DOI:
10.1039/C9SC04501E
(Edge Article)
Chem. Sci., 2019,
10, 11054-11063
Controlled scrambling reactions to polyphosphanes via bond metathesis reactions†
Received
5th September 2019
, Accepted 16th October 2019
First published on 17th October 2019
Abstract
Triphosphanes R′2PP(R)PR′2 (9a,c: R = Py; 9b R = BTz), 1,3-diphenyl-2-pyridyl-triphospholane 9d and pentaphospholanes (RP)5 (13: R = Py; 18: R = BTz) are obtained in high yield of up to 98% from the reaction of dipyrazolylphosphanes RPpyr2 (5: R = Py; 6: R = BTz; pyr = 1,3-dimethylpyrazolyl) and the respective secondary phosphane (R′2PH, R′ = Cy (9a,b), tBu (9c); PhPH(CH2)2PHPh (9d)). The formation of derivatives 9a–d proceeds via a condensation reaction while the formation of 13 and 18 can only be explained by a selective scrambling reaction. We realized that the reaction outcome is strongly solvent dependent as outlined by the controlled scrambling reaction pathway towards pentaphospholane 13. In our further investigations to apply these compounds as ligands we first confined ourselves to the coordination chemistry of triphosphane 9a with respect to coinage metal salts and discussed the observation of different syn- and anti-isomeric metal complexes based on NMR and X-ray analyses as well as quantum chemical calculations. Methylation reactions of 9a with MeOTf yield triphosphan-1-ium Cy2MePP(Py)PCy2+ (10+) and triphosphane-1,3-diium Cy2MePP(Py)PMeCy22+ (112+) cations as triflate salts. Salt 11[OTf]2 reacts with pentaphospholane 13 in an unprecedented chain growth reaction to give the tetraphosphane-1,4-diium triflate salt Cy2MePP(Py)P(Py)PMeCy22+ (19[OTf]2) via a P–P/P–P bond metathesis reaction. The latter salt is unstable in solution and rearranges via a rare [1,2]-migration of the Cy2MeP-group followed by the elimination of the triphosph-2-en-1-ium cation [Cy2MePPPMeCy2]+ (20+) to yield a novel 1,4,2-diazaphospholium salt (21[OTf]).
Introduction
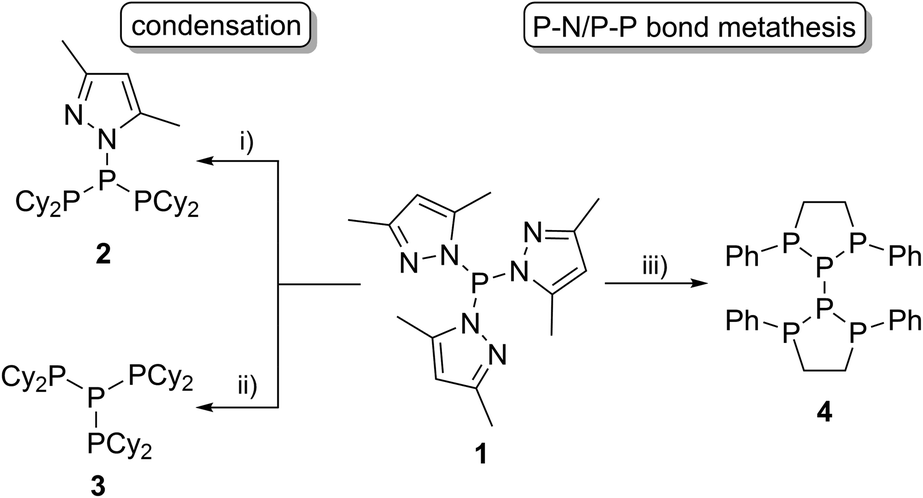
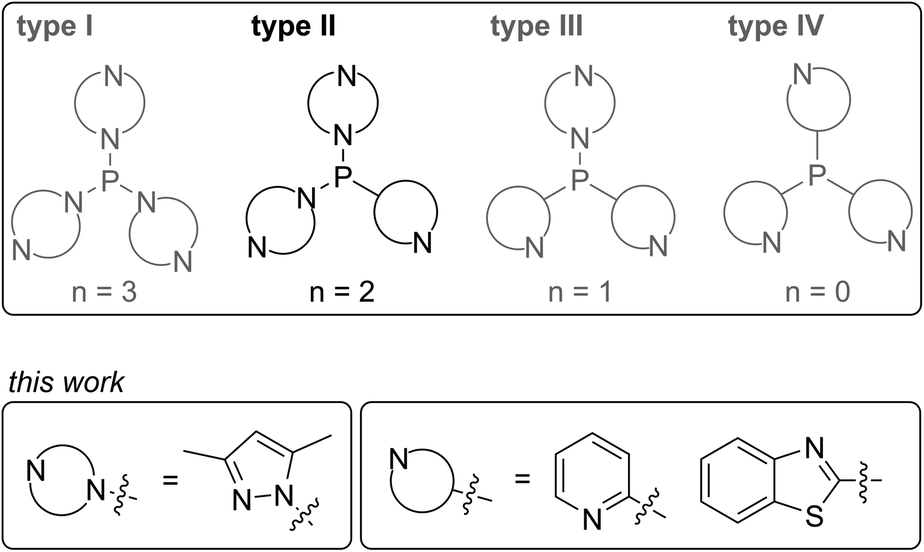
A wide range of polyphosphorus compounds, comprising diverse bonding motifs, have been synthesised in the last few decades.1,2 Many of the observed compounds are often found to be labile in solution and their rearrangement or decomposition reactions are discussed to proceed via intermolecular scrambling reactions.3 Even though there are early reports on controlling such scrambling reactions,4 the selective and high yielding synthesis of certain polyphosphanes is still difficult, hampering their subsequent chemistry. Besides classical approaches for the generation of P–P bonds such as salt metathesis reactions,2a,b dehalosilylation reactions2c or condensation reactions,2d our group focusses on the development of methodologies using tripyrazolylphosphane 1 or derivatives thereof (vide infra) as a P1-building unit for P–P bond forming reactions via condensation or P–N/P–P bond metathesis reactions (Scheme 1).5,6 In this regard, the reaction of 1 with Cy2PH to give triphosphane 2 or iso-tetraphosphane 3 depending on the applied stoichiometry has been reported.5 The reaction of 1 with 1,2-bis(phenylphosphanyl)ethane yields hexaphosphane 4 in a sequence of condensation and P–N/P–P bond metathesis reactions, showing an example of a controlled scrambling reaction.6 In our effort to further control these metathesis reactions and driven by our general interest in azole- and azine-substituted phosphanes, we explored the synthesis of phosphanes with a pyridyl- or benzothiazolyl-substituent bound to phosphorus via the carbon atom. This limits the number of P–N bound pyrazolyl-substituents and therefore the possible reaction pathways by P–N/P–P bond metathesis. For a better overview and in thought of a systematic approach we organised our general types of phosphanes with respect to their number of P–N bound substituents as type I to type IV (Scheme 2). In type I phosphanes all three substituents are bound via P–N bonds whereas a type II phosphane is defined by two P–N bonds and one P–C bond. This order is maintained in type III phosphanes with one P–N and two P–C bonds and is finalised in type IV phosphanes featuring only P–C bonds. Importantly, all substituents at the phosphorus atom have an additional sp2 hybridised donor atom such as nitrogen, being essential as a further reaction or coordination site and for the possible formation of resonance stabilised reaction intermediates. In this contribution, we report the synthesis of the hitherto unknown type II phosphanes carrying either a pyridyl- or benzothiazolyl-substituent and their use for the generation of triphosphanes, a triphospholane and pentaphospholanes and selected one triphosphane to explore its coordination chemistry with respect to coinage metal salts.
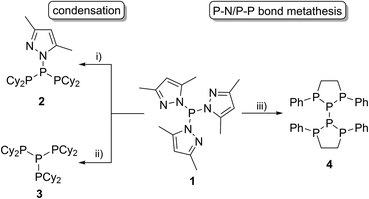 |
| | Scheme 1 Application of tripyrazolylphosphane 1 in P–P bond formation reactions; (i) 2 Cy2PH, –2 pyrH, CH3CN; (ii) 3 Cy2PH, –3 pyrH, and CH3CN; (iii) 5 PhPH(CH2)2PHPh, 4 Ppyr3, –PhP(pyr)(CH2)2P(pyr)Ph, –10 pyrH, CH3CN. | |
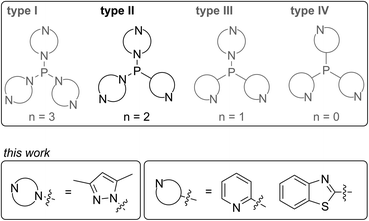 |
| | Scheme 2 Classification of N-heterocyclic substituted phosphanes into type I to type IV phosphanes (n is the number of P–N bonds) with an emphasis on the type II phosphanes presented in this contribution. | |
Results and discussion
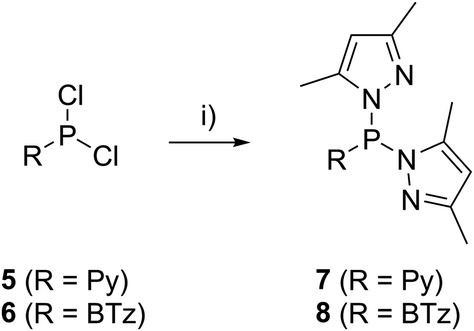
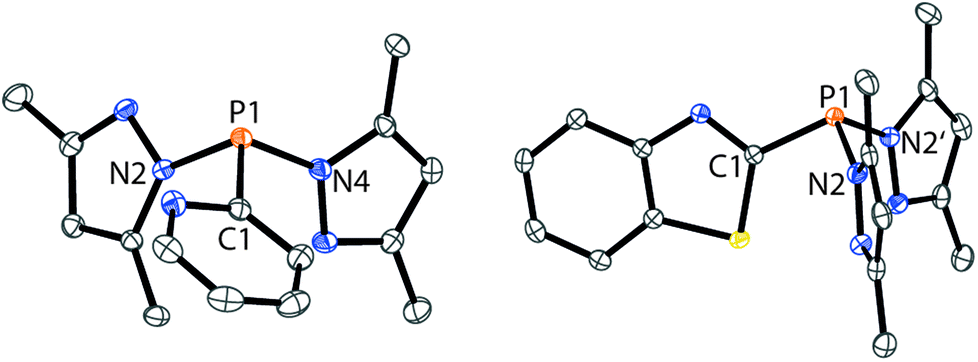
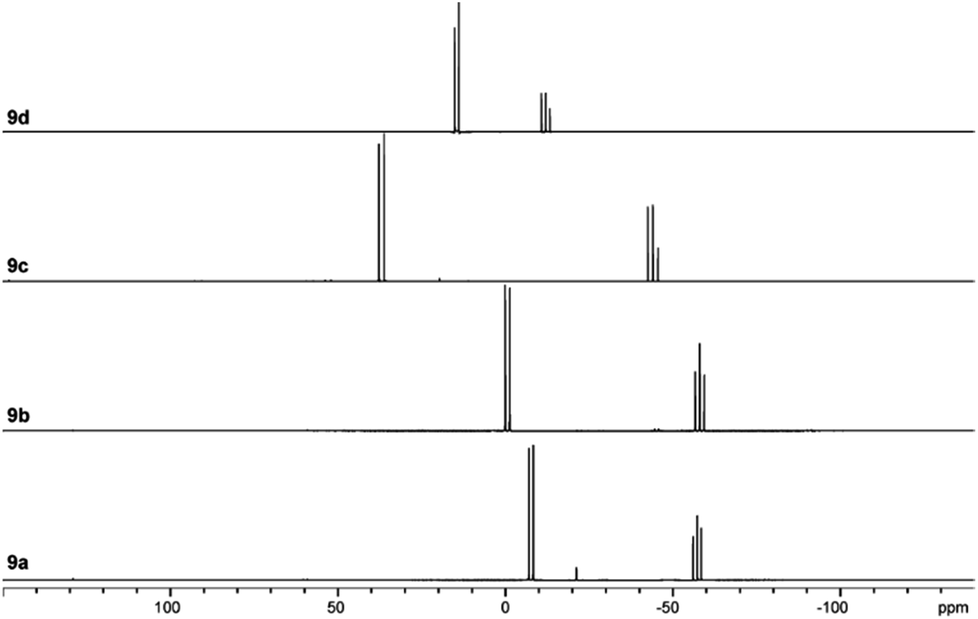
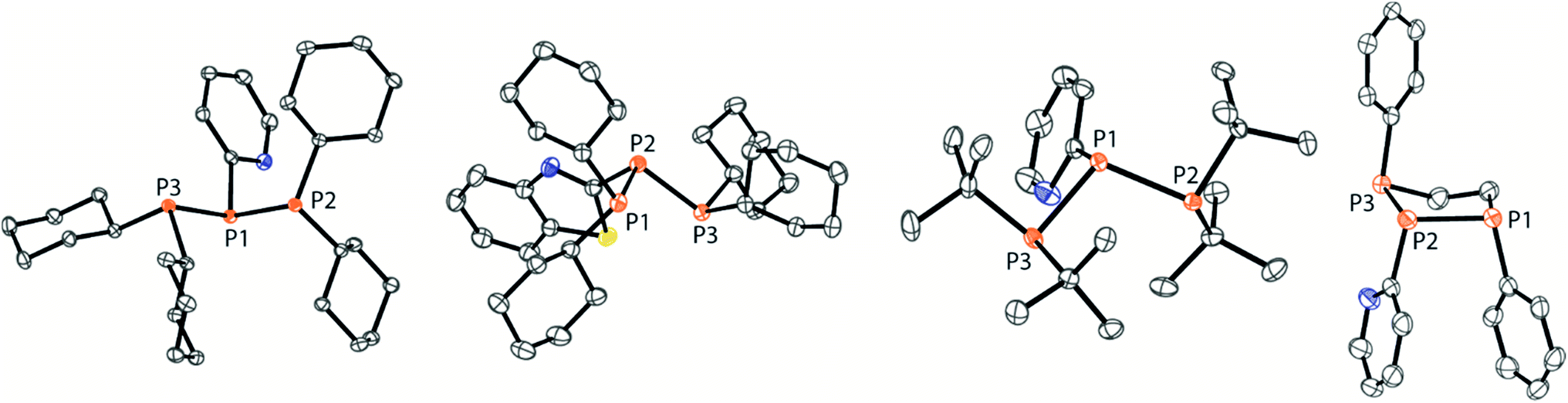
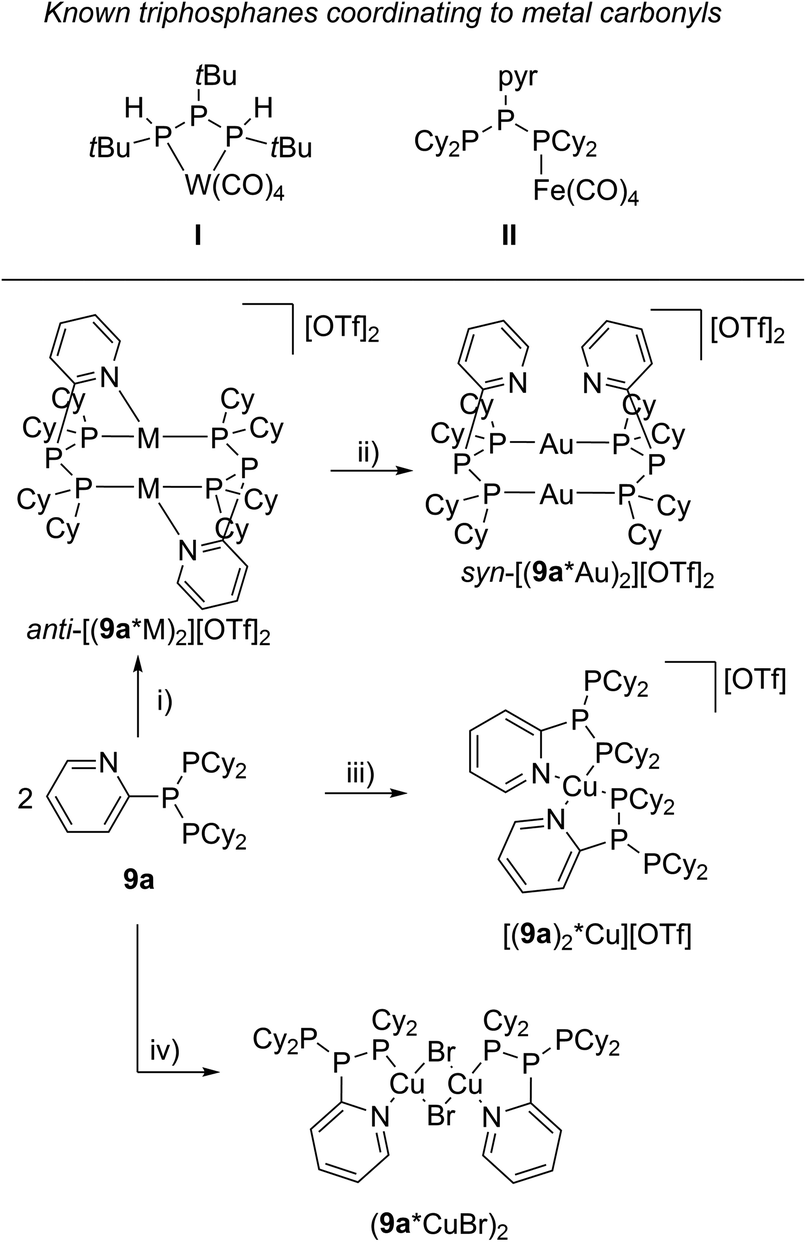
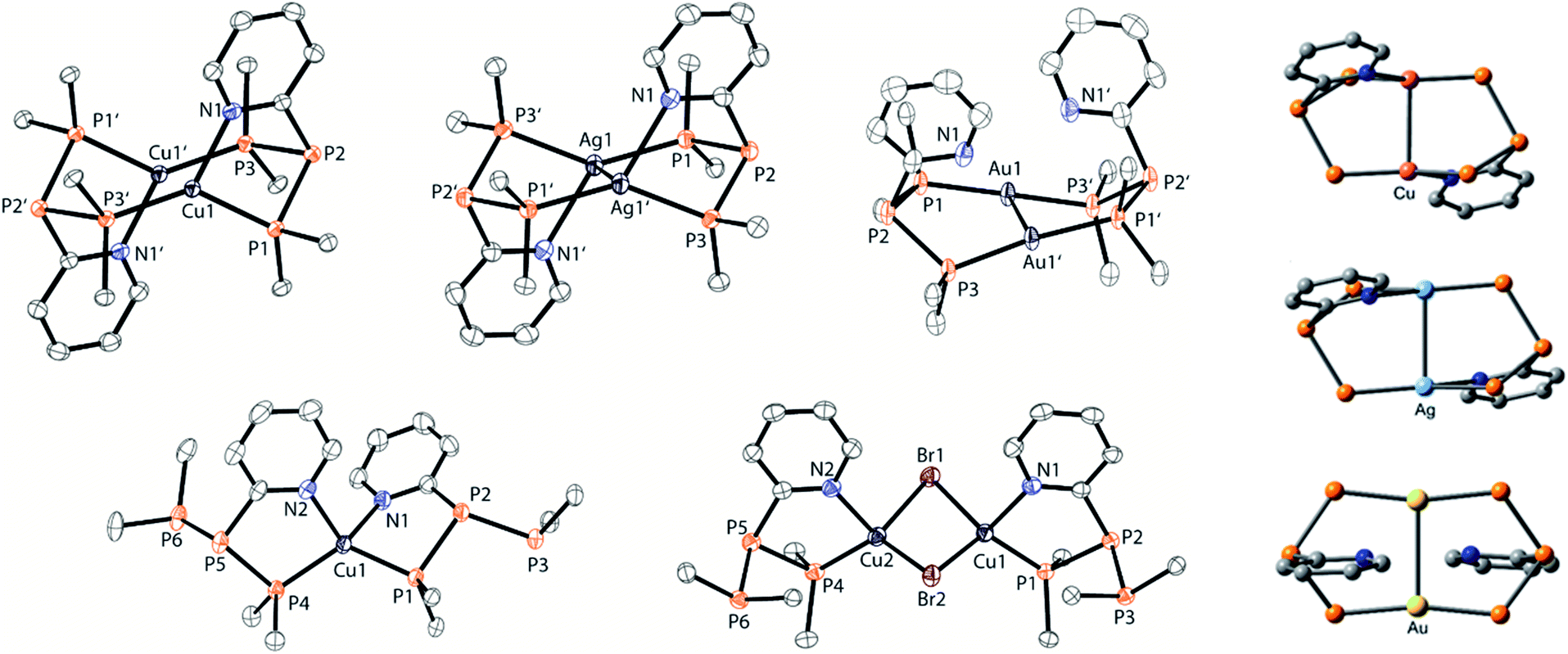
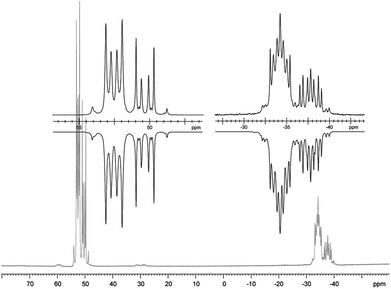
