
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThinking outside the “Blue Box”: from molecular to supramolecular pH-responsiveness†
Arturo
Blanco-Gómez
 a,
Iago
Neira
a,
José L.
Barriada
a,
Manuel
Melle-Franco
b,
Carlos
Peinador
*a and
Marcos D.
García
*a
a,
Iago
Neira
a,
José L.
Barriada
a,
Manuel
Melle-Franco
b,
Carlos
Peinador
*a and
Marcos D.
García
*a
aDepartamento de Química, Centro de Investigacións Científicas Avanzadas (CICA), Facultad de Ciencias, Universidade da Coruña, 15071, A Coruña, Spain. E-mail: carlos.peinador@udc.es; marcos.garcia1@udc.es
bCICECO—Aveiro Institute of Materials Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
First published on 23rd October 2019
Abstract
We present herein the development of a new polycationic cyclophane: the “red box”, second in a series of hydrazone-based analogues of the well-known organic receptor cyclobis(paraquat-p-phenylene)cyclophane (“blue box”). The macrocycle has been prepared in an excellent yield in aqueous media, and shows both a remarkable pH-responsiveness and unusual hydrolytic stability of the two hydrazone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, associated with charge delocalization of the amine lone pair. Whilst in aqueous media the “red box” is able to complex a variety of aromatic substrates, both in its acidic and basic form, in organic media the cyclophane is only able to capture those in the acidic form, resulting in supramolecular pH-responsiveness.
N bonds, associated with charge delocalization of the amine lone pair. Whilst in aqueous media the “red box” is able to complex a variety of aromatic substrates, both in its acidic and basic form, in organic media the cyclophane is only able to capture those in the acidic form, resulting in supramolecular pH-responsiveness.
1. Introduction
Molecular switches (MSs)1 are chemical entities able to perform reversible structural modifications upon the guidance of external stimuli such as light, electrical potential or chemical effectors.2 MSs have started to show their incredible potential not only by themselves,3 but as well when properly implemented in more complex entities,4 as control units within host:guest-based assemblies termed supramolecular switches (SSs)5 or mechanically interlocked molecular switches (MIMSs).6 In these cases, the external stimulus modulates the binding events within the system, leading to controlled catch and release of the guest within SSs, or a large relative movement of the covalent parts of MIMSs.Being the interest in the development of these dynamic molecular entities clearly justified by their wide applicability,5,6 the introduction of switching capabilities into molecular receptors is not trivial.7 This is especially true in the case of macrocyclic hosts,8 which suffer in many cases from challenging kinetically controlled syntheses that hamper not only their preparation,9 but the subsequent fine-tuning of their structure and function en route to SSs and MIMSs.8–11 Traditionally, coordination-driven self-assembly has enormously simplified the synthesis of dynamic cyclophanes, on the basis of thermodynamic control over the cyclization step.12,13 Nevertheless, these metal-containing structures have many potential drawbacks when compared with organic counterparts (e.g. exchange kinetics difficult to lock up, deficient implementation of extra binding abilities in the self-assembled units, potential metal toxicity or, nonetheless, cost-effectiveness). In consequence, the development of efficient synthetic methodologies for the construction of new organic hosts with switching capabilities is undoubtedly at the forefront of current chemical research.14
Recently, we and others have reported the use of imine-based dynamic covalent chemistry15 for the aqueous self-assembly of constitutionally dynamic cyclophanes, wholly organic compounds which can act as binding parts not only in host:guest aggregates,16 but as well within self-threading catenanes and knots.17 In our particular case,18 we have described a new conformationally flexible host, the “white box” (W4+, Scheme 1), an acyl hydrazone-based analogue of the well-known redox-responsive receptor “blue box” (cyclobis(paraquat-p-phenylene)cyclophane) developed by Stoddart and co-workers.10 Our macrocycle shows not only the expected constitutional dynamism in water caused by the imine bonds, but also accessible stimuli-responsiveness induced by the unusual acidity of the amide protons within W4+ (pKa = 6.5). Unfortunately, this new pH-sensitive MS was inappropriate for its implementation in SSs, due to its ability to complex the aromatic substrates tested only in aqueous media, and nearly to the same extent, by both the acidic form (W4+) and conjugate base (W2+) of the cyclophane.
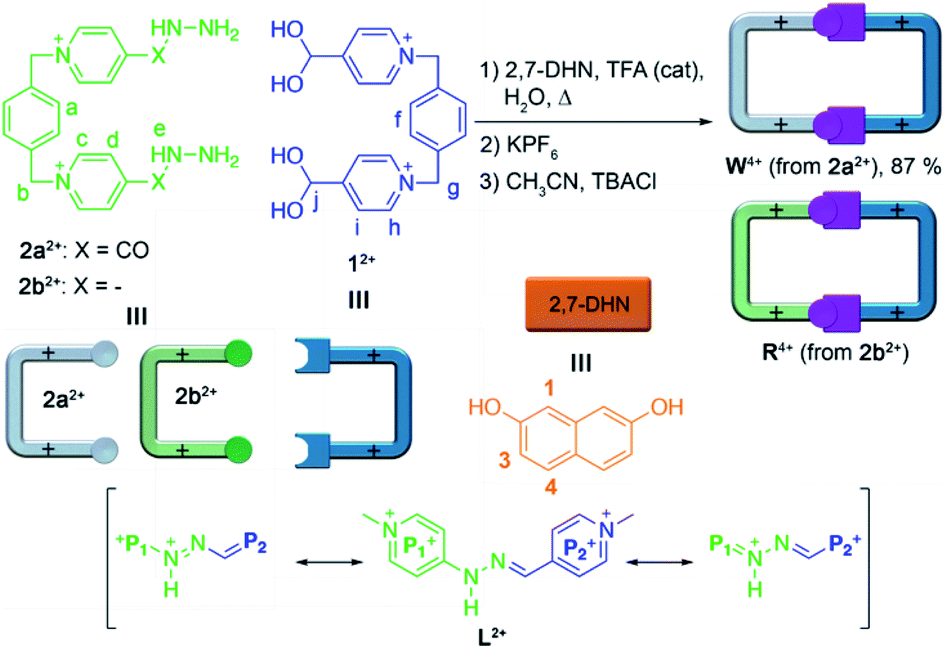 | ||
| Scheme 1 Top: depiction of the synthetic processes for the preparation of the cyclophane “white box” W4+, and its hydrazone-containing analogue “red box” R4+. Bottom: resonant forms for one of the potential tautomeric species of the model compound L2+ (see discussion). | ||
Considering our initial findings, we concluded that the removal of the carbonyl groups within W4+, leading to the hydrazone analogue R4+, would not have a significant effect on the stability of the resulting cyclophane in aqueous media, as it would be mostly determined in both cases by the delocalization of the lone pair of the amide/amine subunits over the neighbouring pyridinium rings. Specifically for R4+, the removal of the acyl groups allows for the potential extension of the resonance stabilization to the two pyridinium moieties on each of the large sides of the molecular rectangle. In consequence, we intuited that R4+ would be quite similar to W4+ in terms of hydrolytic stability, pH-responsiveness and binding ability in aqueous media. Conversely, R4+ would own a more compact cavity and an improved π-acceptor character, consequently increasing its ability to complex aromatic substrates by π–π interactions.
