
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulation of spatiotemporal patterning in artificial cells by a defined protein expression system†
Aoi
Yoshida‡
,
Shunshi
Kohyama‡
 ,
Kei
Fujiwara‡
*,
Saki
Nishikawa
and
Nobuhide
Doi
,
Kei
Fujiwara‡
*,
Saki
Nishikawa
and
Nobuhide
Doi
Department of Biosciences & Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223–8522, Japan. E-mail: fujiwara@bio.keio.ac.jp
First published on 16th October 2019
Abstract
Spatiotemporal patterning is a fundamental mechanism for developmental differentiation and homeostasis in living cells. Because spatiotemporal patterns are based on higher-order collective motions of elements synthesized from genes, their behavior dynamically changes according to the element amounts. Thus, to understand life and use this process for material application, creation of artificial cells with time development of spatiotemporal patterning by changes of element levels is necessary. However, realizing coupling between spatiotemporal patterning and synthesis of elements in artificial cells has been particularly challenging. In this study, we established a system that can synthesize a patterning mechanism of the bacterial cell division plane (the so-called Min system) in artificial cells by modifying a defined protein expression system and demonstrated that artificial cells can show time development of spatiotemporal patterning similar to living cells. This system also allows generation and disappearance of spatiotemporal patterning, is controllable by a small molecule in artificial cells, and has the ability for application in cargo transporters. The system developed here provides a new material and a technique for understanding life, development of drug delivery tools, and creation of molecular robots.
Introduction
Spatiotemporal patterning driven by biological molecules is a fundamental mechanism for maintaining ordered structures in living cells. Among various spatiotemporal regulation systems, intracellular reaction–diffusion coupling (iRD) has a unique characteristic in that proteins move dynamically in a similar manner to waves.1–6 The spontaneous wave movement of iRD is not derived from a power stroke as in the case of molecular machines but is based on coupling of chemical reactions and molecular diffusion similar to the Belousov–Zhabotinsky reaction, a well-known wave patterning exhibited by small molecules.7 Because iRD only appears in states far from equilibrium, artificial cells with dynamic movement and changes of iRD are a key chemical material for emulating living cells.8,9To date, the only iRD reconstituted in vitro and in artificial cells is the Min wave, a spatiotemporal regulator of the bacterial cell division plane.5,6,10–12 The Min wave is a time-dependent propagation of Min proteins (MinC, MinD, and MinE) on membranes similar to a wave.10,13
MinD and MinE are the generators of the Min wave, and MinC, an inhibitor of the initiation assembly of cell division machineries, moves as a cargo of Min waves by interaction with MinD.5 The mechanism for the emergence of the Min wave in cells is as follows (Fig. 1A).5 ATP-bound MinD binds to lipid membranes. Membrane-bound MinD can recruit other MinDs and simultaneously bind to MinE. MinE stimulates ATPase activity of MinD, and the ATPase reaction induces detachment of MinD from membranes. The detached MinD binds ATP again in the cytosol, and this cycle continuously occurs. The balance between these reaction cycles and slow diffusion during membrane binding results in the emergence of Min waves and determines spatiotemporal patterns of Min proteins.
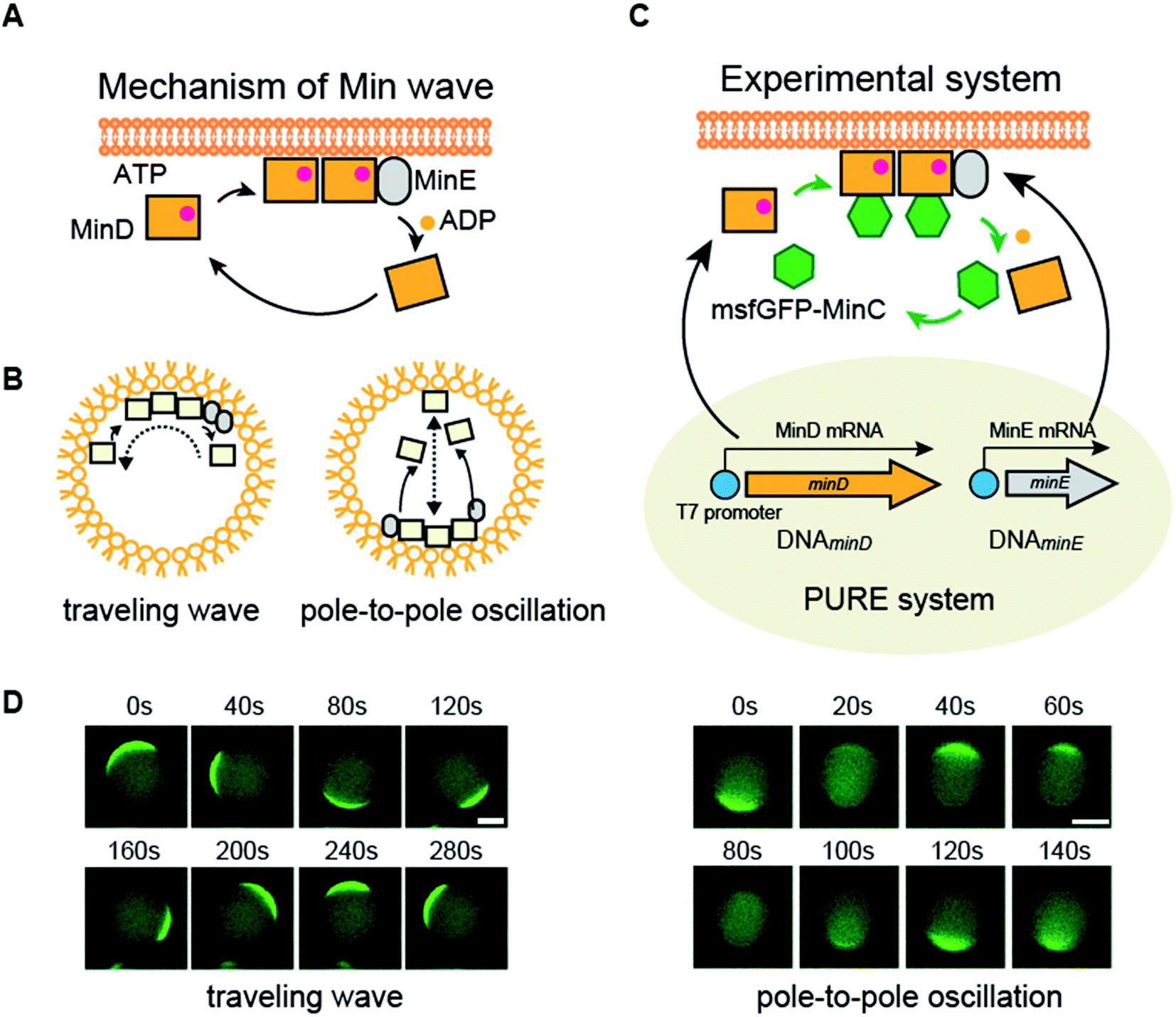 | ||
| Fig. 1 Mechanism of Min waves and experimental system of this study. (A) Molecular mechanism of the Min wave. (B) Two main modes of the Min wave. (C) Representative explanation of the experimental system used in this study. Microdroplets covered with E. coli polar lipids were used as artificial cells. MinD and/or MinE is synthesized by the PURE system in artificial cells, and purified MinDE is supplied with the PURE system if indicated. Spatiotemporal patterns of MinD were tracked by msfGFP-MinC. (D) Min wave generated by purified MinDE with 100 mg mL−1 BSA was tracked by msfGFP-MinC. Scale bars indicate 10 μm. | ||
The Min wave in artificial cells mainly shows two modes of propagation (Fig. 1B): one is traveling waves along the membrane surface and the second is oscillation between poles of the cells (pole-to-pole oscillation).11,14
In cell-sized spaces, Min waves appear in limited concentration ranges of MinDE.14 Hence, a system to change the MinDE concentration confers the ability to change dynamic spatiotemporal patterning to artificial cells. However, in closed spaces such as artificial cells, the concentration of proteins does not change without external systems. As a system to change the protein concentration in a closed space, a protein synthesis system by defined factors, the PURE system,15 has been gaining attention. The PURE system has the ability to synthesize more than 3000 protein species from DNA16 and can reconstitute biological systems such as DNA replication and the membrane insertion system by supplying multiple genes.17–21 The concentration changes due to the protein synthesis from genes emulate a central dogma of molecular biology, and therefore, this process is beneficial to introduce life-like changes of spatiotemporal patterning into artificial cells.
