DOI:
10.1039/C9SC04127C
(Edge Article)
Chem. Sci., 2019,
10, 10562-10565
Enantioselective construction of the tricyclic core of curcusones A–D via a cross-electrophile coupling approach†
Received
16th August 2019
, Accepted 30th September 2019
First published on 1st October 2019
Abstract
Herein we report our recent progress toward the enantioselective total synthesis of the diterpenoid natural products curcusones A–D by means of complementary Stetter annulation or ring-closing metathesis (RCM) disconnections. Using the latter approach, we have achieved the concise construction of the 5–7–6 carbocyclic core embedded in each member of the curcusone family. Essential to this route is the use of a cross-electrophile coupling strategy, which has not previously been harnessed in the context of natural product synthesis.
Introduction
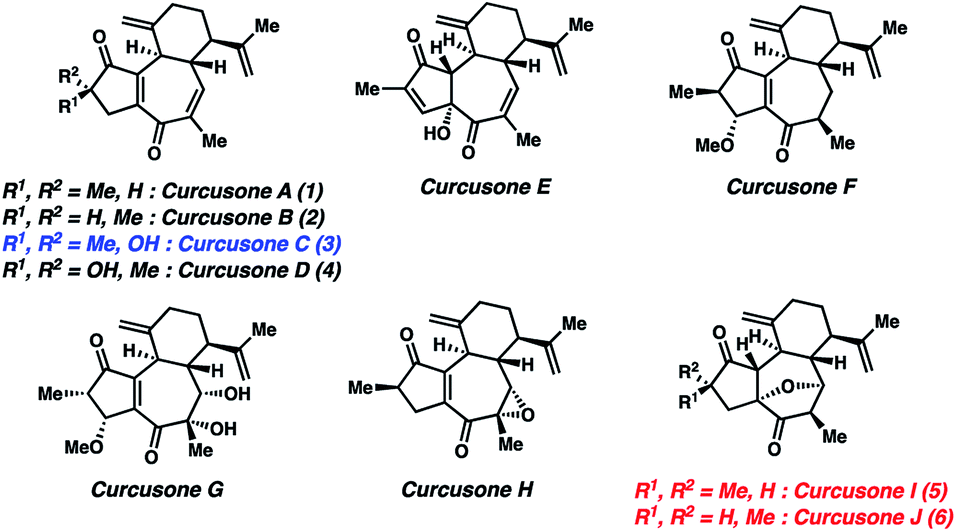
Despite its initial isolation over 30 years ago, the diterpenoid curcusone C (3, Fig. 1)1 and several other structurally related members of the rhamnofolane family of natural products have yet to succumb to total synthesis. This is somewhat surprising, as 3 has demonstrated diverse anti-proliferative properties, notably toward mouse lymphoma (IC50 = 0.26 μM), human promeolycytic leukemia (IC50 = 1.36 μM), human hepatoma (IC50 = 2.17 μM), and human ovarian carcinoma (IC50 = 0.16 μM) cells.2 Salient structural challenges associated with 1–4 include four stereocenters and a 5–7 ring fusion possessing a polarity-dissonant ene-dione motif, the construction of which necessitates the use of a formally Umpolung synthon.3 These combined factors motivated us to pursue a divergent route to 1–4. Notably, the Dai