
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrapping an unprecedented Ti3C3 unit inside the icosahedral C80 fullerene: a crystallographic survey†
Pengyuan
Yu‡
,
Wangqiang
Shen‡
 ,
Lipiao
Bao‡
,
Changwang
Pan
,
Zdenek
Slanina
and
Xing
Lu
*
,
Lipiao
Bao‡
,
Changwang
Pan
,
Zdenek
Slanina
and
Xing
Lu
*
State Key Laboratory of Materials Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan, 430074 China. E-mail: lux@hust.edu.cn
First published on 14th October 2019
Abstract
The sub-nanometer cavity of fullerene cages is an ideal platform to accommodate otherwise unstable species for accurate structural characterization with, for example, rather accurate single crystal X-ray diffraction (XRD) crystallography. Herein, we report the successful entrapment of an isolated Ti3C3 moiety inside the icosahedral-C80 cage to form Ti3C3@Ih-C80via an arc-evaporation process in the gas phase. The single crystal XRD crystallographic results unambiguously reveal that the C3-unit adopts an unprecedented cyclopropane-like structure which coordinates with the three titanium atoms in an unexpected fashion where the triangular C3-unit is nearly perpendicular to the Ti3-plane. The intercalation of a cyclopropanated C3-unit into the titanium layer is thus unambiguously confirmed. The theoretical results reveal that the Ti3C3 cluster transfers six electrons to the Ih-C80 cage so that each titanium atom has a positive charge slightly above +2 and the C3-unit is negatively charged with about −1. It is noteworthy that this is the first observation of the cyclopropane-coordination fashion in any reported organometallic complex, providing new insights into coordination chemistry.
Introduction
Fullerene cages serve as a protector to host a variety of isolated metal atom(s) or otherwise unstable metallic units to form so-called endohedral metallofullerenes (EMFs). By virtue of the solubility of EMFs, high-quality single crystals can be obtained and thus the interplay between the metallic species within the clusters, in addition to the metal–cage interactions, is possible to study by the sophisticated single crystal X-ray diffraction (XRD) approach. Up to now, a collection of EMFs containing one to three pure metal atoms, which are called mono-, di- and tri-EMFs, respectively, has been crystallographically confirmed.1–4 Interesting results such as direct bonding between the repulsive metal ions (e.g., Lu2+–Lu2+, U3+–U3+ or Y2+–Y2+) have been achieved taking advantage of the confinement effect of the fullerene cages.5–7In addition, various metallic clusters, such as metal nitride,8 carbide,9 oxide,10 sulfide11 and even cyanide,12 have been successfully trapped inside fullerene cages. It is noteworthy that only with the protection of the fullerene cages are the metallic clusters able to exist and they transfer a certain number of electrons to the surrounding cages. As typical examples, an improbable monometallic cluster YCN12 was found in Cs(6)-C82 featuring a two-electron-transfer configuration but the Ih-C80 is able to host a variety of metallic species such as La2,13 Sc3N,8 Sc3C2,14 Sc4C2,15 TiSc2C2,16 TiDy2C,17 Sc3CH18 and Sc3C2CN19 which donate six electrons to the cage. It's noted that encapsulation of a carbide cluster consisting of either an odd or an even number of carbon atoms is feasible. Impressively, Popov et al. theoretically revealed Sc4C3@C80 encapsulating a triangular aromatic C3 cluster with two pairs of Sc atoms coordinating with the C3 moiety.20 Recently, Chen and coworkers21 reported the structure of U2C@C80 where a non-bridged uranium-carbon double bond was identified. More interestingly, Popov and coworkers22 reported that a titanium–carbon double bond (Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) exists in TiLu2C@Ih-C80, representing the first example of EMFs with a multiple bond between the metal and the non-metal atom of the endohedral cluster. It is thus expected that novel and even unprecedented bonding features are possibly observed in EMFs containing different metallic clusters.
C) exists in TiLu2C@Ih-C80, representing the first example of EMFs with a multiple bond between the metal and the non-metal atom of the endohedral cluster. It is thus expected that novel and even unprecedented bonding features are possibly observed in EMFs containing different metallic clusters.
Herein, we report the preparation and systematic characterization of a brand-new titanium-containing EMF, namely Ti3C3@Ih-C80, which is one of the rare examples of EMFs that do not contain group-III elements. X-ray crystallographic results reveal that the C3-unit of the Ti3C3 cluster resembles cyclopropane with an average C–C distance of 1.380 Å, perpendicularly coordinated with the Ti3-moiety, exhibiting a totally unexpected coordination fashion in organometallic chemistry.