The targeted type II phosphanes are synthesised by the reaction of the dichlorophosphanes RPCl2 (R = Py (5); R = BTz (6))7,8 with two equivalents of 3,5-dimethyl-1-(trimethylsilyl)pyrazole (Scheme 3).9 The released chloro(trimethyl)silane can be removed under reduced pressure after 16 hours and no further purification is necessary. Both phosphanes 7 and 8 are conveniently prepared on a multi gram scale in excellent yields (97% (7); 96% (8)). The 31P NMR spectra of the dipyrazolylphosphanes show a singlet resonance in each case (7: δ(P) = 44.8 ppm; 8: δ(P) = 38.0 ppm) being significantly shifted to a higher field compared to the corresponding dichlorophosphanes (5: δ(P) = 138.7 ppm; 6: δ(P) = 132.0 ppm). Suitable crystals for X-ray analysis of phosphanes 7 and 8 are obtained by slow vapour diffusion of n-pentane into a saturated CH2Cl2 solution of 7 and 8. Their molecular structures are depicted in Fig. 1. The P–C bond lengths (7: P1–C1 1.827(1) Å; 8: P1–C1 1.828(2) Å) are in the expected range of a P–C single bond (1.83 Å),10 involving a tri-coordinate phosphorus atom. For both phosphanes the P–N bond lengths (7: P1–N4 1.721(1) Å, P1–N2 1.724(1) Å; 8 P1–N2/N2′ 1.719(1) Å) are in accordance with those reported for tri(1H-pyrazol-1-yl)phosphane (1.714(4) Å).11 In continuation, we carried out the reaction of dipyrazolylphosphanes 7 and 8 with secondary phosphanes R′2PH (R′ = Cy, tBu or R′2PH = PhP(H)C2H4P(H)Ph) to give triphosphanes 9a–c and triphospholane 9d with concomitant release of two equivalents of 3,5-dimethylpyrazole (Scheme 4). Compounds 9a–d readily precipitate from the reaction mixture and are isolated by filtration. Subsequent washing with CH3CN and drying in vacuo give 9a–d as colourless solids in good to excellent yield (96% (9a); 93% (9b); 77% (9c); 69% (9d)). The 31P NMR spectra of all compounds show AX2 spin systems which are characteristic of triphosphanes and 1,2,3-triphospholanes (Fig. 2). The NMR spectroscopic parameters are listed in Table 1. Single crystals for X-ray analysis are obtained by slow vapour diffusion of n-pentane into saturated CH2Cl2 solutions of 9a–d. The molecular structures of 9a–d are depicted in Fig. 3. Their structural parameters compare very well with those of similar triphosphanes12 and are thus not discussed in detail. Only the bond angle of the P–P–P moiety in compound 9c is approximately 10° wider which can be explained by the sterically demanding tert-butyl substituents. In recent years numerous examples of the coordination chemistry of anionic13 and zwitterionic14 oligophosphorus compounds towards transition metals have been reported. Also, few examples of neutral triphosphanes coordinated to transition metals are known, however, the triphosphane moiety was assembled while coordinated to at least one metal centre.15 Starting from stable triphosphanes only two complexes coordinating to metal carbonyls have been structurally characterised (Scheme 5, top),5,16 while another example is described in solution.17 In complex I, the triphosphane ligand chelates the W(CO)4 moiety while in complex II the coordination of the Fe(CO)4 moiety proceeds only via the free electron pair of one of the dicylcohexylphosphanyl groups. Featuring an additional nitrogen based donor site we envisioned the formation of multi-dentate coinage metal complexes using triphosphane 9a, exemplarily. This yielded the first triphosphane coordination complexes of Cu(I), Ag(I) and Au(I) (Scheme 5). Reacting 9a with an equimolar amount of Ag[OTf] in fluorobenzene at room temperature leads to the formation of a colourless precipitate which is isolated by filtration to yield the dinuclear complex [(9a*Ag)2][OTf]2 in essentially quantitative yield. The corresponding reaction of 9a with [(MeCN)4Cu][OTf] in CH2Cl2 gives colourless plates upon slow vapour diffusion of n-pentane at −30 °C which can be isolated as [(9a*Cu)2][OTf]2 up to a yield of 94%. Further transmetallation reaction of [(9a*Ag)2][OTf]2 with (tht)AuCl in MeCN gives [(9a*Au)2][OTf]2 in 64% yield by the concomitant precipitation of AgCl. Slow diffusion of n-pentane into a CH2Cl2 solution of 9a and [(MeCN)4Cu][OTf] in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio gives crystalline [(9a)2*Cu][OTf] in 93% yield. Formation of (9a*CuBr)2 is achieved by slow diffusion of n-pentane into a THF solution of equimolar amounts of 9a and (tht)CuBr. The molecular structures of the aforementioned coinage metal complexes are shown in Fig. 4 and the structural parameters are summarised in Table 2. Different from the known coordination motifs for I and II, the molecular structures of [(9a*Cu)2][OTf]2, [(9a*Ag)2][OTf]2 and [(9a*Au)2][OTf]2 reveal dinuclear metal complexes featuring two triphosphane ligands. In addition to the terminal coordinating phosphorus atoms, the nitrogen atoms of the pyridyl-substituents are involved in the coordination resulting in an approximately tetrahedral coordination environment of the respective metal centre. While the molecular structures of the dinuclear complexes [(9a*Cu)2][OTf]2 and [(9a*Ag)2][OTf]2 reveal an anti-arrangement with inversion symmetry, the gold complex [(9a*Au)2][OTf]2 crystallises as a centro-symmetrical syn-isomer. The inversion process within the complexes has been investigated by variable temperature NMR experiments and theoretical calculations (vide infra). The difference in the arrangement in the solid state causes a widening of the P–M–P angles in the copper complex [(9a*Cu)2][OTf]2 (P3′-Cu1-P1 136.52(2)°) compared to the silver complex [(9a*Ag)2][OTf]2 (P1–Ag1–P3′ 143.96(3)°) to an almost linear geometry around the gold atoms in [(9a*Au)2][OTf]2 (P1–Au1–P3′ 171.52(3)°). The distances between the silver atoms in [(9a*Ag)2][OTf]2 and the gold atoms in [(9a*Au)2][OTf]2 indicate the presence of argentophilic and aurophilic interactions, respectively.18 As the distance between the copper atoms in [(9a*Cu)2][OTf]2 is slightly larger than the sum of the van der Waals radii of two copper atoms (2.80 Å),19 significant van der Waals interactions can be