lab recently attempted to synthesize the proposed structures of curcusones I and J (5 and 6) as racemates over 21 steps, only to discover that their putative structures were incorrectly assigned by NMR.4 In contrast to these targets, the structures of 1–4 have been unambiguously defined by X-ray crystallography.2
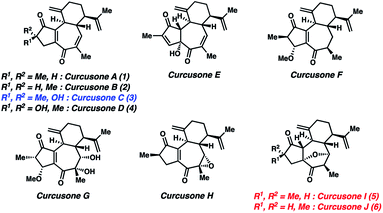 |
| | Fig. 1 Proposed structures of curcusones A–J. | |
Results and discussion
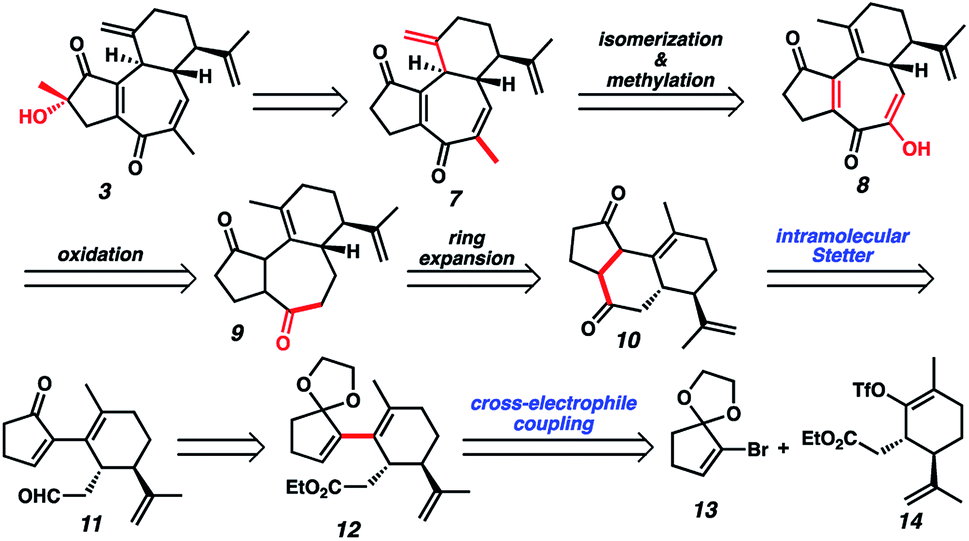
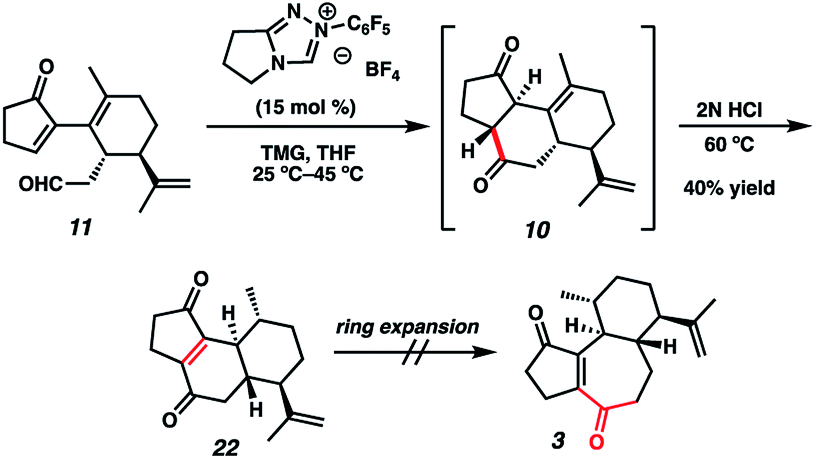
Although we had initially aimed to construct the central seven-membered ring motif in 1–4via a divinylcyclopropane rearrangement,5 we later found this route intractable due to the poor scalability of the key cyclopropanation step and to the oxidative instability of the Frontier material. In a 2nd generation route, we envisioned that curcusone C and its structural relatives could be assembled by α-functionalization of tricycle 7 (Scheme 1). The 2-methylcycloheptadienone moiety embedded in 7 may be derived from methylation and olefin migration of corresponding diosphenol intermediate 8, which itself might be prepared by sequential dehydrogenation and oxygenation of 1,4-dione 9. The central seven-membered ring found in 9 could be constructed via a one-carbon ring expansion of 10. We suspected that the central six-membered ring in 10 may be installed by an intramolecular Stetter reaction of ketoaldehyde 11. Intermediate 11 may itself be accessible by reduction and ketal cleavage of ester 12. The carbon–carbon bond bridging the five- and six-membered ring systems of 12 could be forged by a crucial cross-electrophile coupling of monocyclic fragments 13 and 14.