In this study, we demonstrated that protein synthesis of MinDE using the PURE system with additional elements can regulate Min waves in artificial cells. This achievement enabled us to regulate spatiotemporal patterning in artificial cells including generation and disappearance of Min waves in a time-dependent manner and reproduced spatiotemporal patterning similar to that observed in living cells. Furthermore, this system can be regulated by a small molecule and can be applied for cargo transportation in artificial cells. To our knowledge, this is the first report on Min protein synthesis in artificial cells to produce Min waves and to change spatiotemporal patterning in artificial cells. The system developed here is an important step for elucidating the principle of designing the spatiotemporal pattern in living cells with a DNA sequence and contributes to applications to create molecular robots and novel drug delivery systems.
Results and discussion
Experimental system in this study
In this study, we attempted to synthesize MinDE, which is a component of the Min wave, from genes using a defined transcription–translation system (PURE system) (Fig. 1C). Min waves were visualized by adding msfGFP-MinC, which is a fusion of a fluorescent protein (monomer sfGFP) and MinC (Fig. 1C). Because MinC co-localizes with MinD,5 msfGFP-MinC can track Min waves (Fig. 1D).For artificial cells, we used lipid microdroplets covered with polar lipid extracts from Escherichia coli. We chose E. coli polar lipid extracts because they have previously been used to generate Min waves.10,11,13,14,22 However, from validation using the sfGFP gene, encapsulation in artificial cells formed by polar lipid extracts significantly reduced the efficiency of protein synthesis using the PURE system (ESI Fig. S1A†). However, this problem was solved using a modified PURE system containing a high concentration of BSA (10g L−1). Although this level of BSA slightly decreased protein synthesis levels in tubes, in the case in artificial cells, it recovered protein synthesis similar to those levels observed for tube synthesis (ESI Fig. S1B†). Hence, we employed this modified PURE system for further experiments.
Regulation of spatiotemporal patterning and Min waves in artificial cells by protein synthesis
Next, we attempted to synthesize MinDE from DNA in artificial cells and analyzed its time development of spatiotemporal patterning (Fig. 2A). In a previous study, we found that for MinDE, four spatiotemporal patterns emerge: cytosol (cytosol), homogeneous localization on the membrane (membrane), inhomogeneous localization on the membrane (inhomogeneous), and the Min wave (Fig. 2B).14 Our previous experiments using purified proteins showed that (i) no MinDE (lower than 0.25 μM) or higher MinE concentration than MinD induces localization in the cytosol, (ii) higher MinD concentration than MinE induces homogeneous localization on the membrane, (iii) similar levels of MinD and MinE (>2 μM) induce inhomogeneous localization on the membrane (ESI Fig. S2†), and (iv) similar levels of MinD and MinE (0.25–2 μM) induce Min waves.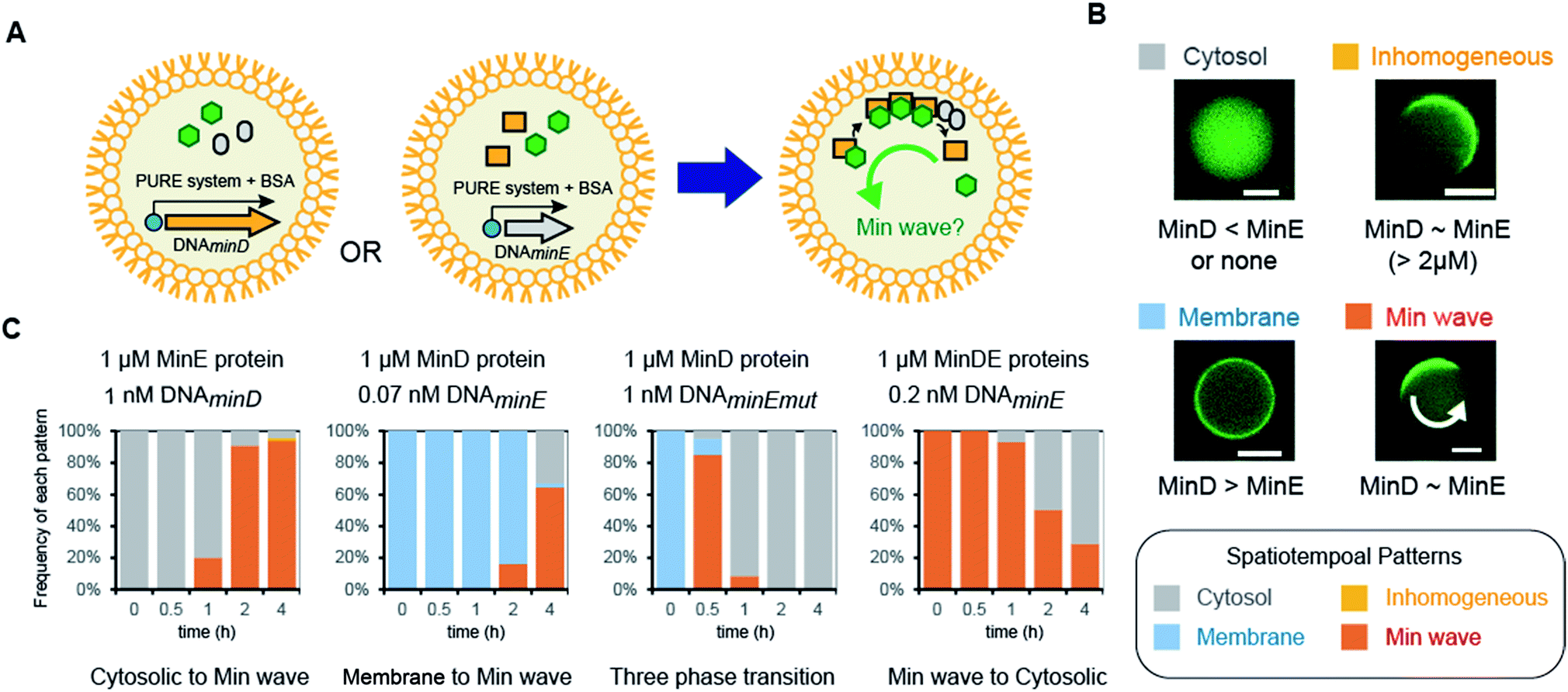 | ||
| Fig. 2 Regulation of spatiotemporal patterning by protein synthesis in artificial cells. (A) Representative illustration of the experimental system. Symbols indicate the same elements shown in Fig. 1C. (B) Patterns of MinC localization and its relationship with the MinDE concentration. Scale bars indicate 10 μm. (C) Time-development of frequencies of each pattern induced by MinD or MinE (or the N-terminal mutant of MinE) synthesized in artificial cells. Concentrations of DNA and proteins used are described in the figure. | ||
The mechanism of spatiotemporal patterning of Min proteins is considered as follows. ATP-bound MinD (MinD-ATP) binds membranes and recruits another MinD-ATP (a positive feedback). ATPase activity of MinD is stimulated by MinE, and ATP-less MinD is dissociated from membranes (a negative feedback). The spatiotemporal patterns of MinD are determined by a tug-of-war between the positive and negative feedbacks. When the positive feedback is stronger than the negative one (namely, MinD > MinE), MinD universally localizes on membranes. When the negative feedback is stronger than the positive one (MinE > MinD), MinD stays in the cytosol. Under a certain balance of the positive and negative feedbacks (MinD ∼ MinE), MinD moves like a wave (Min wave). Under the balance (concentrations of MinDE are similar) but a higher MinDE concentration (>2 μM), MinD inhomogeneously localizes on membranes like a Min wave. However, the localization moves little at the high concentrations (ESI Fig. S2†). Although the reason for the little movement of the inhomogeneous localization is still elusive, the pattern is actually different from the Min wave.