Results and discussion
Soot containing titanium-EMFs was synthesized by an arc discharge method using TiC as the precursor. SnCl4 was first chosen to react with the extract for the enrichment of Ti-EMFs followed by multiple-stage HPLC separation to obtain a pure sample of Ti3C3@C80 (Fig. S1 and S2†). Its MALDI-TOF mass spectrum shows a single peak at m/z 1139.8 (Fig. 1a) which is perfectly consistent with the calculated results of Ti3C83. Interestingly, the retention time of Ti3C3@Ih-C80 in a Buckyprep column is much longer than that of Sc3N@Ih-C80 (Fig. S3†). Previous studies have suggested a correlation between the HPLC retention time and the molecular geometry of fullerenes.23 Generally, the retention mechanism is proportional to the cage polarizability and is dominated by π–π interactions with the stationary phase. This phenomenon may suggest a different electronic configuration of the title compound from that of Sc3N@Ih-C80. Consistently, the absorption spectrum of Ti3C3@Ih-C80 (Fig. 1b) shows a small bandgap of 0.82 eV (onset at 1500 nm) in comparison with the large one of Sc3N@Ih-C80 (1.70 eV),24 suggesting a relatively low stability and unique electronic configuration of Ti3C3@Ih-C80. In addition, no signal is observed in the EPR spectrum of Ti3C3@Ih-C80 even at 100 K (Fig. S4†), indicative of its diamagnetic property with a closed-shell electronic configuration. Compared to the previously reported Ti-containing mixed-metal EMFs, such as TiSc2N@Ih-C80 (ref. 25) and TiY2N@C80,26 which feature obvious EPR signals at low temperatures, it can be concluded that our title compound has a different electronic structure, which is presumably attributed to the difference in the spin distribution of the embedded clusters.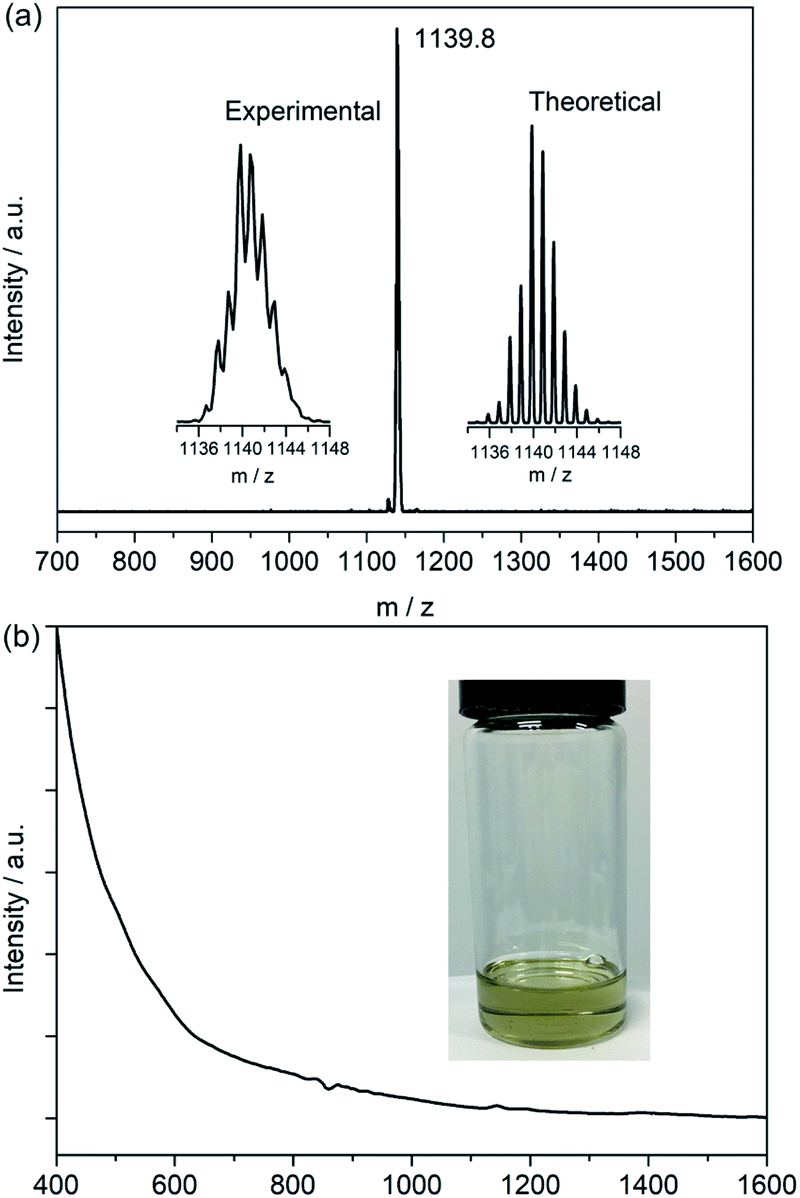 | ||
| Fig. 1 (a) MALDI-TOF mass spectrum of purified Ti3C3@Ih-C80. The insets show the experimental and theoretical isotopic distributions of Ti3C83. (b) Vis-NIR absorption spectrum of Ti3C3@Ih-C80 recorded in CS2. A photograph of the corresponding solution is shown in the inset. | ||
The structure of Ti3C83 is unambiguously established as Ti3C3@Ih-C80, instead of any other form such as Ti3C@C82, by a single crystal XRD study performed on a black crystal obtained by slow diffusion of a benzene solution of NiII(OEP) (OEP is the dianion of octaethyl porphyrin) into the CS2 solution of the EMF. The Ti3C3@Ih-C80·NiII(OEP)·2C6H6 cocrystal falls into the monoclinic C2/m space group, where a half of NiII(OEP) and a half of Ti3C3@Ih-C80 are included in its asymmetric unit cell. The crystallographic mirror plane coincides with one of the symmetric planes of the Ih-C80 cage, resulting in a fully ordered Ih-C80 cage by combining the existing half with its mirror image.
Inside the cage, disorder is observed for the Ti3C3 cluster (see ESI Fig. S5 and Table S1 for details†). Specifically, one carbon atom in the C3-unit exhibits two disordered positions generated by the crystallographic mirror plane, whereas the other two located in the crystallographic mirror plane are fully ordered. Eleven Ti sites are distinguished for the three titanium atoms with occupancy values ranging from 0.127 to 0.506. In fact, three of the disordered sites apart from NiII(OEP) belong to one Ti atom, with Ti2 as the major one (occupancy = 0.505). Meanwhile, two major sites out of the remaining eight disordered Ti-positions close to NiII(OEP), namely Ti1 and Ti1A (occupancy = 0.506), can be assigned to the other two Ti atoms.
Fig. 2 displays the molecular structures of Ti3C3@Ih-C80 showing only the major metal site together with the co-crystallized NiII(OEP) molecule. The porphyrin moiety nestles the fullerene cage with the shortest Ni–cage–carbon distance of 2.950 Å, suggesting substantial π–π interactions. Interestingly, the three carbon atoms stay in the center of the cage to form cyclopropane with an average bond length of 1.380 Å. The Ti–Ti distances among the three titanium atoms are 2.976 Å for Ti1–Ti1A and 3.604 Å for Ti1–Ti2 and Ti1A–Ti2, respectively, which are comparable to the values observed in organotitanium complexes, indicating weak metal–metal interactions between the titanium atoms.27–29 From a geometric point of view, the three titanium atoms intercalate between the cage and the C3-unit, preventing the encapsulated carbon atoms from being merged with the cage framework.
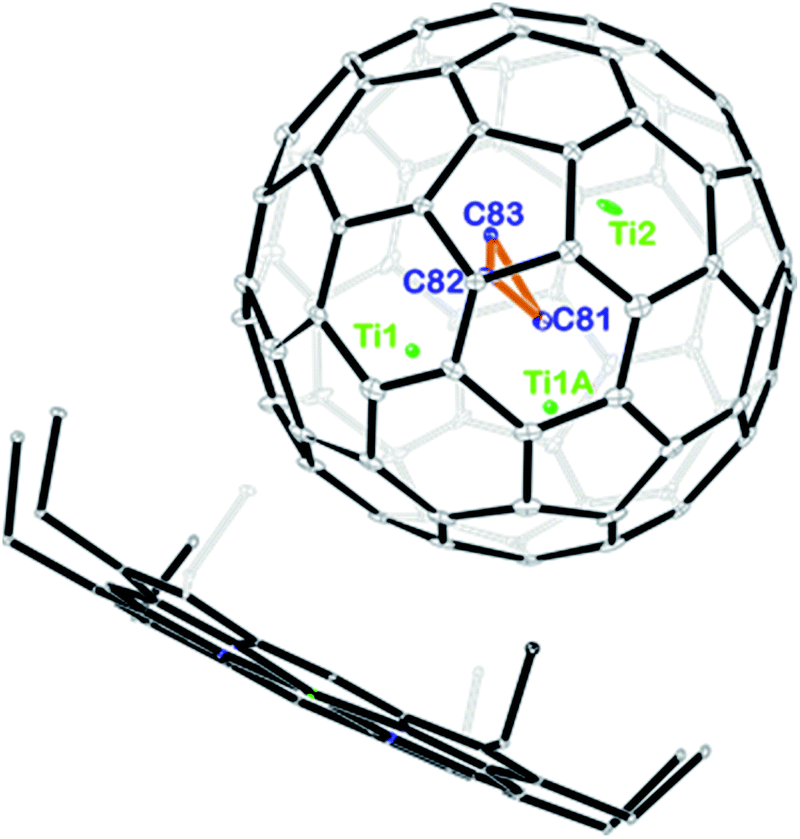 | ||
| Fig. 2 ORTEP drawing of Ti3C3@Ih-C80·[NiII(OEP)] with 15% thermal ellipsoids. Only the major Ti3C3 site is shown. Solvent molecules and H atoms are omitted for clarity. | ||
Fig. 3 illustrates the configuration of the major Ti3C3 site showing its relationship to the adjacent cage frameworks. Each titanium atom is centered above a respective [5,6]-ring junction with average Ti–C (cage) distances of 2.12, 2.12, and 2.13 Å, respectively, which fall in the range of typical Ti–C single bond lengths in reported organotitanium compounds (usually 2.1 Å),30,31 suggestive of strong metal–cage interactions (see also Table S2, ESI†). In contrast, the Ti–C distances within the Ti3C3 cluster are spread in a wide range from 1.838 Å to 3.090 Å, indicative of a rather complicated coordination environment. Interestingly, the plane of the C3-unit is nearly perpendicular to that of the Ti3-moiety with a dihedral angle of 83.43°, presenting a unique coordination fashion between carbon and titanium. To the best of our knowledge, such an unusual coordination pattern for cyclopropane interacting with metal ions has never been reported in any organometallic complexes, despite the above-mentioned theoretical possibility of a triangular C3 unit inside the Sc4C3@C80.20
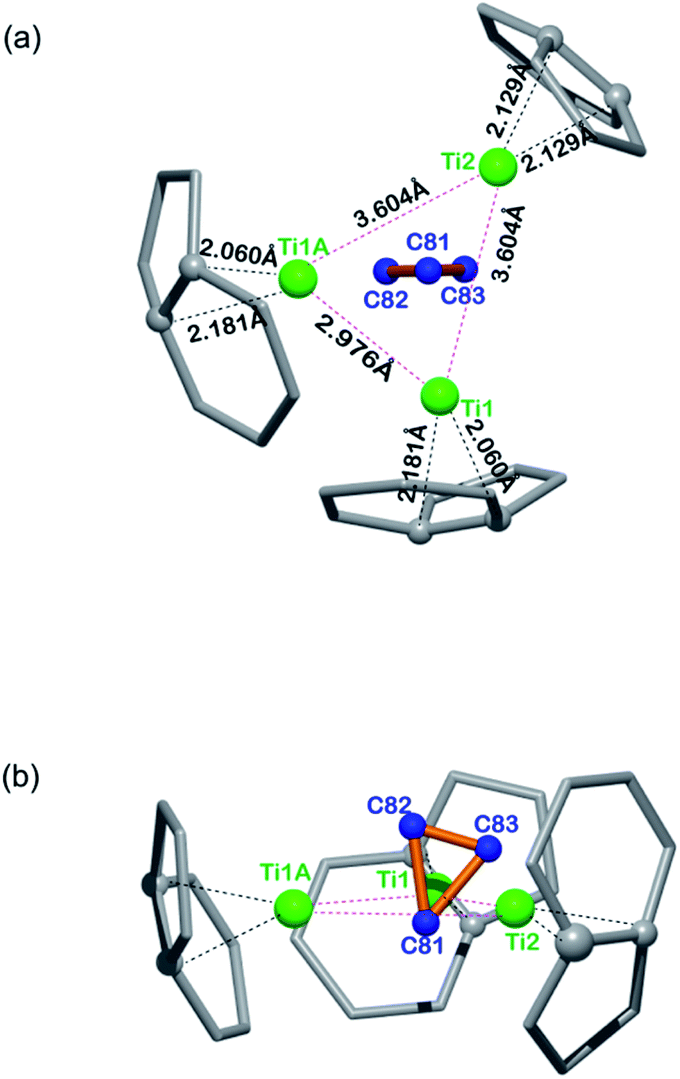 | ||
| Fig. 3 Orthogonal views showing the relative position of the major Ti3C3 site to the closest cage portions. | ||
We have performed density functional theory (DFT) calculations at the B3LYP/3-21G∼SSD level to understand the unique coordination environment in Ti3C3@Ih-C80. The calculated Wiberg indices for the bonds in the cyclopropane ring are 1.18, 0.80, and 0.75 while their calculated lengths are 1.39 Å, 1.58 Å, and 1.62 Å, respectively, a bit longer than the corresponding X-ray results. As for the Ti3-unit, there are six bond indices between the ring carbons and Ti atoms larger than 0.5, namely 0.88, 0.67, 0.64, 0.60, 0.59, and 0.58. There is one (weak) C–C double bond not bridged by Ti, one (weak) single bond bridged by one Ti, and one (still weaker) single bond bridged by two Ti atoms. In this simplified picture (Fig. 4a), each Ti atom is bonded to the ring via two Ti–C bonds, supporting the results of the geometry of the embedded Ti3C3 cluster (Fig. 4b) obtained by the single crystal X-ray method. Moreover, the electron distribution in Ti3C3@Ih-C80 is visualized in Fig. S6, ESI† for more graphical information about bonds between the carbons of the cage and also bonds in the encapsulate, showing a selected isosurface of electron density (i.e., every point of the surface has the same prescribed value of the electron density). At the selected constant electron density of 0.086 a.u., the bonds in the cage and especially in the Ti3C3 cluster are rather well visible, where the near-vertical coordination environment of the Ti3-unit and C3-unit was found. Such a clear picture further provides us with unique bonding information. The B3LYP/3-21G∼SDD Mulliken charges in Ti3C3@Ih-C80 on Ti atoms are 2.77, 2.48, and 2.44 while on the ring carbons they are −0.49, −0.41, and −0.22. The Mulliken-charge choice is supported32 by the fact that it is in good agreement with the available observed charges33 for metallofullerenes. However, unless there are charges deduced from observations for a class of clusterfullerenes, it cannot really be decided which particular charge definition is the best34 for the class. Thus, the results for the isolated Ti3C3@Ih-C80 system demonstrate a charge transfer of about six electrons to the cage, matching the charge state of common (Ih-C80)6– reflected by the Vis-NIR spectrum.