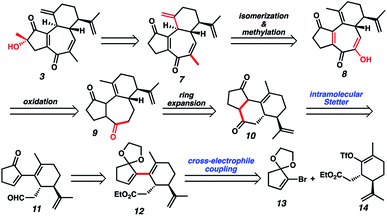 |
| | Scheme 1 2nd generation retrosynthesis of 3. | |
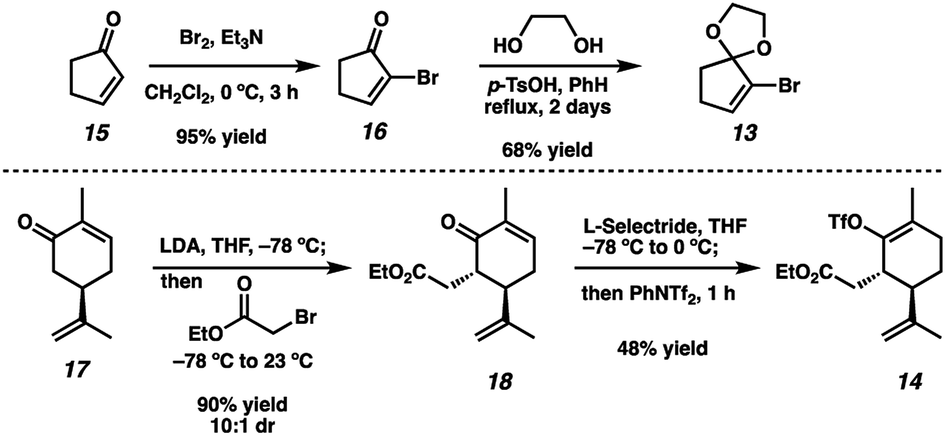
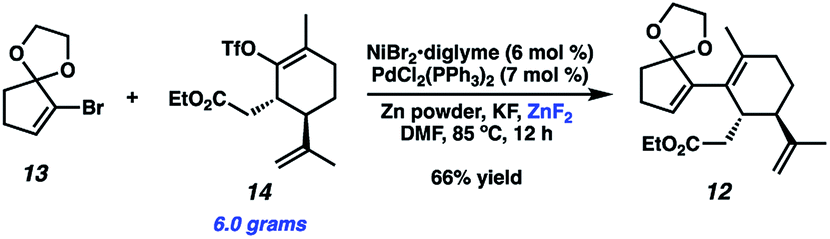
To achieve our first goal of constructing bicycle 12, we set out to perform a reductive cross-coupling of electrophiles 13 and 14. To this end, we synthesized vinyl triflate 14 by a two-step sequence involving α-alkylation of (R)-carvone6 (17, Scheme 2) followed by conjugate reduction of intermediate enone 18 and trapping of the resulting enolate with an electrophilic triflating agent. Numerous other carbon-based halide electrophiles were examined while exploring the α-alkylation step, but all proceeded in unsatisfactory yields and with poor diastereoselectivities. Meanwhile, bromide 13 was readily prepared in two steps according to known methods.7
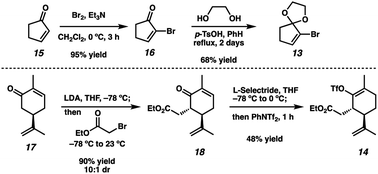 |
| | Scheme 2 Synthesis of coupling partners 13 and 14. | |
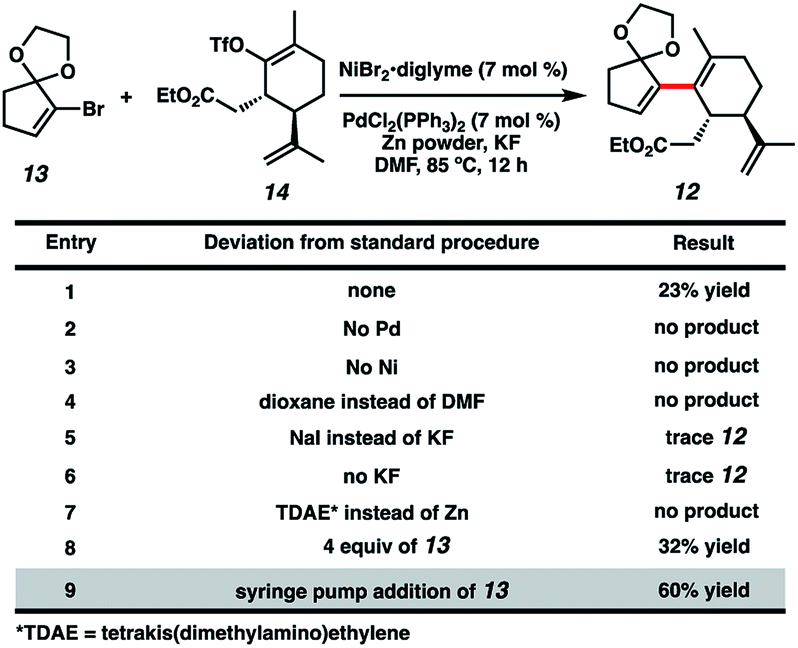
With both coupling partners in hand, focused our attention on the crucial cross-coupling. Although we had initially intended to join these monocyclic fragments via traditional nucleophile–electrophile coupling approaches, we were dismayed to find that these strategies all failed to afford any product. Inspired by pioneering research from the Weix group8 and our colleagues in the Reisman lab,9 we decided to instead employ a reductive coupling strategy using a dual Pd/Ni catalytic system. To our delight, this system offered modest amounts of desired bicycle 12, albeit with rapid decomposition of bromide 13 (Fig. 2, entry 1). Control experiments omitting either the Pd or Ni catalysts resulted in no detectable product formation (entries 2 and 3), revealing that both transition metals were crucial to the reaction. Adjusting the choice of solvent (entry 4), metal halide additive (entries 5 and 6) and terminal reducing agent (entry 7)10 offered no improvement in yields. Increasing the equivalents of bromide 13 slightly benefitted product formation (entry 8), likely due to the instability of 13 under the reaction conditions. Gratifyingly, syringe pump addition of 13 over several hours resulted in markedly better yields with minimal substrate decomposition (entry 9). We suspect this boost in yield is due to the consistently low concentration of 13 in solution, which might discourage undesirable hydrodehalogenation and homodimerization pathways.11
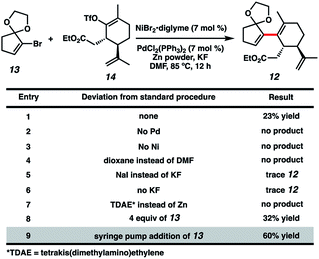 |
| | Fig. 2 Initial optimization of the cross-electrophile coupling. | |
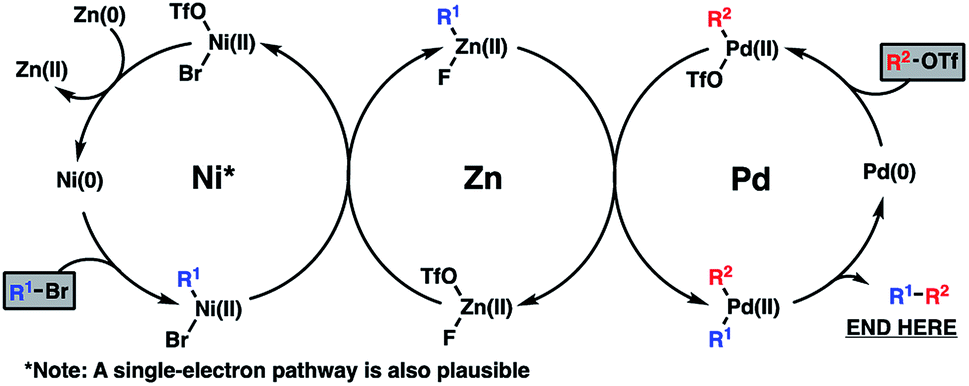
Mechanistically, the reductive coupling is thought to proceed through chemoselective oxidative addition of the active Pd(0) and Ni(0) catalysts with the triflate and bromide partners, respectively (Scheme 3). Based on recent studies from the Weix group,12 a subsequent transmetallation from Ni to Zn is thought to provide a transient organozinc species, which itself will undergo a second transmetallation to Pd to deliver a di-organopalladium species and a Zn(II) salt byproduct. Following this, reductive elimination of the Pd(II) intermediate furnishes the coupled product and regenerates the active Pd(0) catalyst. Meanwhile, reduction of the resultant Ni(II) salt with a stoichiometric Zn(0) reductant will turn over the Ni(0) catalyst.
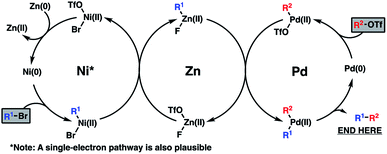 |
| | Scheme 3 Proposed mechanism for reductive cross-coupling. | |
Upon scale-up, yields for this coupling process proved somewhat inconsistent due to a precipitous drop in reaction rates after roughly 50% conversion of triflate 14. We hypothesized this to be caused by competitive coordination of Zn(II) byproduct to the essential fluoride additive, which serves as a bridging ligand during one or both of the proposed transmetallation events. In order to reverse this undesirable effect, we added ZnF2 to the reaction and were pleased to find that this co-additive benefited both reproducibility and scalablity, allowing us to reliably achieve 62–67% yields on multigram scale (Scheme 4). In stark contrast, simply increasing the stoichiometry of KF in the reaction generally lowered yields.
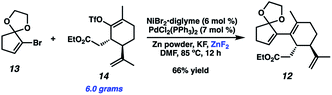 |
| | Scheme 4 Further optimization of the reductive cross-coupling on multigram scale. | |
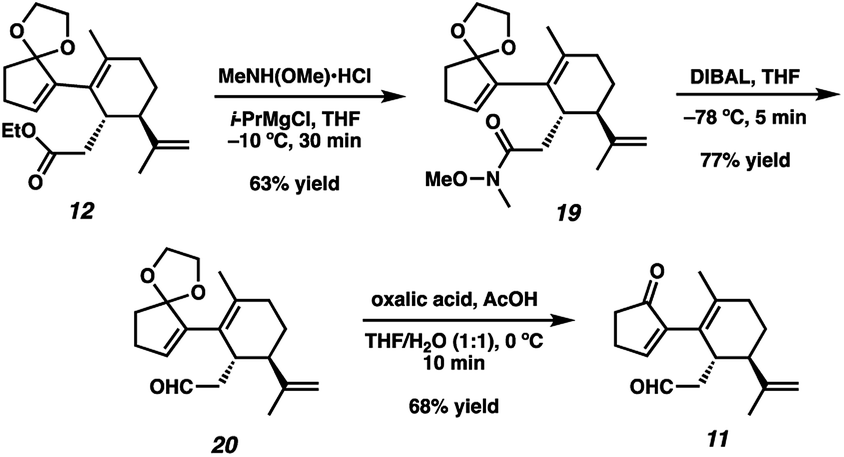
With sufficient quantities of bicycle 12 now in hand, we directed our attention toward the formation of the central seven-membered ring. The 1,4-dione synthon in targets 1–4 enticed us to pursue an Umpolung Stetter disconnection. Although we initially planned to construct the seven-membered ring via one-carbon homologation of the carbonyl tether followed by Stetter annulation, all strategies relying on Wittig–Levine alkoxymethylenation,13 α-alkoxycarbanion addition14 or Van Leusen cyanation15 offered either no reactivity or only trace product. As such, we instead envisioned performing a ring expansion16 of corresponding Stetter adduct 10 (Scheme 1, vide supra). Thus, we eventually found that ester 12 could be readily elaborated to Stetter precursor 11 by means of Weinreb amidation, partial reduction with DIBAL, and acid-promoted ketal cleavage (Scheme 5).
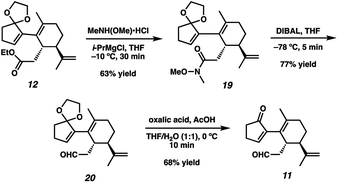 |
| | Scheme 5 Preparation of Stetter precursor 11. | |
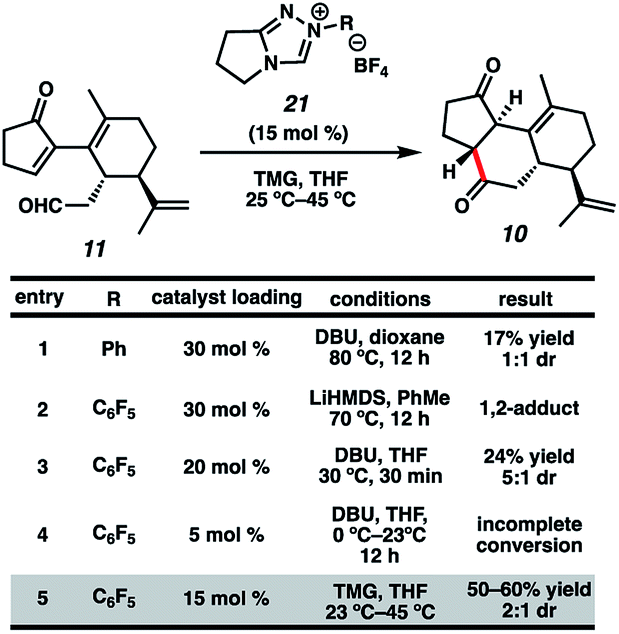
Now having rapid access to ketoaldehyde 11, we explored the crucial Stetter annulation (Fig. 3). Fortuitously, exposure of 11 to NHC scaffold 21 in the presence of DBU did indeed furnish tricycle 10 in low yield and as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers (entry 1).17 Although the absolute stereochemistry of the two diastereomers could not be unambiguously elucidated, a comparison of the relevant J values in the 1H spectrum suggests that they possess the same relative stereochemistry at the 5–6 junction. Substitution of DBU with a metallo-base such as LiHMDS resulted primarily in 1,2-adduct formation (entry 2),18 likely due to the high oxophilicity of the lithium counter-cation. Altering the choice of catalyst only modestly improved yields but dramatically enhanced dr (entry 3). Depression of temperature or catalyst loading resulted in sluggish reactivity and only modest improvements in dr (entry 4). Eventually, a compromise was struck between yield and diastereoselectivity using 1,1,3,3-tetramethylguanidine (TMG) as the catalytic base (entry 5), which provided 10 in fair yield and in 2:1 dr. To the best of our knowledge, this is the first instance of TMG conferring optimal yields and dr in an NHC-catalyzed Stetter reaction.
:1 mixture of diastereomers (entry 1).17 Although the absolute stereochemistry of the two diastereomers could not be unambiguously elucidated, a comparison of the relevant J values in the 1H spectrum suggests that they possess the same relative stereochemistry at the 5–6 junction. Substitution of DBU with a metallo-base such as LiHMDS resulted primarily in 1,2-adduct formation (entry 2),18 likely due to the high oxophilicity of the lithium counter-cation. Altering the choice of catalyst only modestly improved yields but dramatically enhanced dr (entry 3). Depression of temperature or catalyst loading resulted in sluggish reactivity and only modest improvements in dr (entry 4). Eventually, a compromise was struck between yield and diastereoselectivity using 1,1,3,3-tetramethylguanidine (TMG) as the catalytic base (entry 5), which provided 10 in fair yield and in 2:1 dr. To the best of our knowledge, this is the first instance of TMG conferring optimal yields and dr in an NHC-catalyzed Stetter reaction.
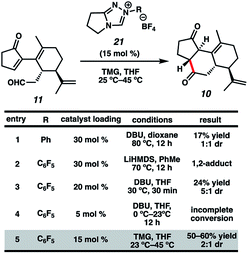 |
| | Fig. 3 Optimization of the catalytic Stetter reaction on ketoaldehyde 11. | |
With the 5–6–6 tricyclic core now installed, we next aimed to effect a one-carbon expansion of the central ring in 10. Unfortunately, all attempts to perform nucleophilic addition into either ketone moiety resulted in no reactivity, perhaps due to competing enolate formation. We eventually discovered that prolonged exposure of the diastereomeric mixture of 10 to strong acid resulted in a gradual olefin migration to stereoconvergently provide 22, bearing the requisite ene-dione motif found in curcusones A–D (Scheme 6). Unfortunately, all attempts to expand the central ring of 22 also failed, motivating us to reconsider our synthetic approach to the construction of the seven-membered ring.
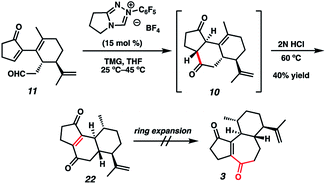 |
| | Scheme 6 Unsuccessful ring expansion of ene-dione 22. | |
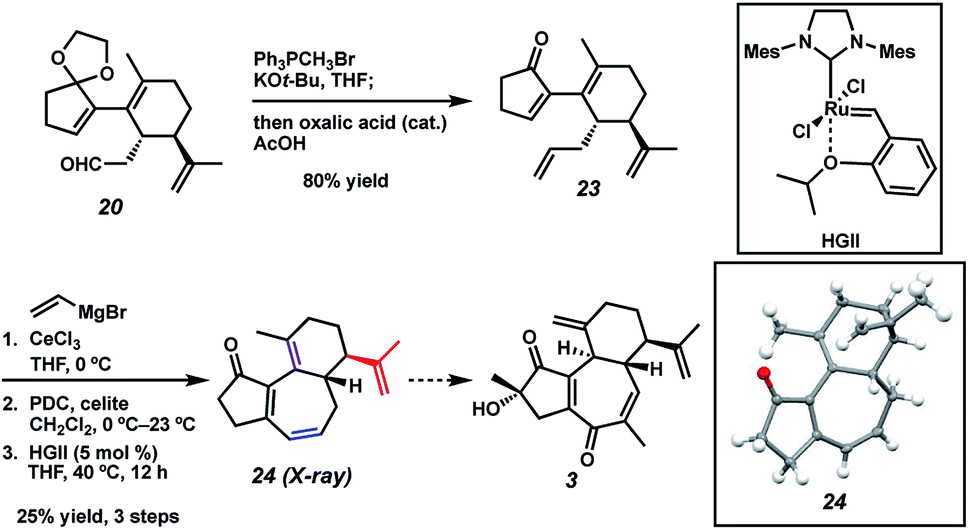
In a revised approach, we anticipated that the central ring might instead arise from an RCM strategy (Scheme 7). Thus, we conducted a one-pot olefination/ketal deprotection on common intermediate 20 to provide enone 23, possessing the first necessary olefin tether. We first hoped to install the second olefin tether by means of Cu-mediated 1,4-addition to the enone of 23, but all attempts to effect this transformation failed. To sidestep this roadblock, we instead chose to exploit a latent element of rotational symmetry in the cyclopentenone moiety. As such, we performed a sequential 1,2-vinylation of 23 to afford an intermediate bis-allylic alcohol, which upon Babler–Dauben oxidative rearrangement19 by the action of PDC provided the desired dienone RCM precursor with good regioselectivity, albeit with minor amounts of a chromatographically inseparable aldehyde isomer.20 We were delighted to find that exposure of semi-crude RCM precursor to catalytic HGII smoothly delivered desired tricycle 24, thereby providing rapid access to the carbocyclic core of curcusones A–D. We were incidentally pleased to find that the isopropenyl group in 24 (highlighted in red) proved to be totally inert toward RCM conditions, ensuring high topological selectivity for the desired ring closure. Interestingly, the crystal structure of 24 reveals that one of the double bonds (highlighted in purple) prefers to lie out of conjugation with the remainder of the pi system, likely due to the considerable strain it imposes on the central ring. Strategies to advance intermediate 24 to curcusone C (3) and its structural congeners are currently under thorough investigation in our group.
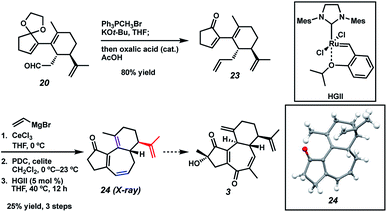 |
| | Scheme 7 Construction of tricycle 24via an RCM approach. | |
Conclusions
In summary, we have presented recent progress toward the synthesis of curcusones A–D. In our efforts, we have notably achieved the first synthesis of the carbocyclic framework embedded in curcusones A–J in only nine steps. Crucial to the success of this route is a cross-electrophile coupling approach, which can be performed on multigram scales. In light of these results, we anticipate that this reductive coupling protocol will find an enduring role in complex molecule synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Caltech and the NSF (1800511) are thanked for supplying funding. Dr Scott Virgil is gratefully acknowledged for purification and crystallization expertise. We thank Dr Michael Takase and Dr Lawrence Henling for collecting X-ray crystallographic data. Dr Steven Loskot and Nicholas Hafeman are acknowledged for helpful discussions.
Notes and references
- W. Naengchomnong, Y. Thebtaranonth, P. Wiriyachitra, K. T. Okamoto and J. Clardy, Tetrahedron Lett., 1986, 27, 2439–2442 CrossRef CAS.
-
(a) G. Chianese, E. Fattorusso, O. O. Aiyelaagbe, P. Luciano, H. C. Schröder, W. E. G. Müller and O. Taglialatela-Scafati, Org. Lett., 2011, 13, 316–319 CrossRef CAS PubMed;
(b) J.-Q. Liu, Y.-F. Yang, X.-Y. Li, E.-Q. Liu, Z.-R. Li, L. Zhou, Y. Li and M.-H. Qiu, Phytochemistry, 2013, 96, 265–272 CrossRef CAS PubMed;
(c) P. Picha, W. Naengchomnong, P. Promratanapongse, E. Kano, S. Hayashi, T. Ohtsubo, S. W. Zhang, H. Shioura, R. Kitai, H. Matsumoto, K. Kawahara, S. Puribhat and P. Phanthumachinda, J. Exp. Clin. Cancer Res., 1996, 15, 177–183 CAS.