By using the modified PURE system, we tested the single synthesis of MinD or MinE from DNA in artificial cells, and in this case, the other protein (not synthesized in the system) was added as a purified protein. First, MinE was added as a purified protein, and MinD was synthesized from DNA by the modified PURE system in artificial cells. Because protein synthesis by the modified PURE system stops before 4 hours (ESI Fig. S1†), we analyzed the changes of spatiotemporal patterning up to 4 hours. In this case, all artificial cells show cytosolic localization, and then, the percentage of artificial cells with the Min wave increases with time; after 4 hours, Min waves were observed in 90% or more of the artificial cells (Fig. 2C).
Second, MinD was added as a purified protein, and MinE was synthesized from DNA using the modified PURE system in artificial cells. However, in the case of 1 nM DNAminE, msfGFP-MinC was transferred to the cytoplasm in almost all artificial cells at 0.5 h. We assumed that this fast change of the localization was due to excess expression of MinE. Therefore, we lowered the concentration of DNAminE. In the case of 0.07 nM DNAminE, membrane localization shifted to Min waves with time and after 4 hours, Min waves were observed in about 65% of the artificial cells (Fig. 2C). Similarly, a MinE mutant, in which the positively charged residues for membrane binding were replaced with neutral or negative ones (R10G/K11E/K12E), was used. In this mutant, the membrane binding ability of MinE was weakened,23 and therefore, it was expected that the mutant generates Min waves at a higher concentration than the wild-type. Actually, by using 1 nM DNA encoding the mutant, more dynamic changes of spatiotemporal patterning were observed. Membrane localization shifted to Min waves in over 80% of artificial cells after 0.5 h, but disappeared with time, and after 4 h synthesis, msfGFP-MinC settled in the cytosol in all artificial cells which indicated that an excess amount of MinE was synthesized (Fig. 2C). Anyway, these results showed that protein synthesis in artificial cells can possibly change among three phases: membrane localization, Min wave, and localization in the cytosol.
Third, 1 μM MinDE was added as a purified protein, and additional MinE was synthesized in artificial cells. In this case, all artificial cells showed Min waves and their frequency decreased with time due to the excess amount of MinE (Fig. 2C). Taken together, either MinD or MinE synthesis in artificial cells can change spatiotemporal patterning in artificial cells with the generation and disappearance of Min waves. Because achievement of both generation and disappearance of dynamic spatiotemporal patterning in artificial cells is difficult in the case of the usage of purified proteins, this point is a remarkable feature of the system using protein synthesis from DNA.
Fourth, we tested Min wave generation by simultaneous protein synthesis MinDE from DNA in artificial cells (Fig. 3A). Estimation by quantitative western blotting demonstrated that the levels of protein synthesis of MinE were more than 6-fold higher than that of MinD. Therefore, to adjust the synthesis levels of each protein, the ratio of DNA concentration of MinD to MinE was altered. We varied DNAMinD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DNAMinE (nM) = 5:2, 5:0.5, and 5:0.2, and the frequency of Min wave generation in artificial cells was tracked with time. As a result, Min waves appeared specifically in the case of DNAMinD:DNAMinE = 5:0.5, and these waves were observed in ∼65% of artificial cells (Fig. 3B). Min waves preferably appeared in artificial cells larger than 15 μm, and therefore, we focused on larger sized artificial cells for further analyses. The ratio of the traveling wave and the pole-to-pole wave was stochastically determined, and almost all waves were traveling waves. The dominance of the traveling wave results from the inherent wave transition from traveling to pole-to-pole in spherical artificial cells as shown in our previous study,14 and it has been indicated that the oval shape of E. coli cells is associated with stable emergence of pole-to-pole oscillation.22 The wave frequency of the Min wave produced by the de novo synthesis of MinDE in the artificial cell was 1/120–1/240 s−1. This value is similar to that of Min waves in artificial cells reconstituted by using purified MinDE.14 However, it is smaller than 1/60–1/120 s−1 in the case of E. coli cells,5 suggesting that some differences such as crowding levels and cell shapes between artificial cells and living E. coli cells are associated with the Min wave frequency. Lower or higher concentration ratios of DNAMinE reduced the frequencies of Min wave emergence in artificial cells (Fig. 3B). These results indicated that control of the MinDE expression ratio is an important factor for Min wave generation in an artificial cell.
:DNAMinE (nM) = 5:2, 5:0.5, and 5:0.2, and the frequency of Min wave generation in artificial cells was tracked with time. As a result, Min waves appeared specifically in the case of DNAMinD:DNAMinE = 5:0.5, and these waves were observed in ∼65% of artificial cells (Fig. 3B). Min waves preferably appeared in artificial cells larger than 15 μm, and therefore, we focused on larger sized artificial cells for further analyses. The ratio of the traveling wave and the pole-to-pole wave was stochastically determined, and almost all waves were traveling waves. The dominance of the traveling wave results from the inherent wave transition from traveling to pole-to-pole in spherical artificial cells as shown in our previous study,14 and it has been indicated that the oval shape of E. coli cells is associated with stable emergence of pole-to-pole oscillation.22 The wave frequency of the Min wave produced by the de novo synthesis of MinDE in the artificial cell was 1/120–1/240 s−1. This value is similar to that of Min waves in artificial cells reconstituted by using purified MinDE.14 However, it is smaller than 1/60–1/120 s−1 in the case of E. coli cells,5 suggesting that some differences such as crowding levels and cell shapes between artificial cells and living E. coli cells are associated with the Min wave frequency. Lower or higher concentration ratios of DNAMinE reduced the frequencies of Min wave emergence in artificial cells (Fig. 3B). These results indicated that control of the MinDE expression ratio is an important factor for Min wave generation in an artificial cell.
