
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of ethanol from aryl methyl ether/lignin, CO2 and H2†
Jingjing
Zhang
 ab,
Qingli
Qian
*ac,
Ying
Wang
ab,
Bernard Baffour
Asare Bediako
ab,
Jiang
Yan
ab and
Buxing
Han
*abcd
ab,
Qingli
Qian
*ac,
Ying
Wang
ab,
Bernard Baffour
Asare Bediako
ab,
Jiang
Yan
ab and
Buxing
Han
*abcd
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: qianql@iccas.ac.cn; hanbx@iccas.ac.cn
bSchool of Chemistry and Chemical Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cPhysical Science Laboratory, Huairou National Comprehensive Science Center, No. 5 Yanqi East Second Street, Beijing 101400, China
dShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 2nd October 2019
Abstract
Currently, ethanol is produced via hydration of ethene or fermentation of foods. Lignin and CO2 are abundant, cheap and renewable feedstocks. Synthesis of ethanol using the lignin or its derivatives is of great importance, but is a great challenge and has rarely been reported. Herein, we propose a route to synthesize ethanol from CO2, H2, and lignin or various aryl methyl ethers, which can be derived from lignin. The reaction could be effectively conducted using Ru–Co bimetallic catalyst and the TON of ethanol could reach 145. Interestingly, ethanol was the only liquid product when lignin was used. A series of control experiments indicate that ethanol was formed via cleavage of aryl ether bond, reverse water gas shift (RWGS) reaction, and C–C bond formation. This protocol opens a way to produce ethanol using abundant renewable resources.
Introduction
Lignin and CO2 are abundant renewable carbon resources.1 Transformation of lignin and/or CO2 into value-added chemicals has attracted great interest.2–15 Lignin is a major component of lignocellulosic biomass, and its valorization remains a challenge because of the complex and robust structure.16 Aryl methyl ethers are commonly used as model compounds to study lignin conversion, which has been successfully transformed into various chemicals, such as benzene, phenol, terephthalic acid, cyclohexanone, cyclohexane, methanol, methane, acetates, acetic acid, etc.17–26 Ethanol is an important bulk chemical and is widely used in human life, especially as an alternative motor fuel. Recently, several important works on converting cellulose to ethanol have been published.27,28 However, synthesis of ethanol using lignin or its derivatives has rarely been reported so far.29,30 Without doubt, synthesis of ethanol using lignin is highly desirable, but is challenging.In this paper, we report the protocol to produce ethanol from lignin or its derivatives, CO2, and H2 (Fig. 1). The reaction involves several key steps including cleavage of ether bond, reverse water gas shift (RWGS) reaction, and C–C bond formation. The reaction could be effectively accelerated by Ru–Co bimetallic catalyst and the TON of ethanol could reach 145.
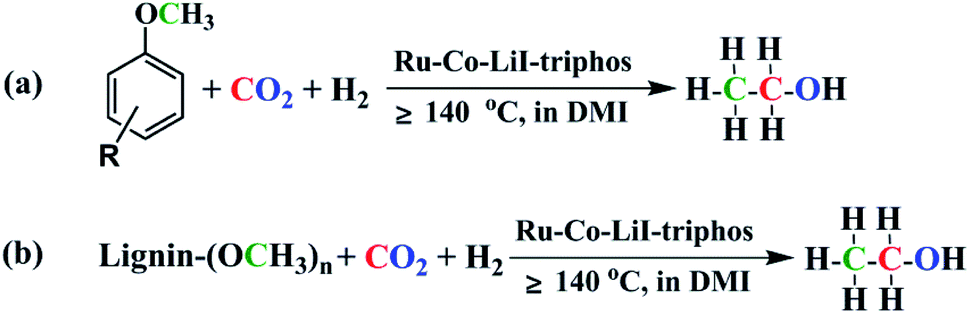 | ||
| Fig. 1 Synthesis of ethanol from aryl methyl ethers (a)/lignins (b), CO2 and H2. | ||
To the best of our knowledge, this is the first work of ethanol synthesis by combining lignin or its derivatives, and CO2, which are abundant renewable feedstocks.
Results and discussion
Catalytic system
We started the catalyst screening with the simplest aryl methyl ether (anisole) as the lignin model compound. The results are revealed in Table 1. The reaction could be effectively accelerated by Ru–Co bimetallic catalyst with 1,1,1-tris(diphenylphosphinomethyl)ethane (triphos) ligand and LiI promoter in 1,3-dimethyl-2-imidazolidinone (DMI) solvent (entry 1). The TON of ethanol based on Ru catalyst was 73. The byproducts of the reaction were phenol, CO, methane and trace propanol. Only minor ethanol could form when single [RuCl2(CO)3]2 was used as catalyst, while no ethanol was observed when Co2(CO)8 catalyst was applied separately (entries 2, 3). This suggested that Ru complex was the major catalyst and Co complex was the cocatalyst. The TON of ethanol rose significantly by combining Ru and Co catalysts, indicating their synergy during the reaction. No ethanol was detected after the reaction when we combined other transition metal complexes (Cu, Ir, Fe) with Co2(CO)8 (entries 4–6). We also combined [RuCl2(CO)3]2 with other transition metal complexes (Fe, Ni), but the reaction rate had little increase or the reaction did not occur at all (entries 7, 8). Thus Ru–Co bimetallic catalyst rendered better catalytic performance. We also tested other combinations of Ru/Co complexes and found that [RuCl2(CO)3]2 and Co2(CO)8 were fit for the reaction (entries 1, and 9–12).| Entry | Catalyst | Promoter | Ligand | Solvent | TONb |
|---|---|---|---|---|---|
| a Reaction conditions: 20 μmol Ru catalyst and 60 μmol Co catalyst (based on the metal), 20 μmol ligand, 2.2 mmol promoter, 2 mL solvent, 3.6 mmol anisole, 3 MPa CO2 and 5 MPa H2 (at room temperature), 190 °C, 10 h. b TON denotes moles of ethanol produced per mole of Ru catalyst. c The yield of ethanol based on anisole was 40.6%. Acronyms: 1,3-dimethyl-2-imidazolidinone (DMI), ruthenium(III) acetylacetonate (Ru(acac)3), 1,1,1-tris(diphenylphosphinomethyl)ethane (triphos), triphenylphosphine (PPh3), 1,2-bis(diphenylphosphino)ethane (dppe), bis(triphenylphosphoranylidene)ammonium chloride (PPNCl), tetrahydrofuran (THF), 1-methyl-2-pyrrolidinone (NMP), 1-ethyl-2-pyrrolidinone (NEP), tetramethylurea (TMU). | |||||
| 1c | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | DMI | 73 |
| 2 | [RuCl2(CO)3]2 | LiI | Triphos | DMI | 3 |
| 3 | Co2(CO)8 | LiI | Triphos | DMI | 0 |
| 4 | CuCl2, Co2(CO)8 | LiI | Triphos | DMI | 0 |
| 5 | IrCl3, Co2(CO)8 | LiI | Triphos | DMI | 0 |
| 6 | Fe2(CO)9, Co2(CO)8 | LiI | Triphos | DMI | 0 |
| 7 | [RuCl2(CO)3]2, Fe2(CO)9 | LiI | Triphos | DMI | 4 |
| 8 | [RuCl2(CO)3]2, NiBr2 | LiI | Triphos | DMI | 0 |
| 9 | Ru(acac)3, Co2(CO)8 | LiI | Triphos | DMI | 36 |
| 10 | Ru3(CO)12, Co2(CO)8 | LiI | Triphos | DMI | 46 |
| 11 | [RuCl2(CO)3]2, Co(PPh3)3Cl | LiI | Triphos | DMI | 46 |
| 12 | [RuCl2(CO)3]2, CoCl2 | LiI | Triphos | DMI | 41 |
| 13 | [RuCl2(CO)3]2, Co2(CO)8 | — | Triphos | DMI | 0 |
| 14 | [RuCl2(CO)3]2, Co2(CO)8 | NaI | Triphos | DMI | 32 |
| 15 | [RuCl2(CO)3]2, Co2(CO)8 | KI | Triphos | DMI | 11 |
| 16 | [RuCl2(CO)3]2, Co2(CO)8 | MgI2 | Triphos | DMI | 1 |
| 17 | [RuCl2(CO)3]2, Co2(CO)8 | ZnI2 | Triphos | DMI | 0 |
| 18 | [RuCl2(CO)3]2, Co2(CO)8 | LiCl | Triphos | DMI | 4 |
| 19 | [RuCl2(CO)3]2, Co2(CO)8 | LiBr | Triphos | DMI | 32 |
| 20 | [RuCl2(CO)3]2, Co2(CO)8 | I2 | Triphos | DMI | 0 |
| 21 | [RuCl2(CO)3]2, Co2(CO)8 | LiBF4 | Triphos | DMI | 0 |
| 22 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | — | DMI | 51 |
| 23 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Dppe | DMI | 59 |
| 24 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | PPh3 | DMI | 57 |
| 25 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Imidazole | DMI | 40 |
| 26 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | PPNCl | DMI | 48 |
| 27 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | NMP | 32 |
| 28 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | NEP | 39 |
| 29 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | Water | 0 |
| 30 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | TMU | 0 |
| 31 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | Toluene | 0 |
| 32 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | THF | 0 |
| 33 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | Acetonitrile | 0 |
| 34 | [RuCl2(CO)3]2, Co2(CO)8 | LiI | Triphos | Squalane | 0 |
The promoter was necessary for the catalysis because no ethanol was produced without it (entry 13). When NaI or KI were utilized instead of LiI, ethanol could also form but the catalytic activity dropped by the order: LiI > NaI > KI (entries 1, 14, 15). When alkaline earth metal iodide (MgI2) was used as promoter the reaction rate further decreased remarkably (entry 16). Whereas the transition metal iodide (ZnI2) could not promote the reaction at all (entry 17). The anionic counterpart of the promoter also played an important role in the reaction. When LiI was substituted by other lithium halides (LiCl, LiBr), the TON of ethanol diminished by the sequence: LiI > LiBr > LiCl (entries 1, 18, 19). Further experiment using iodine (I2) demonstrated that ionic halogen was needed to promote the reaction (entry 20). We also tried LiBF4, but the reaction did not take place (entry 21). The superiority of LiI as the promoter could be ascribed to the stronger Lewis acidity and smaller size of the Li+, and the better nucleophilicity of the I−.31,32
As a tridentate phosphine ligand, triphos could effectively accelerate the reaction activity (entries 1, 22). To study the role of different phosphine ligands, we adopted the bidentate ligand dppe and monodentate ligand PPh3 in the experiment respectively, but they were less effective than triphos (entries 1, 23, 24). The sequence of their promoting effect was as follows: triphos > dppe > PPh3. We also tried other ligands, such as imidazole and PPNCl, but they inhibited the reaction (entries 22, 25, 26). Hence triphos was a suitable ligand for the reaction. It is reported that triphos displayed eminent role in converting renewable molecules including CO2.33,34
Besides DMI, cyclic amides of similar structure (NMP, NEP) could be used as reaction solvent, but they are not as good as DMI (entries 1, 27, 28). Ethanol could not be detected when the reaction was conducted in other solvents, such as water, TMU, toluene, THF, acetonitrile or squalane (entries 29–34). The solvent effect of the cyclic amides has been discussed elsewhere.32 In short, the catalytic system consisting of [RuCl2(CO)3]2, Co2(CO)8, LiI, triphos, and DMI was appropriate for the reaction.
Impact of reaction conditions
Based on the above catalytic system, we studied the impact of the reaction temperature (Fig. 2). The reaction began to occur at 140 °C. The catalytic activity enhanced quickly with elevating temperature. At 190 °C, the TON of ethanol reached 73 and the increase of reaction rate became minor when the temperature was further raised. At 190 °C, we investigated the influence of other reaction parameters, i.e., dosage of each catalyst component and pressure of each reactant gas. The results and detailed discussion were provided in the ESI (Table S1†). The suitable dosages of the catalytic components were 20 μmol Ru catalyst and 60 μmol Co catalyst (based on the metal), 20 μmol triphos, and 2.2 mmol LiI. The appropriate gas pressure was 3 MPa CO2 and 5 MPa H2 (at room temperature). In brief, the condition in entry 1 of Table 1 was the best for the reaction.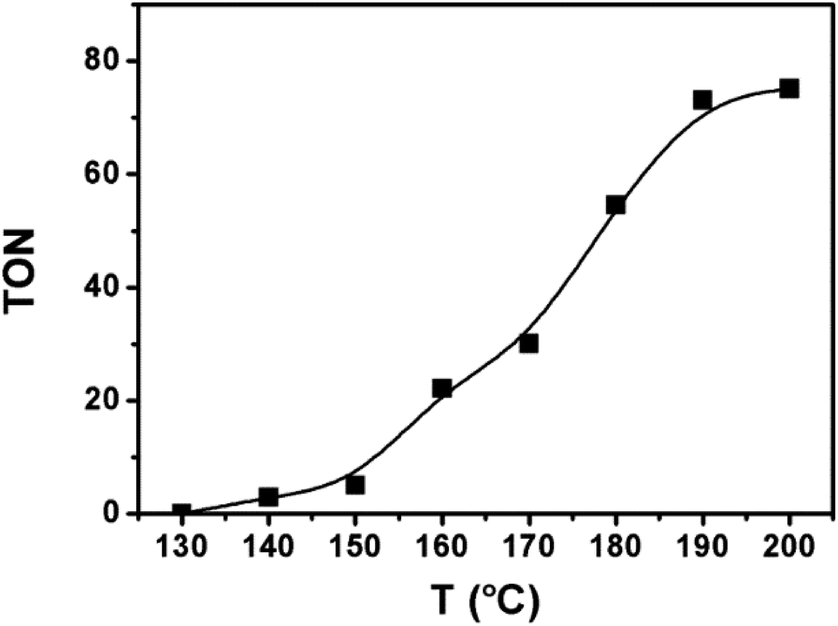 | ||
| Fig. 2 The TON of ethanol at different temperatures. Reaction conditions: 20 μmol [RuCl2(CO)3]2 and 60 μmol Co2(CO)8 (based on the metal), 20 μmol triphos, 2.2 mmol LiI, 2 mL DMI, 3.6 mmol anisole, 3 MPa CO2 and 5 MPa H2 (at room temperature), 10 h. | ||
Reaction mechanism
Fig. 3 displayed the time course of the reaction. CO formed at the beginning of the reaction and it increased quickly with time. With the fast production of ethanol, the CO content in the reactor stopped growing and kept at a constant level, suggesting that CO was an intermediate for ethanol synthesis. This assumption was confirmed by the control experiment using CO instead of CO2 (entry 23 of Table S1†). The CO was generated via RWGS reaction catalyzed by Ru catalyst (Fig. S1†). The Ru catalyzed RWGS reaction has been reported in the literature.35,36 Propanol was also observed after 6 h and it grew very slowly with time. The propanol may be formed via ethanol homologation.37 Methane was observed at the start of the reaction and it increased steadily during the reaction. After 10 h, the rise of ethanol was less obvious, indicating that 10 h was the appropriate reaction time.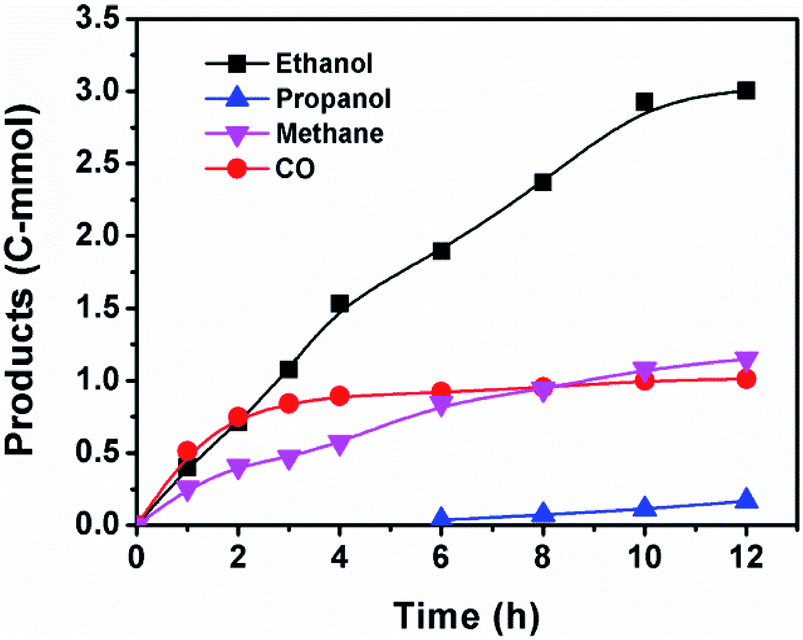 | ||
| Fig. 3 Time course of the reaction. Reaction conditions: 20 μmol [RuCl2(CO)3]2 and 60 μmol Co2(CO)8 (based on the metal), 20 μmol triphos, 2.2 mmol LiI, 2 mL DMI, 3.6 mmol anisole, 3 MPa CO2 and 5 MPa H2 (at room temperature), 190 °C. | ||
As a substrate, anisole was indispensable for the reaction, because little ethanol was detected without it (entry 11 of Table S1†). The anisole decomposed gradually during the reaction and its aryl part was converted to phenol (Fig. S2†). The phenol was stable at reaction condition and did not experience further transformation (Fig. S3†). The stability of phenol was further verified by the control experiment using phenol as substrate instead of anisole (Fig. S4†). The LiI was responsible for the decomposition of the anisole, while other catalytic components and reactant gases could assist this step (Table S2†). The LiI itself could decompose the anisole (entry 1). All the other catalytic components could not decompose the anisole without LiI (entry 2), while they greatly accelerated the decomposition (entries 1, 3). The gases could also markedly help the LiI to decompose the anisole (entries 1, 4). As for the transition metal catalysts, the Ru catalyst was more effective than Co catalyst in promoting the anisole decomposition (entries 4–6). The ligand triphos was also beneficial to the anisole decomposition (entries 3, 7). The LiBF4 was more efficient than LiI in decomposing the anisole (entry 8), but no ethanol was observed after the reaction (entry 21 of Table 1). Therefore the LiI could not only cleave the ether bond of anisole but also was essential to ethanol formation.
After anisole decomposition, trace CH3I was detected and further converted to ethanol in the presence of CO and H2 (Fig. S5 and S6†). CH3I is a common intermediate of ethanol synthesis via methanol homologation with CO and H2.32,38,39 Therefore, in this work CH3I should be a key intermediate generated by decomposing anisole, which subsequently took part in the ethanol formation via reductive carbonylation reaction. This deduction was further supported by the 13CO2 labeling test, where CH313CH2OH was formed in the reaction (Fig. S7†). This suggested that the methyl group (–CH3) in ethanol was from anisole and the –CH2OH group was derived from CO2. Methane in the product was generated by further hydrogenation of CH3I and/or CO (Fig. S8 and S9†). The control experiment using D2 revealed that obvious H–D exchange occurred in the reaction (Fig. S10†). During the transfer of CH3 group from anisole substrate to ethanol and/or propanol, the H atoms on CH3 could be totally substituted by the D atoms. As for the unreacted anisole, the H atoms on the CH3 were intact at the reaction condition. The H–D exchange reactions have been investigated elsewhere.40
Based on above discussion, we proposed the possible reaction pathway (Fig. 4). The generation of ethanol mainly involves three cascade reactions: cleavage of ether bond to form CH3I (1), RWGS reaction to generate CO (2), and subsequent ethanol synthesis via reductive carbonylation (3). It is possible that the carbonyls in the catalyst precursors participated in forming the ethanol, but the impact could be neglected because of the following reasons. Firstly, no ethanol was observed without CO2 or H2 (entries 12, 13 of the Table S1†). Secondly, the 13CO2 labeling test indicated that CH313CH2OH was the product in the reaction (Fig. S7†). This indicated that CO2 rather than the carbonyls in the catalyst precursor formed ethanol. Finally, the HR-ESI-MS characterization of the catalyst after the reaction suggested that the carbonyls were mainly retained on the catalyst (Fig. S11†).
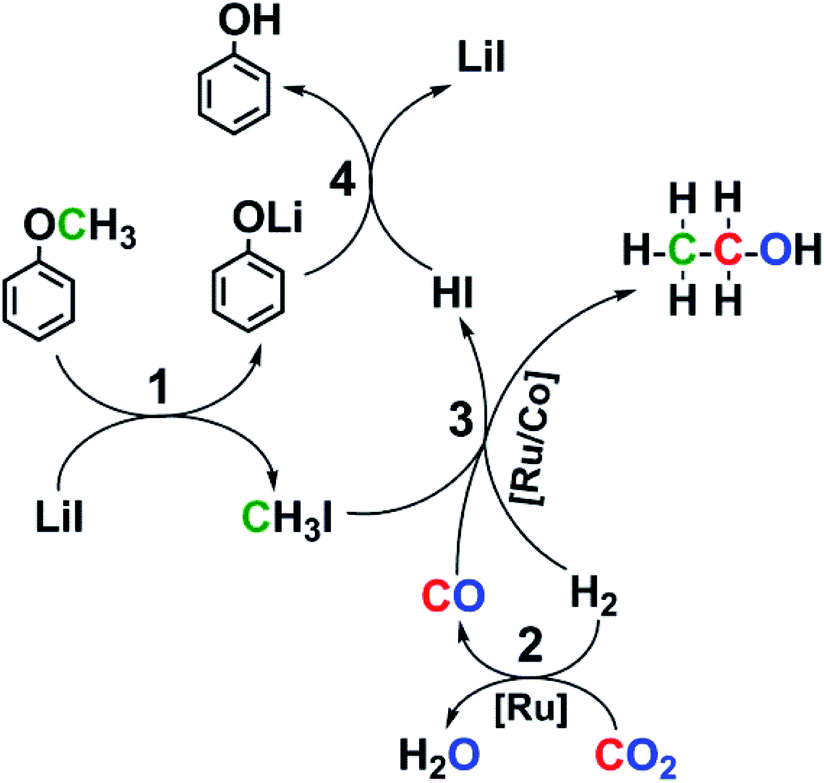 | ||
| Fig. 4 The proposed reaction pathway. | ||
Substrate extension
We also tested other aryl methyl ethers as substrate to produce ethanol (Table 2). Guaiacol and syringol are naturally-occurring organic compound, which can be derived from lignin. They were both suitable substrates for producing ethanol (entries 1 and 2). When another hydroxyl substituted aryl methyl ether, 4-methoxyphenol was tested, the reaction also proceeded well (entry 3). Based on the results of reactions using syringol and 1,4-dimethoxybenzene, we can deduce that two methoxyl groups on each substrate molecule participated in forming ethanol (entries 2, 4). The TON of ethanol using 1,4-dimethoxybenzene as substrate was as high as 145. We also tried aryl methyl ethers with different alkyl substituents, which are electron-donating groups (entries 5–10). The results indicated that the presence of the alkyl groups on the aryl methyl ether slightly decreased the reactivity. Furthermore, the aryl methyl ethers with methyl substituent at different positions had different reactivity, and they followed the order: para > meta > ortho (entries 5–7). The substrates with electron-withdrawing groups, such as 4-trifluoromethyl anisole, 3-trifluoromethyl anisole, and 3,5-difluoroanisole, were also tested in the reaction (entries 11–13). It was found that the electron-withdrawing groups on substrates lowered the reactivity more obviously than electron-donating groups. As for the impact of the position of the trifluoromethyl group, the substrate with the substituent on para position was less reactive than that on meta position (entries 11, 12). When 2-methoxynaphthalene, which had one more fused aromatic ring than anisole, was used as substrate the reaction also occurred efficiently (entry 14). The GC-MS graphs of the reaction solutions in Table 2 are provided in the ESI (Fig. S12–S25†). The results demonstrate that the aromatic part of each substrate was converted into corresponding phenols. The catalytic system could also apply to lignin, where the methoxyl group took part in generating ethanol (entries 15, 16). The contents of methoxyl groups in eucalyptus lignin and willow lignin used in this work were 15.8 wt% and 16.7 wt% respectively, which was determined by iodine stoichiometry titration method.41 The eucalyptus lignin and willow lignin could produce 87.5 and 94.4 mg(ethanol) g(lignin)−1, respectively. Interestingly, ethanol was the only liquid product after the reaction using lignin as substrate.| Entry | Substrates | TON of ethanol | Yield of ethanolc (%) | Yield of propanolc (%) | Conversion of substrated (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 20 μmol [RuCl2(CO)3]2 and 60 μmol Co2(CO)8 (based on the metal), 20 μmol triphos, 2.2 mmol LiI, 2 mL DMI, 3.6 mmol substrate, 3 MPa CO2 and 5 MPa H2 (at room temperature), 190 °C, 10 h. b 0.4 g lignin was added before the reaction. c The yield was calculated based on the mole of methoxyl group in the substrate. d The aryl part of the aryl methyl ether was converted into the corresponding phenol, thus the yield of phenol was equal or very close to the conversion of the corresponding substrate. | |||||
| 1 |
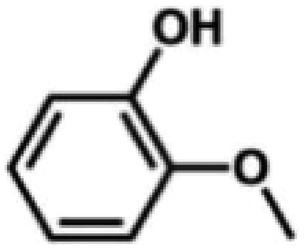
|
74 | 41.1 | 1.4 | 100 |
| 2 |
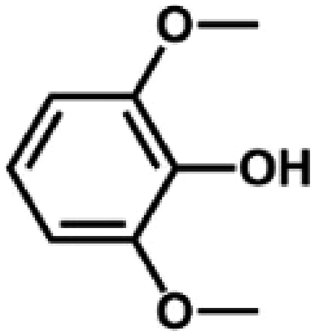
|
126 | 35.0 | 1.3 | 100 |
| 3 |

|
72 | 40.0 | <1 | 71.2 |
| 4 |
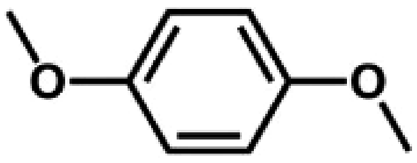
|
145 | 40.3 | 1.0 | 74.3 |
| 5 |
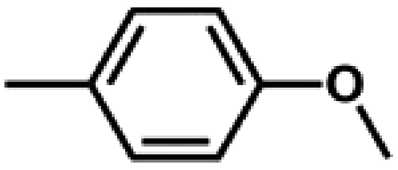
|
72 | 40.0 | 1.2 | 79.2 |
| 6 |
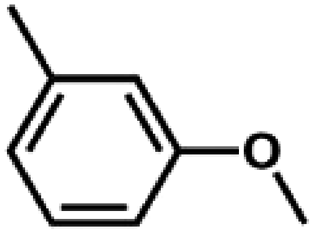
|
69 | 38.3 | <1 | 76.4 |
| 7 |
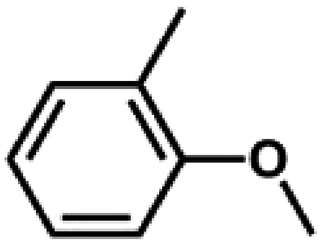
|
61 | 33.9 | <1 | 74.7 |
| 8 |
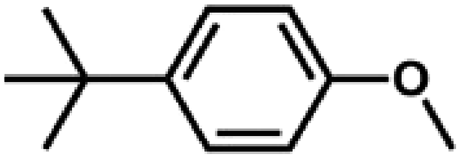
|
71 | 39.4 | <1 | 77.5 |
| 9 |
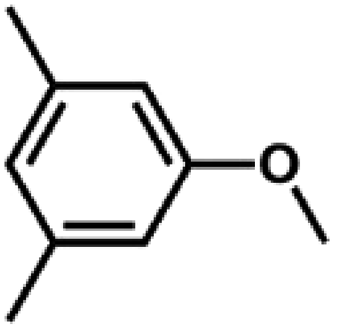
|
68 | 37.8 | <1 | 76.1 |
| 10 |

|
72 | 40.0 | 1.1 | 100 |
| 11 |
|
51 | 28.3 | <1 | 100 |
| 12 |
|
60 | 33.3 | <1 | 100 |
| 13 |
|
58 | 32.2 | <1 | 100 |
| 14 |
|
65 | 36.1 | 1.1 | 83.3 |
| 15b | Eucalyptus lignin | 38 | 37.3 | 0 | 76.5 |
| 16b | Willow lignin | 41 | 38.1 | 0 | 77.8 |
Conclusions
In summary, we have developed the way to synthesize ethanol from Aryl methyl ethers/lignin, CO2 and H2. The reaction could proceed effectively using Ru–Co bimetallic catalyst, LiI promoter and triphos ligand. Ethanol could form at above 140 °C, and at optimized condition the TON of ethanol was as high as 145. The catalytic system has good adaptability to various aryl methyl ethers and lignin of different sources. The reactions using eucalyptus lignin and willow lignin as substrate could produce 87.5 and 94.4 mg(ethanol) g(lignin)−1, respectively, moreover, ethanol was the only liquid product. The generation of ethanol mainly involves three cascade reactions, i.e., cleavage of ether bond to form CH3I, RWGS reaction to generate CO, and subsequent ethanol formation via C–C bond formation. Ru was responsible for the RWGS reaction. Ru and Co cooperatively accelerated the ethanol synthesis. The catalytic system could apply to various aryl methyl ethers and lignin of different sources. To our knowledge, this is the first report of ethanol production using aryl methyl ethers/lignin and CO2. It provides a new strategy of ethanol production, biomass and CO2 valorization. We believe that this work will trigger more research on bulk chemicals synthesis by combining CO2 and biomass.Experimental
Chemicals
Dichlorotricarbonyl ruthenium dimer ([RuCl2(CO)3]2, 98+%), ruthenium(III) acetylacetonate (Ru(acac)3, 98%), ruthenium(III) iodide, iridium(III) chloride, anhydrous (IrCl3, 99.99%), cobalt(III) 2,4-pentanedionate (Co(acac)3, 98%), cobalt(II) chloride (CoCl2, 99.9%), chlorotris(triphenylphosphine)cobalt(I) (Co(PPh3)3Cl, 97%), cobalt(II) bromide (CoBr2, 97%), diiron nonacarbonyl (Fe2(CO)9, 99%), copper(II) chloride (CuCl2, 98%), lithium iodide (LiI, 99.95%), 1,1,1-tris(diphenylphosphinomethyl)ethane (triphos, 97+%), triphenylphosphine (PPh3, 99+%), sodium iodide (NaI, 99.95%), bis(triphenylphosphoranylidene)ammonium chloride (PPNCl, 98+%), potassium iodide (KI, 99.9%), magnesium iodide (MgI2, 99.996%), lithium tetrafluoroborate (LiBF4, 98%), 1-methyl-2-pyrrolidinone (NMP, 99%), imidazole (99%), tetramethylurea (TMU, 99%) and 4-tert-butylanisole (98%) were purchased from Alfa Aesar China Co, Ltd. Dicobalt octacarbonyl (Co2(CO)8, stabilized with 1–5% hexane) and lithium chloride (LiCl, 98%) were supplied by TCI Shanghai Co., Ltd. Lithium bromide (LiBr, 99.5%), 1,4-dimethoxybenzene (99+%), 2-methylanisole (98+%), 3-methylanisole (98+%), 4-methylanisole (99+%), 3,5-difluoroanisole (98+%), 4-methoxybenzotrifluoride (98+%), 3-methoxybenzotrifluoride (98+%) and 1-methoxynaphthalene (97+%) were purchased from Adamas Reagent Co., Ltd. 1,3-Dimethyl-2-imidazolidinone (DMI, 99%), 3,5-dimethylanisole (98+%), 2,6-dimethoxyphenol (98%), 2-methoxy-4-methylphenol (98+%) and guaiacol (98%) were provided by Aladdin Reagent. Zinc iodide (ZnI2, 98%), nickel bromide (NiBr2, 99%), anisole (99%), acetonitrile (99.9%), iodine (I2, 99.5%) and squalane (99%) were bought from Acros Organics. 1-Ethyl-2-pyrrolidinone (NEP, 99%), tetrahydrofuran (THF, 99.5%) and 1,2-bis(diphenylphosphino)ethane (dppe, 97%) were supplied by J&K Scientific Ltd. Toluene (99.5+%) was obtained from Beijing Chemical Works. CO2 (99.99%), H2 (99.99%) and CO (99.99%) were provided by Beijing Analytical Instrument Company. Deuterium gas (D2, 99.999%) was offered by Zhengzhou Xingdao Chemical Technology Co., Ltd. Carbon dioxide-13C (13CO2, 99% 13C) was bought from Beijing Gaisi Chemical Gases Center.Catalytic reaction
All the experiments were carried out in a 16 mL Teflon-lined stainless steel reactor equipped with a magnetic stirrer. The inner diameter of the reactor was 18 mm. In a typical experiment, certain amounts of the Ru–Co catalyst, promoter, ligand, the substrates, and 2 mL solvent were added into the reactor. At room temperature, CO2 and H2 were charged sequentially into the reactor to desired pressure after the reactor was purged with 1 MPa CO2 for three times. The reactor was placed in an air bath of constant temperature, and the magnetic stirrer was set at 800 rpm. After the reaction, the reactor was cooled in an ice-water bath. Then the residual gas was released slowly and collected in a gasbag. Using toluene as the internal standard, the reaction solution was analyzed by GC (Agilent 7890B) equipped with a flame ionization detector and a DB-5 capillary column (0.25 mm in diameter, 30 m in length). The liquid products were identified using GC-MS (Agilent-7890B-5977A) and/or GC-MS (Shimadzu-QP2010) with electron impact (EI) ionization, as well as by comparing the retention times with the standards in the GC traces. The yields of the products were calculated from the GC data. The gaseous samples were detected by a GC (Agilent 4890D) equipped with a TCD detector and a packed column (carbon molecular sieve TDX-01, 3 mm in diameter and 1 m in length) using Argon as the carry gas.Extraction of lignin
The procedure was similar to that reported previously.42 To eucalyptus or willow (200 g) was added 1,4-dioxane (1440 mL) followed by 2 N HCl (160 mL) and the mixture was heated to a gentle reflux under a N2 atmosphere for 1 hour. The mixture was then cooled and the lignin containing liquor was collected by filtration. The collected liquor was partially concentrated in vacuo to give a gummy residue which was taken up in acetone/water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, ∼250 mL) and precipitated by addition to rapidly stirring water (2.5 L). The crude lignin was collected by filtration and dried under vacuum. The dried crude lignin was taken up in acetone/methanol (9:1) and precipitated by dropwise addition to rapidly stirring Et2O (2 L). The precipitated lignin was collected by filtration and dried under vacuum to give a purified eucalyptus or willow lignin (about 20 g). The lignin extracted by above procedure was used in subsequent catalytic reactions without further processing.
:1, ∼250 mL) and precipitated by addition to rapidly stirring water (2.5 L). The crude lignin was collected by filtration and dried under vacuum. The dried crude lignin was taken up in acetone/methanol (9:1) and precipitated by dropwise addition to rapidly stirring Et2O (2 L). The precipitated lignin was collected by filtration and dried under vacuum to give a purified eucalyptus or willow lignin (about 20 g). The lignin extracted by above procedure was used in subsequent catalytic reactions without further processing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (21875262, 21733011, 21533011), Beijing Municipal Science & Technology Commission (Z181100004218004), National Key Research and Development Program of China (2017YFA0403102), and the Chinese Academy of Sciences (QYZDY-SSW-SLH013).Notes and references
- M. Y. He, Y. H. Sun and B. X. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, K. W. Ma, W. A. Elliott, P. Bai, R. L. Johnson, T. W. Walker, B. H. Shanks, R. M. Rioux, M. Neurock and J. A. Dumesic, Nat. Commun., 2019, 10, 1132 CrossRef PubMed.
- Y. Shao, Q. N. Xia, L. Dong, X. H. Liu, X. Han, S. F. Parker, Y. Q. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. H. Yang and Y. Q. Wang, Nat. Commun., 2017, 8, 16104 CrossRef PubMed.
- Y. M. Questell-Santiago, R. Zambrano-Varela, M. T. Amiri and J. S. Luterbacher, Nat. Chem., 2018, 10, 1222–1228 CrossRef CAS PubMed.
- A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226–20229 CrossRef CAS PubMed.
- J. L. Xu, N. Li, X. F. Yang, G. Y. Li, A. Q. Wang, Y. Cong, X. D. Wang and T. Zhang, ACS Catal., 2017, 7, 5880–5886 CrossRef.
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- K. Sordakis, C. H. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- Y. Liu, H. Zhou, J. Z. Guo, W. M. Ren and X. B. Lu, Angew. Chem., Int. Ed., 2017, 56, 4862–4866 CrossRef CAS PubMed.
- Q. Liu, L. P. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5593 Search PubMed.
- Q. Qian, J. Zhang, M. Cui and B. Han, Nat. Commun., 2016, 7, 11481 CrossRef PubMed.
- L. X. Wang, L. Wang, J. Zhang, X. L. Liu, H. Wang, W. Zhang, Q. Yang, J. Y. Ma, X. Dong, S. J. Yoo, J. G. Kim, X. J. Meng and F. S. Xiao, Angew. Chem., Int. Ed., 2018, 57, 6104–6108 CrossRef CAS PubMed.
- S. X. Bai, Q. Shao, P. T. Wang, Q. G. Dai, X. Y. Wang and X. Q. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- Z. Ying, C. Y. Wu, S. Jiang, R. H. Shi, B. Zhang, C. Zhang and F. Y. Zhao, Green Chem., 2016, 18, 3614–3619 RSC.
- Z. H. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- R. C. Runnebaum, R. J. Lobo-Lapidus, T. Nimmanwudipong, D. E. Block and B. C. Gates, Energy Fuels, 2011, 25, 4776–4785 CrossRef CAS.
- Q. L. Meng, M. Q. Hou, H. Z. Liu, J. L. Song and B. X. Han, Nat. Commun., 2017, 8, 14190 CrossRef PubMed.
- Q. Mei, Y. Yang, H. Liu, S. Li, H. Liu and B. Han, Sci. Adv., 2018, 4, eaaq0266 CrossRef PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- Z. C. Luo, Z. X. Zheng, L. Li, Y. T. Cui and C. Zhao, ACS Catal., 2017, 7, 8304–8313 CrossRef CAS.
- C. Hubert, A. Denicourt-Nowicki, J. P. Guegan and A. Roucoux, Dalton Trans., 2009, 36, 7356–7358 RSC.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- H. Wang, Y. F. Zhao, Z. G. Ke, B. Yu, R. P. Li, Y. Y. Wu, Z. P. Wang, J. J. Han and Z. M. Liu, Chem. Commun., 2019, 55, 3069–3072 RSC.
- S. Song, J. G. Zhang, G. Gozaydin and N. Yan, Angew. Chem., Int. Ed., 2019, 58, 4934–4937 CrossRef CAS PubMed.
- J. Chang, T. Danuthai, S. Dewiyanti, C. Wang and A. Borgna, ChemCatChem, 2013, 5, 3041–3049 CrossRef CAS.
- N. Yan and S. P. Ding, Trends in Chemistry, 2019, 1, 457–458 CrossRef.
- M. Yang, H. F. Qi, F. Liu, Y. J. Ren, X. L. Pan, L. L. Zhang, X. Y. Liu, H. Wang, J. F. Pang, M. Y. Zheng, A. Q. Wang and T. Zhang, Joule, 2019, 3, 1937–1948 CrossRef CAS.
- H. L. Wang, Y. Q. Pu, A. Ragauskas and B. Yang, Bioresour. Technol., 2019, 271, 449–461 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709–719 CrossRef CAS PubMed.
- K. Tominaga, Y. Sasaki, T. Watanabe and M. Saito, Stud. Surf. Sci. Catal., 1998, 114, 495–498 CrossRef CAS.
- Y. Wang, J. J. Zhang, Q. L. Qian, B. B. A. Bediako, M. Cui, G. Y. Yang, J. Yan and B. X. Han, Green Chem., 2019, 21, 589–596 RSC.
- I. Mellone, F. Bertini, L. Gonsalvi, A. Guerriero and M. Peruzzini, Chimia, 2015, 69, 331–338 CrossRef CAS PubMed.
- L. L. Zhang, Z. B. Han, L. Zhang, M. X. Li and K. L. Ding, Chin. J. Org. Chem., 2016, 36, 1824–1838 CrossRef CAS.
- K. Tsuchiya, J. D. Huang and K. Tominaga, ACS Catal., 2013, 3, 2865–2868 CrossRef CAS.
- W. L. Li, S. B. Guo and L. Guo, Catal. Surv. Asia, 2017, 21, 185–197 CrossRef CAS.
- M. Cui, Q. L. Qian, Z. H. He, Z. F. Zhang, J. Ma, T. B. Wu, G. Y. Yang and B. X. Han, Chem. Sci., 2016, 7, 5200–5205 RSC.
- M. Roper and H. Loevenich, J. Organomet. Chem., 1983, 255, 95–102 CrossRef.
- M. Roper, H. Loevenich and J. Korff, J. Mol. Catal., 1982, 17, 315–322 CrossRef.
- T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401–406 RSC.
- Q. Q. Mei, H. Z. Liu, X. J. Shen, Q. L. Meng, H. Y. Liu, J. F. Xiang and B. X. Han, Angew. Chem., Int. Ed., 2017, 56, 14868–14872 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03386f |
| This journal is © The Royal Society of Chemistry 2019 |