
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning radical interactions in trisradical tricationic complexes by varying host-cavity sizes†
Kang
Cai
a,
Yi
Shi
a,
Changsu
Cao
b,
Suneal
Vemuri
a,
Binbin
Cui
a,
Dengke
Shen
a,
Huang
Wu
a,
Long
Zhang
a,
Yunyan
Qiu
 a,
Hongliang
Chen
a,
Yang
Jiao
a,
Charlotte L.
Stern
a,
Fehaid M.
Alsubaie
c,
Hai
Xiao
*b,
Jun
Li
b and
J. Fraser
Stoddart
*ade
a,
Hongliang
Chen
a,
Yang
Jiao
a,
Charlotte L.
Stern
a,
Fehaid M.
Alsubaie
c,
Hai
Xiao
*b,
Jun
Li
b and
J. Fraser
Stoddart
*ade
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: stoddart@northwestern.edu
bKey Laboratory of Organic Optoelectronics & Molecular Engineering of the Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: haixiao@tsinghua.edu.cn
cJoint Center of Excellence in Integrated Nano-Systems, King Abdulaziz City for Science and Technology, Riyadh 11442, Kingdom of Saudi Arabia
dInstitute for Molecular Design and Synthesis, Tianjin University, 92 Weijin Road, Tianjin 300072, China
eSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia
First published on 1st November 2019
Abstract
Although host–guest pairing interactions between bisradical dicationic cyclobis(paraquat-p-phenylene) (BB2(˙+)) and the bipyridinium radical cation (BIPY˙+) have been studied extensively, host molecules other than BB2(˙+) are few and far between. Herein, four bisradical dicationic cyclophanes with tunable cavity sizes are investigated as new bisradical dicationic hosts for accommodating the methyl viologen radical cation (MV˙+) to form trisradical tricationic complexes. The structure–property relationships between cavity sizes and binding affinities have been established by comprehensive solution and solid-state characterizations as well as DFT calculations. The association constants of the four new trisradical tricationic complexes are found to range between 7400 and 170![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1, with the strongest one being 4.3 times higher than that for [MV⊂BB]3(˙+). The facile accessibility and tunable stability of these new trisradical tricationic complexes make them attractive redox-controlled recognition motifs for further use in supramolecular chemistry and mechanostereochemistry.
000 M−1, with the strongest one being 4.3 times higher than that for [MV⊂BB]3(˙+). The facile accessibility and tunable stability of these new trisradical tricationic complexes make them attractive redox-controlled recognition motifs for further use in supramolecular chemistry and mechanostereochemistry.
Introduction
Among the various noncovalent bonding interactions1 which have been explored in the context of supramolecular chemistry, radical–radical interactions between conjugated radical cations2 and/or anions3 are relatively new on the chemical scene. The bipyridinium radical cation (BIPY˙+) is a well-known thermally stable species with a strong tendency to undergo π-dimerization2a,4 (pimerization5) in aqueous or confined media. The dimerization constant, however, is usually relatively low2a in organic solvents, a property which limits applications in supramolecular chemistry.Cyclobis(paraquat-p-phenylene) (CBPQT4+), also known as the blue-box4+ (BB4+), in which two BIPY2+ units are connected by two p-xylylene spacers to produce a rigid, box-like conformation with a centroid-to-centroid distance of ca. 6.9 Å between two BIPY2+ units, is an excellent host6 for accommodating BIPY˙+ in its reduced state7 to form a trisradical tricationic complex, [BIPY⊂BB]3(˙+). This complex enjoys enhanced stability,8 on account of the macrocyclic effect,1g rendering the radical host–guest recognition motif attractive for templating the formation of mechanically interlocked molecules9 (MIMs) which otherwise cannot be made. A striking feature of this radical host–guest complex is that it can be switched (Scheme 1a) from radically based attraction to coulombic repulsion7–9 by oxidizing the BIPY˙+ units back to their dicationic states, and so providing10,11 a large driving force for the relative motions of component parts inside MIMs. If the coulombic repulsion-induced motions are restricted by the formation of mechanical bonds, BIPY˙+ radicals with extraordinary stability9a,12 can be produced, affording a new strategy for the design of persistent organic radicals.
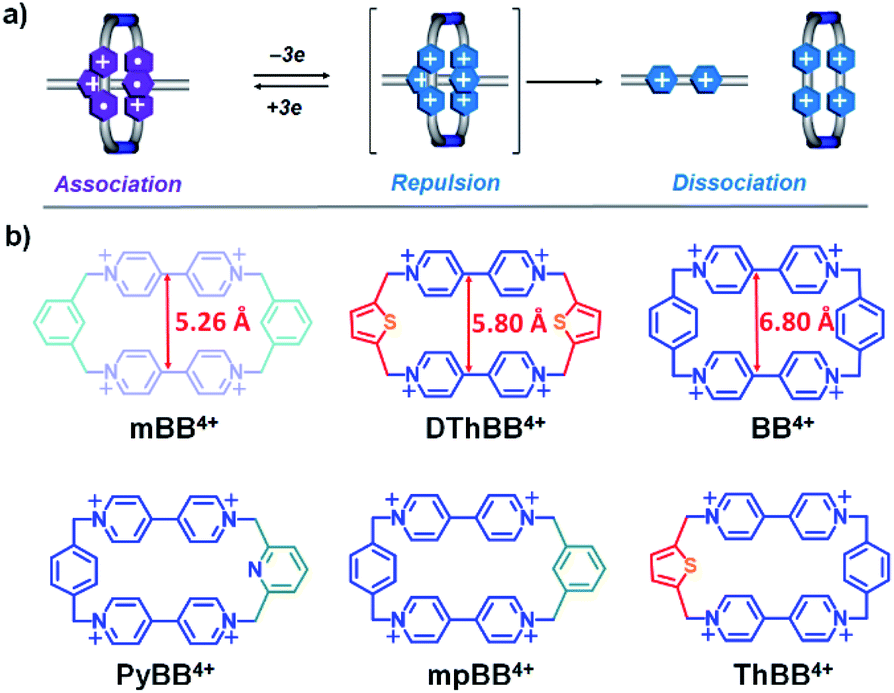 | ||
| Scheme 1 (a) Graphical representations of redox-controlled attraction and repulsion between the bisradical dicationic cyclophane and BIPY˙+; (b) structural formulas of the range of tetracationic cyclophanes. | ||
Despite the attractive properties and applications of trisradical tricationic complexes, most of the current studies7–11 have focused on [BIPY⊂BB]3(+˙), and host molecules13 other than BB2(+˙) have been little investigated. We have a particular interest in developing new BB2(+˙) analogues with smaller cavity sizes, since the decreased separation distance between the two bipyridinium units in cyclophanes is expected to result in increased coulombic repulsion between the guest and different hosts in their oxidized states, which will be beneficial for both the operation of redox-driven molecular machines, such as molecular pumps,10b and the production of mechanically protected persistent organic radicals. In addition, we have observed8 that the centroid-to-centroid distance between two BIPY+˙14 units in free BB2(+˙) (6.9 Å) is ca. 0.5 Å larger than that (6.4 Å) present in [BIPY+˙⊂BB]3(+˙), indicating that a cavity size slightly smaller than that present in BB2(+˙) could be even more favourable for the formation of highly stable trisradical tricationic complexes.
Herein, we describe the syntheses of four tetracationic cyclo-phanes (Scheme 1b), one (mpBB2+) with m- and p-xylylene15 linkers and three (PyBB2+, DThBB2+ and ThBB2+) with either 2,6-pyridinedimethyl16 or 2,5-thiophenedimethyl17 linkers, which retain similar rigidities and conformations to BB4+, while the separation distances between the two BIPY2+ units are gradually decreased (Scheme 1b). Complexation of the methyl viologen radical cation (MV+˙) with these four cyclophanes—mpBB2+, PyBB2+, DThBB2+ and ThBB2+—in their reduced states afforded four new trisradical triscationic complexes—[MV⊂mpBB]3(+˙), [MV⊂PyBB]3(+˙), [MV⊂DThBB]3(+˙) and [MV⊂ThBB]3(+˙)—that were characterised fully by UV-vis-NIR spectroscopy, cyclic voltammetry (CV), and X-ray crystallography. We have discovered that the sizes of the cyclophane cavities exert a large influence on the magnitude of the association constants in MeCN between these four reduced cyclophanes and MV+˙.
Results and discussion
All four tetracationic cyclophanes, namely, mpBB2+, PyBB2+, DThBB2+ and ThBB2+, were prepared—see ESI†—by exploiting the tetrabutylammonium iodide (TBAI) catalysed cyclisation,18 which involves the reaction of the bis(pyridinium) salt precursor (HS·2PF6) with 1,4-bis(bromomethyl)benzene or 2,5-bis(bromomethyl)thiophene in the presence of 20 mol% TBAI in MeCN while stirring under reflux for 4 days. The products were purified by exploiting normal-phase silica gel column chromatography and were obtained in isolated yields (28–52%) which were higher than that (19%) obtained for BB4+ under identical conditions,17 probably because of the decreased ring strain in the four cyclophanes.The electrochemical properties of these four cyclophanes, as their 4PF6− salts, were investigated by cyclic voltammetry (CV) in MeCN at room temperature. The CV curves of these four cyclophanes plus that of BB·4PF6 showed (Fig. S7†) two reversible reduction waves which are characteristic of BIPY2+ units. The positions of the two reduction waves for the four new cyclophanes are similar to those observed for BB·4PF6, with only slight shifts of no more than 0.12 V compared with BB·4PF6. These observations are in accordance with our expectations, since the linkers in these cyclophanes should have no significant influence on the electrochemical properties of their BIPY2+ units.
The formation of the four new trisradical tricationic complexes were investigated by UV-vis-NIR spectroscopy. Methyl viologen (MV2+) was chosen as the representative guest for studying host–guest complexation. The tetracationic cyclophanes and MV2+ were reduced separately in MeCN using Zn dust to yield suspensions which were filtered to remove the reducing agent. The resulting blue solutions displayed (Fig. 1) similar strong absorptions characteristic of BIPY˙+ units in the range 500–700 nm. When the bisradical dicationic cyclophanes were mixed with 1 equiv. MV˙+, new broad peaks appeared at around ca. 1100 nm, characteristic8 of trisradical tricationic complexes. Hence, despite the different constitutions of the linkers, all four cyclophanes retained their ability to host MV˙+ in their reduced states. The association constants (Ka) for the trisradical tricationic complexes were obtained (Fig. 1) by UV-vis-NIR titrations. The absorption changes at ca. 1100 nm were monitored and matched to 1:1 isotherms to give the binding constants (Fig. 1 and Table 1) for all four complexes. In addition, the Ka value for [MV⊂BB]3(˙+) was found, in a control experiment (Fig. S2†), to be (3.9 ± 0.5) × 104 M−1 in agreement with the previously reported8Ka value. For mpBB2(˙+) and PyBB2(˙+), which have very similar constitutions and cavity sizes, Ka values of (8.9 ± 1.3) × 103 M−1 and (7.4 ± 0.6) × 103 M−1 were obtained, respectively. Notably, both the two thiophene-containing compounds DThBB2(˙+) and ThBB2(˙+) display much stronger binding affinities than mpBB2(˙+) or PyBB2(˙+), with their respective Ka values of (6.7 ± 3.8) × 104 M−1 and (1.7 ± 0.6) × 105 M−1, which are also higher than that (Ka = (3.9 ± 0.5) × 104 M−1) of BB2(˙+). These results demonstrate the all-important influence of the linkers on the association constants of these four new trisradical tricationic complexes.
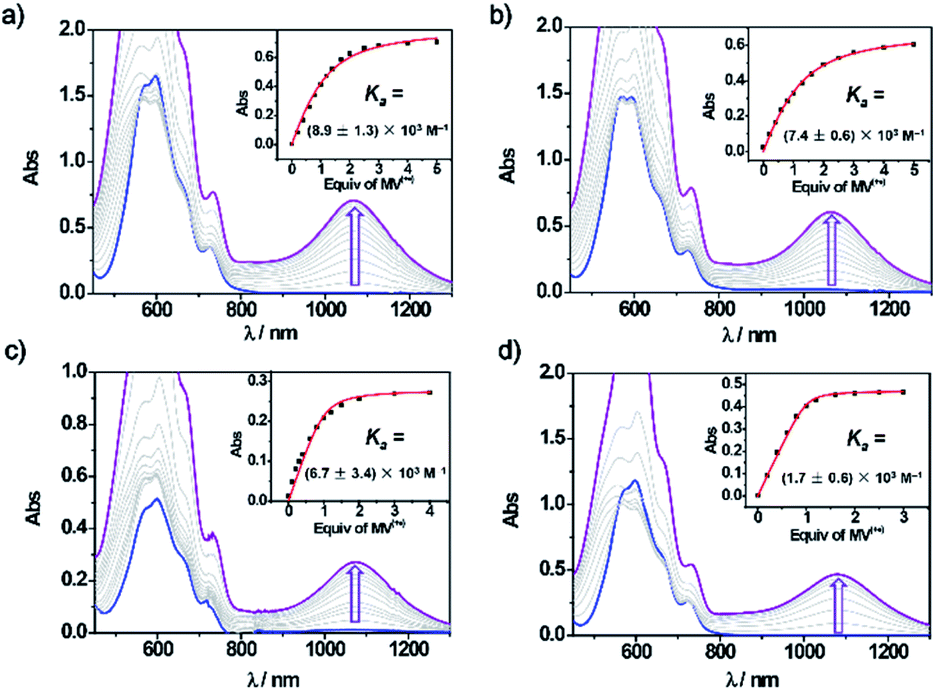 | ||
| Fig. 1 Vis-NIR spectra (MeCN, 2 mm cuvette) on titrating MV+˙ into (a) PyBB2(˙+) (0.25 mM); (b) mpBB2(˙+) (0.25 mM); (c) DThBB2(˙+) (0.20 mM); (d) ThBB2(˙+) (0.25 mM). The inset shows the change in absorption at 1080 nm with titration. The curve fitting is highlighted in red. | ||
| d 1 Å | d 2 Å | K a M−1 | E Exp kJ mol−1 | d Calc | E Calc kJ mol−1 | |
|---|---|---|---|---|---|---|
| a Centroid-to-centroid distances of BIPY2+ units in tetracationic cyclophanes according to solid-state structures. b Centroid-to-centroid distances of BIPY˙+ units in bisradical dicationic cyclophanes according to solid-state structures. c Binding constants of the trisradical triscationic complexes from vis/NIR titrations. d Binding energies of the trisradical triscationic complexes calculated from vis/NIR titrations. e Centroid-to-centroid distances of BIPY˙+ units in bisradical dicationic cyclophanes according to B3LYP-D3-optimized structures solvated in acetonitrile. f B3LYP-D3 method calculated binding energies of the trisradical triscationic complexes. | ||||||
| BB | 6.80 | 6.90 | 39000 ± 5000 |
−26.2 | 6.71 | −102.2 |
| ThBB | 6.10 | 6.72 | 170000 ± 60000 |
−29.9 | 6.35 | −103.7 |
| DThBB | 5.80 | 6.56 | 67000 ± 3800 |
−27.5 | 5.98 | −100.4 |
| mpBB | 5.94 | 6.62 | 8900 ± 1300 | −22.5 | 5.73 | −95.1 |
| PyBB | 5.65 | — | 7400 ± 600 | −22.1 | 5.50 | −91.2 |
In order to gain a better understanding of the reasons behind the different binding strengths of the dicationic diradical cyclophanes towards MV˙+, single crystals of the four trisradical tricationic complexes were grown by vapor diffusion of iPr2O into equimolar solutions in MeCN of the corresponding bisradical dicationic cyclophanes and MV˙+ in an N2-filled glovebox. The four crystal superstructures (Fig. 2) of these trisradical tricationic complexes were similar8 to that of [MV⊂BB]3(˙+), and featured the MV˙+ guests encircled by the cyclophanes, with angles between the cyclophane ring planes and the MV˙+ axis of around 75°. Although the cavity sizes for the individual bisradical dicationic cyclophanes are quite different (Table 1) from each other, the distances between the two BIPY˙+ units in the trisradical tricationic complexes are almost the same, with MV˙+/BIPY˙+ contact distances (Fig. 2) of 3.16–3.19 Å and the centroid-to-centroid distance of two cyclophane BIPY+˙ units of around 6.3 Å. This phenomenon is an example of “induced-fit” binding19—i.e., the cyclophanes adjust their conformations (extend or shrink the cavity sizes) to best accommodate MV˙+ guests and thus optimise the radical-pairing recognition interactions. Accordingly, we assumed that the ideal cavity size for accommodating MV˙+ guests is around 6.3 Å, with smaller or larger sizes resulting in decreased binding affinities towards MV˙+.
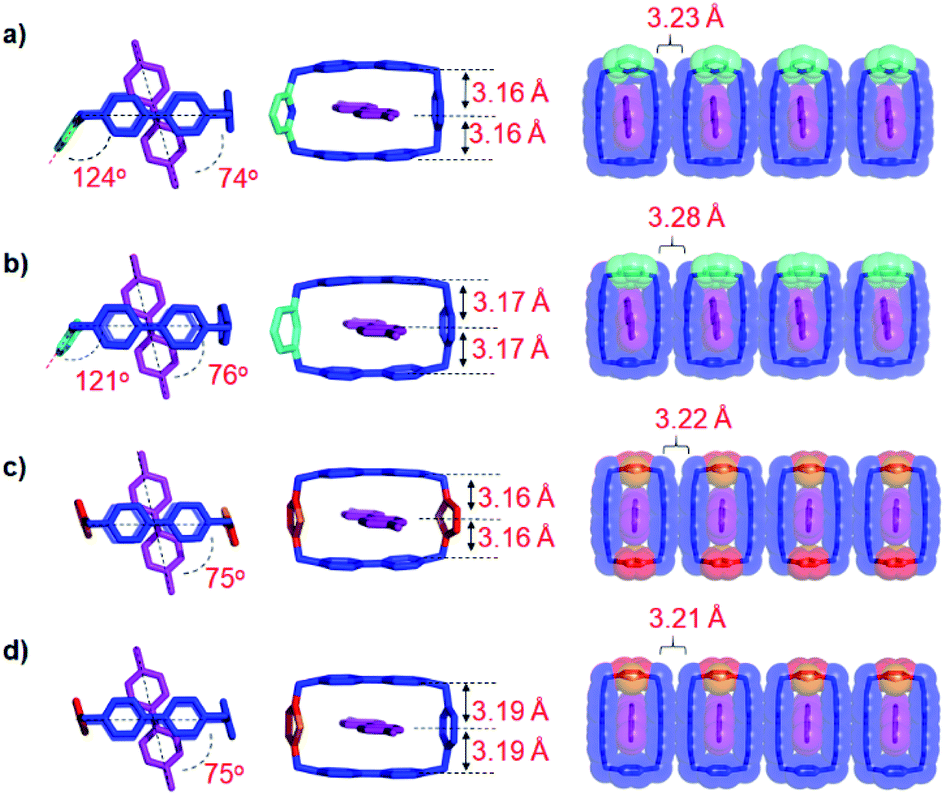 | ||
| Fig. 2 Solid-state superstructures of trisradical triscationic complexes. (a) [MV⊂PyBB]3(˙+); (b) [MV⊂mpBB]3(˙+); (c) [MV⊂DThBB]3(˙+); (d) [MV⊂ThBB]3(˙+). Left: plan views depicted as tubular representations; middle: perspective views depicted as tubular representations; right: 1D packing of the trisradical complexes depicted as tubular superimposed upon space-filling representations. Hydrogen atoms and disorder in the asymmetric cyclophanes are omitted for the sake of clarity. | ||
Subsequently, we analysed the crystal structures of the individual cyclophanes in both their oxidised and reduced forms. All four tetracationic cyclophanes readily formed good crystals which were suitable for X-ray crystallographic analyses. The centroid-to-centroid distance (Fig. 3c) between the two BIPY2+ units of the symmetric tetracationic DThBB4+ is 5.80 Å, which is ca. 0.5 Å smaller than the “ideal size” of 6.3 Å for binding MV+˙ by the bisradical dicationic cyclophanes. PyBB4+ featured (Fig. 3b) a trapezoid-like conformation in view of the different separation distances for the p-xylylene and 2,6-pyridinedimethyl linkers. Consequently, the distance between the two BIPY2+ units gradually deceases upon going from the p-xylylene linker to the m-xylylene linker, and the centroid-to-centroid distance (Fig. 4b) of the two BIPY2+ units is ca. 5.65 Å (Fig. 4), which is about 0.65 Å smaller than 6.3 Å.
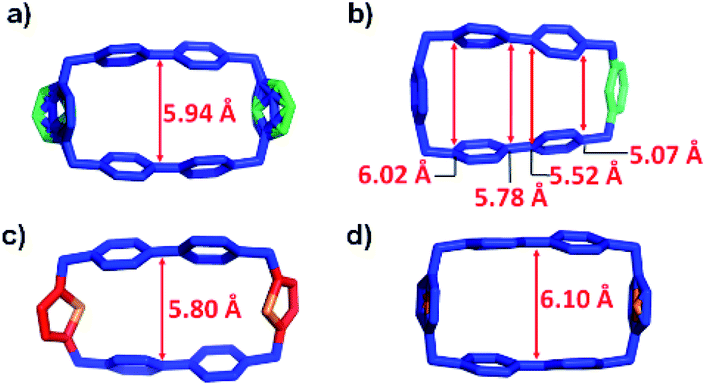 | ||
| Fig. 3 Solid-state structures of (a) mpBB4+; (b) PyBB4+; (c) DThBB4+ and (d) ThBB4+. Hydrogen atoms are omitted for the sake of clarity. Disorder in the linker parts of the asymmetric cyclophanes mpBB4+ and ThBB4+ is displayed in (a) and (d). | ||
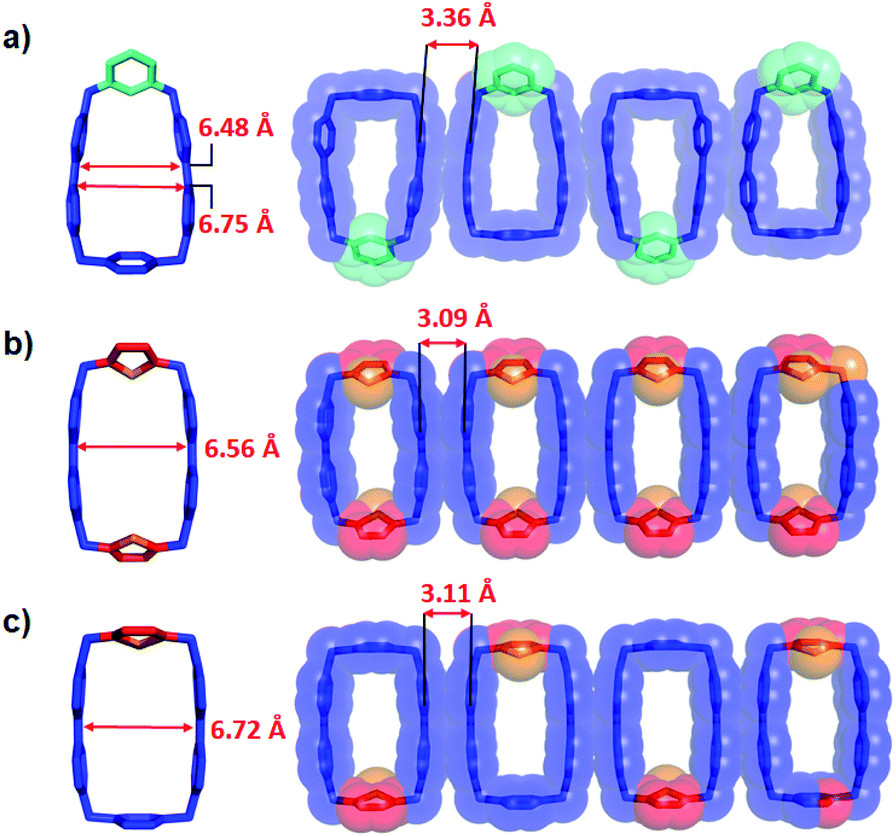 | ||
| Fig. 4 Solid-state structures of (a) mpBB2(˙+); (b) DThBB2(˙+); (c) ThBB2(˙+). Left: plan views depicted as tubular representations; right: 1D packing of the diradical dicationic cyclophanes depicted as tubular superimposed upon space-filling representations. Hydrogen atoms and disorder in the asymmetric cyclophanes are omitted for the sake of clarity. | ||
The crystal structures (Fig. 3a and d, S8, and S10†) of the other two asymmetric cyclophanes, mpBB4+ and ThBB4+, exhibit disorder in the m-xylylene/p-xylylene or p-xylylene/2,5-thiophene-dimethyl linkers. Consequently, both mpBB4+ and ThBB4+ display a symmetric conformation according to X-ray crystallographic analysis with an apparent centroid-to-centroid distance between BIPY2+ units of 5.94 and 6.10 Å, respectively. Since mpBB4+ has an almost identical constitution to that of PyBB4+, the actual conformations and cavity sizes of these two cyclophanes should be similar to each other. Therefore, the apparent centroid-to-centroid distances in mpBB4+ (5.94 Å) and ThBB4+ (6.10 Å), which were deduced from the crystal structures, may not be all that accurate as a consequence of the disorder. In general, these solid-state structures, however, do provide some clues when it comes to interpreting the trend in the host–guest association constants obtained from titration experiments in their reduced states. Since the cavity sizes of DThBB4+ (5.8 Å) and BB4+ (6.8 Å) are either ca. 0.5 Å smaller or larger than 6.3 Å, their binding constants when reduced to bisradical dications for hosting MV+˙ are quite similar, i.e., (6.7 ± 3.8) × 104vs. (3.9 ± 0.5) × 104 M−1). The asymmetric ThBB4+, which can be regarded as an “averaged” version of DThBB4+ and BB4+, displays a binding constant (1.7 ± 0.6) × 104 M−1 for [MV⊂ThBB]3(˙+) that is higher than those for both [MV⊂DThBB]3(˙+) and [MV⊂BB]3(˙+). The other two cyclophanes, PyBB4+ and mpBB4+, which have asymmetric constitutions and much smaller cavities, display much lower binding constants ((7.4 ± 0.6) × 103 M−1 and (8.9 ± 1.3) × 103 M−1, respectively) for [MV⊂PyBB]3(˙+) and [MV⊂mpBB]3(˙+). When the cavity size is decreased even further, as in the case of mBB4+ (5.26 Å) with its two m-xylylene spacers (Scheme 1), no binding towards MV˙+ in its reduced state was observed either in solution or in the solid state.
We also obtained the crystal structures of the bisradical dicationic forms of the cyclophanes, DThBB2(˙+), ThBB2(˙+), and mpBB2(˙+), while PyBB2(˙+) failed to form single crystals suitable enough for X-ray crystallography. Again, disorder also plagued (Fig. 4b and S13†) the crystal structure of ThBB2(˙+) on account of its having two different linkers. No disorder, however, was observed in the case of the other asymmetric cyclophane mpBB2(˙+). Notably, all three bisradical dicationic cyclophanes formed (Fig. 4, S11–S13†) one-dimensional stacked columns in the solid state on account of intermolecular radical–radical interactions between adjacent BIPY˙+ units, in a manner similar to that already reported8 for BB2(˙+), with distances (Fig. 4) between stacked BIPY˙+ units in neighbouring bisradical dicationic cyclophanes of 3.36, 3.09 and 3.11 Å for mpBB2(˙+), DThBB2(˙+) and ThBB2(˙+), respectively. Consequently, these bisradical dicationic cyclophanes display significantly longer centroid-to-centroid BIPY˙+ distances than the corresponding BIPY2+ distances (Fig. 3 and Table 1) in the tetracationic cyclophanes in their solid states, most likely as a result of the strong intermolecular radical–radical interactions enlarging the cavities of the bisradical dicationic cyclophanes in the solid states. In solution, however, the intermolecular stacking interactions between bisradical dicationic cyclophanes will be negligible, and the solution-phase cavity sizes are expected to be smaller than those observed in their solid states.
Finally, density functional theory (DFT) calculations using B3LYP basis set with D3 dispersion correction20 were conducted in order to investigate the binding energies of the trisradical tricationic complexes. The optimised structures of the bisradical dicationic hosts were optimised (Fig. S18†) showing that the predicted cavity sizes—centroid-to-centroid distances between BIPY˙+ units—are in better agreement (Table 1) with the measured distances in the tetracationic cyclophanes in the solid state than those of the bisradical dicationic cyclophanes. The calculations predict (Table 1 and Fig. 5b) the host-guest binding energies to be in the order of |ΔEThBB| < |ΔEBB| < |ΔEDThBB| < |ΔEmpBB| < |ΔEPyBB| in agreement with the experimental results. The binding energies with MV˙+ exhibit strong dependences on the cyclophane cavity sizes. In an attempt to analyse the correlations between the cavity sizes and the binding energies, we decomposed (Table S2†) the binding energies into the contributions21 from dispersion and electrostatic energies. The attractive dispersion interactions (Table S2†) weaken with increasing with host size, while the concomitant decrease (Table S2†) in electrostatic repulsion stabilises host–guest binding. Thus, it is the competition between the two counteracting interactions which results in the optimal host–guest binding (Fig. 5) at a cavity size of ca. 6.3 Å, i.e., as exemplified by ThBB2(˙+).
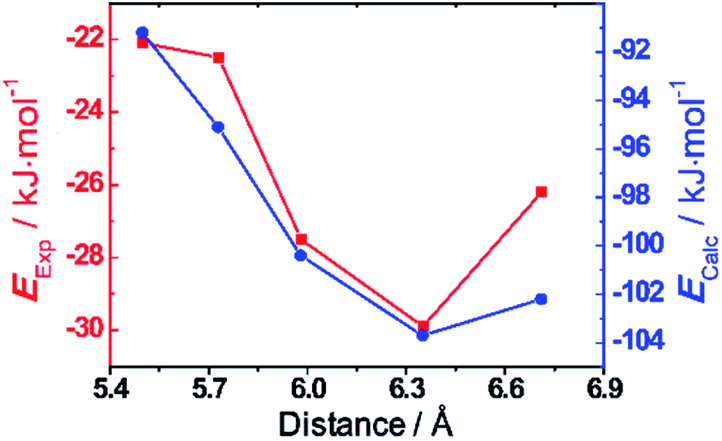 | ||
| Fig. 5 Binding energies (y axis) towards MV˙+ for the five cyclophanes with different cavity sizes (centroid-to-centroid distances between the two BIPY˙+ units, x axis). The x axis presents the DFT-calculated centroid-to-centroid distances22 between the two BIPY˙+ units. The left red y axis indicates the experimental binding energies towards binding with MV˙+ from vis/NIR titration, while the right blue axis presents the DFT-calculated binding energies towards binding with MV˙+. | ||
Conclusions
Four bisradical dicationic cyclophanes were investigated as hosts for binding MV˙+ and found to form trisradical triscationic complexes. UV-vis-NIR titration experiments, X-ray crystallographic characterizations and DFT calculations revealed that the binding affinities of these trisradical triscationic complexes are highly dependent on the cavity sizes of the bisradical dicationic cyclophanes. The structure–property relationship we have established will provide useful information for the design of new radical host–guest pairs in the future. Notably, the asymmetric p-xylylene/2,5-thiophene-dimethyl linked cyclophane ThBB2(+˙) which displayed an association constant of 170000 M−1 in acetonitrile, that is 4.3 times higher than that (39000 M−1) of BB2(+˙) which has two p-xylylene linkers. The facile accessibility, tunable association constants, as well as decreased host cavity sizes associated with these four new trisradical tricationic complexes, make them attractive redox-controlled recognition motifs for further use in supramolecular chemistry and in the template-directed synthesis of mechanically interlocked molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is part of the Joint Center of Excellence in Integrated Nano-Systems (JCIN) at King Abdulaziz City for Science and Technology (KACST) and Northwestern University (NU). The authors thank both KACST and NU for their continued support of this research. This work utilized Northwestern University Micro/Nano Fabrication Facility (NUFAB), which is partially supported by the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205), the Materials Research Science and Engineering Center (DMR-1720139), the State of Illinois, and Northwestern University.Notes and references
- (a) D. C. Sherrington and K. A. Taskinen, Chem. Soc. Rev., 2001, 30, 83–93 RSC; (b) K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC; (c) Z. J. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC; (d) X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101 RSC; (e) Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef PubMed; (f) M. M. Watt, M. S. Collins and D. W. Johnson, Acc. Chem. Res., 2013, 46, 955–966 CrossRef CAS PubMed; (g) Q. Yan, Z. Luo, K. Cai, Y. Ma and D. Zhao, Chem. Soc. Rev., 2014, 43, 4199–4221 RSC.
- For reviews on radical–radical interactions between conjugated radical cations or anions, see: (a) D. W. Zhang, J. Tian, L. Chen, L. Zhang and Z.-T. Li, Chem.–Asian J., 2015, 10, 56–68 CrossRef CAS PubMed; (b) L. Chen, Y.-C. Zhang, W.-K. Wang, J. Tian, L. Zhang, H. Wang, D.-W. Zhang and Z.-T. Li, Chin. Chem. Lett., 2015, 26, 811–816 CrossRef CAS.
- (a) E. M. Fatila, R. A. Mayo, M. Rouzieres, M. C. Jennings, P. Dechambenoit, D. V. Soldatov, C. Mathoniere, R. Clerac, C. Coulon and K. E. Preuss, Chem. Mater., 2015, 27, 4023–4032 CrossRef CAS; (b) X. Zheng, Y. Zhang, N. Cao, X. Li, S. Zhang, R. Du, H. Wang, Z. Ye, Y. Wang, F. Cao and H. Li, Nat. Commun., 2018, 9, 1961 CrossRef; (c) B. Tang, W.-L. Li, Y. Chang, B. Yuan, Y. Wu, M.-T. Zhang, J.-F. Xu, J. Li and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15526–15531 CrossRef CAS PubMed.
- (a) J. W. Park, N. H. Choi and J. H. Kim, J. Phys. Chem., 1996, 100, 769–774 CrossRef CAS; (b) W. S. Jeon, H.-J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828–1829 RSC; (c) A. Iordache, M. Oltean, A. Milet, F. Thomas, B. Baptiste, E. Saint-Aman and C. Bucher, J. Am. Chem. Soc., 2012, 134, 2653–2671 CrossRef CAS; (d) A. T. Buck, J. T. Paletta, S. A. Khindurangala, C. L Beck and A. H. Winter, J. Am. Chem. Soc., 2013, 135, 10594–10597 CrossRef CAS PubMed; (e) L. Zhang, T.-Y. Zhou, J. Tian, H. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Polym. Chem., 2014, 5, 4715–4721 RSC.
- (a) E. M. Kosower and J. Hajdu, J. Am. Chem. Soc., 1971, 93, 2534–2535 CrossRef CAS; (b) W. Geuder, S. Hünig and A. Suchy, Tetrahedron, 1986, 42, 1665–1677 CrossRef CAS.
- B. Odell, M. V. Reddington, A. M. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547–1550 CrossRef.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J.-C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42–49 CrossRef CAS.
- (a) A. C. Fahrenbach, J. C. Barnes, D. A. Lanfranchi, H. Li, A. Coskun, J. J. Gassensmith, Z. Liu, D. Benítez, A. Trabolsi, W. A. Goddard III, M. Elhabiri and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 3061–3072 CrossRef CAS; (b) C. Cheng, T. Cheng, H. Xiao, M. D. Krzyaniak, Y. Wang, P. R. McGonigal, M. Frasconi, J. C. Barnes, A. C. Fahrenbach, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 8288–8300 CrossRef CAS.
- (a) H. Li, A. C. Fahrenbach, S. K. Dey, S. Basu, A. Trabolsi, Z. Zhu, Y. Y. Botros and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8260–8265 CrossRef CAS PubMed; (b) J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS.
- (a) C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714–4723 CrossRef CAS; (b) C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS.
- (a) J. Deng, N. Song, Q. Zhou and Z. Su, Org. Lett., 2007, 26, 5393–5396 CrossRef; (b) A. Iordache, M. Oltean, A. Milet, F. Thomas, B. Baptiste, E. Saint-Aman and C. Bucher, J. Am. Chem. Soc., 2012, 134, 2653–2671 CrossRef CAS; (c) C. Kahlfuss, S. Denis-Quanquin, N. Calin, E. Dumont, M. Garavelli, G. Royal, S. Cobo, E. Saint-Aman and C. Bucher, J. Am. Chem. Soc., 2016, 138, 15234–15242 CrossRef CAS.
- (a) H. Li, Z. Zhu, A. C. Fahrenbach, B. M. Savoie, C. Ke, J. C. Barnes, J. Lei, Y.-L. Zhao, L. M. Lilley, T. J. Marks, M. A. Ratner and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 456–467 CrossRef CAS PubMed; (b) J. Sun, Z. Liu, W.-G. Liu, Y. Wu, Y. Wang, J. C. Barnes, K. R. Hermann, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 12704–12709 CrossRef CAS.
- (a) M. Berville, L. Karmazin, J. A. Wytko and J. Weiss, Chem. Commun., 2015, 51, 15772–15775 RSC; (b) M. Berville, S. Choua, C. Gourlaouen, C. Boudon, L. Ruhlmann, C. Bailly, S. Cobo, E. Saint-Aman, J. Wytko and J. Weiss, ChemPhysChem, 2017, 18, 796–803 CrossRef CAS; (c) K. Cai, M. C. Lipke, Z. Liu, J. Nelson, T. Cheng, Y. Shi, C. Cheng, D. Shen, J.-M. Han, S. Vemuri, Y. Feng, C. L. Stern, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Nat. Commun., 2018, 9, 5275 CrossRef.
- A bolded descriptor denotes a compound, be it free or complexed, and an unbolded descriptor refers to either (i) a component within a molecule or (ii) a component part of a mechanically interlocked molecule.
- M. B. Nielsen, J. G. Hansen and J. Becher, Eur. J. Org. Chem., 1999, 11, 2807–2815 CrossRef.
- H. Scheytza, O. Rademacher and H.-U. Reißig, Eur. J. Org. Chem., 1999, 9, 2373–2381 CrossRef.
- P. R. Ashton, J. A. Preece, J. F. Stoddart, M. S. Tolley, A. J. P. White and D. J. Williams, Synthesis, 1994, 12, 1344–1352 CrossRef.
- J. C. Barnes, M. Juríček, N. A. Vermeulen, E. J. Dale and J. F. Stoddart, J. Org. Chem., 2013, 78, 11962–11969 CrossRef CAS.
- (a) H. J. Schneider, D. Guettes and U. Schneider, J. Am. Chem. Soc., 1988, 110, 6449–6454 CrossRef CAS; (b) C. E. Chang and M. K. Gilson, J. Am. Chem. Soc., 2004, 126, 13156–13164 CrossRef CAS; (c) Y. Shi, K. Cai, H. Xiao, Z. Liu, J. Zhou, D. Shen, Y. Qiu, Q.-H. Guo, C. L. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13835–13842 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- M. R. Geraskina, A. S. Dutton, M. J. Juetten, S. A. Wood and A. H. Winter, Angew. Chem., Int. Ed., 2017, 56, 9435–9439 CrossRef CAS.
- All the four bisradical dicationic cyclophanes have strong intermolecular radical–radical interactions in the solid states, enlarging their cavity sizes significantly. Disorder plagued the crystal structures of the asymmetric PyBB4+ and ThBB4+, making it difficult to determine their cavity sizes accurately. Therefore, DFT-calculated cavity sizes for these bisradical dicationic cyclophanes are probably a better basis for estimating their real cavity sizes.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation. CCDC 1955702–1955711. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04860j |
| This journal is © The Royal Society of Chemistry 2020 |