excluded. The observed P–M bond lengths in [(9a*M)2][OTf]2 as well as in [(9a)2*Cu][OTf] and (9a*CuBr)2 are in good agreement with bond lengths reported for the structurally similar [M2(dcpm)2]2+ cations (M = Cu(I), Ag(I) or Au(I); dcpm = bis(dicylcohexylphosphanyl)methane).20 For (9a*CuBr)2 the formation of the typical Cu2X2 (X = halogen) geometry is found, only marginally differing from the typical planar arrangement reported for many Cu2X2-type structures.21 Compared to free ligand 9a, the P–P bond lengths and the P–P–P angles in the coinage metal complexes are only marginally altered. The room temperature NMR spectra of the dinuclear complexes [(9a*Au)2][OTf]2 and [(9a*Ag)2][OTf]2 show broadened resonances due to dynamic processes in solution.22 The low temperature 31P NMR spectrum of [(9a*Au)2][OTf]2 shows two separate AA′XX′X′′X′′′ spin systems at δ(PA) = −34.2 ppm, δ(PX) = 52.6 ppm and δ(PA) = −37.8 ppm, δ(PX) = 50.3 ppm corresponding to the respective syn- and anti-isomers (Fig. 5). Details of the coupling constants are included in the ESI.† For [(9a*Ag)2][OTf]2 two isomers with two separate AA′XX′X′′X′′′ spin systems at δ(PA) = −48.5 ppm, δ(PX) = 36.1 ppm and δ(PA) = −51.5 ppm, δ(PX) = 36.1 ppm are also observed. However, further line splitting can be observed as a result of the complexation with the Ag107/Ag109 nuclei, which made us refrain from iteratively fitting the corresponding 31P NMR spectrum. The 31P NMR spectrum of the dinuclear copper complex [(9a*Cu)2][OTf]2 shows only the resonances of one AA′XX′X′′X′′′ spin system at δ(PA) = −39.1 ppm and δ(PX) = 15.4 ppm. An additional broadened AX2 spin system (δ(PA) = −37.5 ppm and δ(PX) = 15.3 ppm (1J(PAPX) = −168 Hz)) gives rise to the formation of a monomeric species in solution which was not further investigated. A comparable AX2 spin system is observed for [(9a)2*Cu][OTf] (δ(PA) = −44.8 ppm and δ(PX) = −8.9 ppm (1J(PAPX) = −275 Hz)) as well as for (9a*CuBr)2 (δ(PA) = −46.6 ppm and δ(PX) = −6.2 ppm (1J(PAPX) = −253 Hz)). For both mononuclear copper complexes the resonances in the 31P NMR spectrum are noticeably broadened due to the fast quadrupole relaxation of the 63Cu nucleus.22,23 Theoretical calculations performed at the BP86-D3/def2-TZVP level of theory support the isomerisation in the dinuclear metal complexes [(9a*M)2]2+. For [(9a*Cu)2]2+ we found a relative energy difference between the syn- and the anti-isomer of 5.0 kcal mol−1 which explains that only one isomer is observed in the 31P NMR spectrum. While the two isomers of [(9a*Ag)2]2+ are isoenergetic with a difference of 0.1 kcal mol−1, the syn-isomer of [(9a*Au)2]2+ is favored by 1.7 kcal mol−1 compared to its anti-isomer. The optimised geometries also show the different coordination behaviour of the pyridyl-moieties in the three metal complexes [(9a*M)2]2+. While the free electron pair of the pyridyl nitrogen atom is pointing towards the metal centre in [(9a*Cu)2]2+ and [(9a*Ag)2]2+ it is pointing towards the middle of the Au–Au bond in [(9a*Au)2]2+ (Fig. 4, right). Our calculations furthermore indicate that the isomerisation is not driven by rotation but proceeds via dissociation of the M(I)–N coordination bond or pseudo-coordination in the case of [(9a*Au)2]2+ and subsequent association of the ligand on the opposite side (Fig. 6). This interconversion is the highest in energy for [(9a*Cu)2]2+ (14.7 kcal mol−1), followed by [(9a*Ag)2]2+ (13.2 kcal mol−1) and finally [(9a*Au)2]2+ (9.6 kcal mol−1).23
:1 ratio gives crystalline [(9a)2*Cu][OTf] in 93% yield. Formation of (9a*CuBr)2 is achieved by slow diffusion of n-pentane into a THF solution of equimolar amounts of 9a and (tht)CuBr. The molecular structures of the aforementioned coinage metal complexes are shown in Fig. 4 and the structural parameters are summarised in Table 2. Different from the known coordination motifs for I and II, the molecular structures of [(9a*Cu)2][OTf]2, [(9a*Ag)2][OTf]2 and [(9a*Au)2][OTf]2 reveal dinuclear metal complexes featuring two triphosphane ligands. In addition to the terminal coordinating phosphorus atoms, the nitrogen atoms of the pyridyl-substituents are involved in the coordination resulting in an approximately tetrahedral coordination environment of the respective metal centre. While the molecular structures of the dinuclear complexes [(9a*Cu)2][OTf]2 and [(9a*Ag)2][OTf]2 reveal an anti-arrangement with inversion symmetry, the gold complex [(9a*Au)2][OTf]2 crystallises as a centro-symmetrical syn-isomer. The inversion process within the complexes has been investigated by variable temperature NMR experiments and theoretical calculations (vide infra). The difference in the arrangement in the solid state causes a widening of the P–M–P angles in the copper complex [(9a*Cu)2][OTf]2 (P3′-Cu1-P1 136.52(2)°) compared to the silver complex [(9a*Ag)2][OTf]2 (P1–Ag1–P3′ 143.96(3)°) to an almost linear geometry around the gold atoms in [(9a*Au)2][OTf]2 (P1–Au1–P3′ 171.52(3)°). The distances between the silver atoms in [(9a*Ag)2][OTf]2 and the gold atoms in [(9a*Au)2][OTf]2 indicate the presence of argentophilic and aurophilic interactions, respectively.18 As the distance between the copper atoms in [(9a*Cu)2][OTf]2 is slightly larger than the sum of the van der Waals radii of two copper atoms (2.80 Å),19 significant van der Waals interactions can be excluded. The observed P–M bond lengths in [(9a*M)2][OTf]2 as well as in [(9a)2*Cu][OTf] and (9a*CuBr)2 are in good agreement with bond lengths reported for the structurally similar [M2(dcpm)2]2+ cations (M = Cu(I), Ag(I) or Au(I); dcpm = bis(dicylcohexylphosphanyl)methane).20 For (9a*CuBr)2 the formation of the typical Cu2X2 (X = halogen) geometry is found, only marginally differing from the typical planar arrangement reported for many Cu2X2-type structures.21 Compared to free ligand 9a, the P–P bond lengths and the P–P–P angles in the coinage metal complexes are only marginally altered. The room temperature NMR spectra of the dinuclear complexes [(9a*Au)2][OTf]2 and [(9a*Ag)2][OTf]2 show broadened resonances due to dynamic processes in solution.22 The low temperature 31P NMR spectrum of [(9a*Au)2][OTf]2 shows two separate AA′XX′X′′X′′′ spin systems at δ(PA) = −34.2 ppm, δ(PX) = 52.6 ppm and δ(PA) = −37.8 ppm, δ(PX) = 50.3 ppm corresponding to the respective syn- and anti-isomers (Fig. 5). Details of the coupling constants are included in the ESI.