 | ||
| Fig. 3 Min wave generation in artificial cells using de novo synthesized MinDE. (A) Representative illustration of the experimental system. (B) Time development of frequencies of each pattern induced by MinD and MinE synthesized in artificial cells. The ratio of DNAminD and DNAminE is indicated in the figure. | ||
Time-lapse imaging of spatiotemporal patterning changes in artificial cells by protein synthesis
We showed statistical evidence for the regulation of spatiotemporal patterning in artificial cells by protein synthesis of MinDE. However, the evidence indirectly shows changes of spatiotemporal patterning. To directly show the patterning change, we tracked spatiotemporal patterns of msfGFP-MinC by time-lapse imaging using a heating glass set on the stage of a fluorescence microscope. The time-lapse imaging clearly showed that MinD expression by 1 nM DNAminD in the artificial cells induces the localization shift from the cytosol to the membrane (Fig. 4A and ESI Movie S1†). Similarly, MinD expression by 5 nM DNAminD in the artificial cells supplemented with 1 μM purified MinE induced spatiotemporal patterning shift from membranes to Min waves (Fig. 4B and ESI Movie S2†). Furthermore, MinE expression by 1 nM DNAminE in the artificial cells supplemented with 1 μM purified MinD induced localization shifts from membranes to Min wave, and finally the pattern settled in the cytosol (Fig. 4C and ESI Movie S3†). Namely, the three phases found in mutant expression (Fig. 2C) were also tracked by time-lapse imaging. These results clearly indicated that all types of regulation of spatiotemporal patterning shown in Fig. 2C actually happen.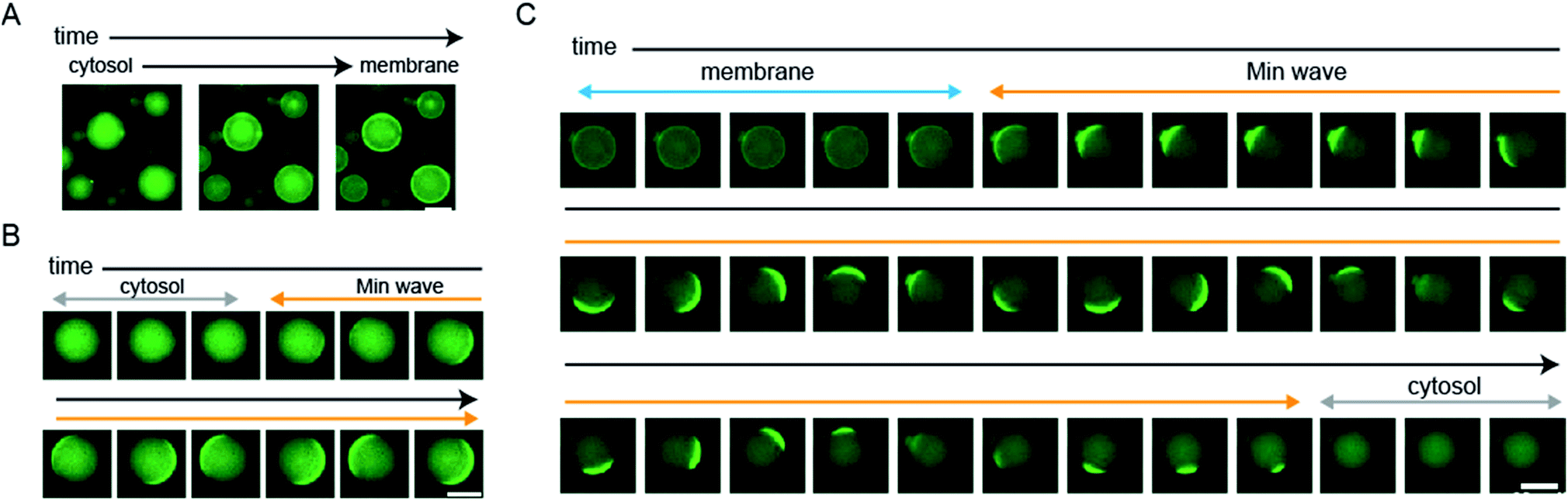 | ||
| Fig. 4 Direct observation of changes in spatiotemporal patterning in artificial cells by protein expression. (A) MinD was synthesized from 1 nM DNAminD in artificial cells, 16 min intervals. (B) MinD was synthesized from 5 nM DNAminD in artificial cells containing 1 μM purified MinE, 20 s intervals. (C) MinE was synthesized from 1 nM DNAminE in artificial cells containing 1 μM purified MinD, 20 s intervals. (A–C) Scale bars indicate 20 μm. | ||
Generation of Min waves in artificial cells by protein synthesis from the minDE operon
In the case of mixtures of multiple DNA species, the ratio of DNA changes among artificial cells due to their small volume inducing fluctuations of numbers of molecules encapsulated. This ratio variation of DNA is not a preferred feature to control generation of Min waves in artificial cells. A method to solve this problem is the conjugation of multiple DNA to synthesize elements from single DNA by constructing an operon DNA structure (Fig. 5A). Although it is not easy to rationally design operon DNA with the ability to synthesize elements at a controlled ratio, the genes of MinD and MinE in the genome of living cells inherently show an operon DNA structure.24 We synthesized a DNA construct to regulate this operon structure under a T7 promoter and examined whether it could be a source of Min wave generation. However, MinDE synthesized in a tube demonstrated that the synthesis levels were 2.3 μM for MinD and 0.4 μM for MinE. Due to this unbalanced synthesis level, encapsulation of the synthesized MinDE in artificial cells did not generate Min waves. Surprisingly, this negative result became positive when MinDE was synthesized in artificial cells. In this case, more than 50% of artificial cells showed Min waves (ESI Movie S4†), and the other 50% led to inhomogeneous localization on membranes (Fig. 5B). Because inhomogeneous localization indicates that the levels of MinDE are similar, but the amounts are higher than those for Min wave conditions, this result strongly indicates that the ratio of MinDE is essentially the same in almost all artificial cells. Although the reason for the variation of the ratio of MinDE between in vitro and artificial cell synthesis is still elusive, the results suggest that synthesis environments such as membranes are associated with control of the synthesis ratio of proteins encoded by operon DNAs.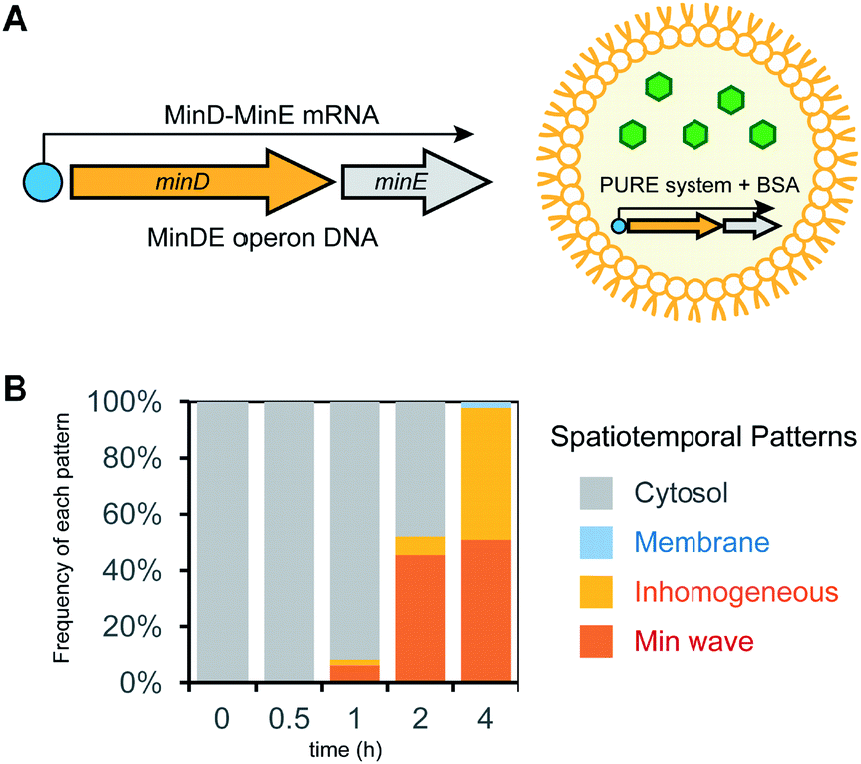 | ||
| Fig. 5 Min wave generation in artificial cells by protein synthesis using a DNA operon structure. (A) DNA operon structure for the synthesis of MinD and MinE from the same DNA. (B) Time development of frequencies of each pattern induced by MinD and MinE synthesized in artificial cells. | ||
Control of Min wave generation in artificial cells by small molecules
Finally, we demonstrated that Min wave generation from DNA in artificial cells can be controlled by a small molecule. We introduced the Lac operator, the function of which involves switching promoter levels by IPTG, to downstream of the DNA operon promoter. Using the Lac operator, the genetic circuit works only in the presence of IPTG (Fig. 6A). Because the commercial PURE system does not have the LacI protein, which is necessary for regulating the Lac operator, we purified LacI as a recombinant protein and added the DNA operon. As expected, in the absence of IPTG, the modified PURE system with LacI did not generate Min waves in artificial cells, and these waves appeared in the presence of IPTG (Fig. 6B). These results indicate that the generation of Min waves by MinDE synthesis from genes in artificial cells can be controlled by an external molecule at the desired timing.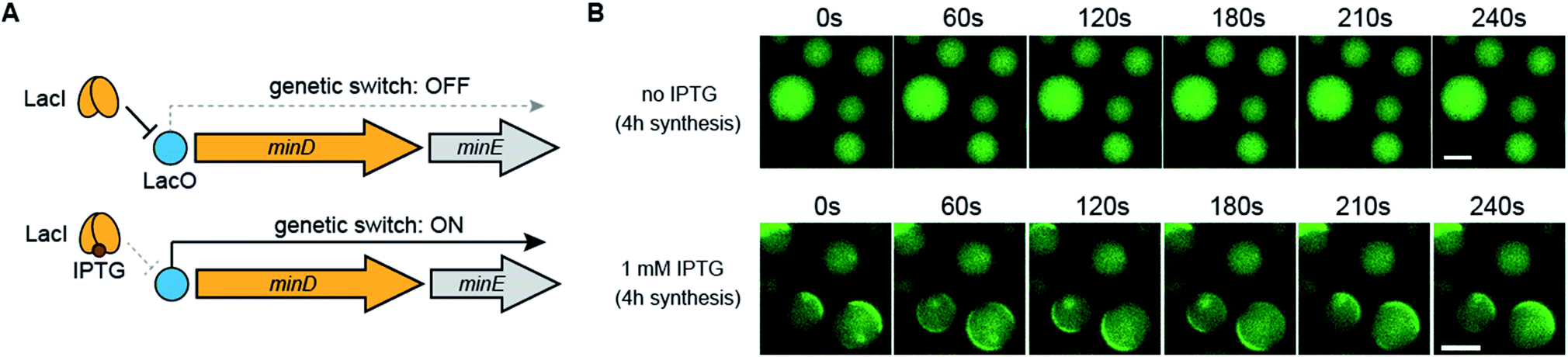 | ||
| Fig. 6 Regulation of Min wave generation via protein synthesis by a small molecule. (A) Representative illustration of the DNA construct regulated by LacI and IPTG. (B) Wide-view figures of Min wave generation in the absence and presence of 1 mM IPTG. Time-lapse images were captured after 4 h of protein synthesis with and without IPTG in artificial cells. In this case, the PURE system supplemented with 3 μM LacI and 10 mg mL−1 BSA was used. The concentration of operon DNA was 0.6 nM. Scale bars indicate 10 μm. | ||
In microdroplets covered with lipids, it is not easy to supply IPTG from an outside solution of artificial cells. By contrast, it is easy to supply IPTG from an outside solution in the case of liposomes. Thus, we prepared liposomes from a mixture of synthetic lipids (PC:PG = 7:3) entrapping the modified PURE system using the droplet-transfer method. We chose the lipid conditions because they have been used for Min wave observation in liposomes in the case of the usage of purified MinDE,12 and the efficiency of liposome formation is better than that of the case of the polar lipids. As expected, protein expression from the Min operon DNA starts by using IPTG from the outside solution, and obvious Min waves were observed (ESI Movie S5†). These results clearly indicate that the regulation system of spatiotemporal patterning developed here works in artificial cells prepared from synthetic lipids.
Effects of lipids as an inhibitor of protein expression by the PURE system in artificial cells
Finally, we investigated the reason why the activity of protein expression was inhibited by encapsulation in microdroplets covered with E. coli polar lipids. Because protein synthesis normally works in microdroplets covered with PC (ESI Fig. S3A†), we suspected that charges of lipids are the cause of the inhibition. In E. coli polar lipids, phosphatidylglycerol (PG) and cardiolipin (CL) are negatively charged and a previous Min wave study has shown that CL is able to be replaced with PG.25 Thus, we tested protein expression activity within microdroplets covered with mixtures of PC and PG. Consequently, elevated PG levels inhibited protein expression levels (ESI Fig. S3A†). The inhibition by PG was restored by BSA addition similar to the case of E. coli polar lipids (ESI Fig. S3B†). These results clearly showed that the inhibition of protein expression resulted from the negative charges of lipids.Next, we tested whether the inhibition by negatively charged lipids is specific to the protein expression inside a cell-sized space or not. To check this point, small unilamellar vesicles (SUVs) made of E. coli polar lipids were added to the reaction mixture for the PURE system. In this experimental system, the PURE system solution is affected by charges of lipids of SUVs. However, the PURE system is not encapsulated in a cell-sized space. Consequently, SUV supplementation showed no effects on levels of sfGFP expression by the PURE system even without BSA addition (ESI Fig. S4†). Thus, we concluded that the inhibition is specific to the case of protein expression inside a cell-sized space.
In our previous study, we found that a high surface-area-to-volume ratio of cell-sized spaces enhances membrane localization of a positively charged protein (MinE) and that BSA or cell extracts cancel the localization change.14 Because the localization shift of MinE was derived from its positive charges, similar localization changes in artificial cells can happen in the case of the components of the PURE system. Especially, MinD also binds membranes via negative charges of lipids, and BSA does not prevent the membrane binding of MinD.14 The difference indicates that molecules weakly interacting with membranes like MinE are associated with the inhibition of protein expression. Therefore, a plausible explanation for the cause of the inhibition is localization changes of some components in the PURE system in cell-sized spaces. Further studies focusing on the localization of each component of the PURE system (nearly 100 species) in artificial cells will identify the detailed molecular mechanism of the inhibition and also will illuminate the mechanism of another membrane effect shown by a previous study, acceleration of protein expression rates in artificial cells.26
Conclusions
Although spatiotemporal patterning in artificial cells has been studied previously,27 its regulation within artificial cells is still a challenge. By suppressing the inhibitory effect of negatively charged lipid membranes on protein expression by the PURE system, here we succeeded in the regulation of spatiotemporal patterning in artificial cells including the generation and disappearance of Min waves via a defined protein expression system to synthesize MinD and MinE from genes. The present results not only develop a way to regulate spatiotemporal patterning in artificial cells but also pave a way to reconstitute the cell division machinery in artificial cells.In addition, we succeeded in controlling the development of Min waves using small molecules. This achievement indicates that spatiotemporal development with timing control such as that in living cells can be reproduced in artificial cells. Finally, we revealed that negatively charged lipids inhibit protein expression by the PURE system in cell-sized spaces. This finding provides novel insights into understanding cell-sized confinement effects,26–29 which contributes to both biology and physics.
By combining recent advances in technologies on cell-free protein expression in artificial cells30–34 and molecular robotics,12,35,36 the ability of artificial cells will be further versatile and intelligent. Furthermore, the achievements in this study will be applied to endow artificial cells with an ability to realize spatiotemporal development by sensing environmental signals like a molecular robot. Hence, the system developed here will provide an important material in the field of molecular robotics with intelligent control and in drug delivery systems in which artificial cells are widely used.
Experimental
Preparation of DNA templates for protein synthesis using the PURE system
The genes of MinD, MinE, and sfGFP under the T7 promoter were amplified from pET15b-MinD, pET29-MinE-His,14 and pET29-sfGFP with a histidine tag by PCR using a primer set (PURE primers: CCCGCGAAATTAATACGACTCAC/CAAAAACCCCTCAAGACCCGT) and ExTaq DNA polymerase (TAKARA, Shiga, Japan). After checking the fragment length of the product by agarose gel electrophoresis, DNA strands were purified using a QIAquick PCR purification kit (Qiagen, Venlo, Netherlands). MinDE operons were constructed by insertion of MinE-His downstream of the MinD gene of pET15b-MinD using PCR and the iVEC method.37 For the construction, KOD One (TOYOBO, Osaka, Japan) was used for fragment amplification. The internal sequence between the terminal codon of MinD and the initiation codon of MinE was GTT, which is the same as the internal sequence of the minDE operon in the Escherichia coli genome. N-terminal mutation of MinE (R10G/K11E/K12E) was introduced using PrimeSTAR Max DNA polymerase (TAKARA) using pET-29-MinE-His as a template and a primer set (TCGGGGGAGGAAAACACAGCCAACATTGCA/GTTTTCCTCCCCCGAGAGAAAGAAATCGAG). After the plasmid purification, DNA sequences were verified using a PRISM3130xl (Applied Biosystems, Foster City, CA, USA). The Lac operator in the pET15b vector was used for LacI-IPTG regulation. These constructs were amplified by PCR using ExTaq DNA polymerase and PURE primers and were used as the DNA template for protein synthesis after purification, similar to the description above.To construct pET15b-msfGFP-MinC, a V206K mutation was introduced into pET15b-sfGFP-MinC14 using PrimeSTAR Max DNA polymerase and a primer set (CAATCTAAACTTTCGAAAGATCCCAACGAA/CGAAAGTTTAGATTGTGTCGACAGGT). The pSUMO vector was constructed by cloning an artificially synthesized His-tagged SUMO gene into the NcoI/XhoI site of pET15b. To construct pSUMO-LacI, the LacI gene was amplified from the genome of E. coli MG1655 strain and was cloned into pSUMO by iVEC. To construct pSUMO-Ulp1, an artificially synthesized Ulp1 gene with a His-tag at the C-terminal was cloned into pSUMO. The DNA sequence except the plasmid backbone of all plasmids was verified using a PRISM3130xl.
Protein expression and purification
E. coli BL21-CodonPlus(DE3) RIPL cells (Agilent Technologies, Santa Clara, CA, USA) were transformed with either pET15-MinD, pET29-MinE-His, pET15-msfGFP-MinC, pSUMO-LacI, or pSUMO-Ulp1-His, and cultivated in LB medium to overexpress proteins. For MinD, MinE, and msfGFP-MinC, the proteins were expressed by 1 mM IPTG at OD600 = 0.2, and the cells were further cultivated at 37 °C for 3–4 h. Cells were collected by centrifugation at 8000 × g for 2 min at 4 °C and suspended in a lysis buffer (50 mM NaH2PO4 pH 7.6, 300 mM NaCl, 1 mM DTT, 0.1 mM PMSF, and 10 mM imidazole). In the case of purification of His-MinD, 0.2 mM ADP was added to the lysis buffer. Then, cells were disrupted by sonication using a Sonifier250 (Branson, Danbury, CT, USA) for 30 min on ice. The crude lysate was fractionated by centrifugation at 20000 × g for 20 min at 4 °C. The soluble fraction was mixed with 1 mL cOmplete His-Tag purification resin (Roche, Basel, Switzerland) with gentle shaking at 4 °C for 1 h. The mixture was loaded onto a Polyprep chromatography column, washed with 15 mL of wash buffer (50 mM NaH2PO4 pH 7.6, 300 mM NaCl, 0.1 mM PMSF, 10% glycerol, 0.1 mM EDTA, and 20 mM imidazole), and then, His-tagged proteins were eluded with an elution buffer (50 mM NaH2PO4 pH 7.6, 300 mM NaCl, 0.1 mM PMSF, 10% glycerol, 0.1 mM EDTA, and 250 mM imidazole). The elution buffer was exchanged with the storage buffer (50 mM HEPES-KOH pH 7.6, 150 mM GluK, 10% glycerol, 0.1 mM EDTA, and 1.0 mM ADP in the case of purification of His-MinD) by ultrafiltration using an AmiconUltra-15 10K centrifugal filter device (Merck Millipore, Billerica, MA, USA). Protein concentrations were estimated by CBB staining after SDS-PAGE. The band intensity was measured using Image J, and the protein concentration was quantified by standard curves obtained from various concentrations of BSA.