- A. Lapworth, J. Chem. Soc., Trans., 1922, 121, 416–427 RSC.
- Y. Li and M. Dai, Angew. Chem., Int. Ed., 2017, 56, 11624–11627 CrossRef CAS PubMed.
- C. W. Lee, B. L. H. Taylor, G. P. Petrova, A. Patel, K. Morokuma, K. N. Houk and B. M. Stoltz, J. Am. Chem. Soc., 2019, 141, 6995–7004 CrossRef PubMed.
- L.-L. Shi, H.-J. Shen, L.-C. Fang, J. Huang, C.-C. Li and Z. Yang, Chem. Commun., 2013, 49, 8806–8808 RSC.
- A. B. Smith III, S. J. Branca, N. N. Pilla and M. A. Guaciaro, Org. Lett., 1982, 47, 1855–1869 Search PubMed.
- L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454–457 CrossRef CAS PubMed.
- K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2017, 139, 5684–5687 CrossRef CAS PubMed.
- L. L Anka-Lufford, K. M. M. Huihui, N. J. Gower, L. K. G. Ackerman and D. J. Weix, Chem.–Eur. J., 2016, 22, 11564–11567 CrossRef PubMed.
- D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed.
- A. M. Olivares and D. J. Weix, J. Am. Chem. Soc., 2018, 140, 2446–2449 CrossRef CAS PubMed.
- S. G. Levine, J. Am. Chem. Soc., 1958, 80, 6150–6151 CrossRef CAS.
- W. C. Still, J. Am. Chem. Soc., 1978, 100, 1481–1487 CrossRef CAS.
- O. H. Oldenziel, D. Van Leusen and A. M. Van Leusen, J. Org. Chem., 1977, 42, 3114–3118 CrossRef CAS.
- E. J. Kantorowski and M. J. Kurth, Tetrahedron, 2000, 56, 4317–4353 CrossRef CAS.
- N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed.
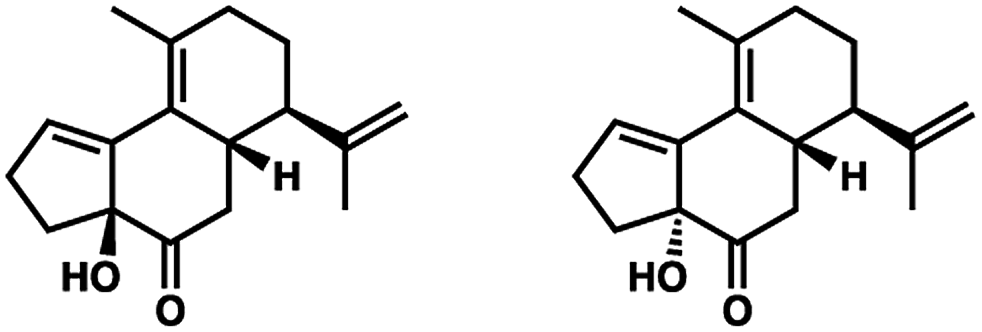
- The following 1,2-adducts were obtained in roughly equal proportions:
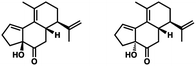 .
.
-
(a) J. H. Babler and M. J. Coghlan, Synth. Commun., 1976, 6, 469–474 CrossRef CAS;
(b) W. G. Dauben and D. M. Michno, J. Org. Chem., 1977, 42, 682–685 CrossRef CAS.
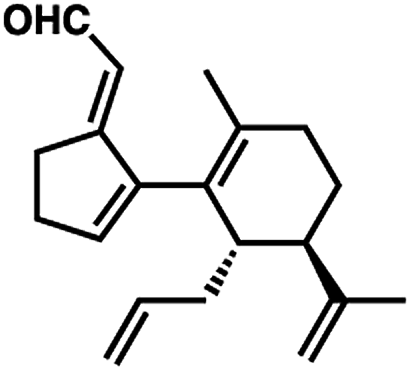
- The following aldehyde isomer was continually observed as a minor byproduct from the oxidative transposition:
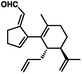 .
.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR and IR spectra, and X-ray crystallography data. CCDC 1955394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04127c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*