† For [(9a*Ag)2][OTf]2 two isomers with two separate AA′XX′X′′X′′′ spin systems at δ(PA) = −48.5 ppm, δ(PX) = 36.1 ppm and δ(PA) = −51.5 ppm, δ(PX) = 36.1 ppm are also observed. However, further line splitting can be observed as a result of the complexation with the Ag107/Ag109 nuclei, which made us refrain from iteratively fitting the corresponding 31P NMR spectrum. The 31P NMR spectrum of the dinuclear copper complex [(9a*Cu)2][OTf]2 shows only the resonances of one AA′XX′X′′X′′′ spin system at δ(PA) = −39.1 ppm and δ(PX) = 15.4 ppm. An additional broadened AX2 spin system (δ(PA) = −37.5 ppm and δ(PX) = 15.3 ppm (1J(PAPX) = −168 Hz)) gives rise to the formation of a monomeric species in solution which was not further investigated. A comparable AX2 spin system is observed for [(9a)2*Cu][OTf] (δ(PA) = −44.8 ppm and δ(PX) = −8.9 ppm (1J(PAPX) = −275 Hz)) as well as for (9a*CuBr)2 (δ(PA) = −46.6 ppm and δ(PX) = −6.2 ppm (1J(PAPX) = −253 Hz)). For both mononuclear copper complexes the resonances in the 31P NMR spectrum are noticeably broadened due to the fast quadrupole relaxation of the 63Cu nucleus.22,23 Theoretical calculations performed at the BP86-D3/def2-TZVP level of theory support the isomerisation in the dinuclear metal complexes [(9a*M)2]2+. For [(9a*Cu)2]2+ we found a relative energy difference between the syn- and the anti-isomer of 5.0 kcal mol−1 which explains that only one isomer is observed in the 31P NMR spectrum. While the two isomers of [(9a*Ag)2]2+ are isoenergetic with a difference of 0.1 kcal mol−1, the syn-isomer of [(9a*Au)2]2+ is favored by 1.7 kcal mol−1 compared to its anti-isomer. The optimised geometries also show the different coordination behaviour of the pyridyl-moieties in the three metal complexes [(9a*M)2]2+. While the free electron pair of the pyridyl nitrogen atom is pointing towards the metal centre in [(9a*Cu)2]2+ and [(9a*Ag)2]2+ it is pointing towards the middle of the Au–Au bond in [(9a*Au)2]2+ (Fig. 4, right). Our calculations furthermore indicate that the isomerisation is not driven by rotation but proceeds via dissociation of the M(I)–N coordination bond or pseudo-coordination in the case of [(9a*Au)2]2+ and subsequent association of the ligand on the opposite side (Fig. 6). This interconversion is the highest in energy for [(9a*Cu)2]2+ (14.7 kcal mol−1), followed by [(9a*Ag)2]2+ (13.2 kcal mol−1) and finally [(9a*Au)2]2+ (9.6 kcal mol−1).23
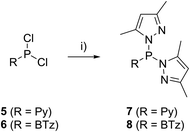 |
| | Scheme 3 Synthesis of dipyrazolylphosphanes 7 and 8; R = Py; (i) 2 eq. pyrSiMe3, neat, –2 Me3SiCl; R = BTz; (i) 2 eq. pyrSiMe3, Et2O, –2 Me3SiCl. | |
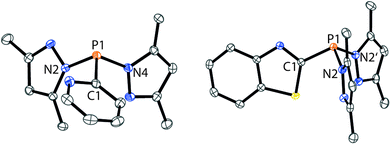 |
| | Fig. 1 Molecular structures of 7 (left) and 8 (right) (hydrogen atoms are omitted for clarity and thermal ellipsoids are displayed at 50% probability). Selected bond lengths (Å) and angles (°): (7) P1–C1 1.827(1), P1–N4 1.721(1), P1–N2 1.724(1), C1–P1–N4 101.28(5), N4–P1–N2 103.26(5), N2–P1–C1 100.18(5); (8) P1–C1 1.828(2), P1–N2 1.719(1), C1–P1–N2 101.26(5), N2–P1–N2′ 103.84(7). | |
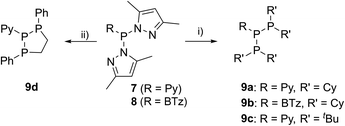 |
| | Scheme 4 Synthesis of triphosphanes 9a–c: (i) +2 eq. R′2PH, CH3CN, –2 pyrH; and triphospholane 9d: (ii) 1 eq. PhPH(CH2)2PHPh, CH3CN, –2 pyrH. | |
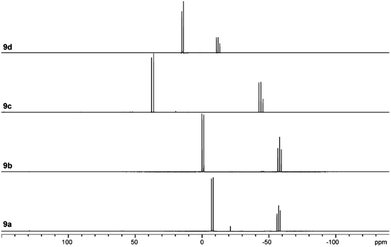 |
| | Fig. 2
31P NMR spectra of compounds 9a–d. | |
Table 1
31P NMR parameters of compounds 9a–d
| Compound |
Chemical shifts (ppm) |
1
JPP (Hz) |
|
9a
|
−57.3 |
−7.7 |
−251 |
|
9b
|
−58.1 |
−0.7 |
−268 |
|
9c
|
−44.1 |
37.0 |
−307 |
|
9d
|
−12.1 |
14.5 |
−260 |
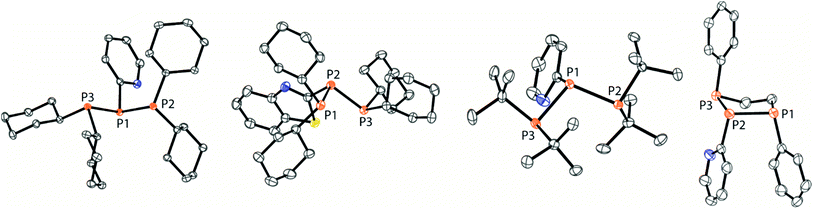 |
| | Fig. 3 Molecular structures of triphosphanes 9a–c and triphospholane 9d (hydrogen atoms are omitted for clarity and thermal ellipsoids are displayed at 50% probability). Selected bond lengths (Å) and angles (°) (9a): P1–P2 2.2301(3), P1–P3 2.2371(3), P3–P1–P2 97.38(1) (9b); P1–P2 2.2377(5), P2–P3 2.2234(5), P1–P2–P3 98.11(2) (9c); P1–P2 2.2181(3), P1–P3 2.2485(3), P2–P1–P3 107.64(1) (9d); P1–P2 2.270(2), P2–P3 2.197(2), P1–P2–P3 97.16(6). | |
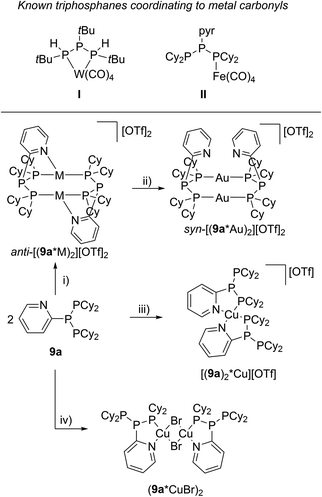 |
| | Scheme 5 Known transition metal complexes featuring a neutral triphosphane ligand (top); synthesis of coinage metal coordination complexes starting from triphosphane 9a (bottom); (i) 2 eq. Ag[OTf], PhF (M = Ag); 2 eq. [(MeCN)4Cu][OTf], CH2Cl2, –8 MeCN (M = Cu); (ii) 2 eq. (tht)AuCl, MeCN, –2 tht, –2 AgCl; (iii) 1 eq. [(MeCN)4Cu][OTf], CH2Cl2, –4 MeCN; (iv) 2 eq. (tht)CuBr, THF, –2 tht. | |
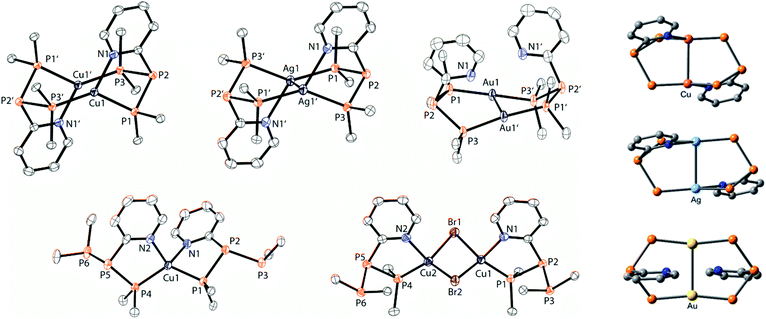 |
| | Fig. 4 Molecular structures of the coinage metal complexes [(9a*Cu)2][OTf]2, [(9a*Ag)2][OTf]2, [(9a*Au)2][OTf]2, [(9a)2*Cu][OTf] and (9a*CuBr)2 (hydrogen atoms, anions and solvate molecules are omitted for clarity, cyclohexyl groups are represented only by their phosphorus bound carbon atoms, and thermal ellipsoids are displayed at 50% probability; left). Selected geometrical parameters are given in Table 2; Optimised geometries of [(9a*M)2][OTf]2 using the BP86-D3/def2-TZVP functional, showing the different coordination arrangement of the pyridyl-moieties towards the metal atoms (M = Cu, Ag, and Au; right). | |
Table 2 Selected geometrical parameters of crystallographically characterised coinage metal complexes [(9a*Cu)2][OTf]2, [(9a*Ag)2][OTf]2, [(9a*Au)2][OTf]2, [(9a)2*Cu][OTf] and (9a*CuBr)2
|
|
[(9a*Cu)2][OTf]2 |
[(9a*Ag)2][OTf]2 |
[(9a*Au)2][OTf]2 |
[(9a)2*Cu][OTf]a |
(9a*CuBr)2a |
|
Average bond lengths and angles are given.
|
| M1–M1′ in Å |
2.8225(5) |
2.9013(4) |
2.880(3) |
— |
2.942 |
| M1–P1 in Å |
2.2755(4) |
2.3818(8) |
2.3033(8) |
2.243 |
2.191 |
| M1–P3′ in Å |
2.2176(5) |
2.4468(7) |
2.3198(8) |
|
|
| M–N in Å |
2.048(1) |
2.404(3) |
2.948(3) |
2.106 |
2.116 |
| P–Pa in Å |
2.225 |
2.224 |
2.226 |
2.212 |
2.214 |
| P1-M1-P3 in °′ |
136.52(2) |
143.96(3) |
171.52(3) |
— |
— |
| P–P–P in ° |
98.26(2) |
100.16(4) |
98.29(5) |
104.43 |
102.43 |
 |
| | Fig. 5
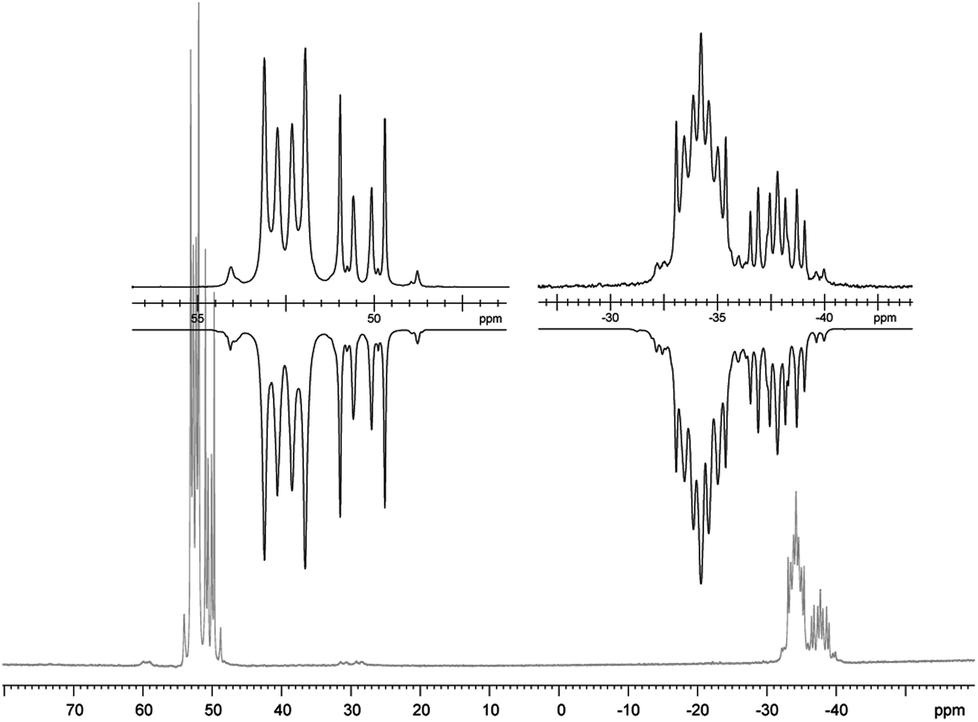
31P NMR spectrum of [(9a*Au)2][OTf]2 (CD2Cl2, 243 K, insets show the two AA′XX′X′′X′′′ spin systems of the experimental (upwards) and fitted spectra (downwards)). | |
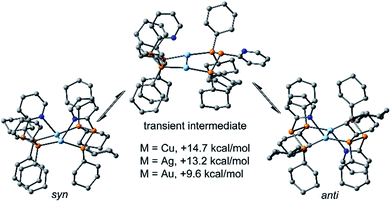 |
| | Fig. 6 Interconversion mechanism of the syn- and anti-isomers of [(9a*M)2]2+; (M = Cu, Ag, and Au). | |
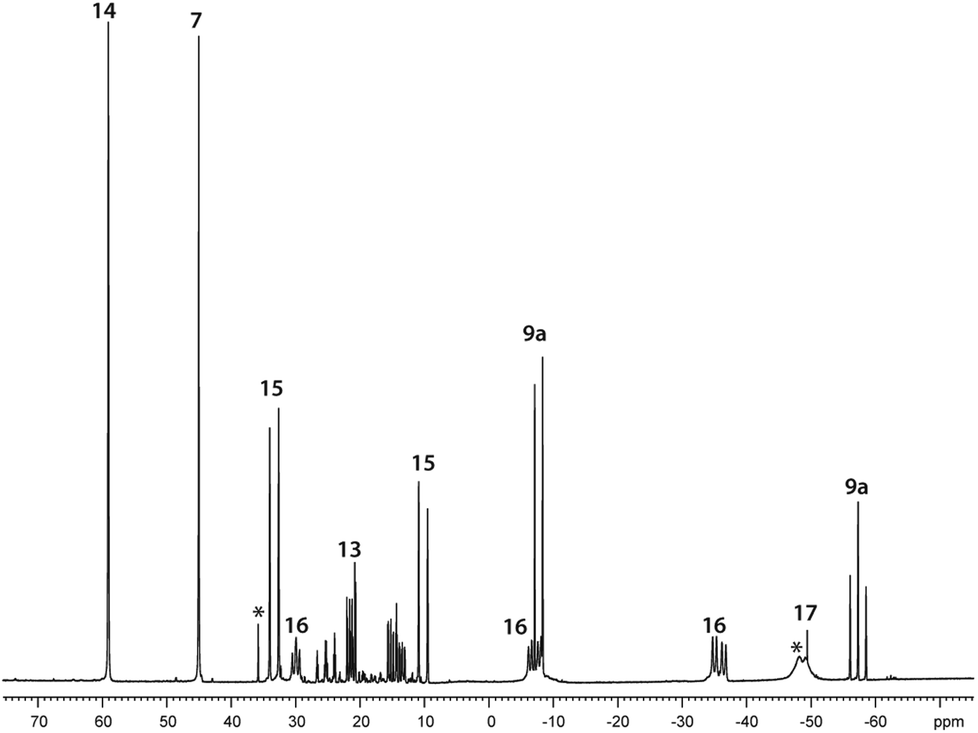
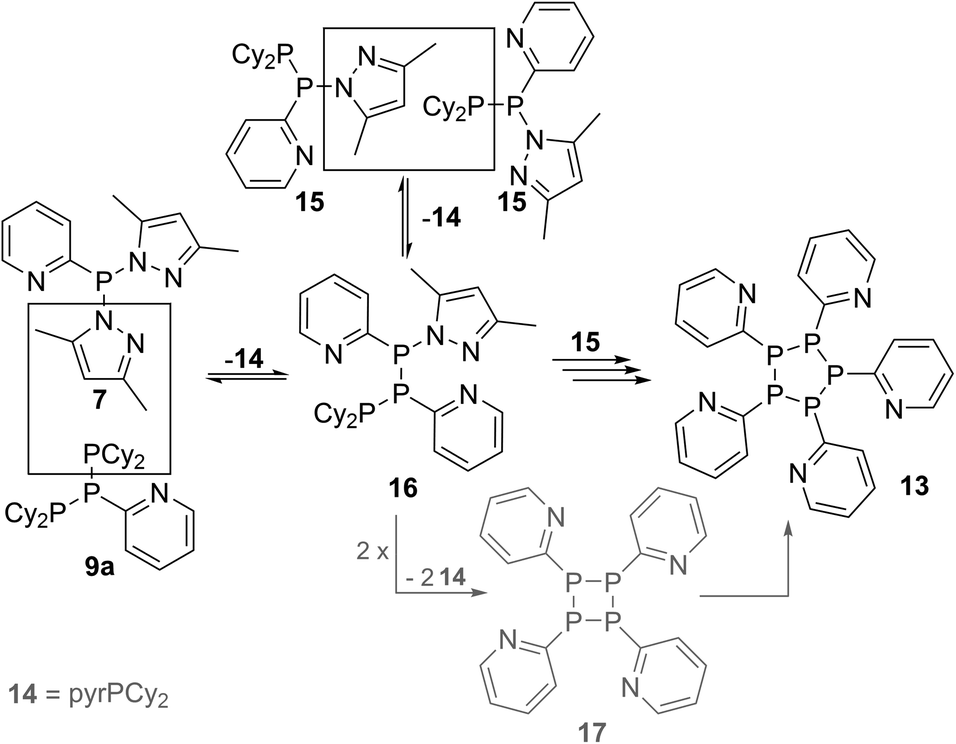
Further reactivity studies on triphosphane 9a revealed that this compound readily rearranges in solution by means of a P–P/P–P bond metathesis reaction. After 24 h the CH2Cl2 solution of 9a shows an additional singlet resonance at δ(P) = 21.0 ppm which we assign to tetracyclohexyldiphosphane (12)24 and an AA′BB′C spin system at δ(P) = 14.0–25.2 ppm being characteristic of pentaphospholanes of type (RP)5.24,25 As this scrambling reaction is in a state of equilibrium the isolation of 13 is hampered. However, the reaction of pure 13 with Cy4P2 yielding 9a illustrates that pentaphospholane 13 might be used as a PyP-synthon as it inserts into the P–P bond of Cy4P2. The selective synthesis of pentaphospholane 13 is achieved by the equimolar reaction of 7 with Cy2PH in Et2O as a non-polar solvent (Scheme 6). The formed precipitate is filtered and dried in vacuo giving 13 in 98% yield as a colourless powder which shows the AA′BB′C spin system in the 31P NMR spectrum (Fig. 7). Dicyclohexyl(3,5-dimethylpyrazolyl)phosphane (14) is identified as a side product in the supernatant solution by its singlet resonance in the 31P NMR spectrum (δ(P) = 58.4 ppm) indicating a P–N/P–P bond metathesis reaction.6 To further understand the formation of 13 we studied the reaction in CD2Cl2 by means of 31P NMR spectroscopy (Fig. 8). After 3.5 h compound 7 is still present in the solution while Cy2PH is fully consumed by the condensation reaction with 7 to form triphosphane 9a (vide supra) and diphosphane 15 which is identified by an AX spin system (δ(PA) = 10.2 ppm and δ(PX) = 33.4 ppm; 1J(PAPX) = −281 Hz). The AMX spin system at δ(PA) = −35.6 ppm, δ(PM) = −7.2 ppm and δ(P)X = 29.9 ppm is assigned to triphosphane 16 which is formed via a P–N/P–P bond metathesis reaction of two equivalents of 15 and via a P–N/P–P bond metathesis reaction of 7 and 9a (Scheme 7). In both cases phosphane 14 is liberated. We propose a further chain growth in which triphosphane 16 reacts with 15 with the release of phosphane 14 in a P–N/P–P oligomerisation reaction which ultimately yields pentaphospholane 13. We furthermore propose the formation of tetraphosphetane 17 (δ(P) = −49.4 ppm) which can be formed via the reaction of two equivalents 16 with concomitant release of 14 (Scheme 7). Similar to the known ring expansion reactions of certain cyclophosphanes,2617 might react to form pentaphospholane 13, being the thermodynamically favoured product. In analogy to 13 we are able to isolate the corresponding benzothiazolyl substituted pentaphospholane 18.27 Crystals suitable for X-ray analysis are obtained for both compounds by the slow diffusion of n-pentane into saturated CH2Cl2 solutions at −30 °C (Fig. 9).
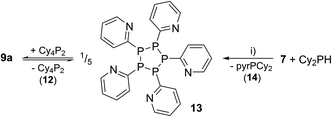 |
| | Scheme 6 Equilibrium reaction of 9a with 13 and 12 and selective synthesis of 13 by the reaction of 7 with Cy2PH; (i) Et2O, –pyrH. | |
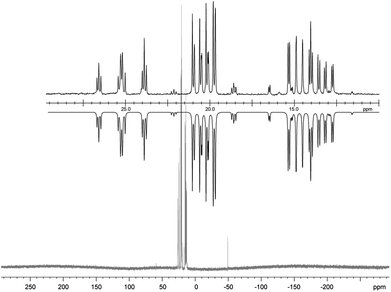 |
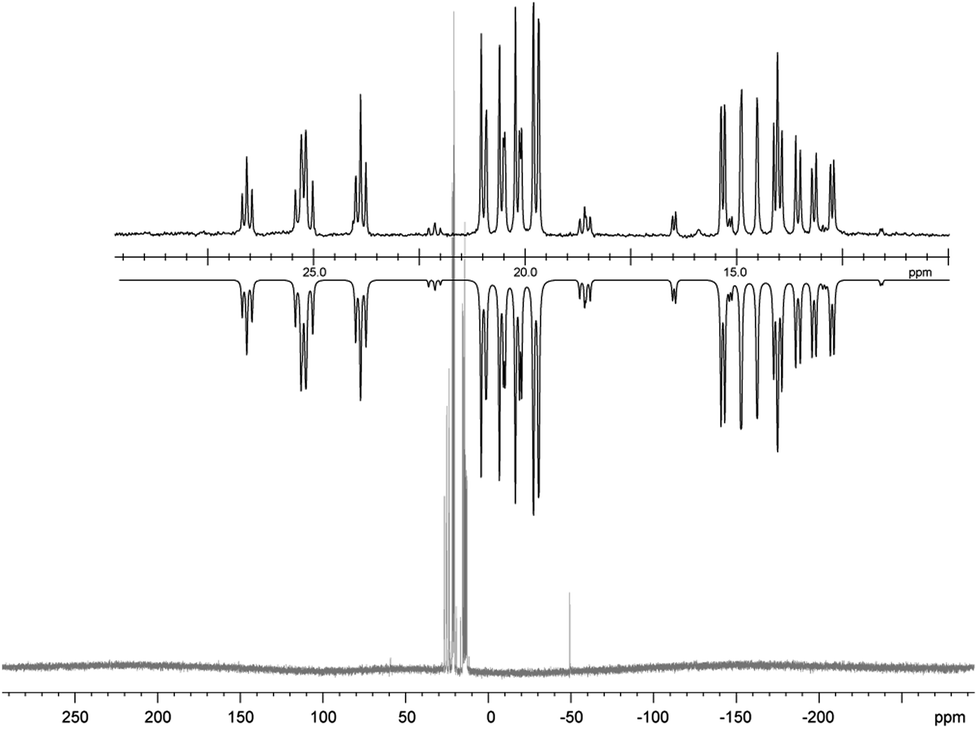
| | Fig. 7
31P NMR spectrum of pentaphospholane 13 (CD3CN, 300 K, the inset shows the AA′BB′C spin system of the experimental (upwards) and fitted spectra (downwards)). | |
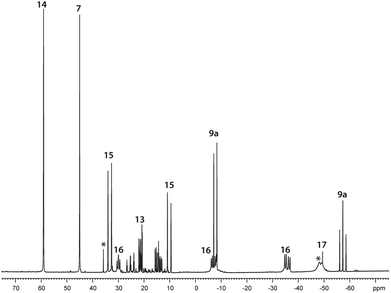 |
| | Fig. 8
31P NMR spectrum of the reaction of 7 with Cy2PH in a 1:1 ratio in CD2Cl2 after 3.5 h; unidentified products are marked with asterisks. | |
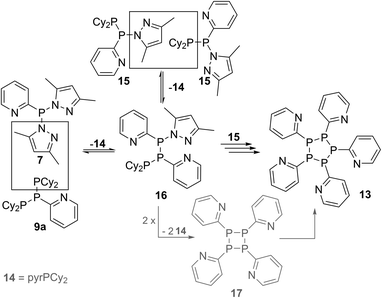 |
| | Scheme 7 Possible formation of 16via P–N/P–P bond metathesis reactions. | |
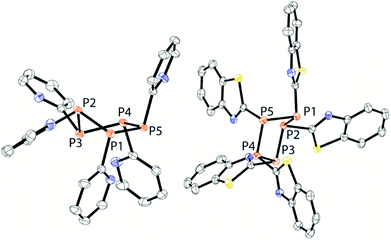 |
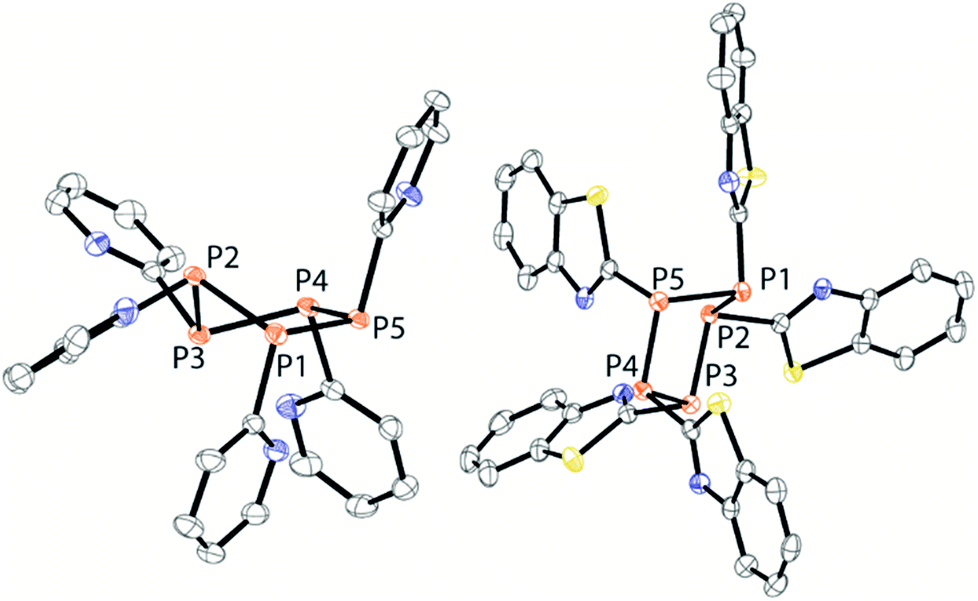
| | Fig. 9 Molecular structures of the pentaphospholanes 13 (left) and 18 (right) (hydrogen atoms and solvate molecules are omitted for clarity and thermal ellipsoids are displayed at 50% probability). Selected bond lengths (Å) and angles (°) (13): P1–P2 2.2222(4), P2–P3 2.2200(5); P3–P4 2.22200(5), P4–P5 2.2078(4), P1–P2–P3 97.25(2), P2–P3–P4 95.59(2), P3–P4–P5 102.27(2), P4–P5–P1 106.77(2), P5–P1–P2 97.14(2) (18); P1–P2 2.2336(6), P2–P3 2.2232(6), P3–P4 2.338(6), P4–P5 2.2069(6), P5–P1 2.2288(6), P1–P2–P3 93.54(2), P2–P3–P4 100.29(2), P3–P4–P5 107.27(2), P4–P5–P1 96.32(2), P5–P1–P2 96.85(2). | |
Both compounds reveal the typical envelope conformation of the P5-ring featuring the substituents in the all-trans position. The P–P bond lengths observed for both pentaphospholanes 13 (av. P–P 2.218 Å) and 18 (av. P–P 2.246 Å) are in a range known for pentaphospholanes such as (PhP)5 (av. 2.217 Å); also the P–P–P angles are in good accordance with the reported data for (PhP)5.28 We subsequently envisioned the use of 13 as a PyP-synthon for insertion reactions into P–P bonds which was already indicated by the aforementioned scrambling reaction between 9a, Cy4P2 and 13 (see Scheme 6).
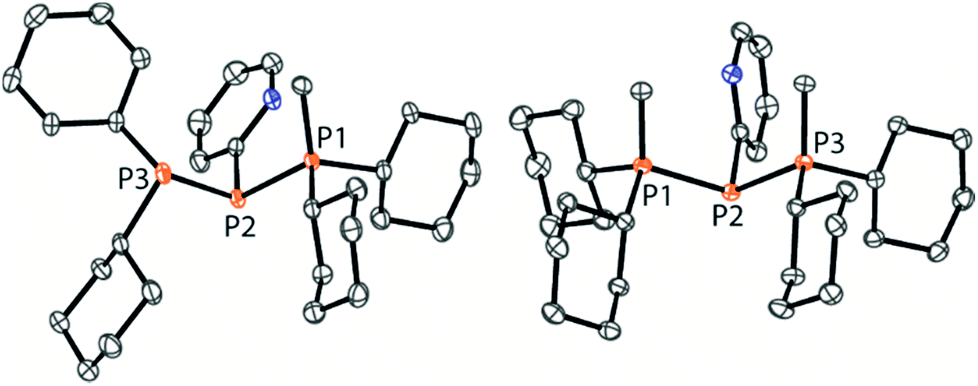
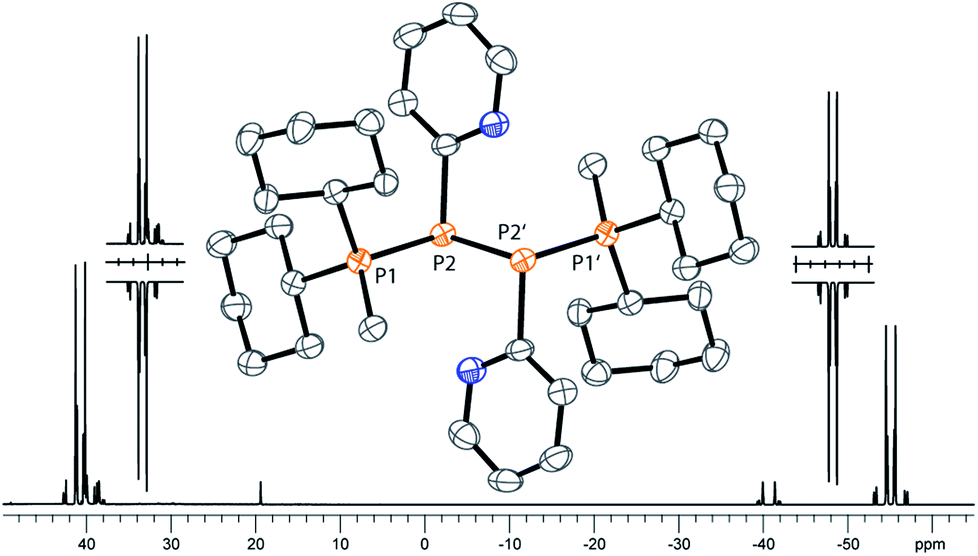
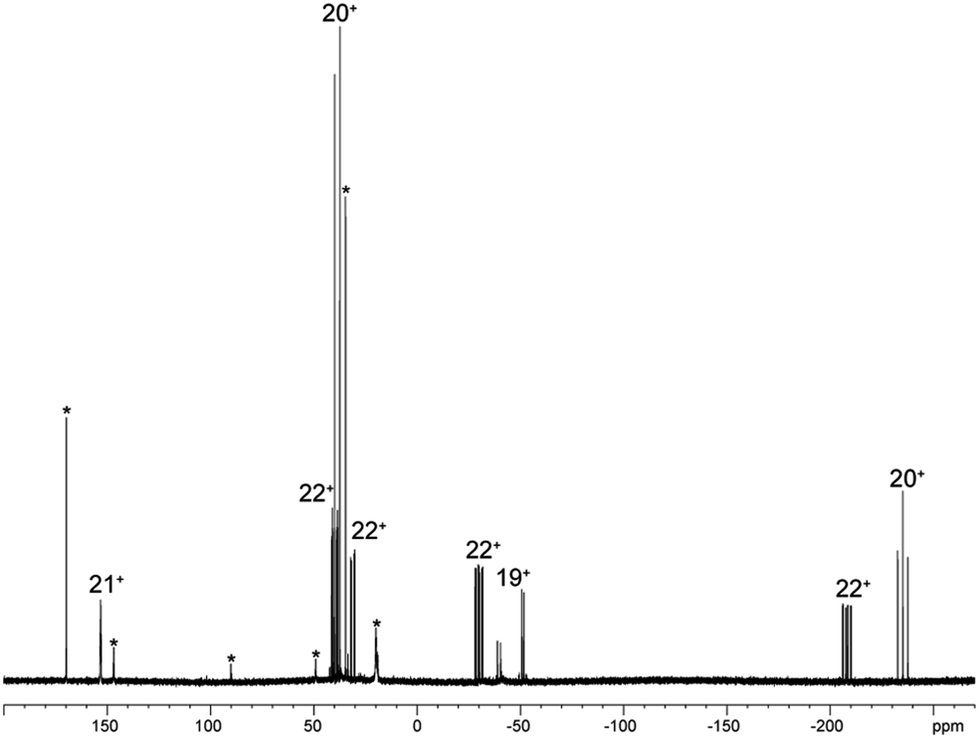
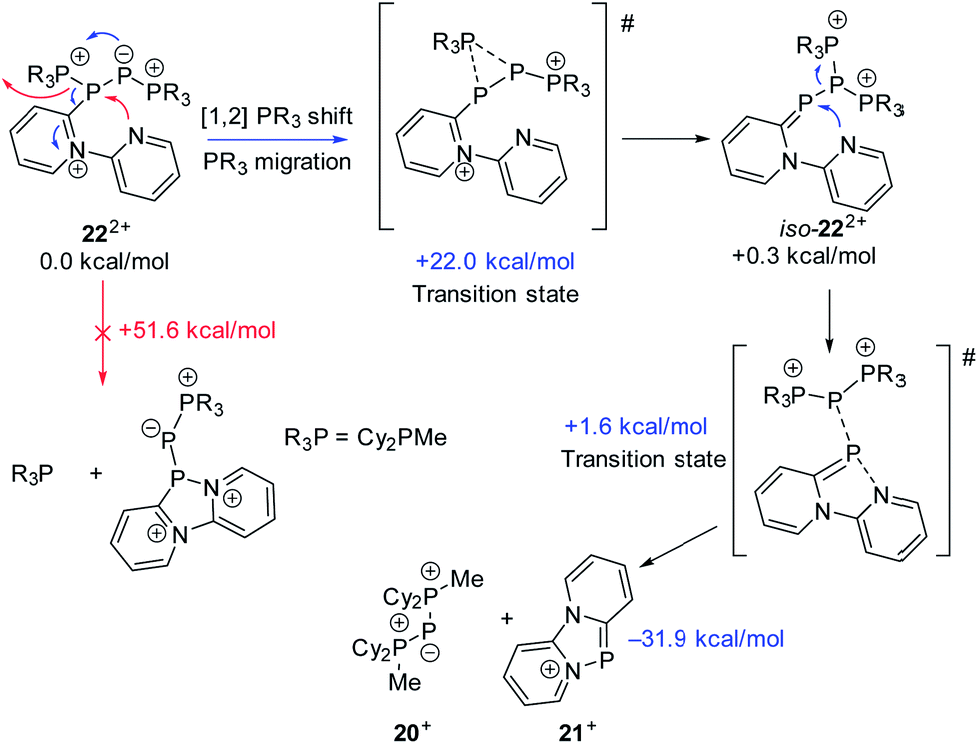
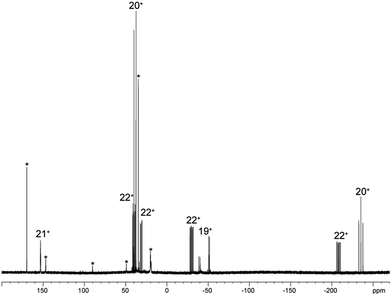
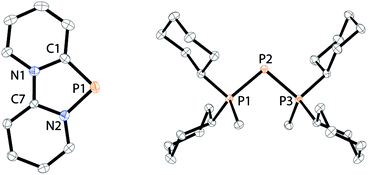
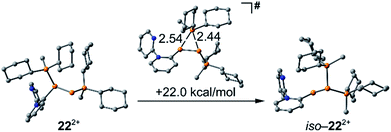
As similar scrambling reactions might be expected for triphosphanium and triphosphanediium salts, we methylated triphosphane 9a with different amounts of MeOTf. The 31P NMR spectrum of the 1:1 reaction of 9a with MeOTf shows an AMX spin system (δ(PA) = −46.2 ppm, δ(PM) = −12.9 ppm and δ(PX) = 34.2 ppm (1J(PAPM) = −290 Hz, 1J(PAPX) = −281 Hz, and 2J(PMPX) = 58 Hz)), suggesting the methylation of the phosphorus atom rather than the nitrogen atom of the pyridyl-substituent. After the removal of all volatiles in vacuo triphosphan-1-ium triflate salt 10[OTf] can be isolated in quantitative yield as a colourless powder. A second methylation is achieved when 9a is reacted with an excess of MeOTf under solvent free conditions (Scheme 8). Similar to the aforementioned reaction, only the phosphorus atoms are methylated giving triphosphane-1,3-diiumtriflate salt 11[OTf]2. As both dicyclohexylphosphanyl moieties are methylated, the 31P NMR spectrum of 10[OTf]2 shows an AX2 spin system with resonances at δ(PA) = −67.9 ppm and δ(PX) = 44.0 ppm (1J(PAPX) = −315 Hz). The molecular structures of cation 10+ and dication 112+ are depicted in Fig. 10. Note that a small number of triphosphanediium salts are reported in the literature and compound 10[OTf] extends the library of cationic, catenated phosphorus compounds as a structurally characterised triphosphan-1-ium salt.29–31 The P1–P2 bond length in cation 10+ (P1–P2 2.1956(5) Å) is shortened whereas the P2–P3 bond (P2–P3 2.2362(5) Å) is comparable to those in 9a (P1–P2 2.2301(3) Å, P1–P3 2.2371(3) Å) or similar diphosphanium cations (compare [Ph2P–PPh3]+: 2.230(1) Å).29b The P–P–P bond angle in 10+ (97.89(2)°) is similar to that in compound 9a (97.16(6)°). The P–P bond lengths in 112+ (P1–P2 2.2152(4) Å and P2–P3 2.2182(4) Å) are marginally shorter compared to those in 9a but in accordance with bond lengths reported for similar triphosphane-1,3-diium cations (compare [Me3P–P(Cy)-PMe3]2+: 2.1979(5) Å and 2.1976(6) Å).29b The P–P–P bond angle in 112+ (107.43(1)°) is significantly wider compared to those in 9a and [Me3P–P(Cy)-PMe3]2+ (103.11(2)°). In order to investigate the envisioned PyP-transfer into a P–P bond, we reacted 13 with 5 equivalents of 11[OTf]2 in CH3CN (Scheme 9). Upon addition of 13 to a colourless CH3CN solution of 11[OTf]2 the reaction mixture turns from deep red to pale yellow. The 31P NMR spectrum of the reaction mixture after 24 h shows two AA′XX′ spin systems which are attributed to two isomers of tetraphosphane-1,4-diium cations 192+ (Fig. 11), indicating the successful PyP-transfer via a P–P/P–P bond metathesis reaction. The A part of the prominent AA′XX′ spin system at δ(PA) = −55.1 ppm is assigned to the inner pyridyl-substituted P nuclei and the X part at δ(PX) = 40.7 ppm to the tetra-coordinate phosphorus atoms which is similar to known tetraphosphane-1,4-diium salts.32 The minor spin system resonates at δ(PA) = −40.1 ppm and δ(PX) = 39.8 ppm, respectively. After work-up compound 19[OTf]2 can be isolated analytically pure in 51% yield. X-ray analysis revealed that the centrosymmetric meso-isomer 192+ crystallized showing a central torsion angle of 180° (Fig. 11). The P–P bond lengths in 192+ (P1–P2 2.234(1) Å and P2–P2′2.232(2) Å) are in good agreement with the reported data for comparable tetraphosphane-1,4-diium cations (compare [Ph3P–(PPh)2–PPh3]2+: P1–P2 2.258(1) Å, P2–P2′ 2.221(1) Å).32 Similar to the reported PhP-transfer from (PhP)5 to a NHC,3313 can be used as a PyP-synthon, thus featuring an additional reaction or coordination site. When compound 19[OTf]2 was kept in a CD3CN solution for 14 days, the 31P NMR spectrum shows three rearrangement products (Fig. 12). Next to an AX2 spin system (δ(PA) = −235.3 ppm and δ(PX) = 38.8 ppm (1J(PAPX) = −501 Hz)), a pseudo triplet resonance (δ(P) = 153.1 ppm; 1J(PN) = 50 Hz) and an AMXY spin system (δ(PA) = −208.2 ppm, δ(PM) = −30.0 ppm, δ(PX) = 31.1 ppm, and δ(PY) = 39.8 ppm; 1J(PAPy) = −490 Hz, 1J(PMPX) = −342 Hz, 1J(PAPM) = −298 Hz, 2J(PMPY) = 91 Hz, 2J(PAPX) = 45 Hz, 3J(PXPY) = 34 Hz) are observed. The AX2 spin system can be attributed to the 3λ5-triphosph-2-en-1-ium cation (20+), as the spectroscopic parameters are in the range of those observed for [Ph3P–P–PPh3]+ (δ(PA) = −174 ppm and δ(PX) = 30 ppm; 1J(PAPX) = −500 Hz) which was first reported by Schmidpeter and co-workers.34 A variety of similar derivatives has been synthesised and reported by the group of Macdonald.35 Slow vapour diffusion of Et2O into the CD3CN solution yielded crystals suitable for X-ray analysis confirming the structural connectivity of 20+ (Fig. 13). The structural parameters of 20+ are similar to those observed for the cation [Ph3P–P–PPh3]+, i.e. slightly shortened P–P bond lengths (P1–P2 2.134(4), P2–P3 2.1382(4)) and a P–P–P bond angle of 103.85(2)°.34 By co-crystallisation, a further product was identified as the triflate salt of diazaphospholium cation 21+ (Fig. 13). We assign this species to the pseudo triplet resonance observed in the 31P NMR spectrum. The P–C bond distance of 1.733(1) Å slightly exceeds the upper limit of typical P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths (1.61–1.71 Å),36 while the P–N bond length of P1–N2 1.741(1) Å indicates a P–N single bond (P–N: 1.78 Å).37 Furthermore, the structure of 21+ reveals an acute angle around the P atom (C1–P1–N2 87.08(6)°) and a planar arrangement, suggesting a delocalized π-system. We assign the AMXY spin system to asymmetric tetraphosphanediium dication 222+, most likely formed by an intramolecular aromatic substitution reaction of 192+. This can be attributed to be the first step of
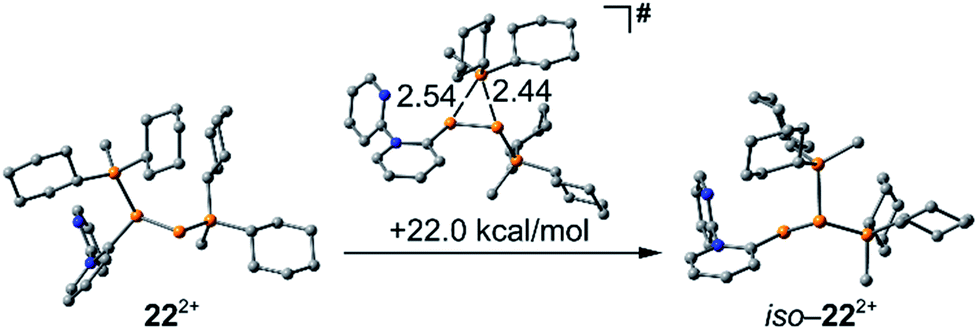
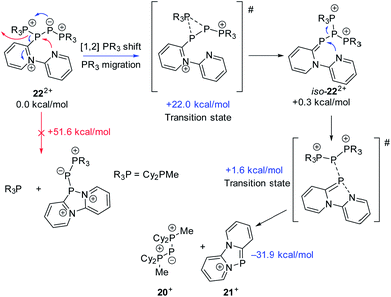