For other proteins, the proteins were expressed by 0.1 mM IPTG at OD600 = 0.2, and the cells were further cultivated at 30 °C overnight. After collection, the cells were dissolved with STI buffer (20 mM Tris–HCl pH 8.0, 500 mM NaCl, and 40 mM imidazole) and disrupted by sonication. The supernatant of the cell extract after centrifugation at 20000 × g for 10 min at 4 °C was loaded onto a HisTrap column (GE healthcare, Chicago, IL, USA). The column was washed with STI buffer and His-tagged proteins were eluted with EL buffer (20 mM Tris–HCl pH 8.0, 500 mM NaCl, and 400 mM imidazole). The eluted fractions were diluted 10-fold with 20 mM Hepes-KOH pH 7.6, and then, the target proteins were purified with a HiTrap Q column (GE healthcare) using 20 mM Hepes-KOH pH 7.6 with 100–400 mM KCl. For SUMO-tagged proteins, the SUMO-tag was cleaved using laboratory purified Ulp1. The His-SUMO fragment and Ulp1 were removed with cOmplete His-Tag purification resin.
BSA was purchased from Sigma-Aldrich (Cohn Fraction V, A6003). Prior to usage, BSA was washed with ultrapure water by ultrafiltration using an AmiconUltra-15 10K centrifugal filter device.
Observation of Min waves in artificial cells
E. coli polar lipid extract (Avanti, Alabaster, AL, USA) in chloroform at 25 mg mL−1 was dried under an argon flow and then, dissolved with mineral oil (Nacalai Tesque, Kyoto, Japan) to 1 mg mL−1. The lipid mixture was sonicated for 90 min at 60 °C and mixed by vortexing. 2 μL of the inner solution (positive control or the PURE system before or after reaction) was added to 100 μL of the prepared lipid mixture. Water-in-oil microdroplets as the artificial cells were prepared by tapping, following previous studies.14,38 This process encapsulates 100% of the inner solution into the microdroplets.In the case of positive control of Min wave generation, the inner solution consisted of 1 μM MinD, 1 μM MinE, 0.3 μM msfGFP-MinC, 2.5 mM ATP, and 100 mg mL−1 BSA in Min buffer (25 mM Tris–HCl pH 7.5, 150 mM GluK, and 5 mM GluMg). To observe the Min wave in artificial cells, 10 μL of the artificial cell solution was placed into two glass coverslip slit with a double-sided tape spacer. In the case of protein synthesis in tubes, the mixture described below was encapsulated similarly. In the case of protein synthesis in artificial cells, the encapsulation protocol is the same; however, a glass based dish was used instead of the two glass coverslip slits. All movements of MinC in artificial cells larger than 15 μm diameter were tracked using a fluorescence microscope (Axiovert observer Z1, Carl Zeiss). Images were taken at 10 or 20 s intervals for 5 or 10 minutes. Judgement of the presence or absence of the Min wave was based on the changes of localization of Min waves over 45 degrees in artificial cells within 5 min. The frequency of each spatiotemporal pattern of MinC localization was determined from 150–700 artificial cells at each time.
SUV preparation
Small unilamellar vesicles (SUVs) are prepared by an extruder method. Briefly, 2.5 mg E. coli polar lipids or POPC dissolved in chloroform were dried under an argon gas flow. To obtain multilamellar vesicles, 500 μL of HG buffer (50 mM Hepes-KOH pH 7.6, and 200 mM GluK) was added to the dried film. The obtained multilamellar vesicles were converted to SUVs using an extruder (Avanti) using a 400 nm pore filter and 50 nm filter.Protein synthesis by the PURE system in test tubes and in artificial cells
For protein synthesis from genes in tubes and in artificial cells, PUREfrex ver. 1.0 (Gene Frontier, Chiba, Japan) was used as the PURE system. For in vitro synthesis, msfGFP-MinC (0.3 μM), 10 mg mL−1 BSA, and 1 nM DNA encoding sfGFP, or the MinDE operon, were added to PUREfrex ver. 1.0 in test tubes on ice. Protein synthesis was carried out for 4 h at 37 °C. Concentrations of elements for cell-free protein synthesis and temperature were at the optimum conditions of PUREfrex ver. 1.0. After the 4 h reaction, concentrations of the protein synthesized were quantified by western blotting or fluorescence imaging. The maximum levels of MinDE expression by the modified PURE system we observed were 3 μM MinD and 20 μM MinE. Because no fluorescence probe was conjugated to MinDE, the precise protein expression levels in artificial cells were not quantified. However, the similar levels of sfGFP expression in tubes and in artificial cells by the modified PURE system (ESI Fig. S1†) indicate that the expression levels of MinDE are less than the maximum levels found in the tube reactions. Primary antibodies used in western blotting were prepared from a rabbit polyclonal antibody obtained by immunization using purified 6xHis-MinD or MinE-6xHis, respectively (Scrum Inc., Sumida-ku, Tokyo). Anti-rabbit IgG HRP-linked whole antibody (Sigma-Aldrich, St. Louis, MO, USA) was used as a secondary antibody, and proteins were detected using Chemi-Lumi One Ultra (Nacalai Tesque). Band intensities were measured using ImageJ, and the protein concentration was estimated from the standard curves obtained from the purified proteins. To check the ability of the protein synthesized to generate the Min wave, the mixture after 4 h of reaction was encapsulated into artificial cells as described above.For protein synthesis in artificial cells, 0.3 μM msfGFP-MinC, 10 mg mL−1 BSA, 0.07 to 5 nM DNA encoding MinD, MinE, MinE N-terminal mutant, or MinDE operon was added to PUREfrex ver. 1.0. If necessary, purified 1 μM MinD, 1 μM MinE, or 3 μM LacI was added. For the Lac operator experiment, DNA was pre-incubated with LacI for 10 min at room temperature to minimize leakage of transcription. After mixing all elements on ice, the mixture was encapsulated in artificial cells as described above. The solution prepared was spotted on glass based dishes (AGC TECHNO GLASS, Shizuoka, Japan), and protein synthesis in artificial cells was carried out at 37 °C. Change of dynamics in artificial cells was observed using a fluorescence microscope 0.5, 1, 2, and 4 h after the time to start incubation at 37 °C.
To obtain time-lapse images of spatiotemporal patterning within artificial cells, artificial cells entrapping the modified PURE system and purified Min proteins if indicated were spotted on a cover glass and incubated on a heat glass (Tpi-SQFTX, Tokai Hit, Shizuoka, Japan) set on the fluorescence microscope at 37 °C. Fluorescence images of msfGFP-minC were captured at a 20 second interval for 1 h of incubation.
To convert microdroplets to liposomes, a small scale droplet-transfer method38 was applied. Briefly, the microdroplet solution prepared using the POPC/DOPG mixture (7:3) entrapping the PURE system with the DNA encoding for minDE under the Lac operon, 10 g L−1 BSA, and 100 mM sucrose was layered on the external solution [1/2 Solution A of the PUREfrex kit and 100 mM IPTG], and liposomes were formed by centrifugation at 20000 × g for 30 s at room temperature. Liposomes were collected from the aqueous phase of the tube centrifuged. Because of the transfer efficiency, less than 5% of the microdroplets were converted into liposomes. After 4 h of incubation at 37 °C, liposomes were examined using the fluorescence microscope.
For the analysis of the effects of lipids on cell-free protein expression, E. coli polar lipids and a series of PC/PG mixtures (POPC:DOPG = 10:0, 7:3, and 5:5) were used to prepare artificial cells. To prepare the PC/PG mixtures, PC and PG in minerals were individually prepared similar to the case of E. coli polar lipids, and the resultant lipids were mixed. For the analysis, 1 nM DNA encoding sfGFP was added to PUREfrex ver. 1.0 with and without 10 mg mL−1 BSA.
Author contribution
K. F. conducted and designed the research. S. K., A. Y., and K. F. conceived and performed wet experiments. S. N. partially performed experiments with the aid of S. K. All authors analyzed and discussed the data. K. F. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. N. Yoshinaga (Tohoku University) and Prof. M. Yanagisawa (The University of Tokyo) for helpful discussion. We acknowledge the financial support from JSPS KAKENHI Grant Number JP15KT0081, JP15H00826, JP16H00809, 17H02103, 17H05235, JP18H04565 for K. F.Notes and references
- S. Adachi, K. Hori and S. Hiraga, J. Mol. Biol., 2006, 356, 850–863 CrossRef CAS PubMed.
- Y. Arai, T. Shibata, S. Matsuoka, M. J. Sato, T. Yanagida and M. Ueda, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12399–12404 CrossRef CAS PubMed.
- C. H. Huang, M. Tang, C. Shi, P. A. Iglesias and P. N. Devreotes, Nat. Cell Biol., 2013, 15, 1307–1316 CrossRef CAS PubMed.
- A. B. Goryachev and A. V. Pokhilko, FEBS Lett., 2008, 582, 1437–1443 CrossRef CAS PubMed.
- L. Rothfield, A. Taghbalout and Y. L. Shih, Nat. Rev. Microbiol., 2005, 3, 959–968 CrossRef CAS PubMed.
- V. W. Rowlett and W. Margolin, Curr. Biol., 2013, 23, R553–R556 CrossRef CAS PubMed.
- M. Dolnik, A. M. Zhabotinsky, A. B. Rovinsky and I. R. Epstein, Chem. Eng. Sci., 2000, 55, 223–231 CrossRef CAS.
- S. Soh, M. Byrska, K. Kandere-Grzybowska and B. A. Grzybowski, Angew. Chem., Int. Ed., 2010, 49, 4170–4198 CrossRef CAS PubMed.
- I. R. Epstein and B. Xu, Nat. Nanotechnol., 2016, 11, 312–319 CrossRef CAS PubMed.
- M. Loose, E. Fischer-Friedrich, J. Ries, K. Kruse and P. Schwille, Science, 2008, 320, 789–792 CrossRef CAS PubMed.
- K. Zieske, G. Chwastek and P. Schwille, Angew. Chem., Int. Ed., 2016, 55, 13455–13459 CrossRef CAS PubMed.
- T. Litschel, B. Ramm, R. Maas, M. Heymann and P. Schwille, Angew. Chem., Int. Ed., 2018, 57, 16286–16290 CrossRef CAS PubMed.
- M. Loose, E. Fischer-Friedrich, C. Herold, K. Kruse and P. Schwille, Nat. Struct. Mol. Biol., 2011, 18, 577–583 CrossRef CAS PubMed.
- S. Kohyama, N. Yoshinaga, M. Yanagisawa, K. Fujiwara and N. Doi, eLife, 2019, e44591 CrossRef PubMed.
- Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda, Nat. Biotechnol., 2001, 19, 751–755 CrossRef CAS PubMed.
- T. Niwa, B. W. Ying, K. Saito, W. Jin, S. Takada, T. Ueda and H. Taguchi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4201–4206 CrossRef CAS PubMed.
- H. Asahara and S. Chong, Nucleic Acids Res., 2010, 38, e141 CrossRef PubMed.
- K. Fujiwara, T. Katayama and S. M. Nomura, Nucleic Acids Res., 2013, 41, 7176–7183 CrossRef CAS PubMed.
- H. Matsubayashi, Y. Kuruma and T. Ueda, Angew. Chem., Int. Ed., 2014, 53, 7535–7538 CrossRef CAS PubMed.
- J. Li, W. Haas, K. Jackson, E. Kuru, M. C. Jewett, Z. H. Fan, S. Gygi and G. M. Church, ACS Synth. Biol., 2017, 6, 1327–1336 CrossRef CAS PubMed.
- R. Mizuuchi and N. Ichihashi, Nat. Ecol. Evol., 2018, 2, 1654–1660 CrossRef PubMed.
- K. Zieske and P. Schwille, Angew. Chem., Int. Ed., 2013, 52, 459–462 CrossRef CAS PubMed.
- C. W. Hsieh, T. Y. Lin, H. M. Lai, C. C. Lin, T. S. Hsieh and Y. L. Shih, Mol. Microbiol., 2010, 75, 499–512 CrossRef CAS PubMed.
- P. A. de Boer, R. E. Crossley and L. I. Rothfield, Cell, 1989, 56, 641–649 CrossRef CAS PubMed.
- E. Mossessova and C. D. Lima, Mol. Cell, 2000, 5, 865–876 CrossRef CAS PubMed.
- A. Kato, M. Yanagisawa, Y. T. Sato, K. Fujiwara and K. Yoshikawa, Sci. Rep., 2012, 2, 283 CrossRef PubMed.
- M. Yanagisawa, T. Sakaue and K. Yoshikawa, Int. Rev. Cell Mol. Biol., 2014, 307, 175–204 CAS.
- M. Miyazaki, M. Chiba, H. Eguchi, T. Ohki and S. Ishiwata, Nat. Cell Biol., 2015, 17, 480–489 CrossRef CAS PubMed.
- C. Watanabe and M. Yanagisawa, Phys. Chem. Chem. Phys., 2018, 20, 8842–8847 RSC.
- P. van Nies, I. Westerlaken, D. Blanken, M. Salas, M. Mencia and C. Danelon, Nat. Commun., 2018, 9, 1583 CrossRef PubMed.
- G. Rampioni, F. D'Angelo, L. Leoni and P. Stano, Front. Bioeng. Biotechnol., 2019, 7, 1 CrossRef PubMed.
- S. Berhanu, T. Ueda and Y. Kuruma, Nat. Commun., 2019, 10, 1325 CrossRef PubMed.
- J. Garamella, S. Majumder, A. P. Liu and V. Noireaux, ACS Synth. Biol., 2019, 8, 1913–1920 CrossRef CAS PubMed.
- M. Dwidar, Y. Seike, S. Kobori, C. Whitaker, T. Matsuura and Y. Yokobayashi, J. Am. Chem. Soc., 2019, 141, 11103–11114 CrossRef CAS PubMed.
- M. Hagiya, A. Konagaya, S. Kobayashi, H. Saito and S. Murata, Acc. Chem. Res., 2014, 47, 1681–1690 CrossRef CAS PubMed.
- Y. Sato, Y. Hiratsuka, I. Kawamata, S. Murata and S. M. Nomura, Sci. Robot., 2017, 2, aal3735 CrossRef.
- S. Nozaki and H. Niki, J. Bacteriol., 2019, 201 Search PubMed.
- K. Fujiwara and M. Yanagisawa, ACS Synth. Biol., 2014, 3, 870–874 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S4. Representative images of inhomogeneous localization of msfGFP-MinC on membranes, decrease of protein expression levels in artificial cells and their recovery by BSA, effects of SUVs on protein expression by the PURE system, and movies for spatiotemporal patterning regulation by de novo synthesis of Min proteins in artificial cells and Min wave generation by the MinDE DNA operon. See DOI: 10.1039/c9sc02441g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |