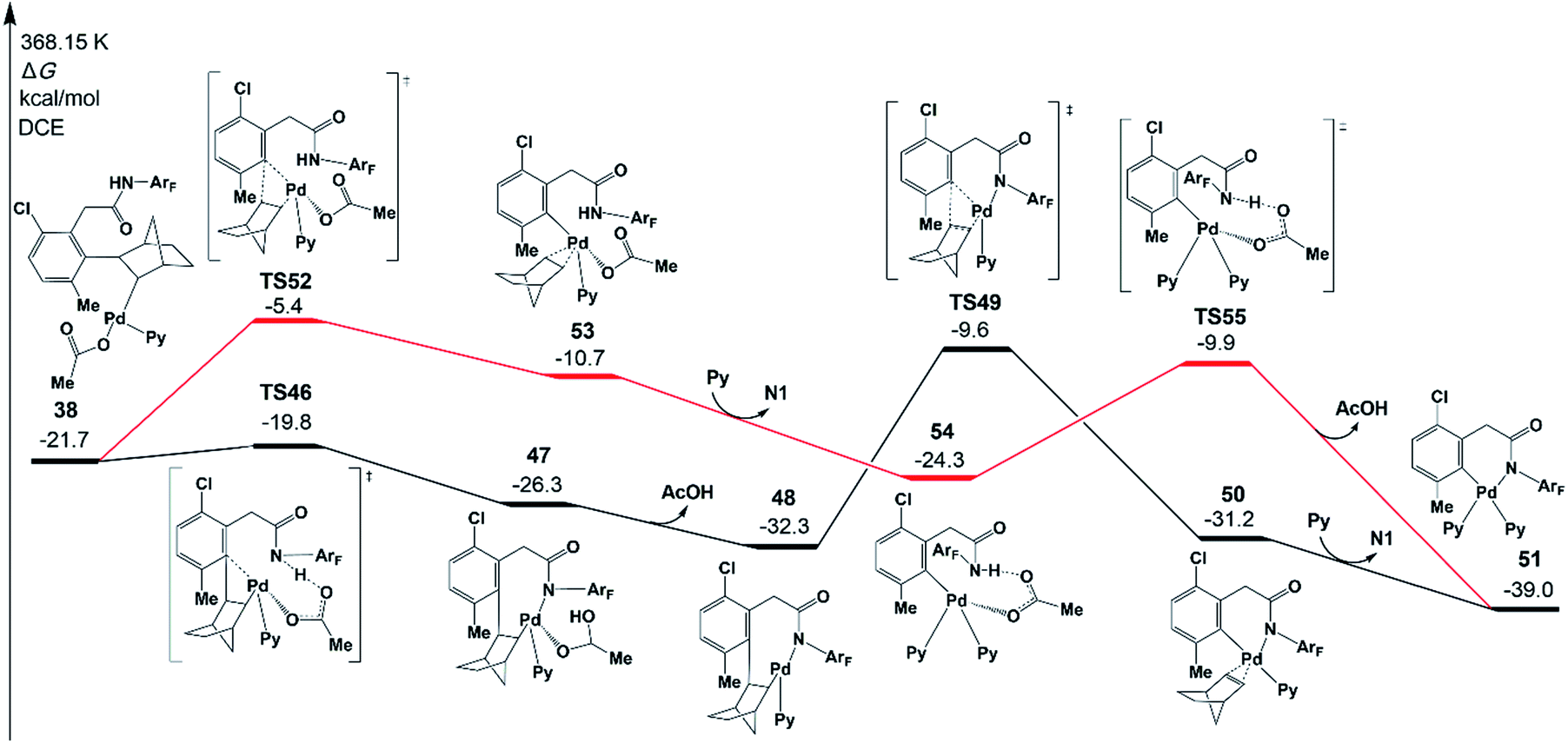
To complete the catalytic cycle, the Pd catalyst has to leave the reaction system to release norbornene and the meta-functionalized product, which occurs through β-carbon elimination and protodemetallation. We will first focus on the β-carbon elimination starting from 38. To obtain the six-membered-ring palladacycle IV (Scheme 2), there are two key steps: (a) ortho-Cnorbornene bond cleavage and (b) Pd–N bond formation. Two pathways are possible in terms of the order of those two elementary steps.In one pathway, the Pd–N bond formation takes place first, as shown in Fig. 9 (black line). Via transition state TS46, the resultant intermediate 47 was located, which is an eight-membered-ring palladacycle coordinated with acetic acid. The dissociation of acetic acid generates an intermediate 48. The second step is the β-carbon elimination viaTS49 to afford an intermediate 50. This intermediate undergoes ligand exchange to form six-membered-ring palladacycle 51. In the other pathway, as shown by the red line in Fig. 9, the ortho-Cnorbornene bond is cleaved via transition state TS52, followed by the deprotonation reaction (TS55) of the amide auxiliary. The highest energy TS in the second pathway is TS52 (−5.4 kcal mol−1), whose energy is a bit higher than that of TS49 (−9.6 kcal mol−1) involved in the first pathway. These results demonstrate that the first pathway more preferably occurs than the second one.
 | ||
| Fig. 9 Gibbs free energy profile for the two possible pathways of β-carbon elimination. | ||
The activation energy of norbornene de-insertion viaTS49 is as low as 22.7 kcal mol−1 relative to 48, suggesting that it can occur easily under the experimental conditions. On the other hand, the β-H elimination from 38 needs a much larger activation energy of 64.3 kcal mol−1. As mentioned above, the norbornene insertion viaTS23 also needs a low activation energy of 22.2 kcal mol−1 relative to 14. The easy norbornene de-insertion, along with norbornene insertion, is of high importance in completing the catalytic meta-functionalization reaction.
Fig. 10 depicts free energy profile of the protodemetallation step starting from the intermediate 51. The protonation reaction occurs on the meta-C viaTS57 (−11.6 kcal mol−1), leading to an intermediate 58. This intermediate undergoes acetic acid coordination, protonation of the amide auxiliary, and product dissociation to yield the meta-functionalized product 62 with the Pd(II) catalyst. The large activation energy of the above steps is 27.4 kcal mol−1 (TS57 relative to 51), which is acceptable under the experimental conditions.
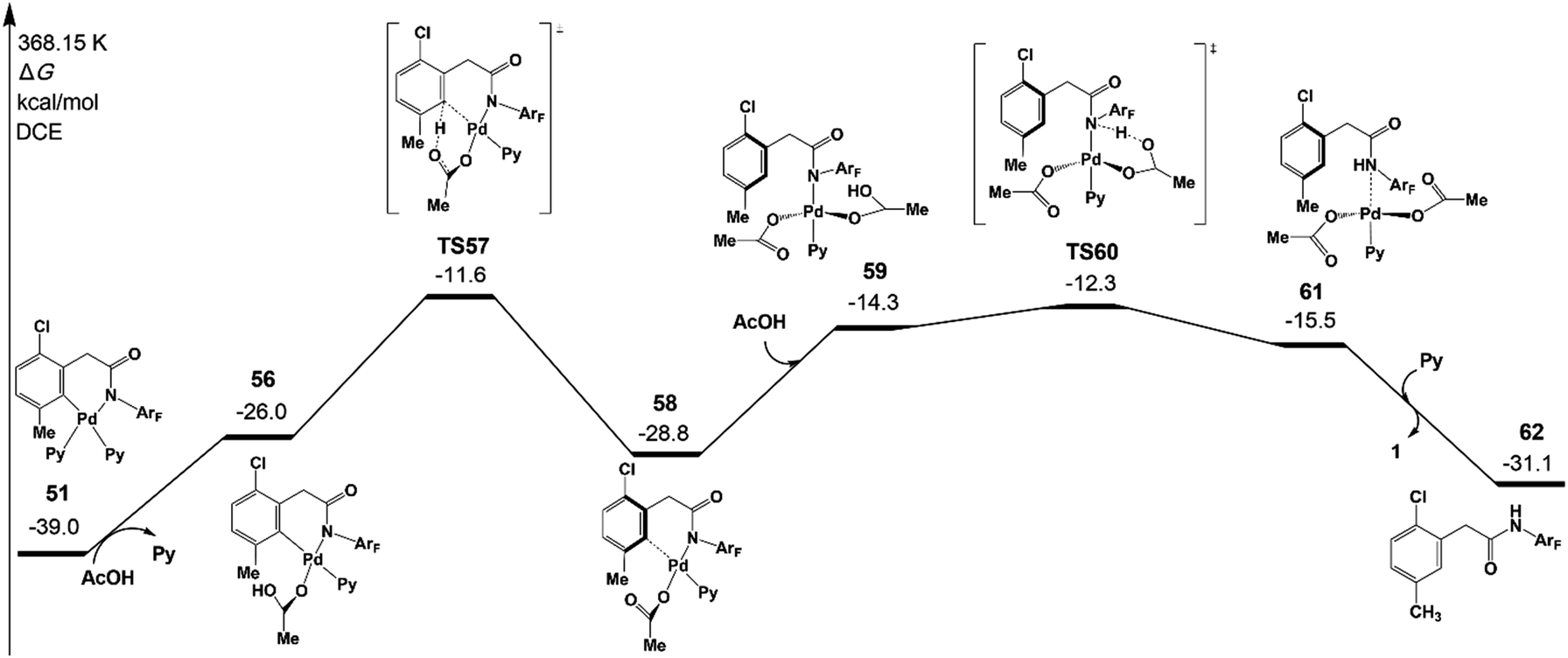 | ||
| Fig. 10 Gibbs free energy profile for the protodemetallation step of intermediate 51 leading to the meta-functionalized product 62. | ||
6. Formation pathways of three byproducts
Besides the meta-functionalized product, three other byproducts could also be generated, as shown in Scheme 2; they are benzocyclobutene, ortho-functionalized and ortho,meta-difunctionalized products. Interestingly, the meta-functionalized product and benzocyclobutene were observed experimentally with pyridine-based ligands such as pyridine, L1, and L2. To disclose the reason for this experimental result, the formation reactions of benzocyclobutene, ortho-functionalized and ortho,meta-difunctionalized products are investigated theoretically in the present work.As shown in Fig. 11, one possible reaction pathway starting from intermediate 30 is Cnorborneyl–Cphenyl bond formation via reductive elimination to afford benzocyclobutene 64. However, the reductive elimination (TS63) with two ligands (red line in Fig. 11) needs a large activation energy of 41.2 kcal mol−1 relative to 30. Then, the pathway starting from 31 after the ligand dissociation was examined. The reductive elimination from 31viaTS65 (28.2 kcal mol−1) forms a product complex of benzocyclobutene 66. The benzocyclobutene 64 will be formed when PdPy2 is dissociated. The possibility of benzocyclobutene formation through the C–C reductive elimination from 33 is also examined. Starting from 32, the C–C reductive elimination (TS67) is 19.0 kcal mol−1 in energy and results in benzocyclobutene 64 and CH3Pd(I)Py2. However, starting from the intermediate 39 that comes from the abstraction of iodide from 33, the energy barrier of C–C reductive elimination (TS69) is only 15.0 kcal mol−1, which is quite close to the activation energy of C–C reductive coupling viaTS40 (15.4 kcal mol−1), in excellent agreement with the experimental observation of the same yields of meta-functionalization and benzocyclobutene products. CH3Pd(OAc)Py2 will be generated in this pathway.
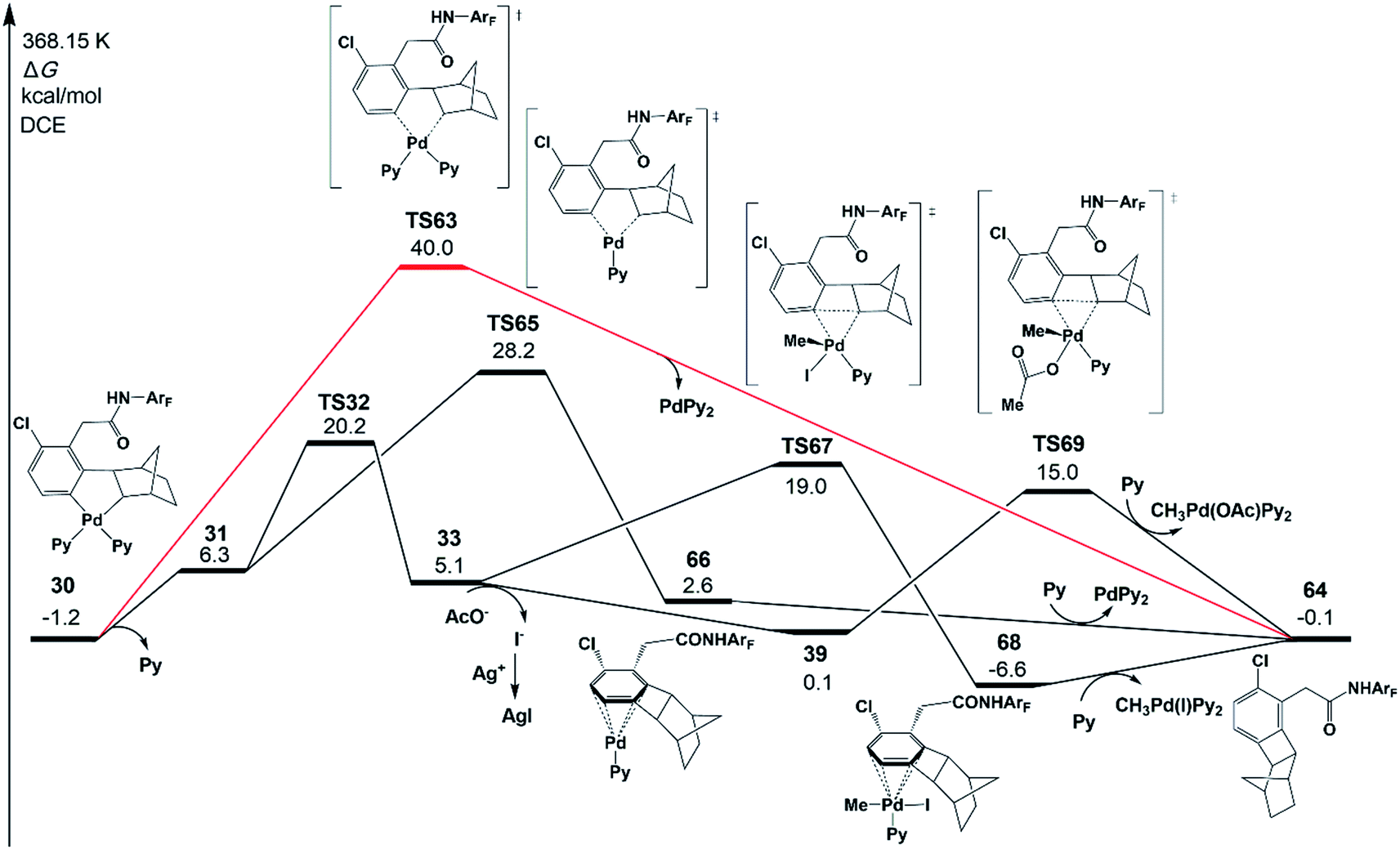 | ||
| Fig. 11 Gibbs free energy profile for the formation mechanism of the benzocyclobutene product 64 starting from intermediate 30. Only selected chemical structures are shown for clarity. | ||
Further examinations revealed that the activation energies of the formation pathways of ortho-functionalized and ortho,meta-difunctionalized products are as high as 32.7 and 38.0 kcal mol−1, respectively, suggesting that these pathways could not compete with meta-functionalization and benzocyclobutene formation, which agrees well with the experimental observation. Please see Fig. S6 and S7 in the ESI for details.†
7. Ligand and norbornene effects on the selectivity of meta-C–H alkylation (or arylation) and benzocyclobutene products
The previous experiments showed that the nature of the ligand, norbornene, and alkyl iodide significantly influence the selectivity between meta-functionalized and benzocyclobutene products.10,11 For the reaction of 1a with methyl iodide, the meta-functionalized product is more favored than benzocyclobutene when N1 is used as norbornene and either L1 or L2 is used as the ligand, but this catalytic system is not effective with ethyl iodide. Further modification of N1 to N2 favors the ethylation pathway over the cyclobutene pathway, where L2 is used as the ligand and 1b (Scheme 5) is used as the amide substrate. The presence of N2 also favors other meta-alkylation products using various alkyl iodides.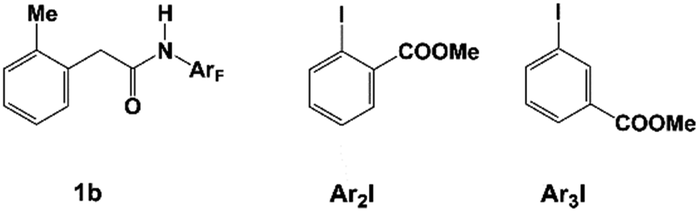 | ||
| Scheme 5 Amide substrate (1b), methyl 2-iodobenzoate (Ar2I), and methyl 3-iodobenzoate (Ar3I). | ||
As discussed above, several important steps control the selectivity between meta-functionalized and benzocyclobutene products. The Pd(IV) pathway via oxidative addition TS32 (or TS35) and the C–C reductive elimination step TS65 lead to the Pd(IV) intermediate 33 and benzocyclobutene product, respectively. If the Pd(IV) pathway viaTS32 (or TS35) is preferred over TS65, the C–C reductive coupling TS36 (or TS40) and C–C reductive elimination TS67 (or TS69) will become the selectivity-determining steps for meta-functionalized and benzocyclobutene products, respectively.
To discuss the ligand effect, the reaction of 1a with methyl iodide is investigated for consistency with the experiment. As shown in Table 1, the calculated results show excellent agreement with the experimental observation. The concerted oxidative addition pathway viaTS35 always could not compete with the SN2 mechanism viaTS32 in the oxidative addition step. Although the catalyst Pd(acridine)2 has possibly been formed together with the benzocyclobutene viaTS65, which would switch on the Pd(II) pathway viaTS44 with a low activation energy of 17.4 kcal mol−1, benzocyclobutene formation is still preferred to meta-alkylation, revealing that the Pd(IV) pathway and the C–C reductive elimination viaTS65 are the dominant pathways under the experimental conditions.
| Charges | Pd(IV) pathway | ΔΔG°‡(a)a | meta-Methylation | Benzocyclobutene | ΔΔG°‡(b)b | Pd(II) pathway | Experimental yields10 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pd in 31 | TS32 | TS35 | TS65 | TS36 | TS40 | TS67 | TS69 | TS44 | meta-Methylation:benzocyclobutene | |||
| a ΔΔG°‡(a) = ΔΔG°‡(TS32–TS65). b ΔΔG°‡(b) = ΔΔG°‡(min{TS36,TS40}–min{TS67,TS69}). | ||||||||||||
| Py | +0.329 | 20.2 | 26.0 | 28.2 | −8.0 | 17.3 | 15.4 | 19.0 | 15.0 | +0.4 | 22.4 | 15%:15% |
| DMAP | +0.314 | 23.9 | 30.5 | 31.3 | −7.4 | 19.4 | 18.7 | 23.0 | 21.3 | −2.6 | 35.5 | 32%:8% |
| Acridine | +0.294 | 23.4 | 30.2 | 22.9 | +0.5 | 11.0 | 13.8 | 13.7 | 15.6 | −2.7 | 17.4 | 28%:58% |
| L1 | +0.286 | 23.3 | 31.1 | 24.5 | −1.2 | 10.1 | 14.2 | 14.7 | 15.0 | −4.6 | 28.3 | 85%:15% |
| L2 | +0.294 | 24.0 | 33.4 | 24.9 | −0.9 | 10.7 | 7.3 | 15.6 | 13.9 | −6.6 | 23.3 | 80%:20% |
With a small ligand such as Py or DMAP (4-dimethylaminopyridine) as the ligand, the C–C reductive coupling TS36 (or TS40) and C–C reductive elimination TS67 (or TS69) from Pd(IV) intermediate 33 are the selectivity-determining steps. The electron-donating substituent in DMAP stabilizes the Pd(IV) intermediate and further distinguishes the C–C reductive coupling and C–C reductive elimination steps, resulting in the increased yield of meta-alkylation. On the other hand, the presence of a large ligand (acridine, L1, or L2) makes the selectivity-determining step move back to the oxidative addition TS32 and the C–C reductive elimination TS65 starting from the Pd(II) intermediate 31. There are two main reasons: (a) the Pd(IV) intermediate is more sensitive to the ligand size than the Pd(II) intermediate, and thus the C–C reductive elimination from the Pd(IV) intermediate with a large ligand is obviously prohibited; (b) the activation energy of C–C reductive elimination from the Pd(II) intermediate viaTS65 remarkably decreases from 28.2–31.3 kcal mol−1 with a small ligand to 22.9–24.9 kcal mol−1 with a large ligand, which partly comes from the attractive interaction between the large ligand and the 4-(CF3)C6F4 group, as shown by the short F–H distance (2.403 Å) in TS65-N1-L1 in Fig. 12. Further fine tuning of the ligand from acridine to L1 or L2 will disfavor the C–C reductive elimination and change the selectivity between meta-alkylation and benzocyclobutene formation.
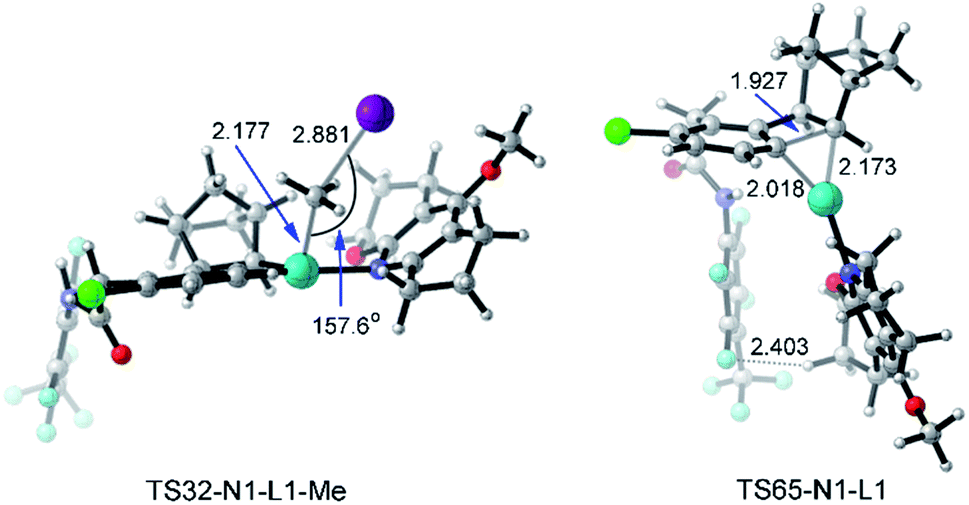 | ||
| Fig. 12 Optimized structures of transition states TS32-N1-L1-Me and TS65-N1-L1 of methyl iodide. The selected distances and angles are given in angstroms and in degrees, respectively. | ||
The NBO charges of the Pd atom in 31 were also calculated for all the reaction systems, as shown in Table 1. By comparing the Pd charge with that of acridine (+0.294) and L2 (+0.294), it is clear that both ligands have similar electron-donating properties; however, the selectivity between the C–C reductive coupling and C–C reductive elimination is reversed from acridine to L2, indicating that both geometric and electronic properties of the ligand play important roles.
Fig. 12 presents the optimized structures of transition states TS32-N1-L1-Me and TS65-N1-L1 with L1 as the ligand, which are the selectivity-determining steps. TS32-N1-L1-Me is a typical SN2 transition state with a Pd–C–I angle of 157.6°. The F–H distance of 2.403 Å in TS65-N1-L1 is shorter than the sum of van der Waals radii of hydrogen (1.10 Å) and fluorine (1.46 Å),46 suggesting the attractive interaction between the ligand and 4-(CF3)C6F4 group.
For the norbornene effect on meta-alkylation, the reaction of 1b with ethyl iodide is studied using N1, N2 and L2 as the ligand, considering experimental conditions. As shown in Table 2, when norbornene N1 is employed, the oxidative addition step viaTS32 is moderately lower in energy (−1.7 kcal mol−1) than the C–C reductive elimination step viaTS65; however, the reductive coupling viaTS36 is disfavored by 0.6 kcal mol−1 in energy than the C–C reductive coupling viaTS67. This result agrees with the experimental observation that the yields of benzocyclobutene and the meta-ethylation products are 85% vs. 10%.
| Charges | Pd(IV) pathway | ΔΔG°‡(a)a | meta-Ethylation | Benzocyclobutene | ΔΔG°‡(b)b | Pd(II) pathway | Experimental yields11 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pd in 31 | TS32 | TS35 | TS65 | TS36 | TS40 | TS67 | TS69 | TS44 | meta-Ethylation:benzocyclobutene | |||
| a ΔΔG°‡(a) = ΔΔG°‡(TS32–TS65). b ΔΔG°‡(b) = ΔΔG°‡(min{TS36,TS40}–min{TS67,TS69}). | ||||||||||||
| N1 | +0.285 | 23.2 | 27.6 | 24.9 | −1.7 | 13.6 | 15.0 | 13.0 | 15.4 | +0.6 | 21.3 | 10%:85% |
| N2 | +0.356 | 22.7 | 23.1 | 30.2 | −7.5 | 23.1 | 19.5 | 33.7 | 31.4 | −11.9 | 30.8 | 97%:0% |
When N2 is used instead of N1, the C–C reductive elimination steps, including TS65, TS67, and TS69, need higher activation energies, leading to an obvious energy difference of 7.5 kcal mol−1 (or 11.8 kcal mol−1) between TS32 (or TS40) and TS65 (or TS69). These results clearly reveal that with N2 as norbornene the meta-ethylation product is preferred over benzocyclobutene, consistent with the experimental result. Because of the repulsion induced by the CO2Me group in N2, the C–C reductive elimination is prohibited, which was also found in our recent theoretical study on Ni-catalyzed (2 + 2 + 2) cycloaddition of 1,6-ene-allenes and alkenes.47 As shown in Fig. 13, the steric repulsion in TS40-N1-L2-Et mainly appears between the ethyl group and substrate (or norbornene), as indicated by the two short H–H distances (2.263 and 2.288 Å). Nevertheless, in TS40-N2-L2-Et, several short H–H distances appear, including 2.217, 2.456, and 2.475 Å, indicating the obvious steric repulsion. The NBO charge of the Pd atom in 31 as shown in Table 2 increases from +0.285 with N1 to +0.356 with N2, which comes from the electron-withdrawing property of the CO2Me group. The presence of the CO2Me group also favors the SN2 pathway viaTS32 with the activation energy slightly decreasing from 23.2 to 22.7 kcal mol−1 and concerted oxidative addition viaTS35 with the activation energy dropping from 27.6 to 23.1 kcal mol−1.45
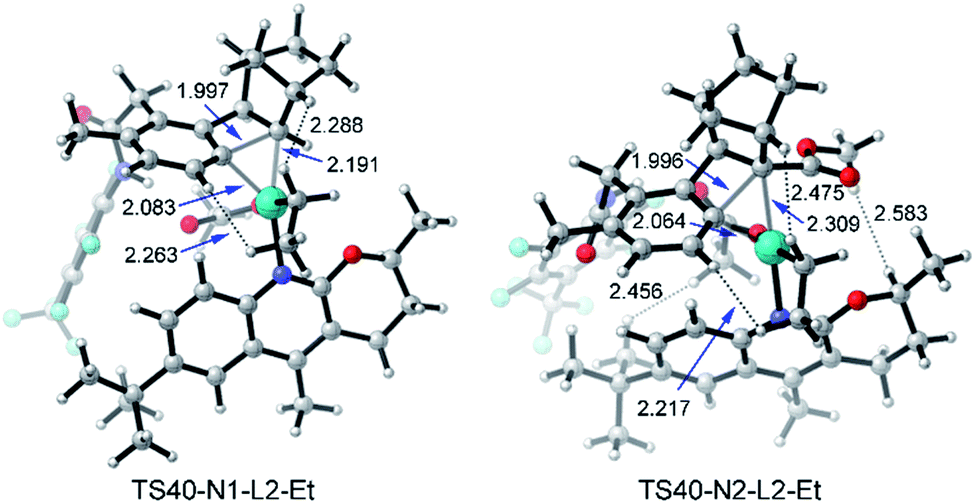 | ||
| Fig. 13 Optimized structures of transition states TS40-N1-L2-Et and TS40-N2-L2-Et of amide substrate 1b and ethyl iodide with the selected distances given in angstroms. | ||
Besides meta-alkylation, meta-arylation producing biaryl compounds can also be achieved with the catalytic systems A (L1 and N1) and B (L2 and N2). Compared to the catalytic system A which is only effective for ortho-substituted aryl iodides, catalytic system B in PhCF3 solvent has been successfully applied to meta-arylation of phenyl iodide and 2-, 3- or 4-substituted aryl iodides.10,11,48 Thus, the key selectivity-determining steps were investigated by employing the catalytic system B with amide substrate 1b and methyl 2-iodobenzoate Ar2I (or methyl 3-iodobenzoate Ar3I, Scheme 5) as the reactant model. Since the concerted oxidative addition transition state has been revealed to be preferred for aryl iodides in the Pd(IV) pathway,20 the SN2 pathway viaTS32 will not be discussed here.
Table 3 shows the computed activation energies of all the selected transition states. The selectivity-determining step is the oxidative addition viaTS35 and C–C reductive elimination viaTS65. For both Ar2I and Ar3I, the activation energy for TS35 is obviously lower than that of TS65, which agrees with the experimental observation that catalytic system B is effective for both ortho-substituted and ortho-unsubstituted aryl iodides. However, ortho-substituted aryl iodide is more easily coupled than ortho-unsubstituted aryl iodide because of the lower activation energy (28.7 kcal mol−1) for oxidative addition with Ar2I than that (30.0 kcal mol−1) with Ar3I, which might be attributed to the attractive interaction between the directing group and CO2Me of Ar2I, as indicated by the short O–H distance (2.059 Å) in Fig. 14. The Pd(IV) pathway viaTS44 does not play an important role, since Pd(L2)2 could not be generated under the experimental conditions. The activation energy (30.2 kcal mol−1) for TS44 with Ar3I is comparable to that of TS35, indicating that the combination of a large ligand and modified norbornene can make the Pd(IV) pathway have similar activation energy, although there is an ortho-substituted group (the directing group) of the phenyl group.
| Pd(IV) pathway | ΔΔG°‡(a)a | meta-Arylation | Benzocyclobutene | ΔΔG°‡(b)b | Pd(II) pathway | Experimental yields10,11 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| TS35 | TS65 | TS36 | TS40 | TS67 | TS69 | TS44 | meta-Arylation:benzocyclobutene | |||
| a ΔΔG°‡(a) = ΔΔG°‡(TS35–TS65). b ΔΔG°‡(b) = ΔΔG°‡(min{TS36,TS40}–min{TS67,TS69}). | ||||||||||
| Ar2I | 28.7 | 34.9 | −6.2 | 26.8 | 21.0 | 34.6 | 29.1 | −8.1 | 37.3 | 100%:0% |
| Ar3I | 30.0 | 34.9 | −4.9 | 24.3 | 17.7 | 39.5 | 34.7 | −17.0 | 30.2 | 100%:0% |
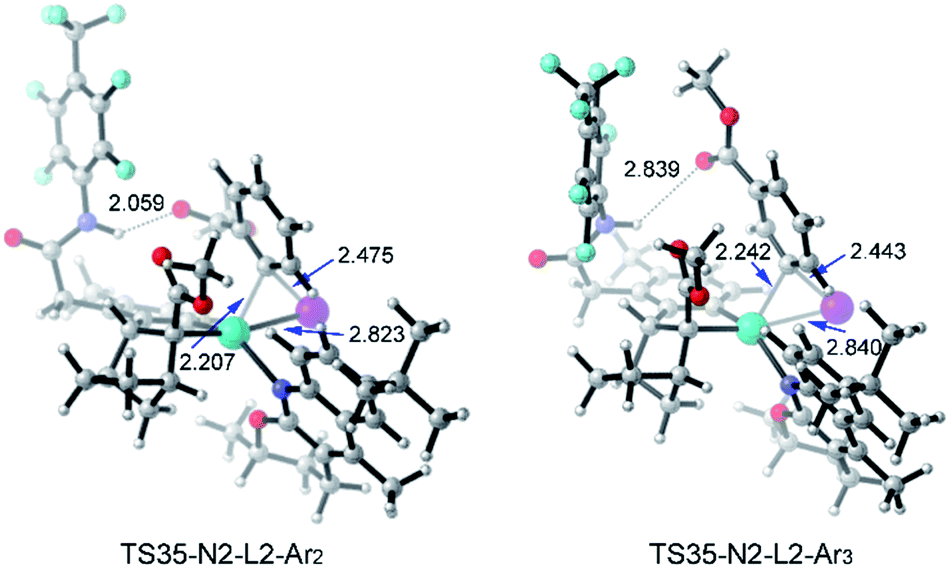 | ||
| Fig. 14 Optimized structures of TS35-N2-L2-Ar2 and T35-N2-L3-Ar3 of amide substrate 1b and aryl iodide (Ar2I and Ar3I) with selected distances given in angstroms. | ||
Conclusions
We have theoretically investigated the detailed mechanism, norbornene and ligand effects, and origins of selectivity in a newly developed Pd/norbornene-catalyzed meta-selective C–H activation and alkylation. The reaction starts initially with the substrate binding to the Pd(II) center through an amidate auxiliary, followed by the ortho-C–H activation via a CMD mechanism, which generates the six-membered-ring cyclopalladate(II) intermediate. This Pd(II) intermediate further undergoes norbornene insertion and meta-C–H activation via a CMD mechanism, forming the five-membered-ring cyclopalladate(II) intermediate. The subsequent meta-functionalization step adopts the Pd(IV) pathway via oxidative addition and C–C reductive coupling rather than the Pd(II) pathway via meta-C–C reductive coupling on dinuclear palladium species because of the low concentration of Pd(0)L2. Finally, to complete the catalytic cycle, norbornene dissociation takes place followed by protodemetallation with the formation of a six-membered-ring palladacycle(II) intermediate.There are two possible pathways for the oxidative addition step, which depends on the nature of the coupling iodide; the alkyl iodide undergoes an SN2 pathway, whereas the aryl iodide prefers concerted oxidative addition rather than the SN2 pathway. For methyl iodide, with norbornene and pyridine (or DMAP), the C–C reductive coupling and C–C reductive elimination from the Pd(IV) intermediate are found to be the selectivity-determining steps of meta-functionalization and benzocyclobutene formation, respectively. However, the use of a large ligand (acridine, L1, and L2) changes the selectivity-determining step to the oxidative addition and the C–C reductive elimination from the Pd(II) intermediate. The geometric and electronic properties of L1 and L2 suppress the benzocyclobutene formation by increasing the energy difference between those two pathways. The combination of 2-carbomethoxynorbornene and L2 utilizes the electronic and steric effects and promotes meta-alkylation and -arylation by disfavoring the C–C reductive elimination steps from the Pd(II) and Pd(IV) intermediates as well as favoring the oxidative addition step.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. Y. acknowledges the “Young Talent Support Plan” of Xi'an Jiaotong University. M. E. acknowledges the financial support from a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) (JP16H06511, JPH04104) and the Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan. This work was also supported by the Joint Funds of the National Natural Science Foundation and China-State Grid Corporation (U1866203), the Fundamental Research Funds for the Central Universities and the World-Class Universities (Disciplines) and the Characteristic Development Guidance Funds for the Central Universities. DFT calculations were partially performed at the HPC Platform of Xi'an Jiaotong University, China and the Research Center for Computational Science, Okazaki, Japan. We are also grateful to Prof. Shigeyoshi Sakaki (Kyoto University) and Prof. Jin-Quan Yu (The Scripps Research Institute) for numerous fruitful discussions.Notes and references
- (a) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003 CrossRef CAS; (b) J. J. Topczewskia and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC; (c) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS PubMed; (d) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS.
- (a) N. Della Ca', M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389 CrossRef; (b) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS; (c) J. Yang, Org. Biomol. Chem., 2015, 13, 1930 RSC; (d) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863 CrossRef CAS; (e) J. Ye, Z. Shi, T. Sperger, Y. Yasukawa, C. Kingston, F. Schoenebeck and M. Lautens, Nat. Chem., 2017, 9, 361 CrossRef CAS; (f) J. Wang, R. Li, Z. Dong, P. Liu and G. Dong, Nat. Chem., 2018, 10, 866 CrossRef CAS.
- (a) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712 CrossRef CAS; (b) M. Bera, S. Agasti, R. Chowdhury, R. Mondal, D. Pal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 5272 CrossRef CAS PubMed; (c) X.-Y. Chen and E. J. Sorensen, Chem. Sci., 2018, 9, 8951 RSC; (d) Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470 CrossRef CAS; (e) T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751 CrossRef CAS; (f) M.-Z. Lu, X.-R. Chen, H. Xu, H.-X. Dai and J.-Q. Yu, Chem. Sci., 2018, 9, 1311 RSC; (g) S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699 CrossRef CAS; (h) B. Ma, Z. Chu, B. Huang, Z. Liu, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2749 CrossRef CAS; (i) M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745 CrossRef CAS; (j) A. Unnikrishnana and R. B. Sunoj, Chem. Sci., 2019, 10, 3826 RSC.
- (a) M. Ye, G.-L. Gao and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 6964 CrossRef CAS; (b) K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 11904 CrossRef CAS.
- (a) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109 CrossRef CAS; (b) M. Font, A. R. A. Spencer and I. Larrosa, Chem. Sci., 2018, 9, 7133 RSC; (c) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef CAS.
- (a) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593 CrossRef CAS; (b) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463 CrossRef CAS; (c) S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595 RSC; (d) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef.
- (a) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS; (b) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS; (c) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed; (d) A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147 RSC.
- (a) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119 CrossRef CAS; (b) M. Lautens and S. Piguel, Angew. Chem., Int. Ed., 2000, 39, 1045 CrossRef CAS; (c) A. Martins, B. Mariampillai and M. Lautens, Top. Curr. Chem., 2010, 292, 1–33 CAS.
- (a) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS PubMed; (b) L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563 CrossRef CAS; (c) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478 CrossRef CAS.
- X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef CAS.
- P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574 CrossRef CAS.
- (a) Q. Ding, S. Ye, G. Cheng, P. Wang, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 417 CrossRef CAS; (b) P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P. X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092 CrossRef CAS; (c) P. Wang, M. E. Farmer, X. Huo, P. Jain, P.-X. Shen, M. Ishoey, J. E. Bradner, S. R. Wisniewski, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 9269 CrossRef CAS PubMed; (d) H. Shi, P. Wang, S. Suzuki, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14876 CrossRef CAS PubMed; (e) P. Wang, M. E. Farmer and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 5125 CrossRef CAS PubMed; (f) G.-C. Li, P. Wang, M. E. Farmer and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 6874 CrossRef CAS; (g) G. Cheng, P. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 8183 CrossRef CAS.
- Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887 CrossRef CAS.
- H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Nature, 2018, 558, 581 CrossRef CAS PubMed.
- (a) F. Ozawa, M. Fujimori, T. Yamamoto and A. Yamamoto, Organometallics, 1986, 5, 2144 CrossRef CAS; (b) F. Ozawa, T. Hidaka, T. Yamamoto and A. Yamamoto, J. Organomet. Chem., 1987, 330, 253 CrossRef CAS; (c) Y. Suzaki, T. Yagyu, Y. Yamamura, A. Mori and K. Osakada, Organometallics, 2002, 21, 5254 CrossRef CAS.
- D. J. Cardenas, B. Martin-Matute and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 5033 CrossRef CAS PubMed.
- (a) M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1988, 346, C27 CrossRef CAS; (b) M. Catellani and M. C. Fagnola, Angew. Chem., Int. Ed., 1995, 33, 2421 CrossRef; (c) A. Dedieu and A. J. Mota, Can. J. Chem., 2009, 87, 838 CrossRef CAS.
- A. Rudolph, N. Rackelmann and M. Lautens, Angew. Chem., Int. Ed., 2007, 46, 1485 CrossRef CAS.
- Q. Liu, Y. Lan, J. Liu, G. Li, Y. D. Wu and A. Lei, J. Am. Chem. Soc., 2009, 131, 10201 CrossRef CAS PubMed.
- G. Maestri, E. Motti, N. Della Ca’, M. Malacria, E. Derat and M. Catellani, J. Am. Chem. Soc., 2011, 133, 8574 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed; (c) S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef CAS PubMed; (d) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS.
- Test calculations were performed on the key intermediates and transition states. Different functionals and basis sets used in optimization and single-point energy calculations gave similar results. Please see the results and discussion in Fig. S14 and Table S1 in the ESI.†.
- D. Andrae, U. Häusermann, M. Dolg, H. Stoll and H. Preus, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- (a) Y. Minenkov, Å. Singstad, G. Occhipinti and V. R. Jensen, Dalton Trans., 2012, 41, 5526 RSC; (b) T. Sperger, I. A. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532 CrossRef CAS PubMed; (c) L. Fang, T. G. Saint-Denis, B. L. H. Taylor, S. Ahlquist, K. Hong, S. Liu, L. Han, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 10702 CrossRef CAS PubMed; (d) Y.-F. Yang, G. Chen, X. Hong, J.-Q. Yu and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 8514 CrossRef CAS PubMed; (e) H. Zheng, K. Semba, Y. Nakao and S. Sakaki, J. Am. Chem. Soc., 2017, 139, 14065 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- In SMD calculations, the dielectric constant and the solute volume of PhCF3 were set as 9.18 and 4.06 angstroms, respectively.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, version 3.1, University of Wisconsin, Madison, WI, 1996 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- C. Y. Legault, CYLView, 1.0b, Universitéde Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
- J. He, S. Li, Y. Q. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed.
- Y. Dang, S. Qu, J. W. Nelson, H. D. Pham, Z.-X. Wang and X. Wang, J. Am. Chem. Soc., 2015, 137, 2006 CrossRef CAS PubMed.
- (a) S. Winstein and T. G. Traylor, J. Am. Chem. Soc., 1955, 77, 3747 CrossRef CAS; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (c) B. Biswas, M. Sugimoto and S. Sakaki, Organometallics, 2000, 19, 3895 CrossRef CAS; (d) W. Guan, F. B. Sayyed, G. Zeng and S. Sakaki, Inorg. Chem., 2014, 53, 6444 CrossRef CAS PubMed.
- The direct-CMD will produce the intermediate 18 directly.
- The transition state of the second norbornene insertion into 30 needs a much higher activation energy (69.2 kcal mol−1). Please see Fig. S8 in the ESI.†.
- The endo-migratory insertion of norbornene viaTS23′ needs a higher activation energy of 30.2 kcal mol−1.
- H. Zhang, H.-Y. Wang, Y. Luo, C. Chen, Y. Cao, P. Chen, Y.-L. Guo, Y. Lan and G. Liu, ACS Catal., 2018, 8, 2173 CrossRef CAS.
- The phenomenon that the SN2 pathway is more favoured than the concerted oxidative addition was also found in the experimental/theoretical study on the Ni-catalyzed coupling reaction of alkyl halides with aryl Grignard reagents in the presence of 1,3-butadiene, see T. Iwasaki, A. Fukuoka, W. Yokoyama, X. Min, I. Hisaki, T. Yang, M. Ehara, H. Kuniyasua and N. Kambe, Chem. Sci., 2018, 9, 2195 RSC.
- Possible transition states for the Cmethyl–Cnorbornene reductive coupling starting from 33 have been examined. The activation energy of those transition states is at least 22.6 kcal mol−1, which is obviously higher than those of TS36, TS40, TS66, and TS68 (Fig. S9 in the ESI†).
- Other possible transition state structures of TS44 with two (or three) ligands have been examined; however, their activation energies are higher than that of TS44 (Fig. S10†).
- The transition state for the Cmethyl–Cnorbornene reductive coupling starting from 43 needs an activation energy of 32.2 kcal mol−1, which is higher than that of TS44 (Fig. S10†).
- The oxidative addition of CH3I on PdPy2, which gives rise to CH3Pd(I)Py2, needs an acceptable activation energy of 26.0 kcal mol−1. Please see Fig. S11 in the ESI.†.
- In Tables 1–3, complex 1 is taken as the energy reference point. If the intermediate 30 is taken as the energy reference point, the activation energies for the TS32 in Table 2 are 24.8 and 27.0 kcal mol−1 for N1 and N2, respectively, which agrees with the fact that positive charge would disfavor the SN2 pathway.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384 CrossRef CAS.
- T. Yang and M. Ehara, J. Org. Chem., 2017, 82, 2150 CrossRef CAS PubMed.
- By tuning directing groups and ligands, meta-arylation has also been reported for ortho-unsubstituted aryl iodides by using Pd and simple norbornene. Please see ref. 12a and J. Han, L. Zhang, Y. Zhu, Y. Zheng, X. Chen, Z.-B. Huang, D.-Q. Shi and Y. Zhao, Chem. Commun., 2016, 52, 6903 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04720d |
| This journal is © The Royal Society of Chemistry 2020 |