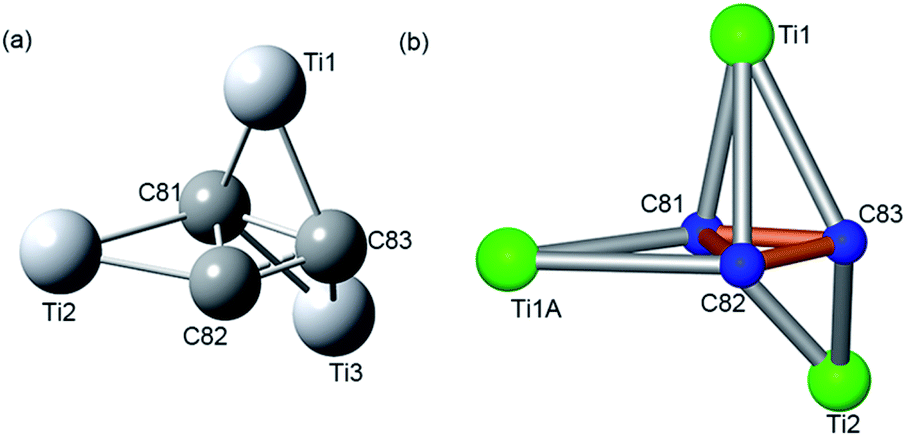 | ||
| Fig. 4 (a) The optimized Ti3C3 configuration at the B3LYP/3-21G∼SDD level and (b) geometry configuration of the major site of the Ti3C3 cluster from X-ray results. | ||
Theoretically, the Ti3C3 encapsulation is also favorable from the calculated energetics point of view.35 The B3LYP/6-31G*∼SDD encapsulation-energy gain per one encapsulated atom for Ti3C3@Ih-C80 is calculated to be about 18 kcal per mol per atom which is even larger than a relative energy gain computed for the recently isolated Y2C2@C1(1660)-C108.36 The stability calculations indeed support the finding that a novel EMF containing a unique Ti3C3 cluster was experimentally obtained.
The electrochemical properties of Ti3C3@Ih-C80 were measured in o-dichlorobenzene (o-DCB) by cyclic voltammetry (CV) and the profile is shown in Fig. 5. In the anodic region, Ti3C3@Ih-C80 displays a reversible oxidation step at 0.30 V and an irreversible one at 0.77 V whereas in the cathodic region, three reversible reduction steps are observed at −0.62 V, −1.22 V and −2.31 V. The electrochemical gap is thus calculated to be 0.92 V which is quite smaller than those of the other Ih-C80 based Ti-containing EMFs, namely, TiLu2C@Ih-C80 (ref. 22), TiSc2C2@Ih-C80 and TiSc2C@Ih-C80,16 suggesting a lower kinetic stability of Ti3C3@Ih-C80 which is also consistent with the Vis-NIR result. In addition, the irreversible oxidation process of Ti3C3@Ih-C80 has not been reported for the other Ti-based EMFs4,17,22,26 (Table 1). These results suggest that we can tune the electronic properties of Ti-EMFs by varying the number of redox-active Ti atoms. Furthermore, the frontier molecular orbital of Ti3C3@Ih-C80 visualized in Fig. S7 (ESI†) shows that the predominant localization of the LUMO is on the Ti3C3 cluster (mainly on the Ti atoms), suggesting that the first reduction step is ascribed to the alteration of the reduction behavior of the inner cluster, similar to the case of Sc2TiC2@Ih-C80.27
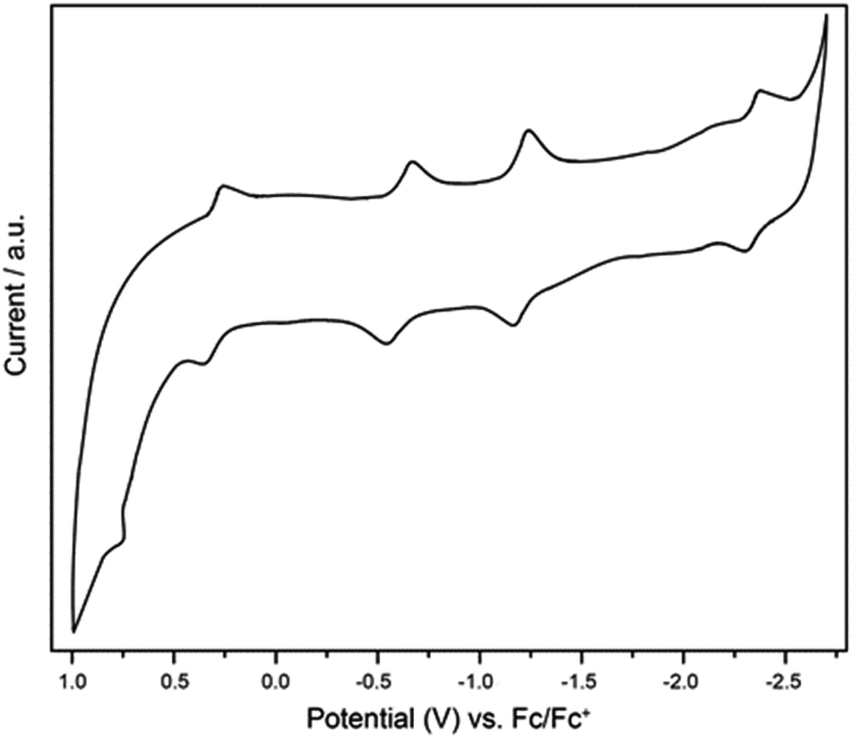 | ||
| Fig. 5 CV curve of Ti3C3@Ih-C80. Conditions: working electrode, glassy carbon electrode; counter electrode, Pt wire; reference electrode, Ag wire; supporting electrolyte, 0.05 M TBAPF6 in o-DCB with ferrocene as the internal standard. Scan rate: 100 mV s−1. | ||
| EMFs | ox E 2 | ox E 1 | red E 1 | red E 2 | red E 3 | ΔEgapb | Ref. |
|---|---|---|---|---|---|---|---|
| a Half-cell values in V versus Fe(Cp)2+/0 unless otherwise noted. b ΔEgap = oxE1 − redE1. c Irreversible peak value. | |||||||
| Ti3C3@Ih-C80 | 0.77c | 0.30 | −0.62 | −1.22 | −2.31 | 0.92 | This work |
| TiLu2C@Ih-C80 | — | 0.64 | −0.91 | — | — | 1.55 | 22 |
| TiSc2C@Ih-C80 | — | 0.66 | −0.67 | −1.51 | −1.66 | 1.33 | 16 |
| Sc2TiC2@Ih-C80 | — | 0.53 | −0.76 | −1.01 | −1.96 | 1.26 | 16 |
| Dy2TiC@Ih-C80 | — | 0.61 | −0.97 | −1.62 | −1.87 | 1.58 | 17 |
| Dy2TiC2@Ih-C80 | — | 0.47 | −1.14 | −1.58 | −2.21 | 1.61 | 17 |
Conclusions
