
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydration of nitriles using a metal–ligand cooperative ruthenium pincer catalyst†
Beibei
Guo
a,
Johannes G.
de Vries
 b and
Edwin
Otten
*a
b and
Edwin
Otten
*a
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: edwin.otten@rug.nl
bLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Strasse 29a, 18059 Rostock, Germany
First published on 7th October 2019
Abstract
Nitrile hydration provides access to amides that are important structural elements in organic chemistry. Here we report catalytic nitrile hydration using ruthenium catalysts based on a pincer scaffold with a dearomatized pyridine backbone. These complexes catalyze the nucleophilic addition of H2O to a wide variety of aliphatic and (hetero)aromatic nitriles in tBuOH as solvent. Reactions occur under mild conditions (room temperature) in the absence of additives. A mechanism for nitrile hydration is proposed that is initiated by metal–ligand cooperative binding of the nitrile.
Introduction
Amides are an important class of compounds that occur in a large variety of biologically active compounds, polymers and synthetic intermediates,1 and a variety of synthetic methods have been developed for their formation.2 Nitrile hydration provides an atom-efficient synthesis of amides and is carried out on an industrial scale, for example in the production of acrylamide3 and nicotinamide.4 The direct nucleophilic addition of water to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond is kinetically slow, and a variety of catalysts have been developed, but it is often difficult to prevent over-hydrolysis to the corresponding carboxylic acids.5 Nitrile hydratase enzymes yield amides with excellent selectivity,6 but the limited substrate scope prevents their widespread use. There has been significant recent interest in the development of nitrile hydration catalysts based on transition metals, but most systems reported to date still require relatively high temperatures to reach appreciable catalytic turnover.7 Recent progress with Rh(I),8 Ru(II),9 Pd(II),10 and Pt(II)11 catalysts has allowed catalytic nitrile hydration under mild conditions, but additives are often required for high activity (e.g., AgOTf in Grubbs' Pt(II)/phosphinous acid catalysts,11 or Sc(OTf)3 in Yin's Pd(II) catalysts10).
N bond is kinetically slow, and a variety of catalysts have been developed, but it is often difficult to prevent over-hydrolysis to the corresponding carboxylic acids.5 Nitrile hydratase enzymes yield amides with excellent selectivity,6 but the limited substrate scope prevents their widespread use. There has been significant recent interest in the development of nitrile hydration catalysts based on transition metals, but most systems reported to date still require relatively high temperatures to reach appreciable catalytic turnover.7 Recent progress with Rh(I),8 Ru(II),9 Pd(II),10 and Pt(II)11 catalysts has allowed catalytic nitrile hydration under mild conditions, but additives are often required for high activity (e.g., AgOTf in Grubbs' Pt(II)/phosphinous acid catalysts,11 or Sc(OTf)3 in Yin's Pd(II) catalysts10).
Metal complexes with pincer ligands have found widespread use in a large variety of catalytic reactions,12 but the incorporation of a reactive fragment in the ligand backbone to enable ‘bifunctional’ or ‘cooperative’ substrate binding/activation has only recently started to emerge.13 Examples include the (reversible) binding of unsaturated fragments such as CO2,14 SO2,15 carbonyl compounds,16 or nitriles.17 Our group reported that Milstein's dearomatized Ru PNN pincer complex APNN (Scheme 1, left) catalyzes the conjugate addition of alcohols to α,β-unsaturated nitriles via a metal–ligand cooperative (MLC) mechanism by activation of the nitrile CN bond.18 Very recently, similar reactivity was observed with a related Mn catalyst.19 This mode of activation reduces the bond order from 3 in the nitrile (CN) to 2 in the MLC intermediate (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–Ru), which significantly alters the reactivity profile of the substrate.
N–Ru), which significantly alters the reactivity profile of the substrate.
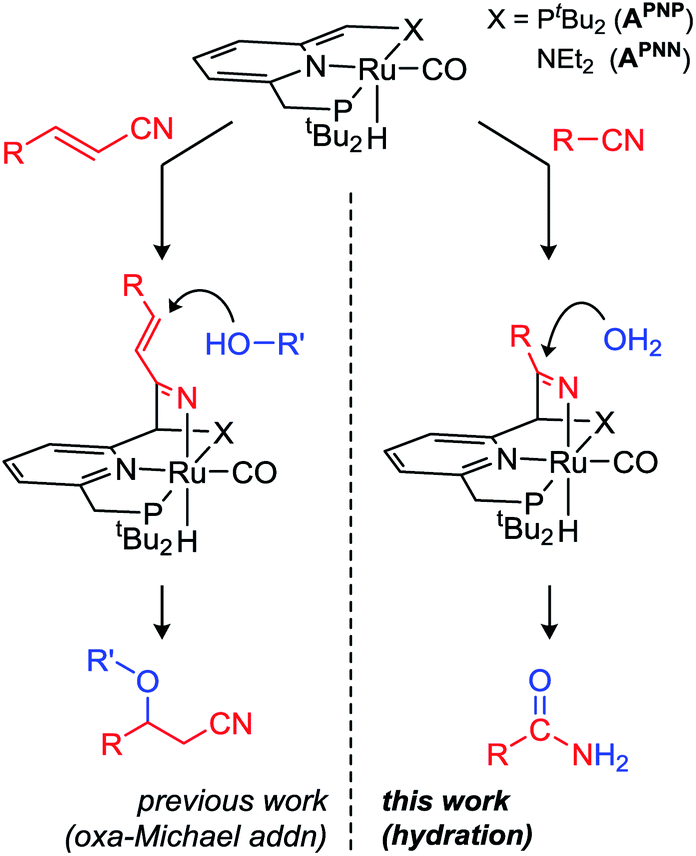 | ||
| Scheme 1 MLC activation of nitriles towards addition of O-nucleophiles. | ||
Having established that conjugate (1,4-) addition of weak alcohol nucleophiles is enabled by metal–ligand cooperation, we hypothesized that also 1,2-additions to MLC-activated nitriles may be feasible. Herein we describe our results on catalytic nitrile hydration using Ru complexes with dearomatized pyridine-based pincer ligands, and demonstrate that a large variety of aliphatic and (hetero)aromatic nitriles is selectively converted to the corresponding amides under very mild conditions.
Results and discussion
Catalyst development and reaction scope
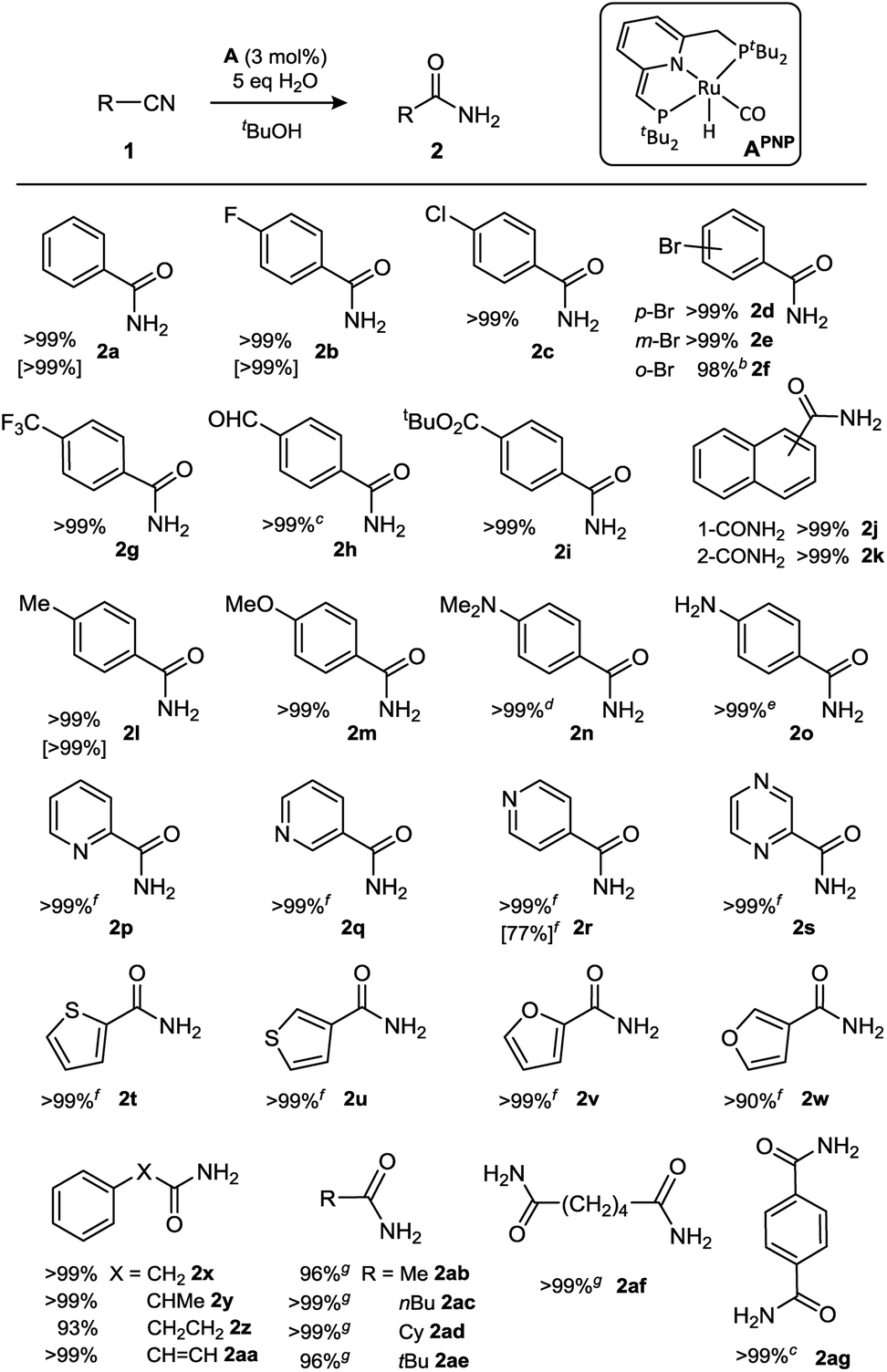
Previously it was found that the direct conjugate addition of H2O to α,β-unsaturated nitriles was sluggish using catalyst APNN.18c However, for some of the substrates tested we noticed that amides were obtained in low yield by hydration of the nitrile moiety. Encouraged by these observations, we initiated a screening of reaction conditions for the hydration of acetonitrile. These initial results (see ESI†) showed that both PNN complex APNN and the symmetrical PNP analogue APNP are active, and performed best in tBuOH solvent. An optimum in catalyst activity was found when 5 equivalents of H2O were added, which gave 63% conversion to acetamide after 24 h at room temperature (3 mol% catalyst APNP). Repeating these reactions with either 2 or 8 equiv. of H2O present gave lower nitrile conversions of 44 and 42%, respectively, and the reaction is almost completely suppressed in the presence of 20 equiv. of H2O (4% conversion after 24 h). In view of the propensity of complexes such as A to heterolytically cleave OH bonds (including H2O) in a reversible manner,20 it is likely that the nitrile, H2O and other components in the reaction mixture compete for reaction with the dearomatized complexes A, and the presence of increased amounts of water will result in a larger equilibrium concentration of Ru-hydroxides (vide infra). The decrease in catalyst activity at H2O amounts beyond the optimum (5 equiv.) indicates that in the present catalyst system, Ru-hydroxides are likely not the active species. Although a variety of transition metal hydroxide species have been reported to catalyze nitrile hydration, these often require elevated temperatures.21 Control experiments carried out in the absence of the dearomatized Ru pincer complexes, either with 3 mol% KOtBu or the ruthenium precursor to APNP (the aromatic (PNP)Ru(H)(Cl)(CO) complex) led to no conversion, indicating that the metal–ligand cooperative character of A is important in this catalytic conversion.We subsequently focused on examining the scope of substrates that underwent hydration to the amide using APNP as catalyst (Table 1). A selection of substrates was also subjected to catalysis by the nonsymmetrical APNN catalyst, which resulted in similar results (Table 1, entries in brackets).
a Isolated yield after reaction at room temperature for 1 day, unless noted otherwise. Conversions using catalyst APNN under the same conditions are given in square brackets.
b Reaction at 50 °C for 2 days.
c Reaction in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of THF/tBuOH due to poor solubility of the starting material.
d Reaction at 50 °C for 20 hours.
e Reaction at 80 °C for 16 hours.
f Reaction at room temperature for 1 day with 0.5 mol% catalyst loading.
g Reaction at 50 °C for 24 hours. :1 mixture of THF/tBuOH due to poor solubility of the starting material.
d Reaction at 50 °C for 20 hours.
e Reaction at 80 °C for 16 hours.
f Reaction at room temperature for 1 day with 0.5 mol% catalyst loading.
g Reaction at 50 °C for 24 hours.
|
|---|
|
Substrates with halogen substituents (1b–f) as well as electron-withdrawing trifluoromethyl (1g) or aldehyde groups (1h) at the para-position all afforded the corresponding amides in quantitative isolated yields. In case of aldehyde 1h, its poor solubility in tBuOH resulted in slow conversion under the standard conditions, but addition of THF as co-solvent afforded a homogeneous solution and resulted in facile amide formation. For the three bromo-substituted substrates (1d–f), conversion of the hindered ortho-isomer was sluggish at room temperature, but at 50 °C the reaction went to completion within 2 days. The hydration reaction also occurred with naphthylnitriles (1j,k). The presence of an electron-withdrawing para-nitro substituent completely suppressed the reaction at room temperature; heating to 80 °C restored some catalytic activity, but the reaction stalled at ca. 25% conversion under those conditions. Catalyst deactivation by the nitro-group does not seem to take place, as hydration of 1b in the presence of an equimolar amount of nitrobenzene gives full conversion to 2b at a rate that is similar to that in the absence of nitrobenzene (see ESI†). A likely explanation is that the p-nitro benzamide product, which has a relatively acidic N–H bond, competes with the nitrile substrate for binding with the catalyst and leads to product inhibition (vide infra). Benzonitriles with electron-donating substituents (p-Me, -OMe, -NR2) are also hydrated, although the aniline derivatives required heating to achieve full conversion (1n: 50 °C for 20 h; 1o: 80 °C for 16 h). In the case of the free aniline 1o, this may be due to competing (reversible) N–H bond activation to form Ru-anilido species.22 Consistent with this hypothesis is the observation that 4-hydroxybenzonitrile is not converted even at 80 °C, indicating that the acidic Ar–OH moiety deactivates the catalyst in an irreversible manner.18a,23
Unhindered esters such as 4-acetoxybenzonitrile and ethyl 4-cyanobenzoate also do not form the amide products, likely because of competing ester hydrolysis to the carboxylic acids. For more hindered esters, however, the reaction works well as demonstrated by full conversion of tert-butyl 4-cyanobenzoate (1i).
Heteroaromatic nitriles are also converted, with even shorter reaction times under the standard conditions (<1 h with 3 mol% catalyst APNP). Consequently, this class of substrates reaches full conversion within a day at room temperature using a catalyst loading of 0.5 mol% (TON = 200). The ortho-, meta- and para-isomers of cyanopyridine (1p–r), as well as the pyrazine derivative (1s) are converted to the amide products in quantitative yields. Moreover, substrates with oxygen- (furan, 1t,u) or sulfur-containing rings (thiophenes, 1v,w), structural motifs that are often considered catalyst poisons, underwent this ruthenium-catalyzed process with remarkable ease.
Substrates with sp3-C substituents adjacent to the CN group react more slowly but nevertheless are converted with complete selectivity to the amide. For example, benzyl cyanide (1x) and its α-methylated analogue (1y) show full conversion to the amide within 1 day at room temperature, and the same applies to 3-phenylpropionitrile (1z) and the sp2-substituted cinnamonitrile (1aa). While purely aliphatic nitriles also react at room temperature, these substrates require longer reaction times to reach synthetically useful conversions (65–83% after 2 days). However, gentle heating of the reaction mixtures to 50 °C for 1 day affords amide products in >99% isolated yield for primary, secondary as well as tertiary alkylnitriles (1ab–ae). Substrates with two nitrile moieties were selectively converted to the corresponding diamides (1af/1ag).
The substrates shown in Table 1 are converted by catalysts A already at room temperature (and many reach full conversion within 24 h), but, with the exception of heteroaromatic nitriles, turnover numbers (TONs) for the majority of entries are only modest due to the presence of 3 mol% catalyst (maximum TON = 33). To examine whether higher turnover numbers can be obtained, we tested the reaction at elevated temperature (70 °C) for aromatic nitrile 1b and aliphatic dinitrile 1af. For 1b, hydration is initially fast (63% conversion in 1 h), but then gradually slows down to reach 98% conversion in 20 h under those conditions (TON = 196). Dinitrile 1af also gave >98% conversion of the starting material in 24 h at 70 °C to afford a mixture of mono- and diamide products in a 57:43 ratio, which corresponds to a total nitrile TON of 307. Moreover, with 3-cyanopyridine (1q) the catalyst reaches 1000 turnovers within a day at 70 °C, demonstrating the robustness of the catalyst at elevated temperature.
Mechanistic considerations
To obtain insight in the species present during turnover, we monitored a catalysis reaction mixture (3 mol% APNP, 5 eq. H2O in tBuOH) with the fluoro-substituted benzonitrile 1b by 19F NMR spectroscopy. This showed the presence of three distinct 19F-containing species. Two of these correspond to the nitrile starting material and the amide product by comparison to authentic samples. A third species was present throughout the course of the reaction in minor amount (19F NMR: δ −118 ppm, ca. 2.5% based on integration). This resonance was also observed in the 19F NMR of a catalysis mixture with APNN, suggesting that APNP and APNN lead to similar speciation under the reaction conditions.To characterize this species and develop an understanding of the individual steps that might be involved in the catalysis, we carried out stoichiometric NMR scale experiments in THF-d8 between the dearomatized pincer complexes A and the components present in the catalysis reaction mixture (see ESI for details†). Analysis of a 1:1 mixture of complex APNP and nitrile 1b by 1H NMR spectroscopy showed a substantial broadening of the Ru–H signal which is additionally shifted downfield by ca. 7 ppm, indicating a rapidly exchanging equilibrium between the starting materials and the Ru-nitrile adduct BPNP (Scheme 2).18b
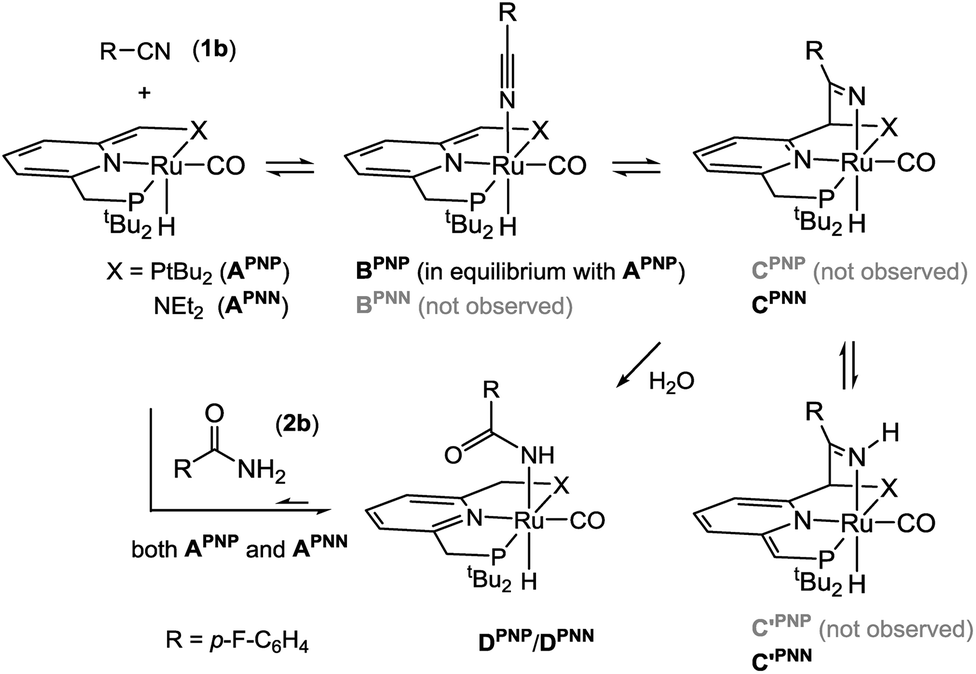 | ||
| Scheme 2 Stoichiometric reactions between A and the components present in the reaction mixture. | ||
The sterically less hindered PNN analogue APNN reacts with nitrile 1b to an equilibrium mixture of CPNN and C′PNN according to NMR spectroscopy. In this mixture, a characteristic 1H NMR resonance is observed at δ 11.65 (singlet) for the NH fragment in C′PNN. Additionally, signals at δ −10.47/−13.75 ppm (doublets, JPH = 25.7/32.8 Hz) appear for the Ru–H groups in C′PNN/CPNN. The NMR features are very similar to those observed previously by us for the reaction of APNN and benzonitrile,18b and confirm that the latter products arise from MLC binding of the CN bond. The different outcomes of the reaction between 1b and APNN or APNP indicates that the divergent steric demands of the PNP vs. PNN ligand affects the equilibria between species A, B and C. Although a detailed comparison between PNN- and PNP-based catalysts is beyond the scope of this research, preliminary DFT calculations indicate that MLC-binding of benzonitrile is exergonic at the N-arm (−2.7 kcal mol−1) whereas it is endergonic at the P-arm (PNN: +9.4 kcal mol; PNP: +7.2 kcal mol−1). It should be noted that, even though the relative stability of the various intermediates is sensitive to steric effects, metal–ligand cooperative reaction pathways are involved in complexes with both these ligands.24
Regardless of the different equilibrium compositions, addition of H2O (1 equiv.) to a THF-d8 solution of both the PNN- or PNP-based mixture resulted in the slow appearance of a new species (D) with a 19F NMR shift (ca. δ −118 ppm) that agrees well with the one observed during catalysis in tBuOH. Compounds D were also obtained cleanly by treatment of the dearomatized Ru pincer complexes A with amide 2b, and are formulated as Ru-carboxamides (DPNN/DPNP, Scheme 2) on the basis of their NMR spectra (see ESI†).
The observation that D is the dominant Ru-containing species during turnover suggests that it may be a dormant catalyst state, and that catalysis could be subject to product inhibition. Indeed, when a mixture of nitrile substrate and amide product (33 and 10 equiv., respectively, relative to APNP) is present at the start, the reaction rate is decreased but full conversion is still obtained (see ESI†). Also, when DPNP is prepared independently and tested in catalysis, hydration of 1b occurs with a rate that is qualitatively similar to that with APNP.
These observations indicate that the coordinatively saturated complex D is able to generate the active species via a rapid equilibrium with A. This was further confirmed by the observation that DPNN reacts with nitrile 1b to regenerate the equilibrium mixture of CPNN and C′PNN, a reaction that presumably involves APNN as an intermediate (Scheme 2). For the PNP analogue, the concentration of DPNP decreases upon addition of H2O, and NMR resonances attributable to the free amide 2b appear. From these data it is clear that the catalyst speciation in this system is complex: the dearomatized pincer complexes A are involved in dynamic equilibria with H2O, nitrile, as well as amide. Thus, even though under the reaction conditions most of the Ru is present in a coordinatively saturated, inactive form (D), access to catalytically active species is kinetically facile and allows turnover of the nitrile substrate.
Mechanistic proposals for catalytic nitrile hydration in the literature often involve Lewis acid activation of the nitrile, sometimes in conjunction with ligand-mediated (‘bifunctional’) deprotonation of H2O,25,26 or nucleophilic attack of a reactive ligand–OH fragment (e.g., in catalysts with phosphinous acid ligands).27 For bimetallic complexes, a reactive metal-hydroxide group can be generated adjacent to a metal-nitrile adduct, which provides a low-energy pathway to hydration.28
On the basis of the results discussed above, we propose that the mechanism of nitrile hydration by A is distinct and follows the steps illustrated in Scheme 3. Under the reaction conditions, complexes A react reversibly with amide or H2O to form the off-cycle species D and E, respectively. Although these species are not catalytically relevant, a rapid equilibrium between these dormant states and A ensures an entry into the catalytic cycle. We propose that catalysis is initiated by MLC binding of the nitrile substrate (1) to form C. The MLC mode of CN bond activation results in a reduced CN bond order of 2, and transfers the basicity ‘stored’ in the pincer framework in A onto the nitrile-derived Ru–N moiety in C. This allows deprotonation of the pro-nucleophile H2O and attack at the electrophilic C-atom of the activated nitrile to form F, either directly or facilitated by a hydrogen-bond network involving additional H2O or tBuOH as proton-shuttles.29,27b The coordinated hydroxyamido fragment in intermediate F may be liberated from the metal complex by a retro-cycloaddition to form the iminol, which tautomerizes in solution to the final amide product (2). Alternative pathways that directly convert F to the Ru-carboxamide D cannot be ruled out at present. The proposed reaction pathway accounts for several key experimental observations, including (i) the requirement for a reactive ligand site for catalytic activity, (ii) an optimum in catalyst activity as function of the amount of water added, (iii) and the occurrence of product inhibition.
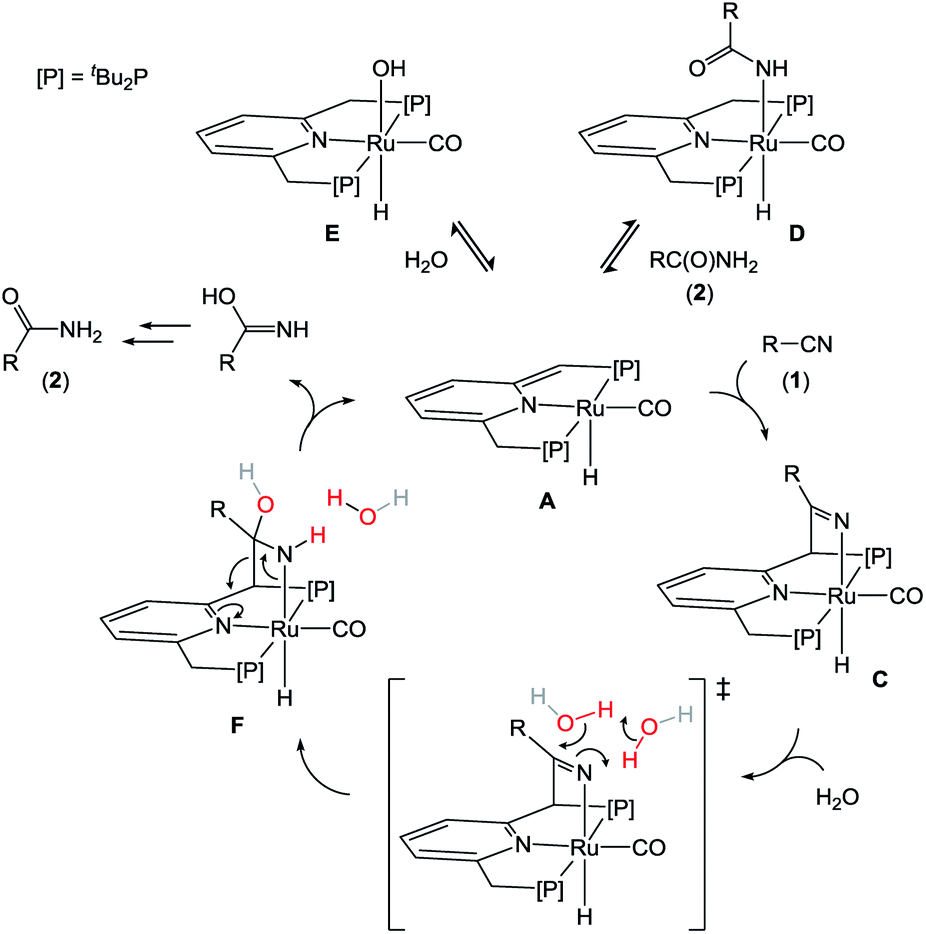 | ||
| Scheme 3 Proposed catalytic cycle. | ||
Conclusions
In summary, this work describes an efficient homogeneous catalyst for the hydration of nitriles. Complexes based on dearomatized PNP or PNN ligands are shown to be active under very mild reaction conditions (ambient temperature, additive-free). The PNP Ru pincer catalyst is tolerant to a variety of functional groups, and allows the hydration of a broad range of aliphatic, aromatic and heteroaromatic nitriles. On the basis of stoichiometric experiments, a mechanism is proposed that involves metal–ligand cooperative activation of the nitrile CN bond. The results suggest that the generation of intermediates with a CN moiety (i.e., a reduced bond order in comparison to the nitrile starting material) via this mode of nitrile activation significantly increases its reactivity and leads to facile attack by (pro)nucleophiles as weak as H2O. We anticipate that this strategy may be more broadly applicable and lead to novel reactivity of nitriles and other unsaturated organic compounds. A more detailed examination of the catalytic reaction mechanism, as well as modification of the catalyst to increase productivity (e.g., by minimizing product inhibition) are ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Netherlands Organisation for Scientific Research (NWO) (VIDI grant to EO) and the China Scholarship Council (grant to BG) is gratefully acknowledged. We would like to thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high performance computing cluster.Notes and references
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (b) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- (a) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed.
- T. Ohara, T. Sato, N. Shimizu, G. Prescher, H. Schwind, O. Weiberg, K. Marten and H. Greim, in Ullmann's Encyclopedia of Industrial Chemistry, 2011, DOI:10.1002/14356007.a01_161.pub3.
- R. Blum, in Ullmann's Encyclopedia of Industrial Chemistry, 2015, pp. 1–9 Search PubMed.
- (a) C. O'Connor, Q. Rev., Chem. Soc., 1970, 24, 553–564 RSC; (b) T. Tu, Z. Wang, Z. Liu, X. Feng and Q. Wang, Green Chem., 2012, 14, 921–924 RSC.
- (a) M. Kobayashi and S. Shimizu, Nat. Biotechnol., 1998, 16, 733 CrossRef CAS PubMed; (b) P. K. Mascharak, in Molecular Design in Inorganic Biochemistry, ed. D. Rabinovich, Springer-Verlag, Berlin, Heidelberg, 2014, pp. 89–113 Search PubMed; (c) J.-S. Gong, J.-S. Shi, Z.-M. Lu, H. Li, Z.-M. Zhou and Z.-H. Xu, Crit. Rev. Biotechnol., 2017, 37, 69–81 CrossRef CAS PubMed.
- (a) S.-I. Murahashi and H. Takaya, Acc. Chem. Res., 2000, 33, 225–233 CrossRef CAS PubMed; (b) V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef CAS; (c) T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949–974 CrossRef CAS; (d) R. García-Álvarez, J. Francos, E. Tomás-Mendivil, P. Crochet and V. Cadierno, J. Organomet. Chem., 2014, 771, 93–104 CrossRef; (e) E. L. Downs and D. R. Tyler, Coord. Chem. Rev., 2014, 280, 28–37 CrossRef CAS.
- A. Goto, K. Endo and S. Saito, Angew. Chem., Int. Ed., 2008, 47, 3607–3609 CrossRef CAS PubMed.
- M. Nirmala, G. Saranya and P. Viswanathamurthi, Inorg. Chim. Acta, 2016, 442, 134–144 CrossRef CAS.
- S. Zhang, H. Xu, C. Lou, A. M. Senan, Z. Chen and G. Yin, Eur. J. Org. Chem., 2017, 2017, 1870–1875 CrossRef CAS.
- X. Xing, C. Xu, B. Chen, C. Li, S. C. Virgil and R. H. Grubbs, J. Am. Chem. Soc., 2018, 140, 17782–17789 CrossRef CAS PubMed.
- (a) G. Van Koten and D. Milstein, Organometallic Pincer Chemistry, Springer Berlin Heidelberg, 2013 Search PubMed; (b) D. Morales-Morales, Pincer Compounds: Chemistry and Applications, Elsevier, 2018 Search PubMed.
- (a) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (c) H. Li, T. P. Gonçalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS; (d) J. I. van der Vlugt, in Pincer Compounds: Chemistry and Applications, ed. D. Morales-Morales, Elsevier, 2018, pp. 599–621 Search PubMed.
- (a) C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643–4645 CrossRef CAS; (b) M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem.–Eur. J., 2012, 18, 9194–9197 CrossRef CAS PubMed; (c) M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6445–6454 CrossRef CAS PubMed; (d) M. Feller, E. Ben-Ari, Y. Diskin-Posner and D. Milstein, J. Coord. Chem., 2018, 71, 1679–1689 CrossRef CAS.
- R. Stichauer and M. Vogt, Organometallics, 2018, 37, 3639–3643 CrossRef CAS.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Chem. Commun., 2013, 49, 7147–7149 RSC.
- (a) A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CrossRef CAS PubMed; (b) M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed; (c) G. A. Filonenko, E. Cosimi, L. Lefort, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2014, 4, 2667–2671 CrossRef CAS.
- (a) S. Perdriau, D. S. Zijlstra, H. J. Heeres, J. G. de Vries and E. Otten, Angew. Chem., Int. Ed., 2015, 54, 4236–4240 CrossRef CAS PubMed; (b) L. E. Eijsink, S. C. P. Perdriau, J. G. de Vries and E. Otten, Dalton Trans., 2016, 45, 16033–16039 RSC; (c) B. Guo, D. S. Zijlstra, J. G. de Vries and E. Otten, ChemCatChem, 2018, 10, 2868–2872 CrossRef CAS PubMed.
- S. Tang and D. Milstein, Chem. Sci., 2019 10.1039/c9sc03269j.
- (a) S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS PubMed; (b) C. L. Mathis, J. Geary, Y. Ardon, M. S. Reese, M. A. Philliber, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2019, 141, 14317–14328 CrossRef PubMed.
- (a) J. Chin and J. H. Kim, Angew. Chem., Int. Ed., 1990, 29, 523–525 CrossRef; (b) N. H. Anderson, J. M. Boncella and A. M. Tondreau, Organometallics, 2018, 37, 4675–4684 CrossRef CAS.
- E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef CAS PubMed.
- S. Perdriau, M.-C. Chang, E. Otten, H. J. Heeres and J. G. de Vries, Chem.–Eur. J., 2014, 20, 15434–15442 CrossRef CAS PubMed.
- (a) H. Li and M. B. Hall, ACS Catal., 2015, 5, 1895–1913 CrossRef CAS; (b) C. Hou, Z. Zhang, C. Zhao and Z. Ke, Inorg. Chem., 2016, 55, 6539–6551 CrossRef CAS PubMed.
- (a) R. García-Álvarez, J. Díez, P. Crochet and V. Cadierno, Organometallics, 2010, 29, 3955–3965 CrossRef; (b) S. M. M. Knapp, T. J. Sherbow, R. B. Yelle, J. J. Juliette and D. R. Tyler, Organometallics, 2013, 32, 3744–3752 CrossRef CAS; (c) W.-C. Lee, J. M. Sears, R. A. Enow, K. Eads, D. A. Krogstad and B. J. Frost, Inorg. Chem., 2013, 52, 1737–1746 CrossRef CAS PubMed; (d) S. M. M. Knapp, T. J. Sherbow, R. B. Yelle, L. N. Zakharov, J. J. Juliette and D. R. Tyler, Organometallics, 2013, 32, 824–834 CrossRef CAS; (e) M. K. Rong, K. van Duin, T. van Dijk, J. J. M. de Pater, B.-J. Deelman, M. Nieger, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Organometallics, 2017, 36, 1079–1090 CrossRef CAS PubMed; (f) R. González-Fernández, P. Crochet and V. Cadierno, ChemistrySelect, 2018, 3, 4324–4329 CrossRef.
- (a) T. Oshiki, H. Yamashita, K. Sawada, M. Utsunomiya, K. Takahashi and K. Takai, Organometallics, 2005, 24, 6287–6290 CrossRef CAS; (b) P. Daw, A. Sinha, S. M. W. Rahaman, S. Dinda and J. K. Bera, Organometallics, 2012, 31, 3790–3797 CrossRef CAS; (c) K. Singh, A. Sarbajna, I. Dutta, P. Pandey and J. K. Bera, Chem.–Eur. J., 2017, 23, 7761–7771 CrossRef CAS PubMed.
- (a) T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657–8660 CrossRef CAS; (b) E. Tomás-Mendivil, V. Cadierno, M. I. Menéndez and R. López, Chem.–Eur. J., 2015, 21, 16874–16886 CrossRef PubMed; (c) R. González-Fernández, P. Crochet, V. Cadierno, M. I. Menéndez and R. López, Chem.–Eur. J., 2017, 23, 15210–15221 CrossRef PubMed.
- (a) C. J. McKenzie and R. Robson, J. Chem. Soc., Chem. Commun., 1988, 112–114 RSC; (b) E. C. Wilkinson, Y. Dong and L. Que, J. Am. Chem. Soc., 1994, 116, 8394–8395 CrossRef CAS; (c) F. Meyer, E. Kaifer, P. Kircher, K. Heinze and H. Pritzkow, Chem.–Eur. J., 1999, 5, 1617–1630 CrossRef CAS; (d) P. J. Zinn, T. N. Sorrell, D. R. Powell, V. W. Day and A. S. Borovik, Inorg. Chem., 2007, 46, 10120–10132 CrossRef CAS PubMed.
- (a) M. A. Iron, E. Ben-Ari, R. Cohen and D. Milstein, Dalton Trans., 2009, 9433–9439 RSC; (b) J. Li, Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2009, 131, 13584–13585 CrossRef CAS PubMed; (c) S. Qu, Y. Dang, C. Song, M. Wen, K.-W. Huang and Z.-X. Wang, J. Am. Chem. Soc., 2014, 136, 4974–4991 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04624k |
| This journal is © The Royal Society of Chemistry 2019 |