2. Results and discussion
2.1. Synthesis and characterization of the “Red Box”
Following our previously reported protocol for the synthesis of W4+,18 we successfully accomplished the quantitative self-assembly in aqueous media of the inclusion complex 2,7-DHN⊂R4+ (DHN = dihydroxynaphthalene). This was achieved by performing the TFA-d (10%) catalysed reaction between equimolar 1.5 mM D2O solutions of the complementary tweezers 1·2Br and 2b·2Br,19 and 1.5 eq. of the aromatic template.20 Hence, after 24 hours at 60 °C, the 1D/2D NMR experiments recorded for the sample nicely matched those expected for the host:guest aggregate as a sole new species (Fig. 1c and Table 1). In essence, the signals assigned to 2,7-DHN are consistently shielded as a result of the inclusion of the substrate within the cavity of R4+, with H3 being particularly affected due to the occurrence of [C–H⋯π] interactions with the phenylene rings of the host (ΔδH3 = −1.61 ppm, ΔδHa = 0.3 ppm and ΔδHf = 0.3 ppm).21 Furthermore, resonances assigned to Hj (δHj = 7.34 ppm) and Cj (δCj = 138.9 ppm) on 2,7-DHN⊂R4+ are also compatible with the formation of the imine bonds on R4+.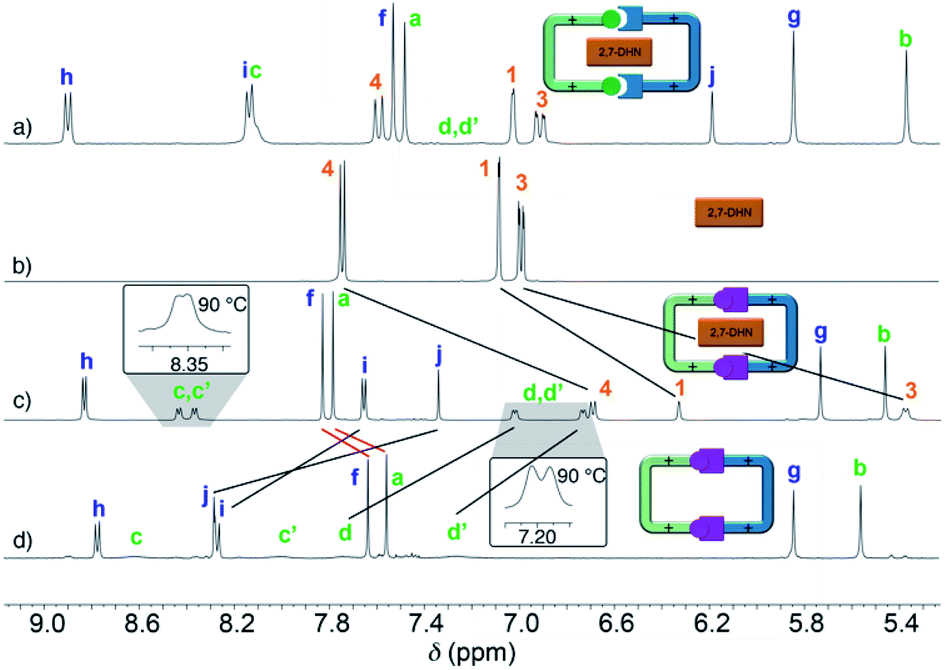 | ||
| Fig. 1 1H NMR (D2O, 500 MHz) of: (a) equimolar 1.5 mM mixture of 1·2Br, 2b·2Br and 2,7-DHN (1.5 eq.) at r.t. and t = 0; (b) 1.5 mM 2,7-DHN; (c) mixture (a) after 24 hours at 60 °C with TFA-d (10%); (d) equimolar 1.5 mM mixture of 1·2Br and 2b·2Br after 24 hours at 60 °C with TFA-d (10%). Proton numbering in Scheme 1. | ||
|
|
|||||
|---|---|---|---|---|---|
| Guest | R 4+ | ||||
| Δδ (D2O, ppm) | K a (M−1) | ||||
| H1 | H2 | H3 | H4 | ||
| a 1H NMR recorded in D2O. b 1H-NMR recorded in CD3CN. c Not calculated because of the inappropriate solubility of the substrate under the conditions of the titration | |||||
| 1,5-DHNa | −1.45 | −2.23 | −1.03 | 1.34 × 104 | |
| 1,5-DHNca | −1.26 | −1.99 | −1.24 | 1.61 × 104 | |
| 2,7-DHNa | −0.74 | −1.60 | −1.04 | 2.05 × 104 | |
| Carbazoleb | −2.12 | −2.56 | −1.66 | −0.94 | |
| Pyreneb | −1.47 | −1.50 | −1.14 | ||
| Guest | R 2+ | ||||
|---|---|---|---|---|---|
| Δδ (D2O, ppm) | K a (M−1) | ||||
| H1 | H2 | H3 | H4 | ||
| 1,5-DHNca | −1.20 | −1.87 | −1.41 | 1.72 × 104 | |
Interestingly, due to either the well-known prototropic tautomerism of the 4-hydrazinyl-1-alkylpyridinium moieties, or simply by delocalization of each of the two amine lone pairs over the corresponding pyridinium rings (Scheme 1),22 restricted rotation around the (P1+)C–NHN bonds is observed as in structurally related hydrazones (vide infra),23 resulting in the chemical inequivalence between Hc–c′ and Hd–d′, positioned on the upper and lower side of those rings within the host. This inequivalence was clearly identified in the corresponding EXSY NMR (Fig. S44†), which shows the expected cross peaks between the above-mentioned nuclei. VT 1H NMR experiments also confirmed this end, showing a faster exchange regime between Hc–c′ and Hd–d′ on increasing the temperature, and resulting in the collapse of the four signals initially observed at r.t. into two (see insets in Fig. 1c), a fact that enabled the estimation of ΔG# = 16.2 kcal mol−1 for the hindered rotation.19,24 Finally, DOSY NMR also supported the formation of 2,7-DHN⊂R4+ (Fig. S45†), with all the resonances on the aggregate diffusing as a whole in the corresponding spectrum.
Surprisingly, the synthesis of the empty cyclophane R·4Br could be achieved in a template-free fashion on a preparative scale (1.5 mM), using the very same reaction conditions as for the self-assembly of 2,7-DHN⊂R4+ (Fig. 1d). Once the reaction was finished, addition of excess KPF6 to the corresponding aqueous solution, followed by filtration and washing of the obtained solid with water, allowed the isolation of virtually pure R·4PF6 in an 83% yield. 1D/2D NMR experiments in CD3CN showed a sole main species, in good agreement with the expected cyclophane (Fig. 2c). On this occasion, protons within the P1+ moieties completely coalesce on the NMR at r.t, and VT-NMR showed a change to a situation of slow exchange for Hc–c′ and Hd–d′ upon cooling of the sample. The estimation of ΔG# = 15.2 kcal mol−1 from these experiments is in good agreement with the influence of the complexed substrate on the restricted rotation previously discussed for the (P1+)C–NHN bonds within 2,7-DHN⊂R4+ in D2O. Further evidence on the identity of the compound was obtained by ESI-MS, with the spectrum showing both the typical loss of PF6− counterions on the salt and that of HPF6 fragments resulting from the increased acidity of the amine protons within the cationic macrocycle (Fig. 2a).25
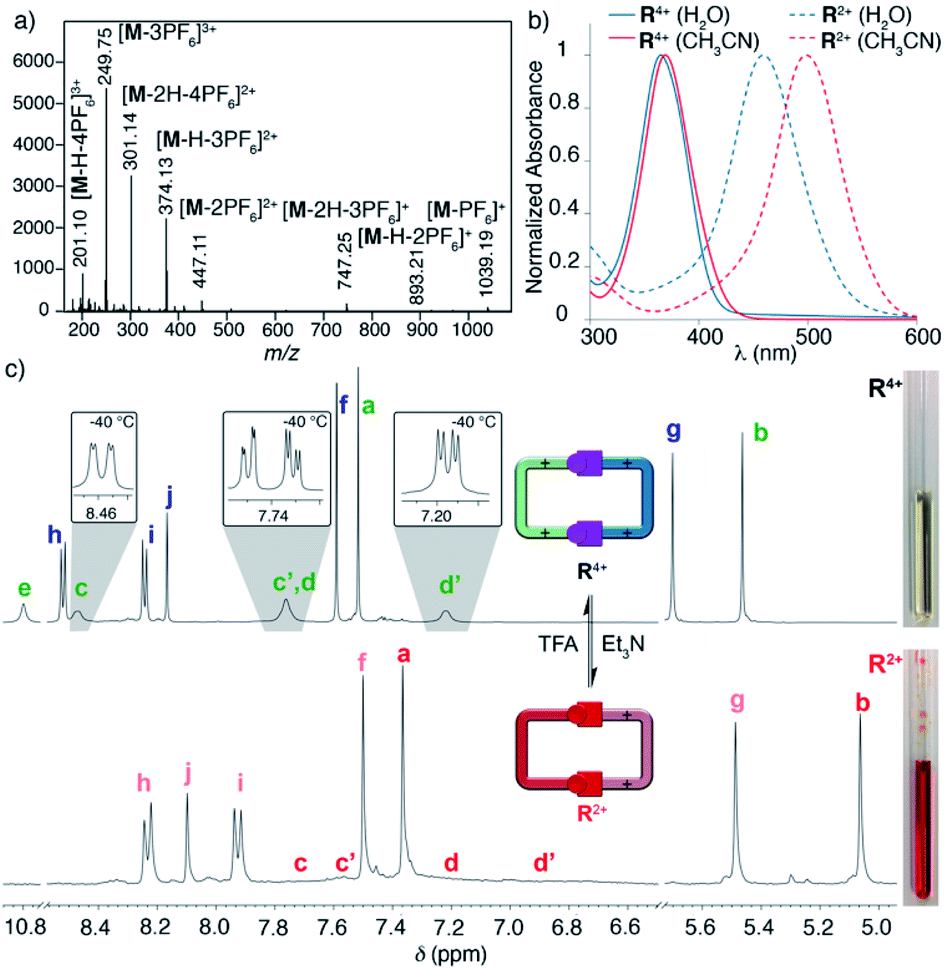 | ||
| Fig. 2 (a) LR ESI-MS spectrum, for R·4PF6 showing the loss of PF6− and HPF6 fragments. (b) Normalized UV-vis spectra of R·4Cl in H2O (blue solid line), R·4PF6 in CH3CN (red solid line), R·2Cl in H2O (blue dotted line), and R·2PF6 CH3CN (red dotted line). (c) 1H NMR (CD3CN, r.t., 400 MHz): (c1), 1.5 mM R·4PF6 (insets, VT-NMR, −40 °C), (c2): 1.5 mM R·2PF6 (obtained by addition of 1 eq. of Et3N to (c1)). | ||
Finally, the water-soluble salt R·4Cl was also easily obtained in an 89% yield by ion metathesis of R·4PF6 dissolved in CH3CN.19 Again, 1D/2D NMR data compiled for the empty receptor in D2O matched that expected for the compound. In this case, a value of ΔG# = 15.5 kcal mol−1 was estimated from VT-NMR experiments for the restricted rotation within R4+.19
2.2. Kinetic and thermodynamic stability of the “red box”
Puzzled by obtaining R·4Br in a template-free fashion, we performed a series of experiments to clarify the kinetic and thermodynamic stability of our cyclophane.19 Firstly, we observed how the TFA-d catalyzed reaction of 1·2Br and 2b·2Br in D2O produced R4+ as the main product within the 1.5–20 mM concentration window, accounting for the preference of the reaction in aqueous media to produce the cyclic receptor over potential oligomeric structures. In contrast, the very same reaction performed in DMSO-d6 yielded after 1 day heating at 60 °C a complex mixture of R4+ and new imine-containing species. Interconversion, between those species tentatively assigned as the potential oligomeric products and R4+, was not observed under all the reaction conditions tested (i.e. dilution, increased temperature or reaction times, addition of an excess of 2,7-DHN to the reaction mixture or, crucially, by solvent swapping to D2O).19 These results clearly point out to the “red box” as being the main product of a kinetically controlled process in water but, interestingly enough, not in organic media. Additional evidence of the extraordinary kinetic stability of the imine bonds was observed in aqueous media, where the proton signals for R·4Cl dissolved in buffered 1.5 mM D2O solutions at pD = 1.5 were not altered, even after the addition of a 10-fold excess of the highly reactive aldehyde hydrate 4-(dihydroxymethyl)-1-methylpyridin-1-ium.As a corollary of these surprising observations, we inferred an increased kinetic stability of the imine bonds within R4+ compared to W4+, a prediction that could be verified by the irreversible transformation of the later into the former (Scheme 2), by heating for 24 hours a mixture of the “white box” and 2b·2Br in D2O with a catalytic amount of TFA-d (10%).19,26
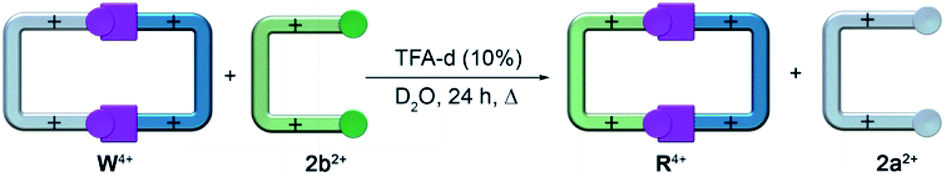 | ||
| Scheme 2 Irreversible transformation of W4+ into R4+. | ||
2.3. pH-Responsiveness of the “red box”
To validate our second prediction on the “red box”, its pH-responsiveness, we firstly performed an UV-vis acid-base titration for the macrocycle in water. On increasing the pH, the appearance of the conjugated base R2+ clearly results in a substantial decrease of the originally observed main absorption for R4+ (λmax = 365 nm, ε = 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 054 L mol−1 cm−1, associated with π–π* transitions), and the concomitant manifestation of a new band at λmax = 464 nm (ε = 61092 L mol−1 cm−1), which clearly indicates an increased intramolecular charge-transfer over the pyridinium rings upon deprotonation (Fig. 2b). Although the presence of two very close isosbestic points on the titration experiment precludes the precise determination of the pKa for R·4Cl by UV-vis (Fig. S95†), an approximate value of 8.3 could be estimated. This assessment is in decent agreement with the experimental data obtained for the model compound L·2I by UV-vis (pKa = 9.0), and potentiometric (pKa = 8.8) titrations (Fig. 3b and c).19,27
054 L mol−1 cm−1, associated with π–π* transitions), and the concomitant manifestation of a new band at λmax = 464 nm (ε = 61092 L mol−1 cm−1), which clearly indicates an increased intramolecular charge-transfer over the pyridinium rings upon deprotonation (Fig. 2b). Although the presence of two very close isosbestic points on the titration experiment precludes the precise determination of the pKa for R·4Cl by UV-vis (Fig. S95†), an approximate value of 8.3 could be estimated. This assessment is in decent agreement with the experimental data obtained for the model compound L·2I by UV-vis (pKa = 9.0), and potentiometric (pKa = 8.8) titrations (Fig. 3b and c).19,27
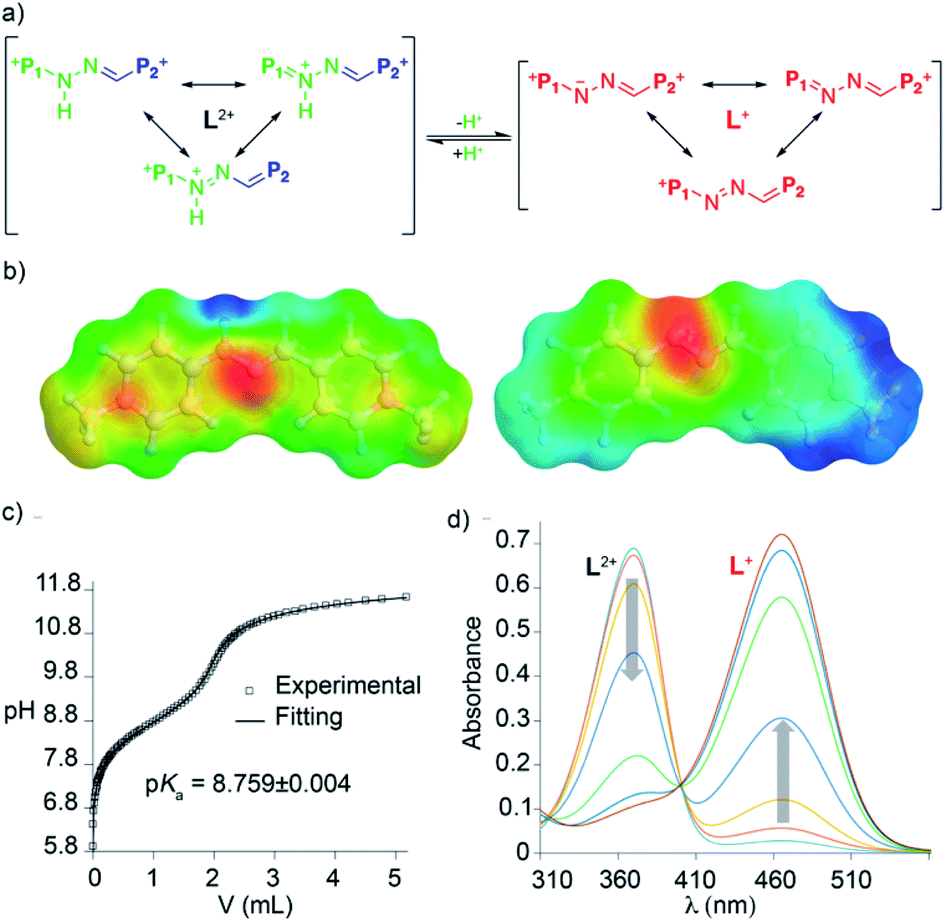 | ||
| Fig. 3 (a) Schematic depiction of the acid–base equilibrium of L·2I in water. (b) Color-coded EPS of L2+ (left, coloured from 0.35 eV to 0.59 eV), and L1+ (right, coloured from −0.03 eV to 0.34 eV). (c) Fitting of the potentiometric titration data of 8.3 mM L·2I with 40 mM KOH solution. (d) UV-vis spectra for the titration of 20 μM L·2I with aliquots of appropriate solutions of NaH2PO4/Na2HPO4 and NaHCO3/Na2CO3 buffers of increasing pH. | ||
The pH-responsiveness of R4+ was qualitatively assessed as well in organic media (Fig. 2c). Addition of 1 eq. of Et3N to a 1.5 mM solution of R·4PF6 in CD3CN produced substantial changes in the 1H NMR of the macrocycle, in good agreement with its deprotonation (i.e. disappearance of the amine signal He, and substantial shielding of the remaining resonances due to the increased electronic density on the chromophores). The observed changes could be efficiently restored by addition of 1 eq. of TFA-d to the organic solution (see photographs in Fig. 2). Finally, the pH-responsive behaviour of the compound in CH3CN was also monitored by UV-vis (Fig. 2b), showing a similar behaviour to that observed in water. Accordingly, the main absorption band for R·4PF6 at λmax = 369 nm (ε = 73826 L mol−1 cm−1) significantly disappears upon deprotonation, resulting in the appearance of a new main band centered at λmax = 498 nm (ε = 92606 L mol−1 cm−1).
In order to shed some light on the intriguing features of the “red box”, we performed systematic DFT calculations at the M06-2X-6-31g(d,p) level in water on R4+ and the simplified model L2+. Firstly, DFT energies reproduced well the lack of substantial prototropic tautomerism at r.t. for the species.19 Secondly, the conformational analysis showed very similar results for R4+ and R2+, identifying conformations of minimum energy for those sharing “blue box”-like configurations with the two imines in an (E)-configuration (i.e. quadrangular structures with alignment of the π-deficient pyridinium moieties on each side of the rectangle, Fig. 4). In addition, activation energies for all the rotatable bonds within the model compound L2+ were computed, with the results being in agreement with the observed NMR spectra. In particular, the barrier that restricts the rotation around the (P1+)C–NHN bond, with an estimated value of 15.2 kcal mol−1 from VT-NMR experiments on L2+, was well reproduced, showing a computed value of 14 kcal mol−1.
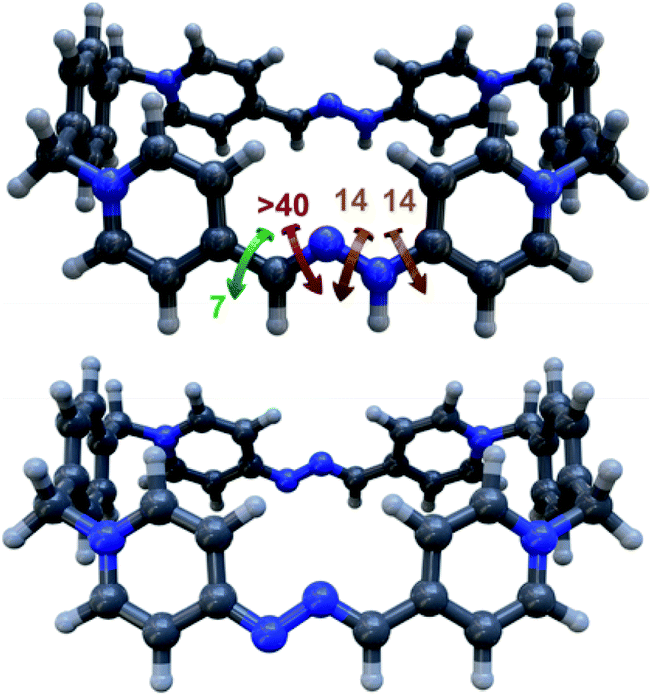 | ||
| Fig. 4 Predicted conformations of minimum energy, according to DFT calculations at the M06-2x-6-31g(d,p) level of theory in water, for: top; R4+ (also shown the estimated activation energies in kcal mol−1 for all the corresponding rotatable bonds for the model compound L2+); bottom: R2+. | ||
The pKa values for R4+ and L2+ in water were also computed, yielding values for L2+ and R4+ of 7.8 and 7.4, respectively, in reasonable agreement with the experimental values. The simulated UV-vis absorption spectra within the TD-DFT approach also matched the experimental observations (Table S19†). This allowed us to assign the intense absorption peak for the acid and basic forms of “red box” and the model compound L2+ to π electron transitions between the frontier orbitals in the “L” fragments. Interestingly, L2+ and L+ showed quite similar electronic densities for the frontier orbitals (Fig. S114†), whilst the Electrostatic Potential on the Solvent-accessible-surfaces (EPSs) showed an enhanced localized positive potential in the proton area for the acid form, which has a partial positive charge of 0.39 (Fig. 3 and S115†).
2.4. The “red box” as a molecular receptor: from molecular to supramolecular pH responsiveness
As the final part of our study, we proceeded to test the hosting ability of the “red box”. Consequently, 1D/2D NMR experiments were firstly recorded in D2O for R·4Cl and a series of selected electron-rich aromatic substrates (1,5-DHN, carbazole, and pyrene).19 In essence, the complexation induced chemical shifts obtained for the guests within the aggregates (Table 1) were fully consistent with the host being able to sequester those from the aqueous media. In all cases, the substrates showed the expected shielding induced by the concurrence of π–π and C–H⋯π interactions.21 Regarding the host part of the assembly, all the studied cases exhibited fast or near coalescence exchange regimes on the NMR timescale.19 Moreover, those nuclei on the macrocyclic part of the complexes showed among them very similar patterns of relative shifts (comparing the chemical shifts for the different guest⊂R4+ species with those of R4+). Finally, association constants for the complexes of R4+ with the DHN derivatives could be determined in water by NMR titrations, yielding Ka values in the 104 M−1 range (Table 1).In order to compare the differences between R4+ and its conjugate base R2+on the complexation of a given substrate in aqueous media, 1,5-DHNc (see the structure in Fig. 5) was used as an appropriate water-soluble and pH-insensitive substrate. In both cases, 1H-NMR titrations in buffered aqueous media showed a similar ability of both forms of the macrocycle to complex the model substrate, Ka = (1.61 ± 0.14) × 104 M−1 (pD = 6) and (1.72 ± 0.20) × 104 M−1 (pD = 10). These results account for the hydrophobic effect, and not the net charge of the host, being crucial for the host–guest association in aqueous media.
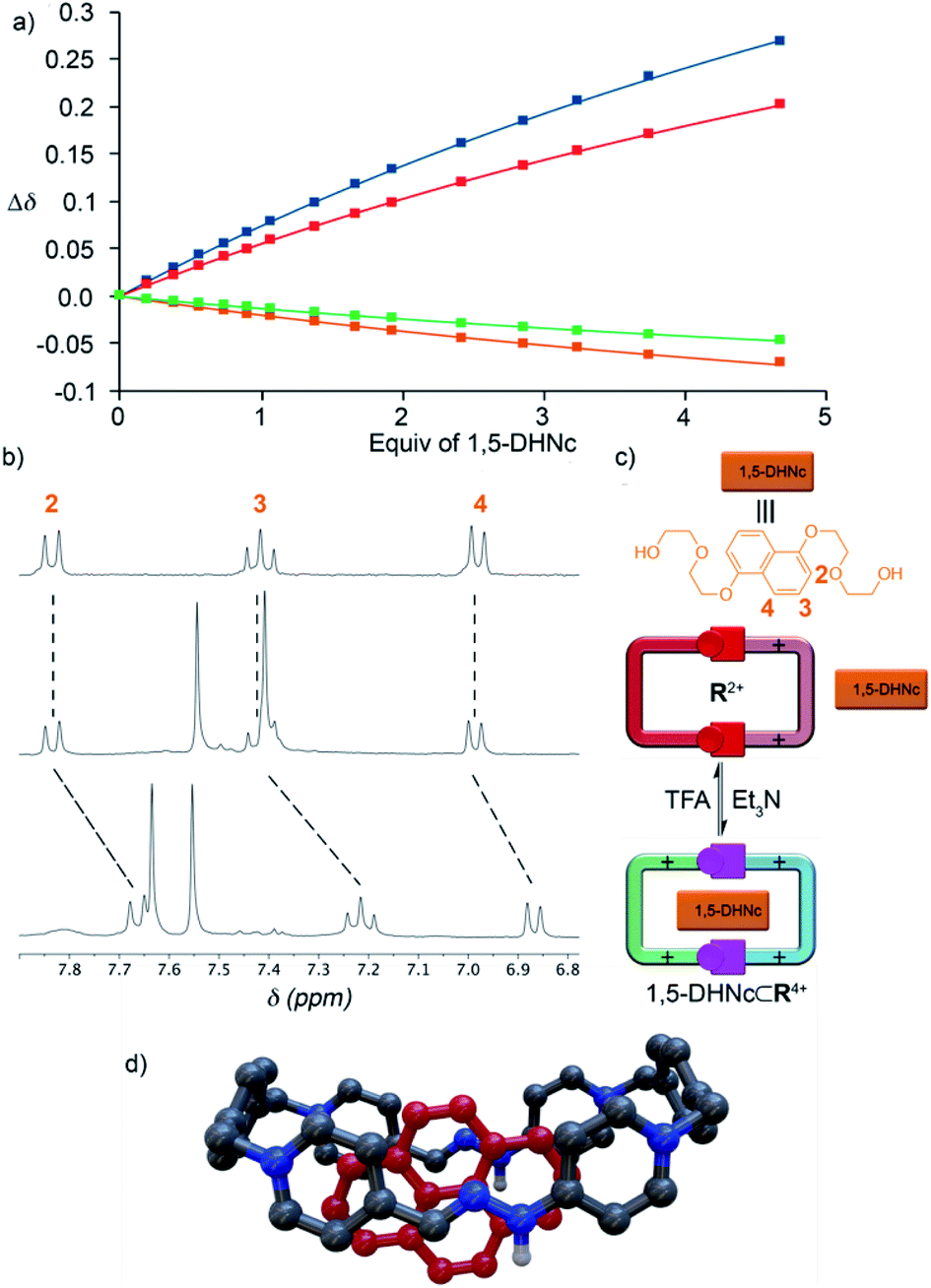 | ||
| Fig. 5 (a) Fitting of the 1H-NMR titration data for 1,5-DHNc⊂R·4PF6, signals Hh (blue), Hj (red), Hf (green) and Ha (brown). (b) Partial spectrum 1H NMR (CD3CN, r.t., 300 MHz) of 1,5-DHNc (top), equimolar mixture of R·4PF6, 1,5-DHNc and Et3N (middle), and equimolar mixture of R·4PF6, 1,5-DHNc and TFA-d (bottom). (c) Schematic depiction of the supramolecular switch. (d) DFT optimized structure of pyrene⊂R4+ at the M06-2x-6-31g(d,p) level in acetonitrile (all hydrogen atoms, except those of the amine groups, were removed for clarity). | ||
On the other hand, the results obtained for the complexation of the model substrates by R·4PF6 in organic media were more interesting (Table 1). Contrarily to the previously reported results for W·4PF6,18R·4PF6 was found to be able to sequester selected aromatic guests in CD3CN, with complexation induced shifts being in good agreement with those expected for the aggregates, and small Ka values matching the lack of hydrophobic effect. In contrast, the conjugate base R2+ (prepared by dissolving R·4Cl in buffered water at pH = 10, and isolated by precipitation with excess KPF6 as R·2PF6) was found to be unable to complex the aromatic substrates, accounting for the decreased ability of the macrocycle in its basic form to engage in π–π interactions. As exemplified in Fig. 5 for 1,5-DHNc⊂R4+, the host:guest complexes can be conveniently assembled/disassembled in CD3CN, simply by consecutive addition of Et3N as a base or TFA-d as an acid.
Finally, in order to shed some light on the structural features of the host–guest aggregates, the geometries of the different potential configurations for pyrene⊂R4+ were explored using a combination of semiempirical molecular dynamics and DFT methods.19 The results obtained for the most stable geometry of the assembly showed minor differences between the conformation of free and complexed macrocycles (Fig. 5d), supporting the longitudinal insertion mode of the guest and the coplanar conformation of R4+. Additionally, the distance between the mean plane of pyrene and each of the long sides of the guest, 3.3–3.4 Å (Fig. S117†), was found to be optimal for the establishment of π–π interactions as reported for similar systems.12,13,18,21
3. Conclusions
In summary, we have reported herein the successful development of a new stimuli-responsive polycationic cyclophane, the “red box”. Synthesized in water in an excellent yield following a kinetically controlled process involving the acid-catalysed hydrazone bonding between suitable complementary tweezers, the cyclophane shows a remarkable hydrolytic stability and accessible pH-responsiveness (pKa ∼ 8.3 for the amine protons). In both its acidic (R4+) and basic (R2+) form, the macrocycle exhibits an appropriate behaviour as a host for a series of selected aromatics in aqueous media. In contrast, in organic media, the receptor is able to complex selected model compounds only in its acidic form, a fact attributed to the decreased π-deficient character of the host in its basic form. The solvent dependent complexation behaviour of this new host opens the door, for instance, for its use as a recyclable scavenger of polycyclic aromatic hydrocarbons and other relevant aromatic compounds.10 Furthermore, its adequate pH-responsiveness in organic media would potentially allow for the implementation of the molecular receptor in a wide variety of SSs and MIMSs.5,6Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Ministerio de Economía y Competitividad (MINECO FEDER, Grant CTQ2016-75629-P) and Xunta de Galicia (ED431C 2018/39). MMF acknowledges support from the Portuguese Foundation for Science and Technology (FCT), under the projects PTDC/FIS-NAN/4662/2014, IF/00894/2015, and FCT Ref. UID/CTM/50011/2019 for CICECO-Aveiro Institute of Materials. I. N. thanks the MECD (FPU program) for financial support. Dedicated to Professor José María Quintela on the occasion of his retirement.References
- Molecular switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed.
- For selected reviews on molecular switches, see: (a) R. J. Mortimer, Chem. Soc. Rev., 1997, 26, 147–156 RSC; (b) L. Fabbrizzi, M. Licchelli and P. Pallavicini, Acc. Chem. Res., 1999, 32, 846–853 CrossRef CAS; (c) B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed; (d) J. L. Segura and N. Martin, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS; (e) D. Bleger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef CAS PubMed.
- For selected reviews on the applications of molecular switches, see: (a) M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed; (b) N. Fuentes, A. Martin-Lasanta, L. Alvarez de Cienfuegos, M. Ribagorda, A. Parra and J. M. Cuerva, Nanoscale, 2011, 3, 4003–4014 RSC; (c) Y. Wang and Q. Li, Adv. Mater., 2012, 24, 1926–1945 CrossRef CAS PubMed; (d) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed; (e) J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- According to Stoddart et al., these dynamic entities can be classified as molecular, supramolecular or mechanically interlocked switches: R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC.
- For selected reviews on supramolecular switches and their applications, see: (a) S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed; (b) N. Songa and Y.-W. Yang, Chem. Soc. Rev., 2015, 44, 3474–3504 RSC; (c) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed; (d) X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed; (e) E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. García, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS PubMed.
- For selected reviews on mechanically interlocked molecular switches and their applications, see: (a) W. Yang, Y. Li, H. Liu, L. Chi and Y. Li, Small, 2012, 8, 504–516 CrossRef CAS PubMed; (b) M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935–1949 CrossRef CAS PubMed; (c) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (d) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC; (e) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed; (f) N. Pairault and J. Niemeyer, Synlett, 2018, 29, 689–698 CrossRef CAS.
- See, for instance: M. Natalia and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- The “blue box”, and by extension the whole family of related molecular receptors ExnBoxm4+, is the classic example of successful implementation of (redox)-responsive MSs in MIMSs: E. J. Dale, N. A. Vermeulen, M. Juríček, J. C. Barnes, R. M. Young, M. R. Wasielewski and J. F. Stoddart, Acc. Chem. Res., 2016, 49, 262–273 CrossRef CAS PubMed.
- Although the use of thermodynamically controlled conditions, on the key SN2 macrocyclization, has recently emerged as an enabling methodology for the synthesis of “blue box” analogues,10 C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714–4723 CrossRef CAS PubMed the fine-tuning of the properties of this type of receptor, for instance by the exo-functionalization of the receptor’s scaffold, is nothing but trivial. See for instance: .
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- For an account by our research group on the metal-directed self-assembly of “blue box” analogues, see: M. D. García, C. Alvariño, E. M. López-Vidal, T. Rama, C. Peinador and J. M. Quintela, Inorg. Chim. Acta, 2014, 417, 27–37 CrossRef.
- For recent examples of host-based supramolecular stimuli-responsiveness, see: (a) M. Frasconi, I. R. Fernando, Y. Wu, Z. Liu, W.-G. Liu, S. M. Dyar, G. Barin, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 11057–11068 CrossRef CAS PubMed; (b) K. Kurihara, K. Yazaki, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2017, 56, 11360–11364 CrossRef CAS PubMed; (c) X. Chi, W. Cen, J. A. Queenan, L. Long, V. M. Lynch, N. M. Khashab and J. L. Sessler, J. Am. Chem. Soc., 2019, 141, 6468–6472 CrossRef CAS PubMed; (d) H. Wu, Y. Chen, L. Zhang, O. Anamimoghadam, D. Shen, Z. Liu, K. Cai, C. Pezzato, C. L. Stern, Y. Liu and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1280–1289 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- (a) Y. Zhang, X. Zheng, N. Cao, C. Yang and H. Li, Org. Lett., 2018, 20, 2356–2359 CrossRef CAS PubMed; (b) X. Zheng, Y. Zhang, G. Wu, J.-R. Liu, N. Cao, L. Wang, Y. Wang, X. Li, X. Hong, C. Yang and H. Li, Chem. Commun., 2018, 54, 3138–3141 RSC; (c) G. Wu, C.-Y. Wang, T. Jiao, H. Zhu, F. Huang and H. Li, J. Am. Chem. Soc., 2018, 140, 5955–5961 CrossRef CAS PubMed; (d) L. Shen, N. Cao, L. Tong, X. Zhang, G. Wu, T. Jiao, Q. Yin, J. Zhu, Y. Pan and H. Li, Angew. Chem., Int. Ed., 2018, 57, 16486–16490 CrossRef CAS PubMed.
- (a) H. Li, H. Zhang, A. D. Lammer, M. Wang, X. Li, V. M. Lynch and J. L. Sessler, Nat. Chem., 2015, 7, 1003–1008 CrossRef CAS PubMed; (b) C.-Y. Wang, G. Wu, T. Jiao, L. Shen, G. Ma, Y. Pan and H. Li, Chem. Commun., 2018, 54, 5106–5109 RSC; (c) F. B. L. Cougnon, K. Caprice, M. Pupier, A. Bauza and A. Frontera, J. Am. Chem. Soc., 2018, 140, 12442–12450 CrossRef CAS PubMed; (d) K. Caprice, M. Pupier, A. Bauza, A. Frontera and F. B. L. Cougnon, Angew. Chem., Int. Ed., 2019, 58, 8053–8057 CrossRef CAS PubMed.
- A. Blanco-Gómez, Á. Fernández-Blanco, V. Blanco, J. Rodríguez, C. Peinador and M. D. García, J. Am. Chem. Soc., 2019, 141, 3959–3964 CrossRef PubMed.
- See the ESI† for further details.
- As observed in Fig. 1a, signals for the aromatic template in the initial mixture of components, at r.t. and t = 0, appear shielded if compared with free 2,7-DHN, accounting for a weak interaction with both tweezers and demonstrating the ability of the substrate to preorganize the reacting building blocks.
- This type of [C–H⋯π] interaction has been extensively reported by our research group in similar host–guest systems. See, for instance: (a) C. Alvariño, E. Pía, M. D. García, V. Blanco, A. Fernández, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2013, 19, 15329–15335 CrossRef PubMed; (b) T. Rama, E. M. López-Vidal, M. D. García, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2015, 21, 9482–9487 CrossRef CAS PubMed.
- B. D. Batts and E. Spinner, Aust. J. Chem., 1969, 22, 2611–2626 CrossRef CAS.
- (a) M. Prinz, S. Parlar, G. Bayraktar, V. Alptuzun, E. Erciyas, A. Fallarero, D. Karlsson, P. Vuorela, M. Burek, C. Forster, E. Turunc, G. Armagan, A. Yalcin, C. Schiller, K. Leuner, M. Krug, C. A. Sotriffer and U. Holzgrabe, Eur. J. Pharm. Sci., 2013, 49, 603–613 CrossRef CAS PubMed; (b) S. Parlar, Y. Erzurumlu, R. Ilhan, P. Ballar Kırmızıbayrak, V. Alptüzün and E. Erciyas, Chem. Biol. Drug Des., 2018, 92, 1198–1205 CrossRef CAS PubMed.
- H. Kessler, Angew. Chem., Int. Ed., 1970, 9, 219–235 CrossRef CAS.
- As it would be expected for the cyclophane experiencing acid–base reactions on the gas phase: C. A. Schalley, Mass Spectrometry and Gas-Phase Chemistry of Non-Covalent Complexes, Wiley, Hoboken/NJ, 2009 Search PubMed.
- The well-known decreased lability in water of hydrazone compared to acyl hydrazone bonds is altered on this occasion because of the enlarged charge delocalization of the imine bonds within R4+ compared to W4+: J. Kalia and R. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- The calculation of the pKa value for R·4Cl in aqueous media, by the corresponding potentiometric titration, was precluded by precipitation of the conjugate base due to the highly saline medium required for the experiment.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04489b |
| This journal is © The Royal Society of Chemistry 2019 |