the rearrangement reaction of 192+ to form 20+ and 21+. We further studied this rearrangement reaction using the TURBOMOLE 7.0 program at the PB86-D3/def2-TZVP level of theory (see ESI for details†) and taking into consideration solvent effects by using the Conductor-like Screening Model (COSMO). Our calculations started from 22+ and show that the cleavage of Cy2PMe is energetically quite costly. This is in accordance with the 31P NMR spectrum as Cy2PMe is not observed (δ(P) = −19.8 ppm).38 More likely and energetically favoured is a [1,2]-shift of the Cy2PMe group to form iso-222+ with a reaction barrier of +22.0 kcal mol−1. Fig. 14 shows the optimised structures for this [1,2]-Cy2PMe shift reaction. It is noteworthy that similar [1,2]- and [1,3]-PR3 shifts have been reported.39 Compared to 222+, iso-222+ is almost isoenergetic and readily reacts in an exergonic step (−31.9 kcal mol−1) to form cations 20+ and 21+ with a low barrier of only 1.9 kcal mol−1 (see Scheme 10 and ESI for details†).
C bond lengths (1.61–1.71 Å),36 while the P–N bond length of P1–N2 1.741(1) Å indicates a P–N single bond (P–N: 1.78 Å).37 Furthermore, the structure of 21+ reveals an acute angle around the P atom (C1–P1–N2 87.08(6)°) and a planar arrangement, suggesting a delocalized π-system. We assign the AMXY spin system to asymmetric tetraphosphanediium dication 222+, most likely formed by an intramolecular aromatic substitution reaction of 192+. This can be attributed to be the first step of
the rearrangement reaction of 192+ to form 20+ and 21+. We further studied this rearrangement reaction using the TURBOMOLE 7.0 program at the PB86-D3/def2-TZVP level of theory (see ESI for details†) and taking into consideration solvent effects by using the Conductor-like Screening Model (COSMO). Our calculations started from 22+ and show that the cleavage of Cy2PMe is energetically quite costly. This is in accordance with the 31P NMR spectrum as Cy2PMe is not observed (δ(P) = −19.8 ppm).38 More likely and energetically favoured is a [1,2]-shift of the Cy2PMe group to form iso-222+ with a reaction barrier of +22.0 kcal mol−1. Fig. 14 shows the optimised structures for this [1,2]-Cy2PMe shift reaction. It is noteworthy that similar [1,2]- and [1,3]-PR3 shifts have been reported.39 Compared to 222+, iso-222+ is almost isoenergetic and readily reacts in an exergonic step (−31.9 kcal mol−1) to form cations 20+ and 21+ with a low barrier of only 1.9 kcal mol−1 (see Scheme 10 and ESI for details†).
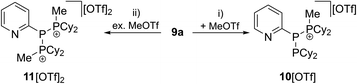 |
| | Scheme 8 Methylation reactions of 9a; (i) 1 eq. MeOTf, Et2O; (ii) 5 eq. MeOTf, neat. | |
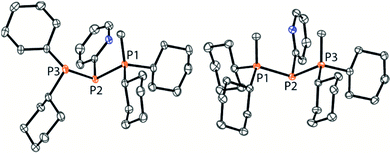 |
| | Fig. 10 Molecular structures of 10+ in 10[OTf] (left) and 112+ in 11[OTf]2 (hydrogen atoms and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability). Selected bond lengths (Å) and angles (°) (10+): P1–P2 2.1956(5), P2–P3 2.2362(5), P1–P2–P3 97.89(2) (112+); P1–P2 2.2152(4), P2–P3 2.2182(4), and P1–P2–P3 107.43(1). | |
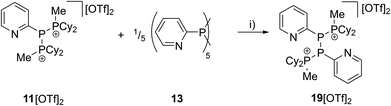 |
| | Scheme 9 Synthesis of tetraphosphane-1,4-diium triflate salt 19[OTf]2; (i) MeCN. | |
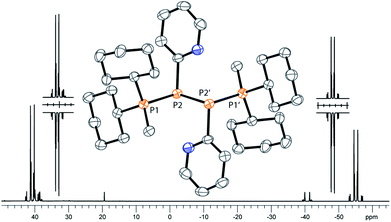 |
| | Fig. 11
31P NMR spectrum of dication 19[OTf]2 (CD2Cl2, 300 K, the inset shows the AA′XX′ spin system of the experimental (upwards) and fitted spectra (downwards)); molecular structure of 192+ in 19[OTf]2 (hydrogen atoms and anions are omitted for clarity; thermal ellipsoids are displayed at 50% probability). Selected bond lengths (Å) and angles (°):P1–P2 2.234(1), P2–P2′ 2.232(2), and P1–P2–P2′ 97.60(6). | |
 |
| | Fig. 12
31P NMR spectrum of a solution of 19[OTf]2 in CD3CN after 14 days. | |
 |
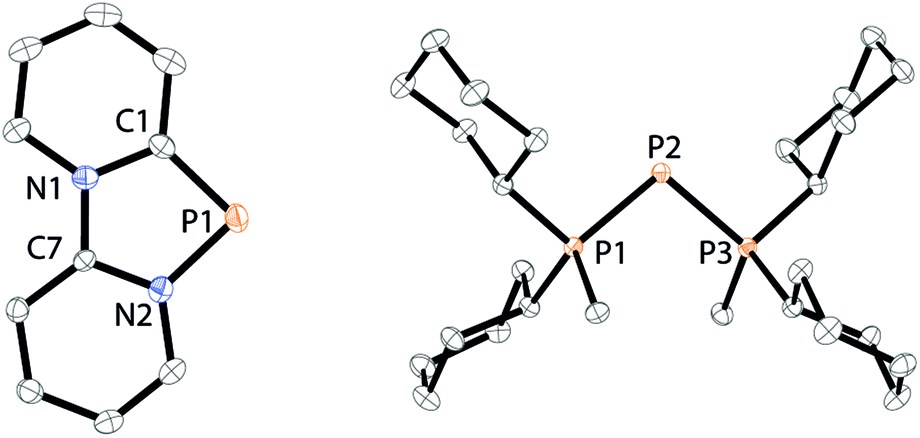
| | Fig. 13 Molecular structure of 1,4,2-diazaphospholium 21+ in 21[OTf] (left) and 3λ5-triphosph-2-en-1-ium 20+ in 20[OTf] (right) (hydrogen atoms and anions are omitted for clarity and thermal ellipsoids are displayed at 50% probability). Selected bond lengths (Å) and angles (°) (20+): P1–C1 1.733(1), P1–N2 1.741(1), C1–P1–N2 87.08(6) (20); P1–P2 2.134(4), P2–P3 2.1382(4), P1–P2–P3 103.85(2). | |
 |
| | Fig. 14 Optimised structures of 222+, iso-222+ and the transition state. | |
 |
| | Scheme 10 Rearrangement reaction of 222+ to 20+ and 21+ calculated at the PB86-D3/def2-TZVP level of theory. | |
Conclusions
In summary we have shown that phosphanes which we denote as type II phosphanes (two P–N bonds and one P–C bond) can be used to build polyphosphorus compounds featuring a further nitrogen donor functionality at the C bound substituent. The reactions of type II phosphanes 7 and 8 with secondary phosphanes of type R′2PH in a 1:2 ratio in MeCN give triphosphanes 9a–c and triphospholane 9dvia protolysis reactions. With the multi-gram scale synthesis of 9a we were able to conveniently investigate its coordination chemistry towards coinage metal salts. The synthesised coordination complexes are studied by X-ray analysis, multi nuclear NMR spectroscopy and quantum chemical calculations. Further reactivity studies of 9a focused on the reaction with the electrophilic MeOTf. Depending on the amount of MeOTf, the reaction yields triphosphane-1,3-diium triflate salt 11[OTf]2 or triphosphan-1-ium 10[OTf] featuring a hitherto unknown structural motif. Pentaphospholanes 13 and 18 are synthesised by reacting type II phosphane 7 or 8 with Cy2PH in a 1:1 ratio in Et2O or CH2Cl2, respectively. The reaction proceeds via a controlled scrambling reaction showing the susceptible solvent dependency of the reaction of 7 and 8 with Cy2PH. By reacting 11[OTf]2 with 13 we have shown that pentaphospholane 13 is a suitable PyP-synthon in a P–P/P–P bond metathesis reaction to yield tetraphosphane-1,4-diium triflate 19[OTf]2. In an interesting rearrangement reaction 19[OTf]2 forms triphosph-2-en-1-ium triflate salt 20[OTf] and 1,4,2-diazaphospholium triflate 21[OTf] via an unprecedented [1,2]-Cy2MeP shift. Based on our findings we are certain that controlled P–N(P)/P–P bond metathesis reactions will allow us to access larger acyclic and cyclic polyphosphanes in a controlled stepwise manner in order to further investigate their fascinating reaction space.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the German Science Foundation (DFG Grant number WE 4621/3-1), the European Regional Development Fund, the Free State of Saxony (ERDF-InfraPro, GEPARD-100326379) and the European Research Council (ERC starting grand, SynPhos-307616) for financial support. A. F. thanks MINECO/AEI of Spain (project CTQ2017-85821-R FEDER funds) for financial support. A. F. thanks the MINECO of Spain for a “Juan de la Cierva” contract. We also thank Philipp Lange for experimental assistance and EA measurements.
Notes and references
- M. Donath, F. Hennersdorf and J. J. Weigand, Chem. Soc. Rev., 2016, 45, 1145 RSC; M. H. Holthausen and J. J. Weigand, Chem. Soc. Rev., 2014, 43, 6639 RSC; S. Gomez-Ruiz and E. Hey-Hawkins, Coord. Chem. Rev., 2011, 255, 1360 CrossRef CAS; M. Scheer, G. Balazs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef PubMed; M. Baudler, Angew. Chem., Int. Ed. Engl., 1987, 26, 419 CrossRef.
-
(a) M. Baudler, Angew. Chem., Int. Ed. Engl., 1982, 21, 492 CrossRefM. Baudler and K. Glinka, Chem. Rev., 1993, 93, 1623 CrossRef CAS; M. Baudler and K. Glinka, Chem. Rev., 1994, 94, 1273 CrossRef;
(b) M. Scheer, St. Gremler, E. Herrmann, U. Grünhagen, M. Dargatz and E. Kleinpeter, Z. Anorg. Allg. Chem., 1991, 600, 203 Search PubMedM. Scheer, St. Gremler, E. Herrmann, M. Dargatz and H.-D. Schädler, Z. Anorg. Allg. Chem., 1992, 619, 1047 CrossRef;
(c) G. Fritz, Comments Inorg. Chem., 1982, 6, 329 Search PubMedG. Fritz, Adv. Inorg. Chem., 1987, 171 CrossRef;
(d) L. Maier, Helv. Chim. Acta, 1966, 49, 1119 Search PubMedE. Niecke, R. Rüger and B. Krebs, Angew. Chem., Int. Ed. Engl., 1982, 21, 544 Search PubMed; A. B. Burg, J. Am. Chem. Soc., 1961, 83, 2226 Search PubMed; K. Jurkschat, C. Mugge, A. Tzschach, W. Uhlig and A. Zschunke, Tetrahedron Lett., 1982, 23, 1345 Search PubMed.
- M. Baudler, G. Reuschenbach and J. Hahn, Z. Anorg. Allg. Chem., 1981, 482, 27 CrossRef CAS; G. Fritz and T. Vaahs, Z. Anorg. Allg. Chem., 1987, 553, 85 CrossRef.
- A. H. Cowley and D. S. Dierdorf, J. Am. Chem. Soc., 1965, 87, 6609 CrossRef CAS; L. R. Avend, R. A. Wolcott, L. V. Cribbs and J. L. Mills, Inorg. Chem., 1989, 28, 200 CrossRef; L. R. Avens, L. V. Cribbs and J. L. Mills, Inorg. Chem., 1989, 28, 211 CrossRef.
- K.-O. Feldmann, R. Fröhlich and J. J. Weigand, Chem. Commun., 2012, 48, 4296 RSC.
- K.-O. Feldmann and J. J. Weigand, J. Am. Chem. Soc., 2012, 134, 15443 CrossRef CAS PubMed.
-
(a) X. Chen, H. Zhu, T. Wang, C. Li, L. Yan, M. Jiang, J. Liu, X. Sun, Z. Jiang and Y. Ding, J. Mol. Catal. A: Chem., 2016, 414, 37 CrossRef CAS;
(b) I. V. Komarov, A. V. Strizhak, M. Y. Kornilov, E. Zraudnitskiy and A. A. Tolmachev, Synth. Commun., 2000, 30, 243 CrossRef CAS.
- We developed a convenient, salt free synthesis of 5 and 6 which is described in the ESI.†.
-
(a) S. Fischer, L. K. Peterson and J. F. Nixon, Can. J. Chem., 1974, 52, 3981 CrossRef CAS;
(b) S. Fischer, J. Hoyano and L. K. Peterson, Can. J. Chem., 1976, 54, 2710 CrossRef CAS.
-
H. Goldwhite, Introduction to phosphorus chemistry, Cambridge University Press, Cambridge, 1981 Search PubMed.
- R. E. Cobbledick and F. W. B. Einstein, Acta Crystallogr., 1975, B31, 2731 CrossRef CAS.
- J. J. Daly, J. Chem. Soc. A, 1966, 1020 RSC.
- S. Gomez-Ruiz and E. Hey-Hawkins, Coord. Chem. Rev., 2011, 255, 1360 CrossRef CAS;
Phosphorus Chemistry: Catalysis and Material Science Applications, ed. M. Peruzzini and L. Gonsalvi, Springer, vol. 37, 2011 Search PubMed.
- J. W. Dube, C. L. B. Macdonald and P. Ragogna, Angew. Chem., Int. Ed., 2012, 51, 13026 CrossRef CAS PubMed; J. W. Dube, C. L. B. Macdonald, B. D. Ellis and R. Paul, Inorg. Chem., 2013, 52, 11438 CrossRef PubMed.
- M. Scheer, C. Kuntz, M. Stubenhofer, M. Zabel and A. Y. Timoshkin, Angew. Chem., Int. Ed., 2010, 49, 188 CrossRef CAS PubMed; M. Baacke, S. Morton, G. Johannsen, N. Weferling and O. Stelzer, Chem. Ber., 1980, 113, 1328 CrossRef; W. S. Sheldrick, S. Morton and O. Stelzer, Z. Anorg. Allg. Chem., 1981, 475, 232 CrossRef; M. Scheer, S. Gremler, E. Herrmann and P. G. Jones, J. Organomet. Chem., 1991, 414, 337 CrossRef.
- D. R. Armstrong, N. Feeder, A. D. Hopkins, M. J. Mays, D. Moncrieff, J. A. Wood, A. D. Woods and D. S. Wright, Chem. Commun., 2000, 2483 RSC.
- C. E. Averre, M. P. Coles, I. R. Crossley and I. J. Day, Dalton Trans., 2012, 41, 278 RSC.
- For a review on argentophilic interactions see: H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746 CrossRef CAS PubMed , for aurophilic interactions see: H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- C.-M. Che, Z. Mao, V. M. Miskowski, M.-C. Tse, C.-K. Chan, K.-K. Cheung, D. L. Phillips and K.-H. Leung, Angew. Chem., Int. Ed., 2000, 39, 4084 CrossRef CAS PubMed; C.-M. Che, M.-C. Tse, M. C. W. Chan, K.-K. Cheung, D. L. Phillips and K.-H. Leung, J. Am. Chem. Soc., 2000, 122, 2464 CrossRef; C.-M. Che, W.-F. Wu, K.-C. Chan and K.-K. Cheung, Chem.–Eur. J., 2001, 7, 4656 CrossRef.
- M. Henary, J. L. Wootton, S. I. Khan and J. I. Zink, Inorg. Chem., 1997, 36, 796 CrossRef CAS; P. Aslanidis, P. J. Cox, S. Divanidis and A. C. Tsipis, Inorg. Chem., 2002, 41, 6875 CrossRef PubMed.
-
H. Friebolin, Ein- und Zweidimensionale NMR-Spektroskopie, Wiley VCH, Weinheim, 2006 CrossRef PubMed; H. Fujii, M. Tomura, T. Kurahashi and M. Kujime, Inorg. Chem., 2007, 46, 541 CrossRef PubMed.
-
(a) The 31P NMR spectra of [(9a*Cu)2][OTf]2, [(9a*Ag)2][OTf]2, [(9a*Au)2][OTf]2, [(9a)2*Cu][OTf] and (9a*CuBr)2 are depicted in the ESI (Fig. 1–5†);
(b) Further information on the theoretical calculations on [(9a*M)2][OTf]2 is also given in the ESI in chapter 4.†.
- K. Schwedtmann, R. Schoemaker, F. Hennersdorf, A. Bauzá, A. Frontera, R. Weiss and J. J. Weigand, Dalton Trans., 2016, 45, 11384 RSC.
-
(a) J. P. Albrand, D. Gagnaire and J. B. Robert, J. Am. Chem. Soc., 1973, 95, 6498 CrossRef CAS;
(b) J. P. Albrand and J. B. Robert, J. Chem. Soc., Chem. Commun., 1974, 644 RSC;
(c) L. R. Smith and J. L. Mills, J. Chem. Soc., Chem. Commun., 1974, 20, 808 RSC.
-
(a) M. Baudler, J. Hahn and E. Clef, Z. Naturforsch., 1984, 39b, 438 CAS;
(b) K. Schwedtmann, J. Haberstroh, S. Roediger, A. Bauzá, A. Frontera, F. Hennersdorf and J. J. Weigand, Chem. Sci., 2019, 10, 6868 RSC.
- For further details see the ESI.†.
- J. J. Daly, J. Chem. Soc., 1964, 6147 RSC.
-
(a) N. Burford, T. S. Cameron and P. J. Ragogna, J. Am. Chem. Soc., 2001, 123, 7947 CrossRef CAS PubMed;
(b) P. A. Gray, Y.-Y. Carpenter, N. Burford and R. McDonald, Dalton Trans., 2016, 45, 2124 RSC.
- For a review on interpnictogen cations see: A. P. M. Robertson, P. A. Gray and N. Burford, Angew. Chem., Int. Ed., 2014, 53, 6050 CrossRef CAS PubMed.
- For cationic, cyclic polyphosphorus compounds see: J. Bresien, K. Faust, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 6926 CrossRef CAS PubMed; S. J. Geier, M. A. Dureen, E. Y. Ouyang and D. W. Stephan, Chem.–Eur. J., 2010, 16, 988 CrossRef PubMed; Y.-Y. Carpenter, N. Burford, M. D. Lumsden and R. McDonald, Inorg. Chem., 2011, 50, 3342 CrossRef PubMed; J. J. Weigand, N. Burford, R. J. Davidson, T. S. Cameron and P. Seeheim, J. Am. Chem. Soc., 2009, 131, 17943 CrossRef PubMed; C. A. Dyker, N. Burford, G. Menard, M. D. Lumsden and A. Decken, Inorg. Chem., 2007, 46, 4277 CrossRef PubMed; N. Burford, C. A. Dyker, M. Lumsden and A. Decken, Angew. Chem., 2005, 117, 6352 CrossRef.
-
(a) C. A. Dyker, N. Burford, M. D. Lumsden and A. Decken, J. Am. Chem. Soc., 2006, 128, 9632 CrossRef CAS PubMed;
(b) Y.-Y. Carpenter, C. A. Dyker, N. Burford, M. D. Lumsden and A. Decken, J. Am. Chem. Soc., 2008, 130, 15732 CrossRef CAS PubMed.
-
(a) A. J. Arduengo III, H. V. R. Dias and J. C. Calabrese, Chem. Lett., 1997, 143 CrossRef;
(b) A. J. Arduengo III, J. C. Calabrese, A. H. Cowley, H. V. R. Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Inorg. Chem., 1997, 36, 2151 CrossRef PubMed;
(c) J. H. Barnard, P. A. Brown, K. L. Shuford and C. D. Martin, Angew. Chem., Int. Ed., 2015, 54, 12083 CrossRef CAS PubMed.
- A. Schmidpeter, S. Lochschmidt and W. S. Sheldrick, Angew. Chem., 1985, 97, 214 CrossRef CAS.
- B. D. Ellis, M. Carlesimo and C. L. B. Macdonald, Chem. Commun., 2003, 1946 RSC; B. D. Ellis and C. L. B. Macdonald, Inorg. Chem., 2006, 45, 6864 CrossRef CAS PubMed; E. L. Norton, K. L. S. Szekely, J. W. Dube, P. G. Bomben and C. L. B. Macdonald, Inorg. Chem., 2008, 47, 1196 CrossRef PubMed.
-
M. Regitz and O. J. Scherer, Multiple Bonds and Low Coordination in Phosphorus Chemistry, Georg Thieme Verlag Thieme Medical Publishers, New York, 1990 Search PubMed.
-
H. R. Allcock, Phosphorus-Nitrogen Compounds, Academic Press, New York, 1972 Search PubMed.
- E. Payet, A. Auffrant, X. F. Le Goff and P. Le Floch, J. Organomet. Chem., 2010, 695, 1499 CrossRef CAS.
- E. Rüba, K. Mereiter, R. Schmid, V. N. Sapunov, K. Kirchner, H. Schottenberger, M. J. Calhorda and L. F. Veiros, Chem.–Eur. J., 2002, 8, 3948 CrossRef CAS; C. Coletti, L. Gonsalvi, A. Guerriero, L. Marvelli, M. Peruzzini, G. Reginato and N. Rem, Organometallics, 2010, 29, 5982 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1950469–1950486. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04501e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Antonio
Frontera
b,
Antonio
Frontera