An unprecedented Ti3C3 cluster has been successfully entrapped inside the Ih-C80 cage to form a stable compound. X-ray results reveal a novel coordination fashion by which the cyclopropane-like C3-unit perpendicularly coordinates to the Ti3-moiety. The electronic configuration of (Ti3C3)6+@C806− was confirmed according to the Vis-NIR spectroscopic and electrochemical characterization in addition to DFT calculations. Successful entrapment of the Ti3C3 cluster inside fullerene cages and identification of the unprecedented cyclopropane-coordination fashion may stimulate intensive interest in creating new hybrid molecules with fullerenes as protectors and discovering new reactivities in coordination chemistry.Experimental
Synthesis and isolation of Ti3C3@Ih-C80
Soot containing titanium-EMFs was produced by evaporating graphite rods packed with mixed TiC and graphite powder (mole ratio of Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C = 1:10) under a 400 mbar He atmosphere and 100 A current. The soot was extracted with CS2, and the filtered extract was treated with SnCl4 for the enrichment of Ti-EMFs, followed by multi-stage HPLC separation conducted on an LC-908 machine (Japan Analytical Industry Co. Ltd.) sequentially employing a 5PBB column (∅ 20 × 250 mm) and a Buckyprep column (∅ 20 × 250 mm). More details about the SnCl4 treatment and HPLC separations are present in the ESI.†
:C = 1:10) under a 400 mbar He atmosphere and 100 A current. The soot was extracted with CS2, and the filtered extract was treated with SnCl4 for the enrichment of Ti-EMFs, followed by multi-stage HPLC separation conducted on an LC-908 machine (Japan Analytical Industry Co. Ltd.) sequentially employing a 5PBB column (∅ 20 × 250 mm) and a Buckyprep column (∅ 20 × 250 mm). More details about the SnCl4 treatment and HPLC separations are present in the ESI.†
Spectroscopic and electrochemical studies
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) (MICROFLEX, Bruker, Germany) was used for the mass spectroscopic characterization. The Vis-NIR spectrum was measured on a PE Lambda 750S spectrophotometer in CS2. Cyclic voltammetry (CV) was performed in o-dichlorobenzene using a CHI-660E instrument with 0.05 M TBAPF6 as the electrolyte. The electron paramagnetic resonance (EPR) spectrum of Ti3C3@Ih-C80 was recorded with a Bruker A300 spectrometer in toluene.Crystallographic characterization
Cocrystals of Ti3C3@Ih-C80 were obtained by slow diffusion of a benzene solution of NiII(OEP) into a CS2 solution of the EMF in a glass tube at room temperature for one month. Single-crystal X-ray data were collected at 100 K using a radiation wavelength of 0.6525 Å with a MarCCD detector at beamline BL17B of the Shanghai Synchrotron Radiation Facility. A multi-scan method was used for absorption corrections. The structures were solved with direct methods and were refined with SHELXL-2014.37Crystal data of Ti3C3@Ih-C80·NiII(OEP)·2(C6H6)
Black block, 0.31 × 0.24 × 0.20 mm, monoclinic, space group C2/m, a = 25.0964(9) Å, b = 15.1544(5) Å, c = 19.7269(7) Å, β = 95.3360(10)°, V = 7470.0(5) Å3, Fw = 1888.20, λ = 0.6525 Å, Z = 4, Dcalc = 1.679 Mg m−3, μ = 0.498 mm−1, T = 100 K, R1[I > 2σ(I)] = 0.1079, wR2 (all data) = 0.2972, GOF (on F2) = 1.068. The maximum residual electron density is 1.953 e Å−3. Crystallographic data have been deposited in the Cambridge Crystallographic Data Center (CCDC number: 1883920).†Computational study of Ti3C3@Ih-C80
The structure optimization started with a combined basis set, the standard 3-21G basis38 for C atoms and SDD basis39 with the SDD effective core potential for Ti atoms, applied within the density functional theory (DFT) approach, namely using Becke's three parameter functional40 with the non-local Lee–Yang–Parr correlation functional41 (denoted as B3LYP/3-21G∼SDD). Herein, analytical harmonic vibrational analysis is then carried out in order to confirm that the local-energy minimum was indeed localized. The B3LYP/3-21G∼SDD optimized structure was further refined by performing re-optimizations with the standard 6-31+G* basis set42 for C atoms (B3LYP/6-31+G*∼SDD). Moreover, in the latter structure the encapsulation energy43 (the energy gain upon encapsulation of three Ti and three C atoms into the C80 cage) was evaluated at the B3LYP/6-31G*∼SDD level with inclusion of the basis set superposition error44 (BSSE/CP7) and the so-called steric correction that reflects the cage distortion upon encapsulation.45 All the computations were carried out with the Gaussian 09 program package.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (51672093, 51602112 and 51602097) is gratefully acknowledged. We thank the staff from the BL17B beamline of the National Center for Protein Science Shanghai at the Shanghai Synchrotron Radiation Facility for assistance during data collection and the Analytical and Testing Center in the Huazhong University of Science and Technology for all related measurements.Notes and references
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- L. Bao, P. Peng and X. Lu, Acc. Chem. Res., 2018, 51, 810–815 CrossRef CAS PubMed.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- S. Yang, T. Wei and F. Jin, Chem. Soc. Rev., 2017, 46, 5005–5058 RSC.
- W. Shen, L. Bao, Y. Wu, C. Pan, S. Zhao, H. Fang, Y. Xie, P. Jin, P. Peng, F.-F. Li and X. Lu, J. Am. Chem. Soc., 2017, 139, 9979–9984 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, R. Morales-Martínez, J. Zhong, C. de Graaf, A. Rodríguez-Fortea, J. M. Poblet, L. Echegoyen, L. Feng and N. Chen, J. Am. Chem. Soc., 2018, 140, 3907–3915 CrossRef CAS PubMed.
- C. Pan, W. Shen, L. Yang, L. Bao, Z. Wei, P. Jin, H. Fang, Y. Xie, T. Akasaka and X. Lu, Chem. Sci., 2019, 10, 4707–4713 RSC.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55 CrossRef CAS.
- C.-R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Angew. Chem., Int. Ed., 2001, 40, 397–399 CrossRef CAS PubMed.
- S. Stevenson, M. A. Mackey, M. A. Stuart, J. P. Phillips, M. L. Easterling, C. J. Chancellor, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2008, 130, 11844–11845 CrossRef CAS PubMed.
- L. Dunsch, S. Yang, L. Zhang, A. Svitova, S. Oswald and A. A. Popov, J. Am. Chem. Soc., 2010, 132, 5413–5421 CrossRef CAS PubMed.
- S. Yang, C. Chen, F. Liu, Y. Xie, F. Li, M. Jiao, M. Suzuki, T. Wei, S. Wang, Z. Chen, X. Lu and T. Akasaka, Sci. Rep., 2013, 3, 1487 CrossRef PubMed.
- T. Akasaka, S. Nagase, K. Kobayashi, M. Wälchli, K. Yamamoto, H. Funasaka, M. Kako, T. Hoshino and T. Erata, Angew. Chem., Int. Ed. Engl., 1997, 36, 1643–1645 CrossRef CAS.
- Y. Yamazaki, K. Nakajima, T. Wakahara, T. Tsuchiya, M. O. Ishitsuka, Y. Maeda, T. Akasaka, M. Waelchli, N. Mizorogi and S. Nagase, Angew. Chem., Int. Ed., 2008, 47, 7905–7908 CrossRef CAS PubMed.
- T.-S. Wang, N. Chen, J.-F. Xiang, B. Li, J.-Y. Wu, W. Xu, L. Jiang, K. Tan, C.-Y. Shu, X. Lu and C.-R. Wang, J. Am. Chem. Soc., 2009, 131, 16646–16647 CrossRef CAS PubMed.
- K. Junghans, K. B. Ghiassi, N. A. Samoylova, Q. Deng, M. Rosenkranz, M. M. Olmstead, A. L. Balch and A. A. Popov, Chem.–Eur. J., 2016, 22, 13098–13107 CrossRef CAS PubMed.
- K. Junghans, C. Schlesier, A. Kostanyan, N. A. Samoylova, Q. Deng, M. Rosenkranz, S. Schiemenz, R. Westerström, T. Greber, B. Büchner and A. A. Popov, Angew. Chem., Int. Ed., 2015, 54, 13411–13415 CrossRef CAS PubMed.
- M. Krause, F. Ziegs, A. A. Popov and L. Dunsch, ChemPhysChem, 2007, 8, 537–540 CrossRef CAS PubMed.
- T. Wang, J. Wu and Y. Feng, Dalton Trans., 2014, 43, 16270–16274 RSC.
- Q. Deng, K. Junghans and A. A. Popov, Theor. Chem. Acc., 2015, 134, 10 Search PubMed.
- X. Zhang, W. Li, L. Feng, X. Chen, A. Hansen, S. Grimme, S. Fortier, D.-C. Sergentu, T. J. Duignan, J. Autschbach, S. Wang, Y. Wang, G. Velkos, A. A. Popov, N. Aghdassi, S. Duhm, X. Li, J. Li, L. Echegoyen, W. H. E. Schwarz and N. Chen, Nat. Commun., 2018, 9, 2753 Search PubMed.
- A. L. Svitova, K. B. Ghiassi, C. Schlesier, K. Junghans, Y. Zhang, M. M. Olmstead, A. L. Balch, L. Dunsch and A. A. Popov, Nat. Commun., 2014, 5, 3568 Search PubMed.
- D. Fuchs, H. Rietschel, R. H. Michel, A. Fischer, P. Weis and M. M. Kappes, J. Phys. Chem., 1996, 100, 725–729 CrossRef CAS.
- M. Krause and L. Dunsch, ChemPhysChem, 2004, 5, 1445–1449 CrossRef CAS PubMed.
- S. Yang, C. Chen, A. A. Popov, W. Zhang, F. Liu and L. Dunsch, Chem. Commun., 2009, 6391–6393 RSC.
- C. Chen, F. Liu, S. Li, N. Wang, A. A. Popov, M. Jiao, T. Wei, Q. Li, L. Dunsch and S. Yang, Inorg. Chem., 2012, 51, 3039–3045 CrossRef CAS PubMed.
- K. V. Zaitsev, Y. F. Oprunenko, A. V. Churakov, J. A. K. Howard, S. S. Karlov and G. S. Zaitseva, J. Organomet. Chem., 2008, 693, 173–179 CrossRef CAS.
- H. Tsurugi, H. Nagae and K. Mashima, Chem. Commun., 2011, 47, 5620–5622 RSC.
- R. H. Duncan Lyngdoh, H. F. Schaefer and R. B. King, Chem. Rev., 2018, 118, 11626–11706 CrossRef CAS PubMed.
- D. Shoken, M. Sharma, M. Botoshansky, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2013, 135, 12592–12595 CrossRef CAS PubMed.
- M. Manßen, N. Lauterbach, J. Dörfler, M. Schmidtmann, W. Saak, S. Doye and R. Beckhaus, Angew. Chem., Int. Ed., 2015, 54, 4383–4387 CrossRef PubMed.
- Z. Slanina, F. Uhlík, S. Nagase, T. Akasaka, L. Adamowicz and X. Lu, Molecules, 2017, 22, 1053 CrossRef PubMed.
- M. Takata, E. Nishibori, M. Sakata and H. Shinohara, New Diamond Front. Carbon Technol., 2002, 12, 271–286 CAS.
- W. J. Hehre, A guide to molecular mechanics and quantum chemical calculations, Wavefunction, Irvine, CA, 2003 Search PubMed.
- Z. Slanina, F. Uhlík, C. Pan, T. Akasaka, X. Lu and L. Adamowicz, Chem. Phys. Lett., 2018, 710, 147–149 CrossRef CAS.
- C. Pan, L. Bao, X. Yu, H. Fang, Y. Xie, T. Akasaka and X. Lu, ACS Nano, 2018, 12, 2065–2069 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2002, 581, 139–147 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- Z. Slanina, F. Uhlík, S.-L. Lee, L. Adamowicz, T. Akasaka and S. Nagase, Int. J. Quantum Chem., 2011, 111, 2712–2718 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- Z. Slanina, F. Uhlík, X. Lu, T. Akasaka, K. H. Lemke, T. M. Seward, S. Nagase and L. Adamowicz, Fullerenes, Nanotubes, Carbon Nanostruct., 2016, 24, 1–7 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1883920. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04315b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |