
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically switchable rotaxanes: recent strides in new directions
Hendrik V.
Schröder†
 * and
Christoph A.
Schalley
* and
Christoph A.
Schalley
Institut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: hendrik.schroeder@fu-berlin.de
First published on 3rd October 2019
Abstract
Are they still electrifying? Electrochemically switchable rotaxanes are well known for their ability to efficiently undergo changes of (co-)conformation and properties under redox-control. Thus, these mechanically interlocked assemblies represent an auspicious liaison between the fields of molecular switches and molecular electronics. Since the first reported example of a redox-switchable molecular shuttle in 1994, improved tools of organic and supramolecular synthesis have enabled sophisticated new architectures, which provide precise control over properties and function. This perspective covers recent advances in the area of electrochemically active rotaxanes including novel molecular switches and machines, metal-containing rotaxanes, non-equilibrium systems and potential applications.
 Hendrik V. Schröder | Dr Hendrik V. Schröder studied chemistry at Freie Universität in Berlin, Germany. Afterwards, he received a scholarship by the German Chemical Industry Fund (FCI) to prepare his doctoral thesis under the guidance of Prof. Dr Christoph A. Schalley. He worked in the fields of switchable rotaxanes and supramolecular electrochemistry. In autumn 2019, he started as postdoc funded by the German Research Foundation (DFG) in the group of Prof. A. James Link at Princeton University, USA, with focus on mechanically interlocked peptides. His current research interests include intertwined molecules, molecular switches and machines, organic synthesis, and electrochemistry. |
 Christoph A. Schalley | Christoph Schalley studied chemistry at the University of Freiburg/Germany and graduated in 1993 from the Technical University Berlin. In his PhD work with Helmut Schwarz at TU Berlin, he received his thorough education as a mass spectrometrist and gas-phase chemist. After postdoctoral work on supramolecular chemistry with Julius Rebek, Jr. at The Scripps Research Institute, La Jolla/USA, he combined both topics when he started his own research group as a habilitand at the University of Bonn. In 2005, he was appointed professor of organic chemistry at Free University Berlin. Meanwhile the Schalley group's research topics are diverse and comprise supramolecular chemistry in the gas phase as well as in solution and at interfaces. |
Introduction
Rotaxanes, a class of mechanically interlocked molecules, consist in their most basic form of a macrocyclic wheel threaded on a linear axle molecule.1 Bulky stopper groups placed at the ends of the molecular thread prevent deslipping of the wheel. Encouraged by synthetic advances made during the last decades, for example in template synthesis,2–4 self-sorting5,6 or dynamic combinatorial chemistry,7–9 complex (supra)molecular architectures in a large structural complexity and variety10,11 have become more easily accessible. Conformational flexibility and dynamic nature of rotaxanes, given by the mechanical bond between their components, make them perfectly suited to undergo substantial structural changes when manipulated by external stimuli.12,13 Based on that, bi- and multi-stable rotaxanes arose as promising candidates for artificial molecular switches and machines over the last decades.14–16Electrochemically switchable rotaxanes bear functional groups, which can undergo reversible redox reactions. These rotaxanes provide (co-)conformational control and can reversibly generate large-amplitude molecular motions by electrochemical stimuli. In other words, they can convert electrochemical energy into directed movements at the molecular scale. Electrochemical energy is considered to be a simple, “clean” (no chemical waste production), selective and fast way to initiate molecular switching processes.17,18 Electrochemistry also offers a rich toolbox of methods to probe structural and electronic properties of even complex supramolecular ensembles.19 Valuable information including a qualitative and quantitative assessment of the available oxidation states,20 electrochemical reaction mechanisms,21 diffusion constants,22 orbital energies,23 and much more can be extracted by electroanalytical methods. Additionally, methods of supramolecular electrochemistry24 provide rare access to thermodynamic and kinetic parameters of intramolecular (co-)conformational changes, which are often out of the scope of classical analytic methods in supramolecular chemistry.25,26
Exactly 25 years ago, the first electrochemically switchable molecular shuttle based on a two-station rotaxane has been reported,27 raising high hopes for the field of molecular switches and machines. Although these rotaxanes are among pioneering examples, systems have evolved from rather simple molecular shuttles to advanced supramolecular architectures bringing multiple functions together. Recent systems come with a precise electrochemical control of molecular motions, properties and functions. Additionally, new types of metal–organic rotaxanes provide potential applications as catalysts, sensors, and single-molecule electronic devices. Undoubtedly, the 2016 Nobel Prize in Chemistry28–30 “for the design and synthesis of molecular machines” marks the outstanding significance of these developments. On the other hand, it redirects the trajectory of the field of rotaxanes towards more functionalized and applicable systems.
Numerous excellent overviews covering switchable rotaxanes,1,31,32 metal-containing rotaxanes2,33,34 and electrochemically active supramolecular systems24,35,36 have been published. This perspective is focused on recent developments of redox-active (pseudo)rotaxanes—especially those reported after the 2016 Nobel Prize—and applications arising from their electrochemical activity on the basis of a non-exhaustive list of illustrative examples. In particular, this summary gives a differentiated view on recent advances beyond classical molecular shuttles. It covers (i) rotaxanes generating molecular motions; (ii) metal-containing rotaxanes; (iii) applications of rotaxanes including sensing, catalysis, molecular electronics and molecular machines; and (iv) non-equilibrium systems.
Rotaxanes generating molecular motions
Fundamentals of redox-controlled molecular shuttles
Archetypical examples of electrochemically switchable rotaxanes are two-station molecular shuttles,27 in which the position of a wheel on a molecular thread can be manipulated by reduction or oxidation (Fig. 1). Electroanalytic methods, such as commonly applied cyclic voltammetry (CV), can provide qualitative evidence for co-conformational changes through altered half-wave (E1/2) or peak (Ep) potentials and can even quantify the distribution of isomers in different redox-states.24,35 The isomer distribution is given by the dimensionless co-conformational equilibrium constant Kx (Fig. 1, eqn (1)) for each redox-state (x). The efficiency of a molecular switch can be expressed as the binding enhancement of a station which reflects the ratio between Kx values in two redox-states (Fig. 1, eqn (2)). In recent years, supramolecular construction motifs have been continuously optimized to gain more control over co-conformational switching.37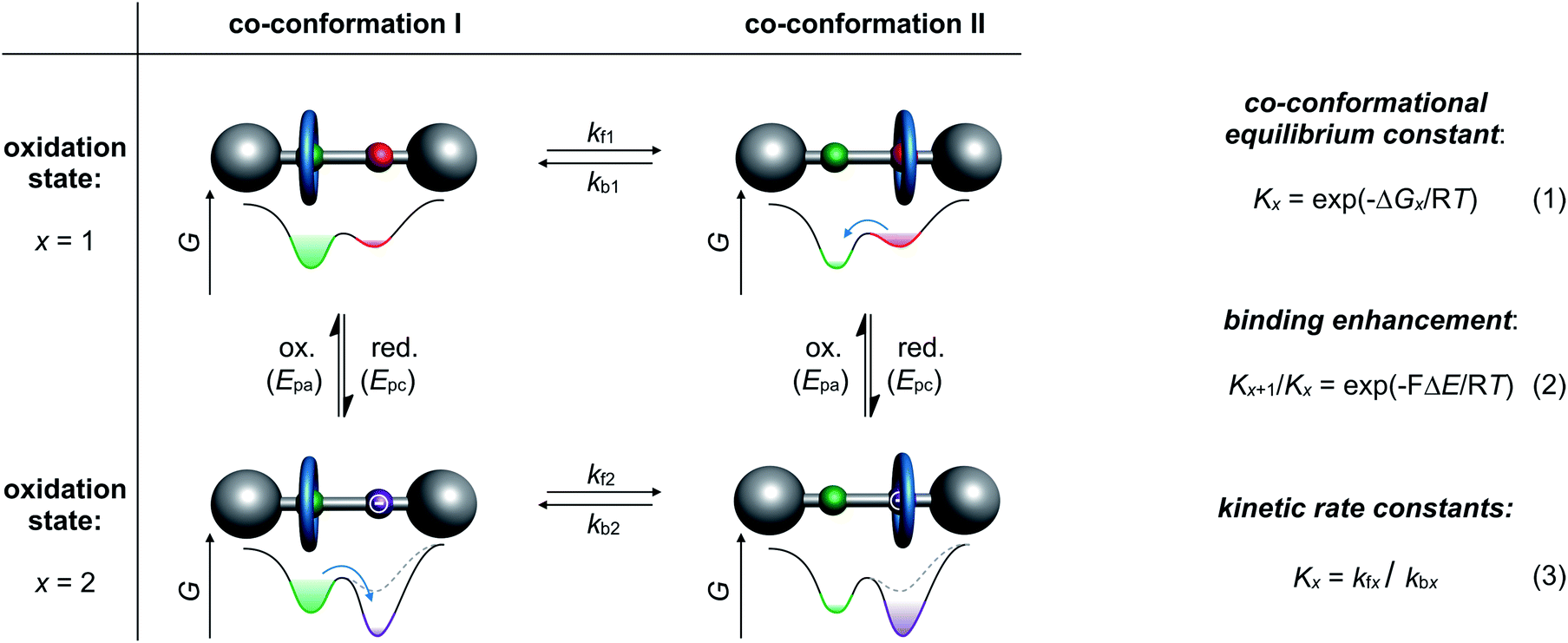 | ||
| Fig. 1 A general square scheme for an electrochemically switchable [2]rotaxane. It illustrates co-conformational isomers and the corresponding energy landscapes of wheel translation in two stable redox-states (x = 1 or 2). The eqn (1)–(3) describe the thermodynamics and kinetics of the switching processes, where Gx is the Gibbs free energy in a given oxidation state, k a first-order rate constant, F the Faraday constant, T the temperature, ΔE a potential difference between two redox-reactions and R the ideal gas constant. | ||
Electrochemical experiments (e.g. CV) at different scan rates or chrono methods can additionally help to identify reversibility, to elucidate reaction mechanisms, and to investigate the kinetics38 of conformational changes. Furthermore, digital simulations39 are often used as a complementary tool not only to predict voltammetric responses of supramolecular systems but also to fit experimental data. Digital simulation provides access to thermodynamic and kinetic parameters (Kx, kfx and kbx) as a self-consistent set of values (Fig. 1, eqn (3)). Naturally, the accuracy of the derived values depends on the correct choice of reaction mechanism, the quality of experimental data and the input variables. Improvement in accuracy can be achieved by comparing with data, for example, diffusion or binding constants derived from independent analytical methods or model systems.
For instance, an electrochemically controllable molecular shuttle with a highly selective co-conformational distribution was reported in a 2003 landmark publication of Leigh, Paolucci and co-workers.40 The thread of hydrogen-bonded [2]rotaxane 1 (Fig. 2) bears a naphthalimide unit, which serves simultaneously as a stopper for the wheel and as a redox-active recognition side. In the neutral state, the tetralactam wheel preferably encircles the diamide over the naphthalimide station. Electrochemical one-electron reduction generates a stable naphthalimide radical anion (Epc = −1.40 V vs. Fc/Fc+, Fc = ferrocene), which is now a more favorable hydrogen bond acceptor. By CV experiments and digital simulations, a binding enhancement of >108 for the naphthalimide station after reduction was determined leading to a translation of the wheel towards it. In comparison to the free thread, re-oxidation (Epa = −0.89 V) is anodically shifted reflecting stabilization of the radical anion by the hydrogen-bonded wheel. Variable-temperature experiments in THF revealed the kinetic parameters ΔH‡ = 59 kJ mol−1 and ΔS‡ = 36 kJ mol−1 for wheel translation from the diamide to the naphthalimide station after reduction, resulting in a shuttling time of ∼50 μs at room temperature. In a recent report, the shuttling mechanism was studied in more detail by transient two-dimensional infrared spectroscopy.41
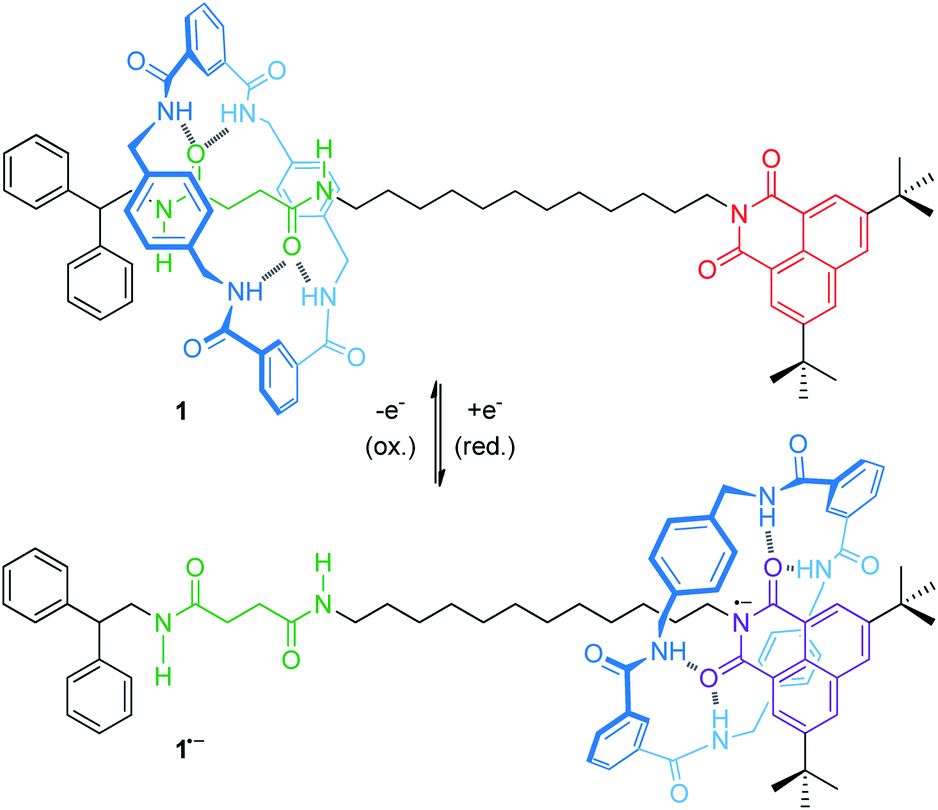 | ||
| Fig. 2 Electrochemically switchable molecular shuttle 1. In the neutral state, the tetralactam wheel preferably encircles the diamide station. One-electron reduction to the 1˙− state moves the wheel to the naphthalimide radical anion site. | ||
Rotaxane shuttles enabling rare interactions
The mechanical bond prevents dissociation of non-covalently bound molecular entities and can stabilize interactions, which are otherwise unfavorable.42 Thus, rotaxanes offer an opportunity to study these rare interactions between subcomponents or even utilize them for (co-)conformational control.For example, Stoddart and co-workers recently reported on the tristable [2]rotaxane 26+ (Fig. 3) with three different recognition sites on the thread, namely two triazoles, two tetrafluorobenzenes and one bipyridinium unit.43 In the initial state, the quadruply charged cyclobis(paraquat-p-phenylene) (CBPQT4+) wheel preferably encircles one of the relatively π-electron-rich triazoles by forming a donor–acceptor complex. The wheel's binding affinity can then be altered by stepwise reduction. Cyclovoltammetry reveals an initial two-electron reduction (Epc1 = −0.14 V vs. Ag/AgCl) to 22˙/4+ closely followed by a one-electron reduction (Epc2 = −0.22 V). The resulting tri-radical species 23(˙+) is stabilized by radical-pairing interactions between the doubly reduced wheel CBPQT2(˙+) and the singly reduced bipyridinium station. A further three-electron reduction (Epc3 = −0.69 V) to 2(0) neutralizes wheel and bipyridinium station. Now, the CBPQT(0) ring migrates to one of the π-electron-poor tetrafluorobenzene moieties. This supramolecular charge-transfer complex—which is enabled through spatial pre-organization of the wheel—is rather weak and cannot be easily studied in “the context of a host–guest complex”,43 unless stabilized by a mechanical bond.
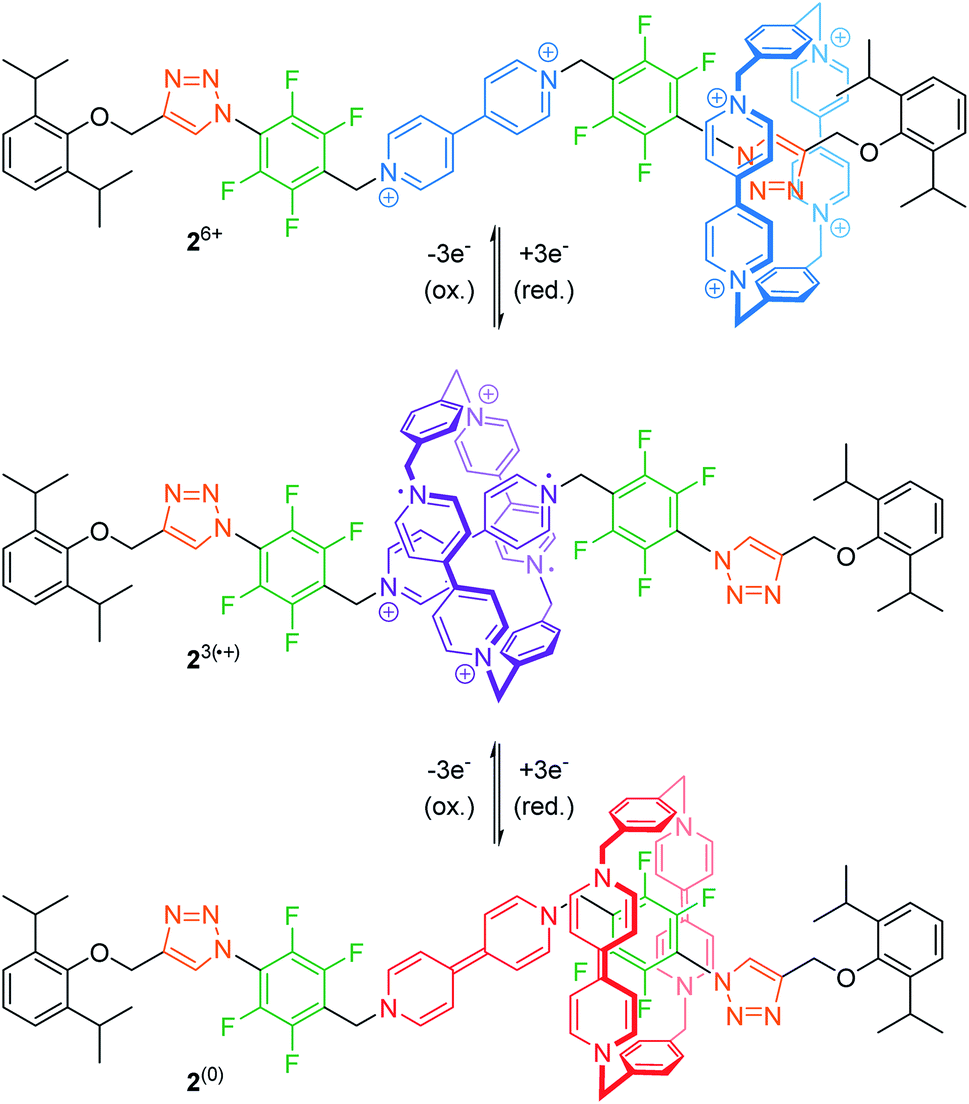 | ||
| Fig. 3 Tristable [2]rotaxane 26+ in its three different redox-states and resulting co-conformations. The unusual CBPQT(0)/tetrafluorobenzene charge-transfer complex is stabilized by the mechanical bond. | ||
Rare through-space interactions also play a role in the orthogonally switchable CBPQT4+-rotaxane recently reported by Jeppesen, Nielsen and co-workers (Fig. 4a).44 The CBPQT4+ wheel of [2]rotaxane 34+ is threaded on a redox-active monopyrrolo-tetrathiafulvalene axle, which is terminated on one side with a 1,1-dicyano-dihydroazulene moiety, a molecular photo- and thermoswitch (Fig. 4b). In both photoisomeric forms, the closed dihydroazulene and open vinylheptafulvene form, deslipping of the wheel is sterically hindered. The presence of the wheel anodically shifts both tetrathiafulvalene (TTF) oxidation potentials as compared to the free thread. However, the first oxidation already induces a movement of the wheel towards the dihydroazulene/phenylene site forming weak interactions between the cyano groups and the wheel's bipyridinium α-protons. These rather weak interactions between wheel and axle significantly accelerate the thermal back-conversion of the dihydroazulene switch (open to closed form) after photoirradiation.
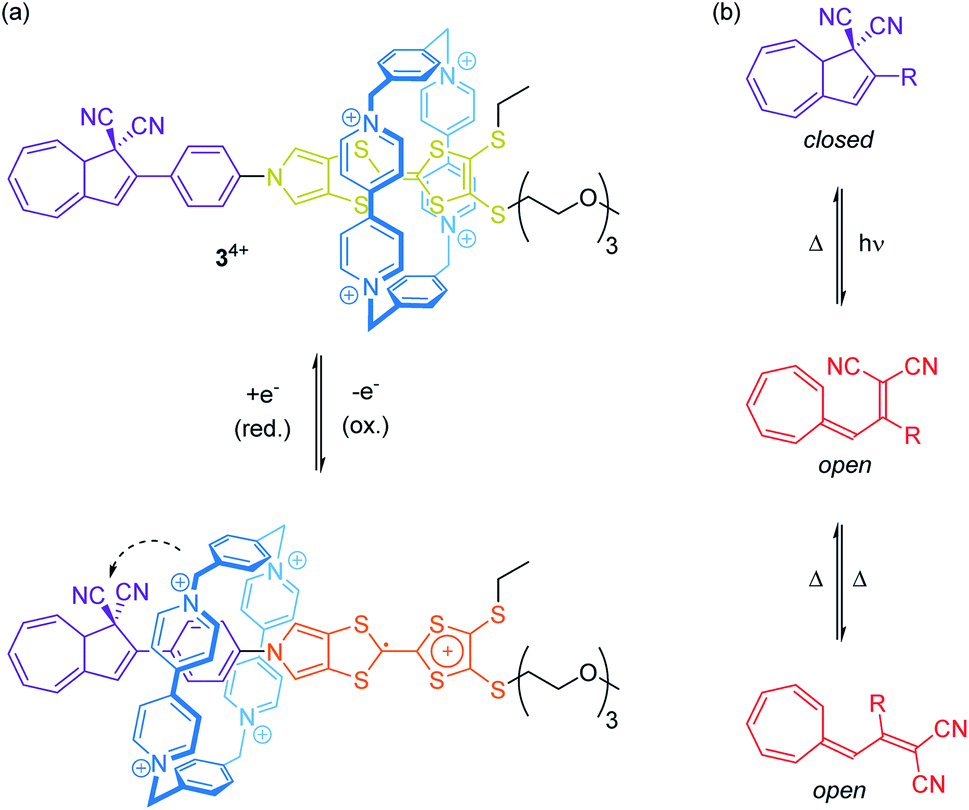 | ||
| Fig. 4 (a) Redox-active [2]rotaxane 34+ bearing a photo- and thermoswitchable dihydroazulene unit as stopper. The dashed arrow illustrates through-space interactions between CBPQT4+ wheel and dihydroazulene site. (b) Photo- and thermoswitching of the 1,1-dicyano-dihydroazulene moiety. | ||
Weak through-space interactions were also found to affect TTF redox-potentials of our recently reported rotaxane shuttles consisting of a TTF-decorated crown ether wheel and a neutral axle.45 The TTF/TTF˙+ oxidation was cathodically shifted compared to the free TTF-wheel, indicating significant stabilization of the TTF˙+ state by interactions between the mechanically bound axle and the TTF˙+ moiety.
Oligorotaxanes and daisy chain rotaxanes
Linear main-chain oligo- and polyrotaxanes, in which several wheels are threaded on an axle, have been used as supramolecular scaffolds to generate large-amplitude motions.46,47 The resulting structural changes are reminiscent of molecular folding and contraction processes of biopolymers, e.g. protein folding or muscle contraction.The Stoddart group presented an oligo[3]rotaxane, in which axle and wheels as a whole undergo an redox-triggered folding/unfolding along the trajectory of the axle.48
Our lab recently reported on oligorotaxanes, in which inter-wheel distances can be controlled by radical-cation interactions between TTF-decorated wheels (Fig. 5a).49 In the initial state, the 24-crown-8 ether wheels of oligo[4]rotaxane 43+ are bound to secondary ammonium stations by hydrogen bonding. Reversible oxidations of the wheels' TTF units weaken the wheel–axle interactions through counteracting Coulomb repulsion. As shown by electrochemistry, UV/Vis-NIR and electron paramagnetic resonance spectroscopy, the TTF˙+ moieties can form inter-wheel mixed-valence (4˙/4+) and radical-cation (43˙/6+) interactions, which consequently lead to a contraction of the ensemble in both redox-states. Further oxidation (TTF˙+ to TTF2+) to the 49+ state creates strong charge repulsion between the TTF2+ units and leads to a maximization of the inter-wheel distances. The resulting accordion-like motion (Fig. 5b) is independent of the molecular thread and, thus, promising for the construction of piston-type rotaxane motors.
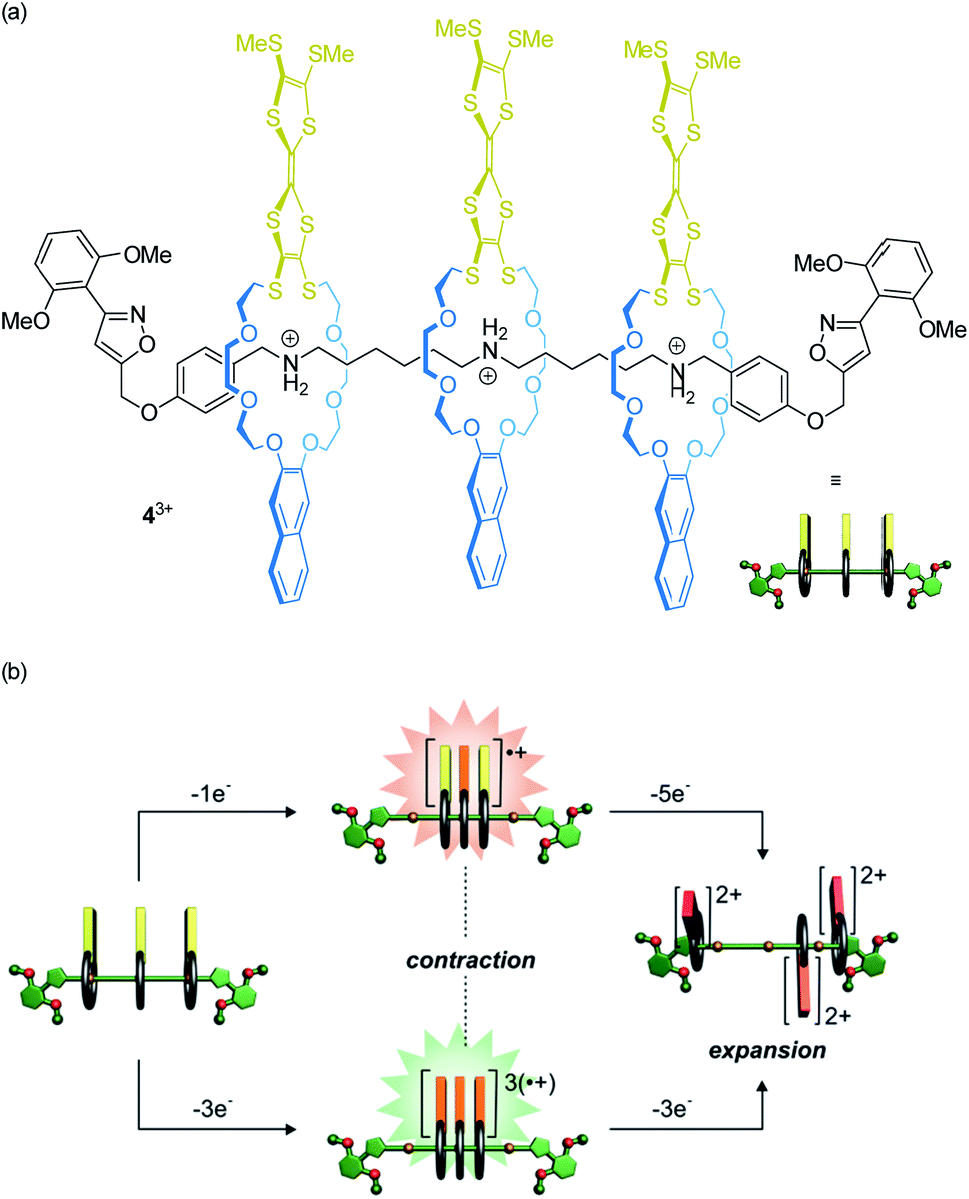 | ||
| Fig. 5 (a) Structure of the electrochemically switchable oligo[4]rotaxane 43+. (b) Graphical summary of the redox-controlled generation of an accordion-like contraction/expansion motion. Partly adapted with permission from ref. 49. Copyright (2019) John Wiley & Sons. | ||
Electrochemically switchable daisy chain rotaxanes are ideal prototypes to generate a muscle-like contraction. Moulin and Giuseppone introduced a [c2]daisy chain rotaxane (54+, Fig. 6) with two different endgroups, where the mechanical actuation is controlled by acid/base addition.50 The crown ether/ammonium rotaxane is decorated with two redox-active stoppers, a 1,7-dipyrrolidinyl-substituted perylene diimide and a triarylamine unit. In the extended form (protonated), three reversible electrochemical processes are observed by cyclovoltammetry: a two-electron reduction (E11/2 = −1.30 V vs. Fc/Fc+) centered on the perylene diimide, an oxidation wave (+0.19 V) resulting from a superposition of two one-electron oxidations centered on the triarylamine and the perylene diimide and a second one-electron oxidation of the perylene diimide unit (+0.31 V). The cyclic voltammogram is a superposition of those of the corresponding individual reference compounds; thus, both redox-active groups are electrochemically unaffected in the extended form. In the contracted form (deprotonated), however, the two-electron reduction wave is broadened and the peak current of the re-oxidation is significantly decreased indicating a lower chemical stability. At the same time, the second oxidation of the perylene diimide unit is shifted to lower potentials. The authors have attributed both effects to the proximity of the crown ethers to the electro-active stoppers after contraction.
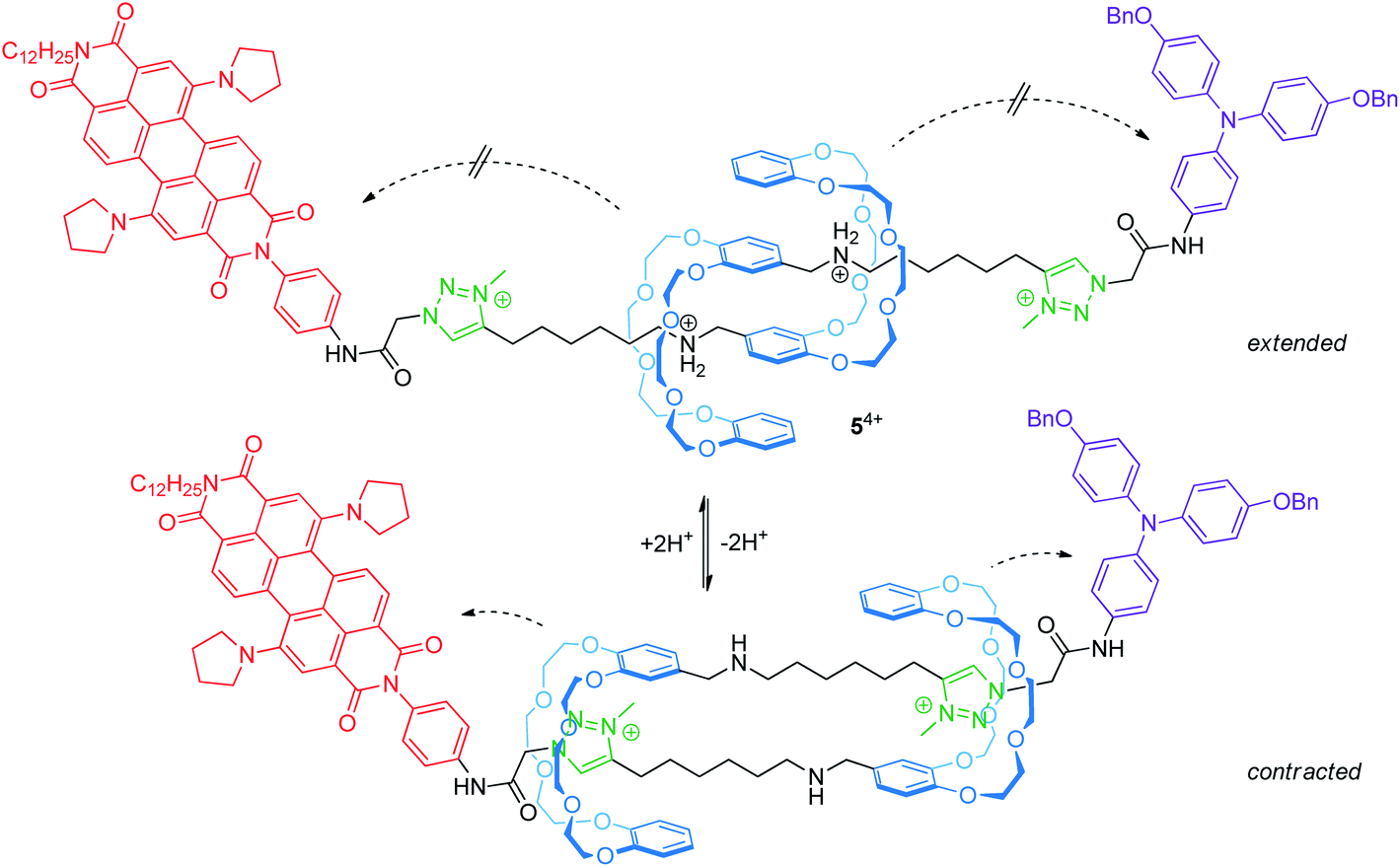 | ||
| Fig. 6 Switching of the acid/base-responsive [c2]daisy chain rotaxane 54+ between its extended and contracted form. Both forms show altered electrochemical properties. The dashed arrows illustrate electronic through-space interactions between the wheels and the redox-centers. | ||
Other types of redox-controlled motion
Electrochemical control of wheel–axle interactions opens up possibilities for other types of controlled motion. For example, electrochemically switchable pseudorotaxanes consisting of redox-active threads and cyclodextrins51 or cucurbiturils52–54 as macrocyclic compounds have been used to create stimuli responsive polymers and materials.Recently, Stoddart and co-workers built the pseudo[1]rotaxane 66+ based on their well investigated CBPQT4+ wheel covalently attached to a viologen thread (Fig. 7a).55 Complex 66+ efficiently switches between a rope-like and a lasso-type conformation upon reduction. CV and differential pulse voltammetry show that the first reduction wave (Epc = −0.33 V vs. Ag/AgCl) is a combination of a two- and a one-electron reduction leading to triradical species 63(˙+). An anodic shift (ΔEpc = +0.10 V relative to a suitable control compound), indicates the stabilization of the self-inclusion conformation to result from the strong, intramolecular radical-pairing interactions between the CBPQT2(˙+) wheel and the reduced thread. Supported by NMR, UV/Vis-NIR and crystallographic data, the authors suggest a “revolving-door mechanism” in which self-entanglement is initiated by rotation of the p-phenylene moiety in the wheel component.
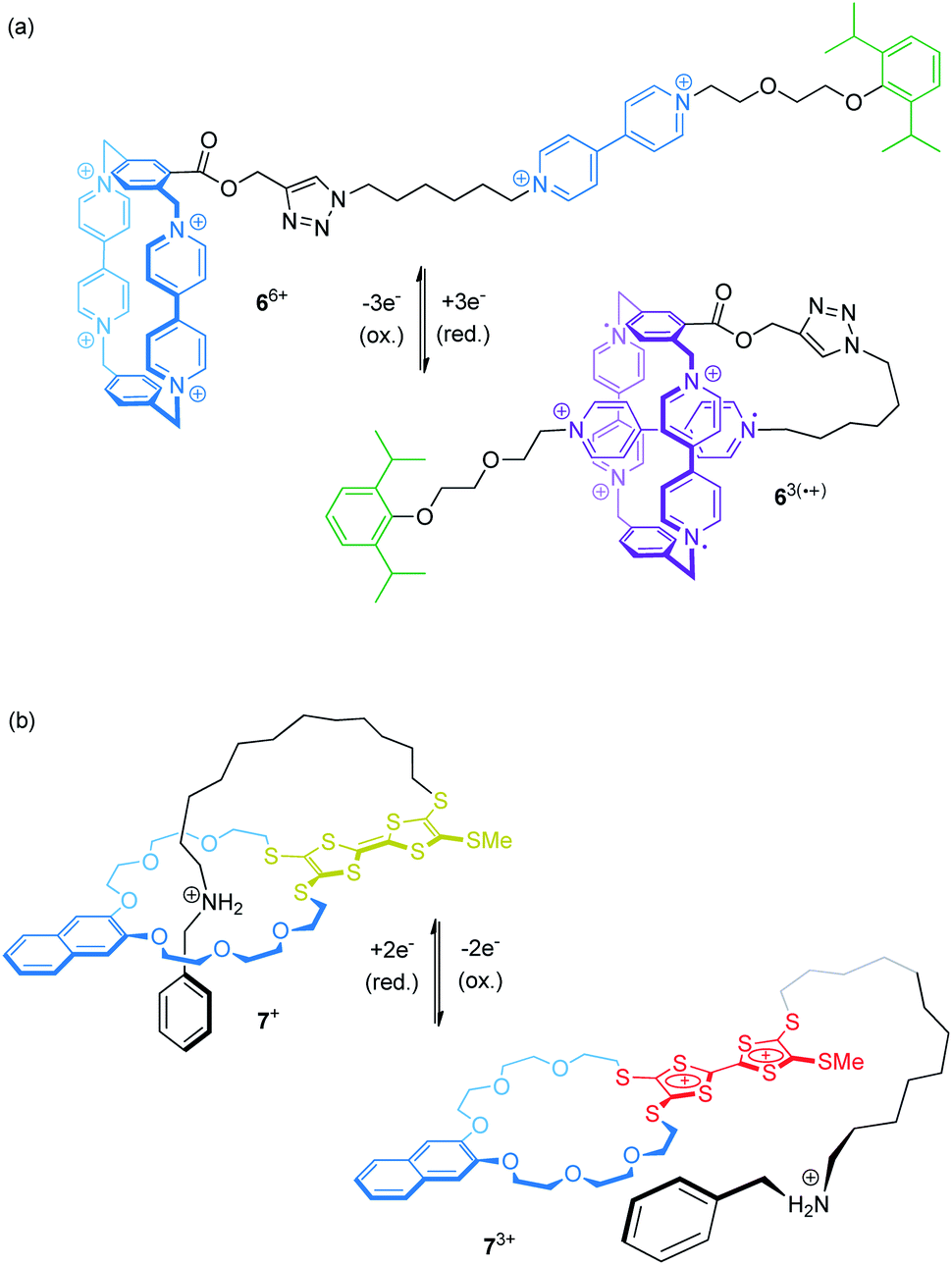 | ||
| Fig. 7 Two examples for electrochemically controlled pseudo[1]rotaxanes which are convertible between a rope-like and a lasso-type conformation: (a) 66+ undergoes self-inclusion after reduction, whereas (b) 7+ is preferentially threaded in the initial state and unthreads upon oxidation. | ||
We recently presented the redox-switchable pseudo[1]rotaxane 7+ (Fig. 7b) based on a crown ether/ammonium motif which adopts a self-inclusion conformation when an incorporated TTF unit is neutral.56 CV experiments and digital simulations demonstrate that stepwise oxidation of the TTF moiety generates charge repulsion in the assembly; thus, stepwise oxidation controls the conformational ratios between the threaded and non-threaded state.
Besides shuttling, controlling the rotation of wheels around an axle (pirouetting) is a worthwhile but challenging goal.33,57,58 Crown ether/ammonium [3]rotaxane 82+ which enables electrochemically controlled pirouetting (Fig. 8) was recently reported.59 The wheels are decorated with redox-switchable TTF units. A structurally similar control with only one TTF-wheel displays two reversible one-electron oxidations at E11/2 = 0.16 V and E21/2 = 0.40 V vs. Fc/Fc+ for the TTF/TTF˙+ and TTF˙+/TTF2+ transition, respectively. In contrast, rotaxane 82+ exhibits a splitting of the TTF/TTF˙+ wave (E11/2 = 0.08 V and E21/2 = 0.19 V) and a two-electron oxidation at E31/2 = 0.48 V. The splitting indicates an intramolecular mixed-valence complex (TTF2)˙+, whose stability is reflected in a comproportionation constant of Kc = 73. The second one-electron oxidation E21/2 yields a stable radical-cation dimer (TTF˙+)2, which was confirmed by electron paramagnetic resonance and UV/Vis-NIR spectroscopy. Supported by digital simulations and density functional theory, it could be shown that the wheels in the oxidation states 2TTF, (TTF2)˙+ and (TTF˙+)2 adopt a syn co-conformation, whereas an anti co-conformation is found for the 2TTF2+ state.
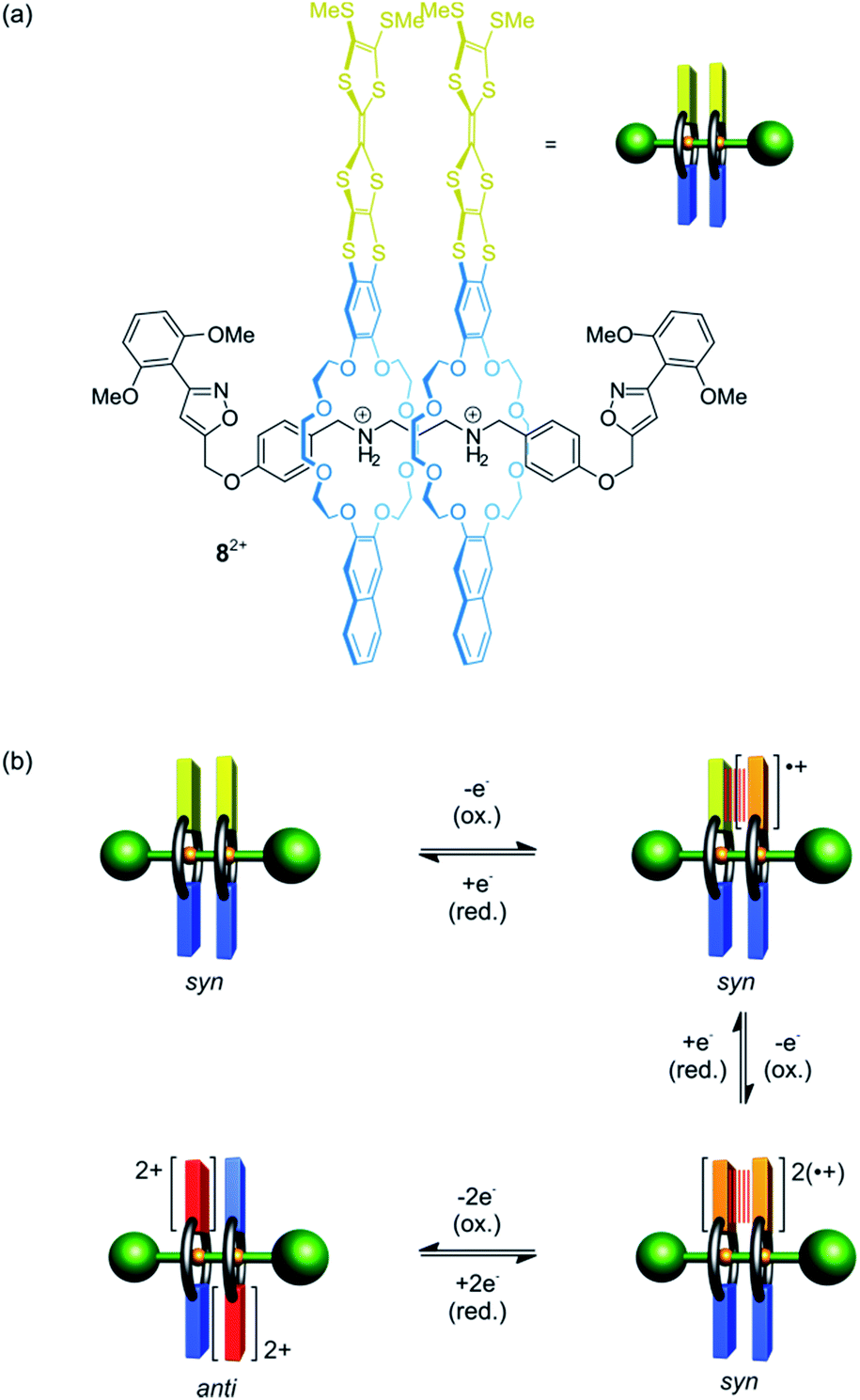 | ||
| Fig. 8 (a) Electrochemically switchable crown ether/ammonium [3]rotaxane 82+, which allows to control relative wheel pirouetting motions. (b) Graphical summary of the most-stable co-conformations of 82+ in its four stable redox-states. | ||
Kinetics of redox-controlled molecular shuttles
Great effort has been made in recent years to understand and control the kinetics of redox-induced translational motion in rotaxane shuttles.38Lately, the dependence of the shuttling kinetics of an electrochemically switchable CBPQT4+-rotaxane on the electrolyte concentration was investigated by Jeppesen and co-workers.60 For electrochemical experiments, supporting electrolytes are frequently required in high concentrations to increase solution conductivity. It was found that tetraalkylammonium salts (R4N+X−) in general lead to an increased switching rate, whereas the size of the R4N+ cation has no significant effect on wheel shuttling. However, a increased rate was observed when using ClO4− instead of PF6− as the electrolyte's anion.
The Stoddart group has recently investigated shuttling rates in a redox-switchable ring-in-ring rotaxane, where the axle itself incorporates a redox-active macrocycle.61 Electrochemically induced shuttling was found to be significantly slower compared to their previous systems.62 The slow motion results in an electrochemical hysteresis for switching as shown by CV and digital simulations. Furthermore, the slow shuttling could be used to characterize a diamagnetic tetraradical complex by 1H NMR spectroscopy, whose lifetime is otherwise too short.
Metal-containing rotaxanes
Since Sauvage's seminal work33,63 on Cu-containing catenanes, metal–ligand interactions have always played a key role in the synthesis of mechanically interlocked molecules, for example in a templating pre-organization of the precursor molecules.34 More recently, the active template approach2 evolved, in which metal ions serve as templates and as mediators for the formation of mechanical bonds. However, metals are also integral subcomponents of rotaxanes bringing with them versatile properties and functions. Metals can serve as persistent redox-centers, which change conformational or optoelectronic properties of a rotaxane with their oxidation state.Rotaxanes bearing covalently bound metal complexes
The group of Korybut-Daszkiewicz reported recent progress on their switchable rotaxanes with a cyclidene/crown ether64 motif. Fig. 9 shows [2]rotaxane 94+ which consists of an axle with a 14- and a 16-membered NiII-complexing tetraazamacrocycle and a threaded dibenzo-24-crown-8 wheel.65 In the initial state, the crown ether preferentially encircles the 14-membered ring since it is a stronger electron acceptor. However, the 16-membered NiII complex shows a less positive oxidation potential at 1.15 V vs. Ag/AgCl as shown by differential pulse voltammetry. One-electron oxidation (NiII/NiIII) converts the 16-membered ring to a better electron acceptor and the wheel translates to the NiIII binding site. Subsequent one-electron oxidation of the 14-membered ring complex at higher potentials induces a reversed shuttling motion of the wheel. Interestingly, a splitting of the signal for the NiII/NiIII oxidation is observed indicating that wheel shuttling takes place on the time scale of the experiment.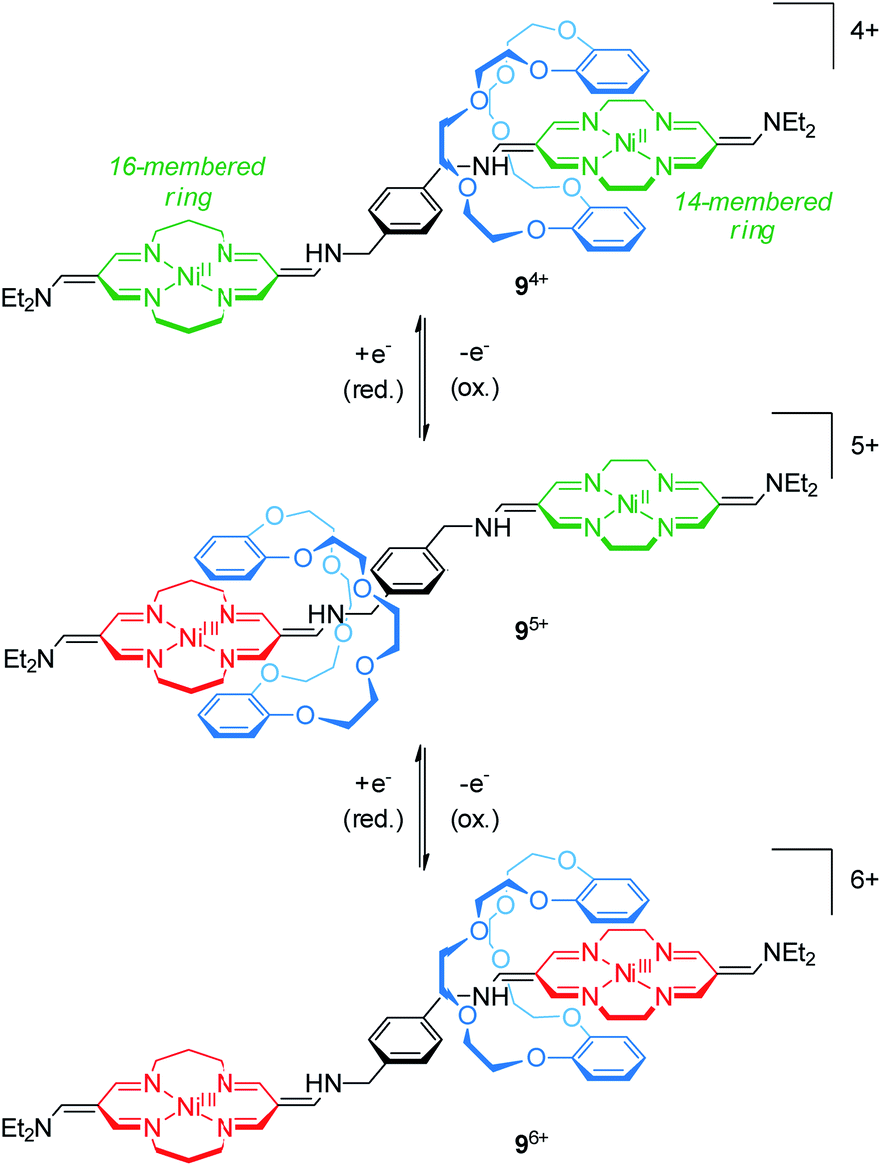 | ||
| Fig. 9 Redox-controlled molecular shuttle 94+ based on the cyclidene/crown ether motif in its three stable oxidation states (NiII/NiII, NiIII/NiII and NiIII/NiIII). | ||
The team of Ma synthesized a pH-controlled molecular shuttle based on a crown ether/ammonium [2]rotaxane.66 The crown ether macrocycle is decorated with a PtII porphyrin moiety, which allows following wheel shuttling by changes of room temperature phosphorescence. Additionally, CV indicates electronic interaction between the PtII porphyrin and a redox-active viologen unit, which is additionally incorporated in the axle.
Delavaux-Nicot, Nierengarten, Maisonhaute and others have reported the pillar[5]arene rotaxane 10 bearing ten peripheral Fc units attached to the wheel to investigate electron transfer processes (Fig. 10).67 One of the stoppers is substituted with two lipoic acid esters, which allow grafting the rotaxane onto gold microelectrodes. In solution, CV and Osteryoung squarewave voltammetry showed a single ten-electron wave (E1/2 = 0.59 vs. SCE, SCE = saturated calomel electrode) due to the Fc/Fc+ oxidations. A set of control experiments confirmed that no other redox-process occurs in this potential window. After grafting the rotaxane onto a gold microelectrode, a self-assembled monolayer of 10 was probed by ultrafast voltammetry revealing the rates of electron transfer kET in dependence of different scan rates and surface coverages. A low coverage of 19 nm2 per molecule leads to a kET of 5.8 × 105 s−1, whereas a denser coverage decreases kET (e.g. 3.1 nm2 per molecule, kET of 1.7 × 105 s−1). The authors suggest that the electron transfer between Fc units (hopping) and from Fc units to the electrode surface (heterogeneous electron transfer) is fast when the molecules are isolated. In the presence of neighboring molecules, however, intermolecular interactions restrict the motion towards the crowded electrode surfaces which decreases the kET values.
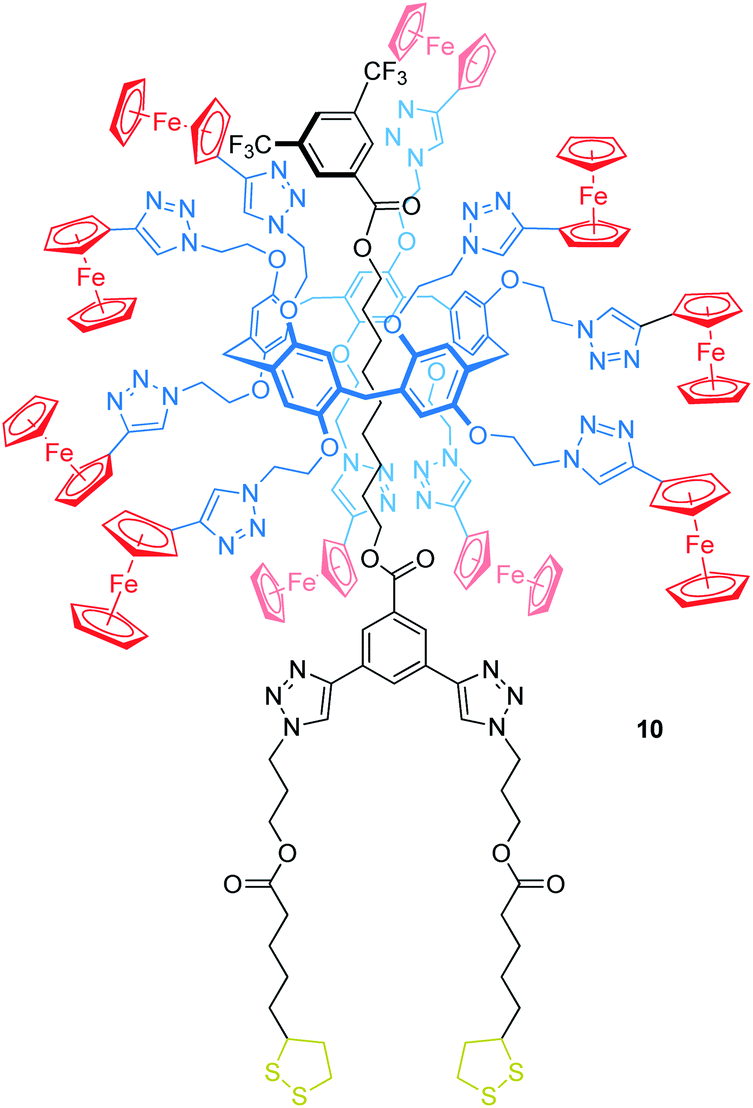 | ||
| Fig. 10 Structure of the ferrocene-decorated rotaxane 10, which has been grafted onto a gold microelectrode. | ||
Ulfkjær et al. described a metal-containing [2]rotaxane, which is stoppered with gold nanoparticles (Fig. 11).22 A diammonium thread with a central Fc unit and cucurbit[7]uril self-assemble into the very stable (Ka = 1013 M−1) pseudo[2]rotaxane 112+ in water. Basic hydrolysis gives 122+, whose thiol groups are able to coordinate onto gold nanoparticles to form a mechanically interlocked assembly. CV experiments were performed with the pseudo[2]rotaxanes 112+ and 122+ indicating typical anodic shifts of the Fc/Fc+ transition after encapsulation by cucurbit[7]uril (ΔE1/2 = +0.17 and +0.19 V, respectively). Scan-rate-depended measurements reveal a diffusion-controlled electrochemical process for 112+. Normally, a diffusion coefficient smaller than that of the free thread would be expected as an indication of the increased bulkiness of the assembly. However, dithiol 122+ displays an abnormal diffusion coefficient and interfacial electron transfer rate constant. The authors suggest that thiol groups bind more strongly to the surface of the used glassy carbon electrode than thiocarbamate groups; thus, pseudorotaxane 122+ displays interfacial rather than diffusion-controlled electron transfer kinetics.
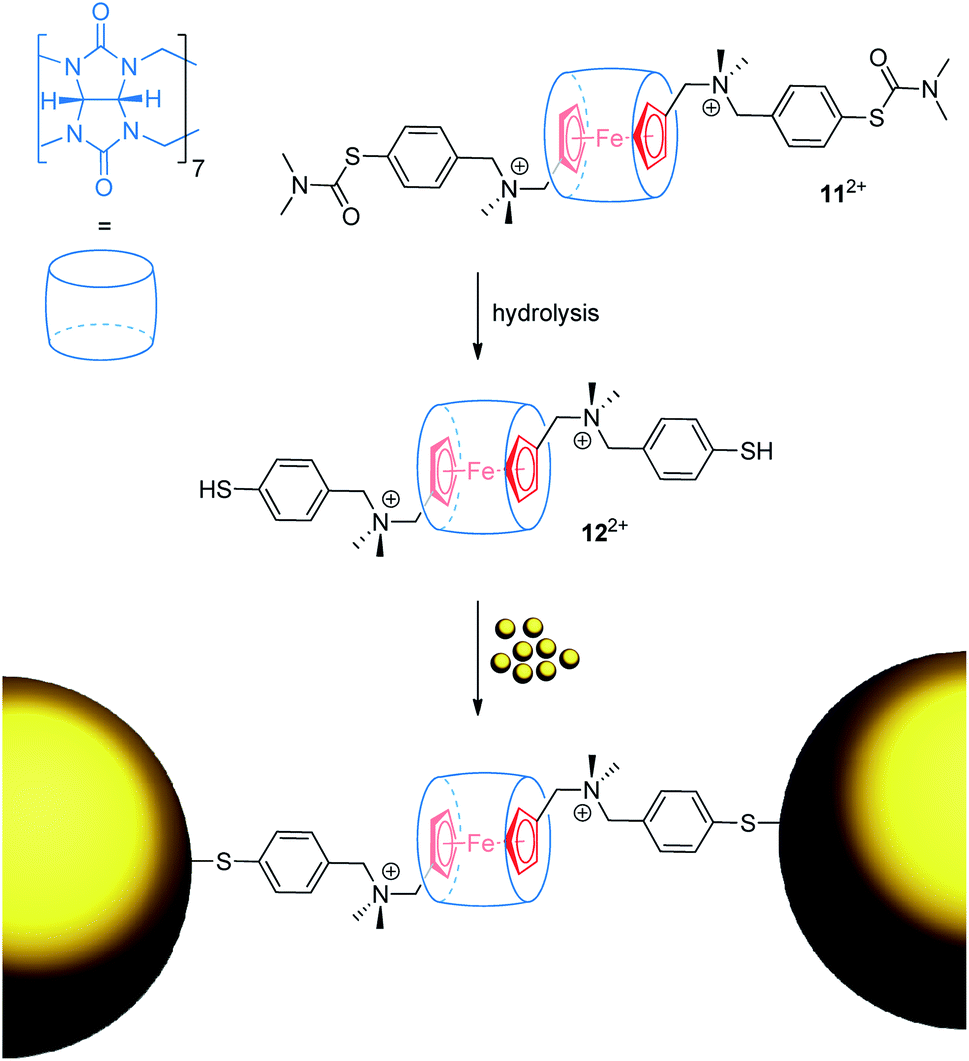 | ||
| Fig. 11 Reaction scheme for the formation of a gold nanoparticle-stoppered rotaxane from the pseudo[2]rotaxane precursor 122+. | ||
Osakada and co-workers described a series of electro-active, Fc-containing [1–4]rotaxanes that were prepared from a common crown ether/ammonium pseudo[2]rotaxane precursor by copper-catalyzed azide–alkyne cycloaddition.68 The triazole groups of the redox-switchable [3]rotaxane can be additionally used for metal complexation. The [3]rotaxane forms 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2 coordination complexes depending on the concentration of added PdII ions.
:1 or 1:2 coordination complexes depending on the concentration of added PdII ions.
An interesting approach of stabilizing sensitive metal complexes by rotaxanation was lately reported by the group of Terao.69 They used an α-cyclodextrin-based [1]rotaxane as insulating shell to stabilize a NiII-bis(dithiobenzoate) complex. CV demonstrates a reversible and quasi-reversible reduction at −1.26 and −1.83 V vs. Fc/Fc+. This represents the first example of reversible bis(dithiobenzoate) transition metal complex reduction. The same strategy was later used to stabilize a redox-active Pt(II) bis(dithiobenzoate) complex.70
Rotaxanes as ligands for metal-coordination
Rotaxanes can provide unique coordination sites for metal ions. Goldup, Roessler and co-workers have described a library of heteroleptic metal/rotaxane complexes (Fig. 12).71 An active template synthesis followed by addition of a transition metal salt yields the heteroleptic metal/rotaxane complexes 13–15. CV and EPR reveal a reversible, metal-centered oxidation (CuI/II) at +0.96 V and +0.56 V vs. SHE (standard hydrogen electrode) and a reversible ligand-centered reduction at −1.50 and −1.44 V for the copper species Cu(14) and Cu(15), respectively. The potential difference for the CuI/II oxidation (ΔE1/2 = 0.40 V) results from the fact that 14 provides an ideal binding site for CuI, whereas 15 provides a flexible enough coordination sphere to sufficiently stabilize CuI and CuII. For Cu(13), only an irreversible process is observed suggesting unstable CuI and CuII complexes. CV control experiments with a non-interlocked complex display two semi-reversible steps for the CuI/II transition caused by ligand disproportionation. Thus, the mechanical bond in 14 and 15 prevents a structural reorganization and allows redox-reactions without degradation of the complexes which would otherwise not be achievable.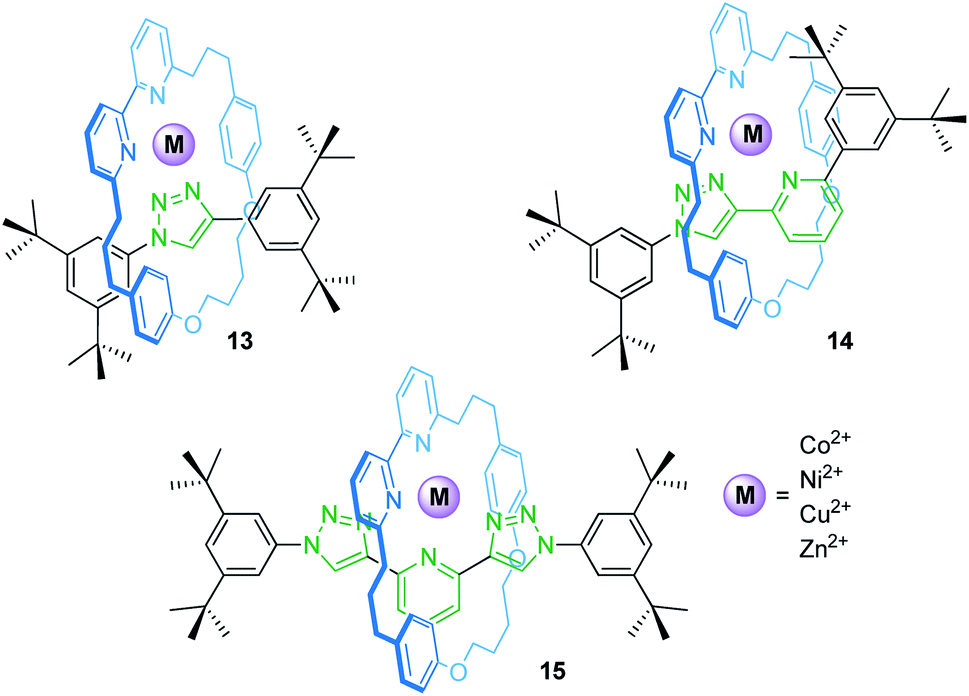 | ||
| Fig. 12 Heteroleptic transition metal/rotaxane complexes 13–15 by Goldup, Roessler and co-workers. | ||
The groups of Armaroli and Diederich synthesized a CuI bis-phenanthroline rotaxane by using tetracyano-p-quinodimethane as the stopper unit in a [2 + 2]cycloaddition-retroelectrocyclization.72 Electrochemical experiments revealed for the resulting rotaxane a reversible CuI/II oxidation and two reversible one-electron reductions for the dicyanoquinodimethane moiety, which was generated during the stoppering reaction.
An efficient operation of multi-ring rotaxanes, such as in the case of palindromic [3]rotaxanes, requires an ingenious design of a switchable supramolecular binding motif. The group of Flood developed a novel construction motif for a CuI-containing pseudo[3]rotaxane (Fig. 13).73 Their multi-component system consists of a phenanthroline-bearing macrocycle, CuI ions, a 5,5′-dimethyl-2,2′-bipyridine ligand, and a redox-switchable 3,6-bis(5′-methyl-2-pyridyl)-1,2,4,5-tetrazine ligand. In the initial state, CuI favors coordination by one macrocycle and one bipyridine ligand. Thus, the potential for single-electron reduction of the tetrazine ligand (Epc = −0.95 V vs. Ag/AgCl) is mainly unaffected. Reduction of the tetrazine to its radical anionic form, however, dramatically enhances its ligand strength and initiates ligand exchange with the bipyridines to form the pseudo[3]rotaxane 16+. As shown by an anodically shifted re-oxidation potential (Epa = −0.25 V), the pseudo[3]rotaxane state is energetically stabilized. Digital simulations and control experiments confirm a stepwise mechanism. The first step (exchange with the first bipyridine/CuI/macrocycle complex) is significantly faster than the second step, due to increased bulkiness of the intermediate pseudo[2]rotaxane. However, the second step displays a higher thermodynamic driving force (19 vs. 25 kJ mol−1) in accordance with a positive thermodynamic cooperativity. Flood recently reported a similar observation for non-covalently coupled “cyanostar” macrocycles in a metal-free pseudo[3]rotaxane.74
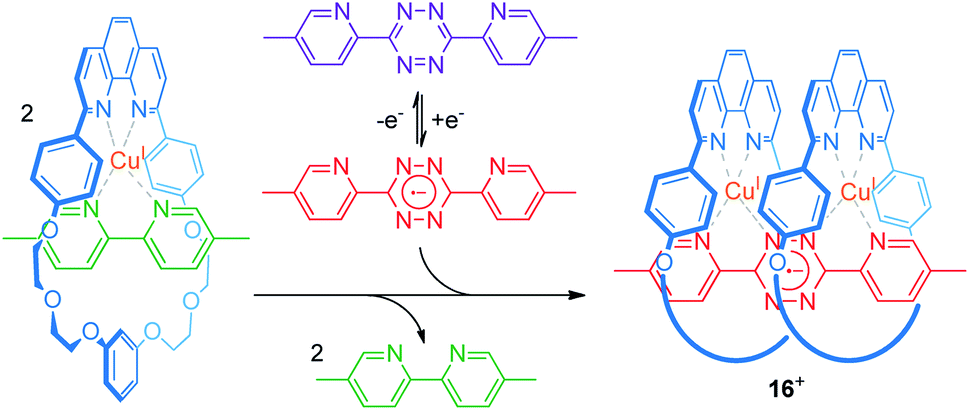 | ||
| Fig. 13 Electrochemical switching of a multi-component system to reversibly form the palindromic pseudo[3]rotaxane 16+. | ||
Applications of redox-active rotaxanes
Electrochemically switchable rotaxanes are obviously appealing for integration into electronic devices, for example by interfacing them with electrodes.75,76 Starting more than a decade ago, switchable rotaxanes on solid supports have been intensively studied in self-assembled monolayers.77–81 This research greatly expanded the fundamental understanding of molecular switching on solid support, but also disclosed that organic structures often suffer from low long-term persistence and structural integrity.82,83 However, in recent years also other applications arose from access to novel construction motifs for redox-active rotaxanes.Beer developed a halogen bonding [2]rotaxane84 which can serve as an electrochemical bromide sensor, even in water-containing media (Fig. 14). Rotaxane 17+ provides a halogen-bonding anion binding cavity and a ferrocene unit as an electrochemical probe. 1H NMR titration experiments in an organic solvent mixture with 10 vol% D2O give association constants for the anions Cl−, Br−, I− and SCN−, which the authors report to be 101, 340, 1125 and 102 M−1, respectively. The halide anions nicely follow the Hofmeister series; however, the SCN− anion, which should exceed I−, displays a rather poor binding indicating that geometry and size complementarity play a crucial role. The electrochemical response to Cl−, Br− and SCN− was further probed by square-wave voltammetry showing cathodic shifts for the Fc/Fc+ transition. Whereas addition of 20 equivalents of Cl− and SCN− display negligibly small shifts (ΔE1/2 < 5 mV), the addition of Br− generates more pronounced shifts (e.g. 20 equiv. result in ΔE1/2 = −10 mV). Control experiments in dry and “wet” solvents indicate that hydration strongly contributes to the observed effects. This is in line with the more pronounced voltammetric response for Br−, since Br− is expected to display lower hydration energy. In another recent report, Beer presented a Fc-decorated [3]rotaxane shuttle which utilizes anion recognition to induce wheel translation.85
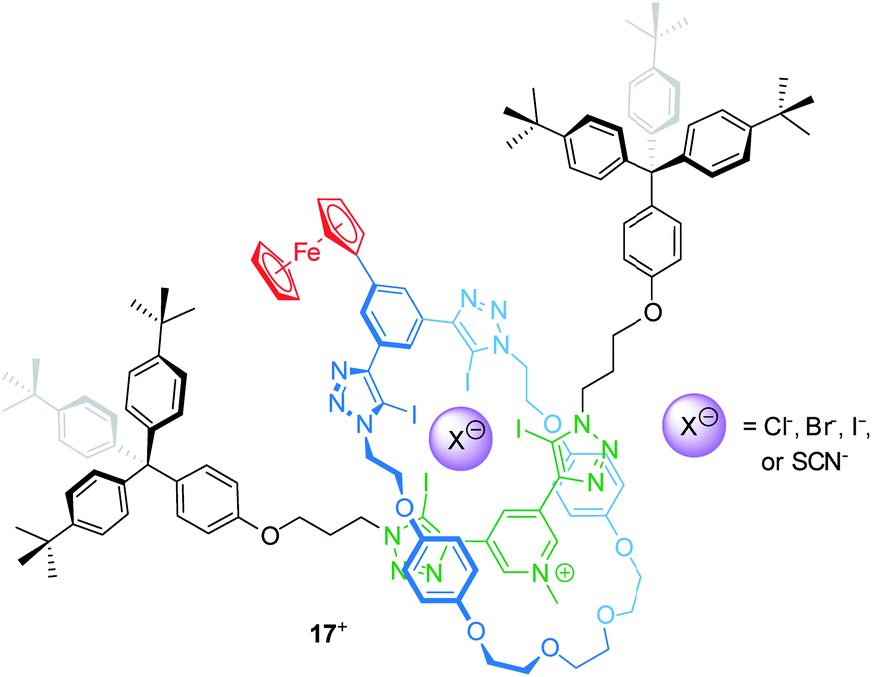 | ||
| Fig. 14 Structure of the bromide-sensing [2]rotaxane 17+, which provides a halogen bonding anion binding cavity. | ||
The Tanaka group has described porphyrin-phthalocyanine catalyst 184+ for O2 to H2O reduction,86 which is based on their previously reported fourfold threaded [2]rotaxane motif87 (Fig. 15a). The heterodimeric scaffold incorporates a μ-oxo-dinuclear iron complex (FeIII–O–FeIII). Experiments using a rotating ring disc electrode showed a fairly high positive onset potential of 0.78 V vs. RHE (reversible hydrogen electrode) indicating a high catalytic activity for oxygen reduction.
 | ||
| Fig. 15 (a) Structure of the fourfold threaded [2]rotaxane catalyst 184+, which incorporates a μ-oxo-dinuclear iron complex. (b) Suggested mechanism for the reduction of oxygen to water. | ||
Furthermore, the determined number of 3.8 transferred electrons (at 0.47 V) and the percentage of H2O generation (88%) confirm that a four-electron reduction of O2 to H2O is the dominant reaction. As shown by CV, the Epc value of a redox-wave at E1/2 = 0.60 V, corresponding to a one-electron reduction of the FeIII phthalocyanine complex, significantly increases in the presence of O2. The proposed mechanism, in which protonation and FeIII phthalocyanine reduction result in an initial water loss, is shown in Fig. 15b. Subsequently, the resulting mixed-valence species and O2 form a peroxo-FeIII complex (FeIII⋯O−–O–FeIII), which releases, after four-electron reduction and protonation, two H2O molecules. The authors attribute the high catalytic activity to the flexible rotaxane scaffold and its inherent structural adaptability. It provides a well suitable environment to accommodate the longer O2 molecule as well as the shorter bridging O2− ligand.
Conjugated molecular chains that conduct electric current—so-called molecular wires—are attracting considerable attention due to their potential use for nanoscale electronic devices. Fig. 16 shows rotaxane 19, consisting of an oligoyne thread with 6 conjugated triple bonds, which is assembled within a scanning tunneling microscope break junction.88 Both, the presence of the 3,5-diphenylpyridine endcapping groups and the encircled phenanthroline containing macrocycle, chemically stabilize the oligoyne by steric shielding and yield a kind of insulated molecular wire. Molecular conductance measurements showed a slight increase of conductance for the rotaxane 19 compared to the free thread suggesting a rather small effect of the wheel on the electronic structure of the thread. Rotaxanation also comes with a drawback: due to the possibility of inhibiting molecular bridging between wheel and gold contacts, the presence of the threaded wheel reduces the probability for successful molecular junction formation.
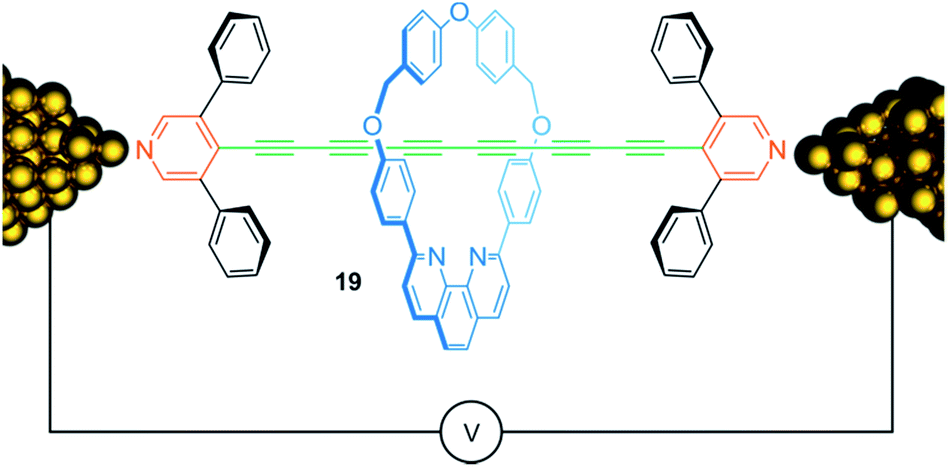 | ||
| Fig. 16 Schematic representation of oligoyne rotaxane 19 within in a single molecule break junction. | ||
A series of heteroleptic CuI pseudo[2]rotaxanes featuring bis[2-(diphenylphosphino)phenyl] ether and macrocyclic phenanthroline ligands have been recently investigated as materials for organic light emitting diodes (OLEDs).89 In a poly(methyl methacrylate) film (1 wt%), they exhibit excellent photophysical properties with photoluminescence quantum yields up to 44% (λexc = 380 nm), which makes them promising for optoelectronic devices. An OLED device based on one of the CuI pseudorotaxanes shows intense green electroluminescence at 10 V with a maximum intensity at ∼520 nm. It is noteworthy that the pseudorotaxane displays a higher electroluminescence and enhanced device stability compared to a reference compound with a non-macrocyclic phenanthroline ligand. The authors attribute the improved performance to the pseudorotaxane structure that prevents complex disassembly and, thus, chemical degradation.
Transport and controlled release of substrates are vital functions of natural molecular machines. Credi and co-workers have reported a synthetic molecular transporter based on the [2]rotaxane 205+ (Fig. 17).90 The crown ether wheel of the rotaxane carries a ruthenium complex, which acts as a molecular cargo. The position of the RuII complex on the thread can be manipulated by acid/base addition. Irradiation with visible light triggers a ligand exchange and the RuII complex dissociates from the rotaxane (unloading). Here, electrochemistry serves as a tool to monitor in situ operation of the transporter. In the protonated state, two one-electron reduction processes for the bipyridinium unit at −0.35 and −0.75 V vs. SCE and a metal-centered oxidation at +1.34 V are observed. Deprotonation leads to wheel shuttling (transport) towards the viologen station and cathodically shifts both reduction potentials of it, whereas the RuII/III oxidation remains unaffected. After irradiation, however, the signal at +1.34 V disappears indicating a successful photodissociation. Although the transport distance is rather small, this proof-of-principle nicely illustrates that artificial systems are capable of reversibly performing transport and cargo loading/unloading processes.
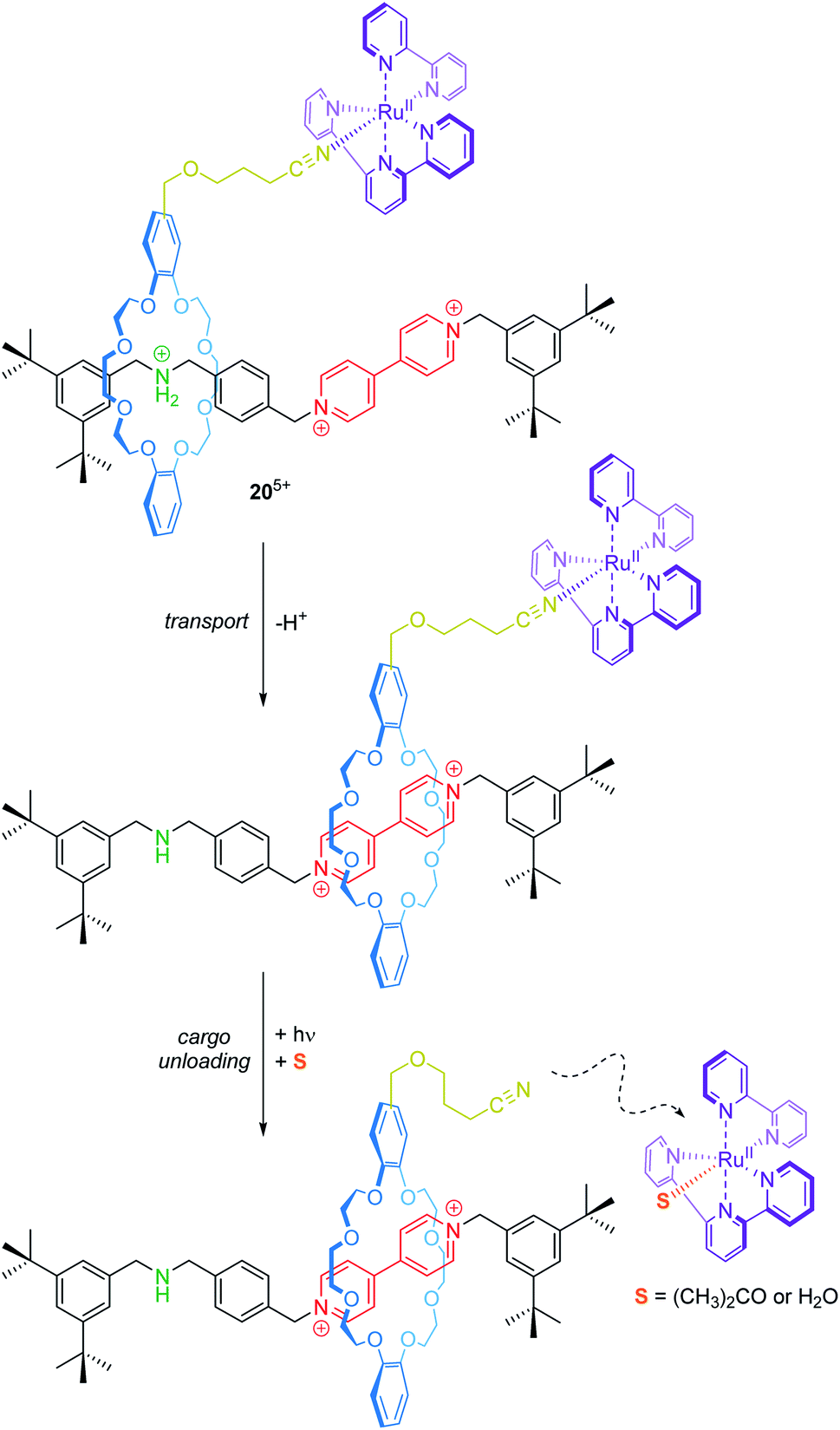 | ||
| Fig. 17 Operation sequence of rotaxane-based molecular transporter 205+ by orthogonal use of acid/base addition and light as external stimuli. The dashed arrow illustrates the release of the formerly bound RuII complex. | ||
Non-equilibrium systems
A fundamental difference between synthetic molecular switches and nature's molecular machines, such as motor proteins, is that the former are found in states of thermodynamic equilibrium whereas the latter are dissipative systems operating far from it.91 Switching as described so far brings the system out of equilibrium, when the stimulus is applied, but the system reacts into a new equilibrium situation, for example by wheel shuttling along the axle.The construction of true non-equilibrium systems, which generate directed molecular motion by consuming energy or fuel, is therefore the next logical step towards autonomously operating artificial molecular machinery.92 Recently, the Leigh group has reported on chemically fueled catenane motors93,94 which generate unidirectional rotary motion. Credi and others described a pseudorotaxane-based molecular pump,95 in which a ring is directionally transported under irradiation of light. Furthermore, the groups of Browne96 and Feringa97 have examined the control of unidirectional rotation in molecular motors by redox-chemistry.
In a series of recent papers, the Stoddart group developed variants of electrochemically driven molecular pumps which operate in non-equilibrium states.98–100 Through an elegantly designed sequence of steric “speed bumps” and redox-switchable binding sites implemented into a molecular axle, macrocycles can be transported along the thread. An energy ratchet mechanism driven by redox oscillations enables a directional transport. If the axle is stoppered at the output side, it is even possible to accumulate multiple wheels on the axle.
Fig. 18 shows the operation sequence of their latest system,100 which involves the symmetric axle 218+ consisting of two molecular pumping units at its ends and a central reservoir to accumulate CBPQT4+ wheels. In the ground state, the tetra-cationic wheel and the 3,5-dimethylpyridinium/viologen moiety repel each other (state I). Upon reduction, however, repulsion is reduced and a CBPQT2(˙+) ring from the bulk solution threads on the axle to form a tri-radical complex with the radical cationic viologen unit (state II). Re-oxidation of the ensemble destabilizes the complex and drives the wheel over the steric barrier given by the isopropylphenyl moiety. The wheel slips into the reservoir domain where it is kinetically trapped (state III). After one redox cycle, it is possible to repeat the process and collect another wheel on each half of the symmetric thread to yield a [5]rotaxane (state IV). Consecutive chemical reduction/oxidation is experimentally difficult due to formation of waste products.100 However, the ratchet mechanism can also be powered by bulk electrolysis cycles in which the working electrode is alternately held at a constant reductive or oxidative potential.
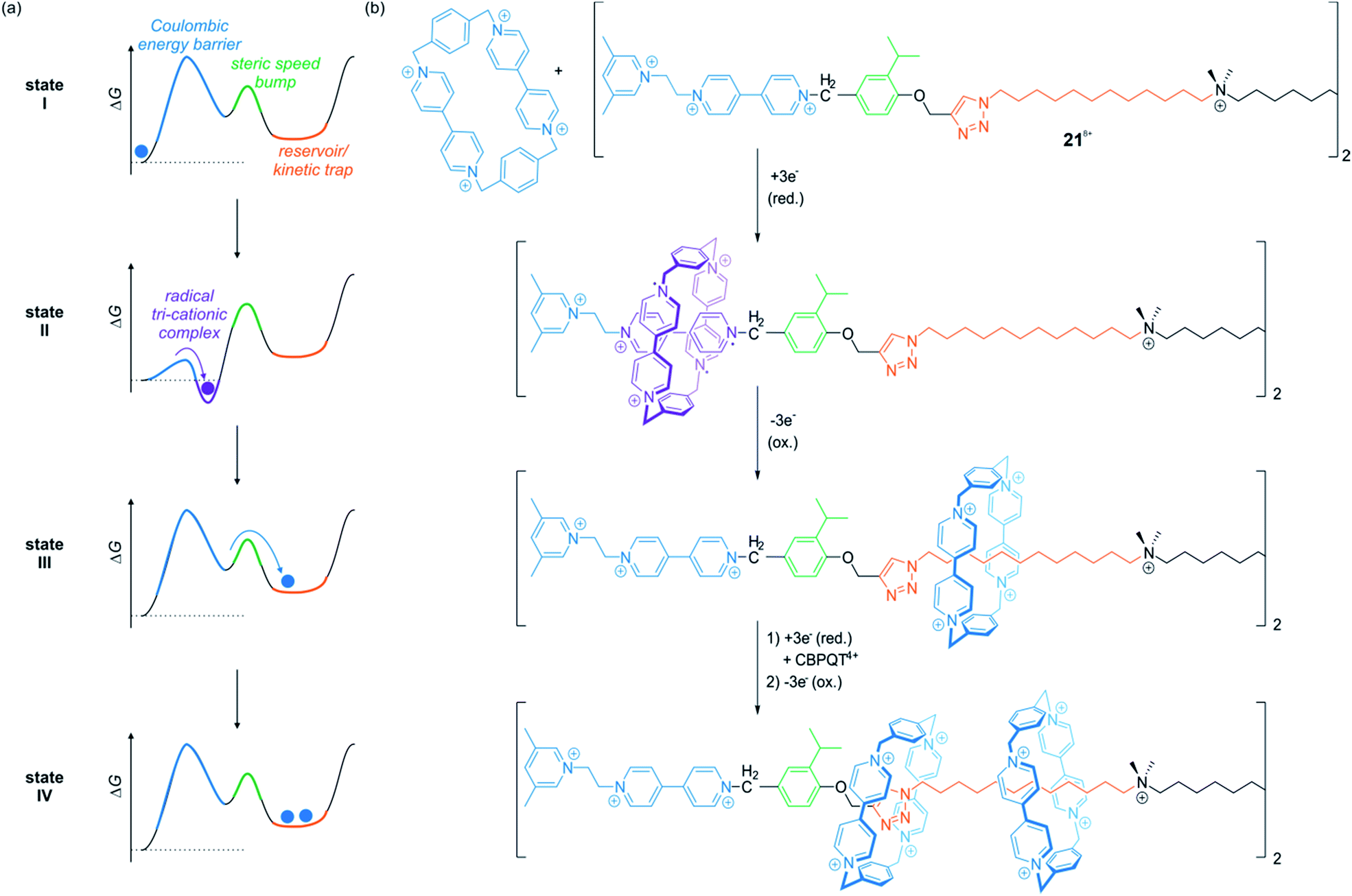 | ||
| Fig. 18 Operation sequence of Stoddart's electrochemically driven molecular pump: (a) simplified energy landscapes of translational wheel motion along axle 218+ in states I–IV. (b) Structure of the axle 218+, the CBPQT4+ wheel and corresponding (pseudo)[3,5]rotaxanes in states I–IV. | ||
Conclusions and perspectives
In their 1994 Nature publication27 on the first electrochemically switchable molecular shuttle, the authors concluded with an appeal to the community of supramolecular chemists: “(…) to improve the molecular properties of these and related systems (…)”. Based on tremendous progress in synthesizing mechanically interlocked molecules, the last 25 years have witnessed this improvement accompanied by an ongoing diversification of rotaxanes towards more complex and functional systems. As shown in this perspective, electrochemically switchable rotaxanes come in a large variety of structure and properties nowadays. The field continues to develop quickly.However, the ongoing growth of complexity of supramolecular architectures also comes with the necessity of improved strategies for subcomponent self-assembly. Advanced synthetic strategies like dynamic combinatorial chemistry or self-sorting will play a pivotal role in the construction of future systems. For example, it has been demonstrated that large, redox-active oligorotaxanes can be efficiently constructed by using dynamic covalent chemistry.101
Furthermore, few supramolecular construction motifs have proven to be particularly reliable for introducing electrochemical switching into rotaxanes, e.g. viologens102 and tetrathiafulvalenes,103 which provide stable and persistent redox-switchable building blocks. The improvement of these functional groups and the development of novel yet functionally similar redox-switches are of major significance for future growth of the field.
What to expect in the future of electrochemically switchable rotaxanes? Regarding electroanalytical methods, it is assumed that optoelectronic properties of switchable molecules will increasingly be probed through single-molecule experiments104 and combined spectroelectrochemical methods,105 which complement and expand classical bulk and interfacial techniques.
Due to the limited long-term stability of organic molecules in solution and on surfaces, a widespread application of rotaxanes, e.g. in hybrid electronic devices, comes with difficulties. However, recent developments in metal–organic rotaxanes or the stabilization of switchable rotaxanes in metal–organic frameworks83 open new pathways towards persistent systems. The use of rotaxanes as unique ligands for metal coordination also comes along with it. Regarding artificial molecular machines, electrochemical energy has already been shown to be an excellent choice to effectively fuel directional molecular motions; thus, we may expect that new generations of redox-driven molecular machines and motors will appear soon. Non-equilibrium self-assembly will also strongly contribute to the future of molecular machines and some upcoming examples of dissipative systems could perhaps surpass their role models from nature in some aspects.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (CRC 765) for financial support. We are also grateful to Chih-Wei Chu, Marius Gaedke and Claudia Kästner for helpful discussions.Notes and references
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, A. L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- C. A. Schalley, F. Vögtle and K. H. Dötz, Templates in Chemistry I, Springer, Heidelberg, Germany, 2004 Search PubMed.
- R. Jäger and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1997, 36, 930–944 CrossRef.
- M. M. Safont-Sempere, G. Fernandez and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- F. B. Cougnon and J. K. Sanders, Acc. Chem. Res., 2012, 45, 2211–2221 CrossRef CAS PubMed.
- R. S. Forgan, J. P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef CAS PubMed.
- O. Lukin, A. Godt and F. Vögtle, Chem.–Eur. J., 2004, 10, 1878–1883 CrossRef CAS PubMed.
- D. Sluysmans and J. F. Stoddart, Trends in Chemistry, 2019, 1, 185–197 CrossRef.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, Hoboken, New Jersey, USA, 2016 Search PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS PubMed.
- H. R. Tseng, S. A. Vignon and J. F. Stoddart, Angew. Chem., Int. Ed., 2003, 42, 1491–1495 CrossRef CAS PubMed.
- A. Credi, M. Semeraro, S. Silvi and M. Venturi, Antioxid. Redox Signaling, 2011, 14, 1119–1165 CrossRef CAS PubMed.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- M. Franz, J. A. Januszewski, D. Wendinger, C. Neiss, L. D. Movsisyan, F. Hampel, H. L. Anderson, A. Gorling and R. R. Tykwinski, Angew. Chem., Int. Ed., 2015, 54, 6645–6649 CrossRef CAS PubMed.
- J. Heinze, Angew. Chem., Int. Ed. Engl., 1984, 23, 831–847 CrossRef.
- A. Ulfkjær, F. W. Nielsen, H. Al-Kerdi, T. Rubeta, Z. K. Nielsen, J. Ulstrup, L. Sun, K. Moth-Poulsen, J. Zhang and M. Pittelkow, Nanoscale, 2018, 10, 9133–9140 RSC.
- H. V. Schröder, H. Hupatz, A. J. Achazi, S. Sobottka, B. Sarkar, B. Paulus and C. A. Schalley, Chem.–Eur. J., 2017, 23, 2960–2967 CrossRef PubMed.
- A. E. Kaifer and M. Gómez-Kaifer, Supramolecular Electrochemistry, Wiley, Weinheim, Germany, 1999 Search PubMed.
- C. A. Schalley, Analytical Methods in Supramolecular Chemistry, Wiley, Weinheim, Germany, 2012 Search PubMed.
- G. Ragazzon, A. Credi and B. Colasson, Chem.–Eur. J., 2017, 23, 2149–2156 CrossRef CAS PubMed.
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- J. P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- H. Tian and Q. C. Wang, Chem. Soc. Rev., 2006, 35, 361–374 RSC.
- S. F. van Dongen, S. Cantekin, J. A. Elemans, A. E. Rowan and R. J. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC.
- J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611–619 CrossRef CAS.
- J. E. M. Lewis, P. D. Beer, S. J. Loeb and S. M. Goldup, Chem. Soc. Rev., 2017, 46, 2577–2591 RSC.
- P. Ceroni, A. Credi and M. Venturi, Electrochemistry of Functional Supramolecular Systems, Wiley, Hoboken, New Jersey, USA, 2010 Search PubMed.
- C. A. Nijhuis, B. J. Ravoo, J. Huskens and D. N. Reinhoudt, Coord. Chem. Rev., 2007, 251, 1761–1780 CrossRef CAS.
- A. C. Fahrenbach, C. J. Bruns, D. Cao and J. F. Stoddart, Acc. Chem. Res., 2012, 45, 1581–1592 CrossRef CAS PubMed.
- A. C. Fahrenbach, C. J. Bruns, H. Li, A. Trabolsi, A. Coskun and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 482–493 CrossRef CAS PubMed.
- D. Britz and J. Strutwolf, Digital Simulation in Electrochemistry, Springer, Basel, Switzerland, 2016 Search PubMed.
- A. Altieri, F. G. Gatti, E. R. Kay, D. A. Leigh, D. Martel, F. Paolucci, A. M. Slawin and J. K. Wong, J. Am. Chem. Soc., 2003, 125, 8644–8654 CrossRef CAS PubMed.
- M. R. Panman, C. N. van Dijk, A. Huerta-Viga, H. J. Sanders, B. H. Bakker, D. A. Leigh, A. M. Brouwer, W. J. Buma and S. Woutersen, Nat. Commun., 2017, 8, 2206 CrossRef PubMed.
- D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Angew. Chem., Int. Ed., 2005, 117, 4633–4640 CrossRef.
- Y. Wang, T. Cheng, J. Sun, Z. Liu, M. Frasconi, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13827–13834 CrossRef CAS PubMed.
- M. D. Kilde, R. Kristensen, G. Olsen, J. O. Jeppesen and M. B. Nielsen, Eur. J. Org. Chem., 2019, 5532–5539 CrossRef CAS.
- H. V. Schröder, S. Sobottka, M. Nössler, H. Hupatz, M. Gaedke, B. Sarkar and C. A. Schalley, Chem. Sci., 2017, 8, 6300–6306 RSC.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed.
- Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H. R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard III, C. M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS PubMed.
- Y. Wang, M. Frasconi, W. G. Liu, J. Sun, Y. Wu, M. S. Nassar, Y. Y. Botros, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, ACS Cent. Sci., 2016, 2, 89–98 CrossRef CAS PubMed.
- H. V. Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 3496–3500 CrossRef PubMed.
- A. Wolf, J. J. Cid, E. Moulin, F. Niess, G. Y. Du, A. Goujon, E. Busseron, A. Ruff, S. Ludwigs and N. Giuseppone, Eur. J. Org. Chem., 2019, 3421–3432 CrossRef CAS.
- Y. Takashima, K. Otani, Y. Kobayashi, H. Aramoto, M. Nakahata, H. Yamaguchi and A. Harada, Macromolecules, 2018, 51, 6318–6326 CrossRef CAS.
- E. R. Janecek, J. R. McKee, C. S. Tan, A. Nykanen, M. Kettunen, J. Laine, O. Ikkala and O. A. Scherman, Angew. Chem., Int. Ed., 2015, 54, 5383–5388 CrossRef CAS PubMed.
- Q. Zhang, D. H. Qu, Q. C. Wang and H. Tian, Angew. Chem., Int. Ed., 2015, 54, 15789–15793 CrossRef CAS PubMed.
- Y. Ahn, Y. Jang, N. Selvapalam, G. Yun and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 3140–3144 CrossRef CAS PubMed.
- Y. Wang, J. Sun, Z. Liu, M. S. Nassar, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2017, 8, 2562–2568 RSC.
- H. V. Schröder, J. M. Wollschläger and C. A. Schalley, Chem. Commun., 2017, 53, 9218–9221 RSC.
- M. R. Panman, B. H. Bakker, D. den Uyl, E. R. Kay, D. A. Leigh, W. J. Buma, A. M. Brouwer, J. A. Geenevasen and S. Woutersen, Nat. Chem., 2013, 5, 929–934 CrossRef CAS PubMed.
- P. Farras, E. C. Escudero-Adan, C. Vinas and F. Teixidor, Inorg. Chem., 2014, 53, 8654–8661 CrossRef CAS PubMed.
- H. V. Schröder, A. Mekic, H. Hupatz, S. Sobottka, F. Witte, L. H. Urner, M. Gaedke, K. Pagel, B. Sarkar, B. Paulus and C. A. Schalley, Nanoscale, 2018, 10, 21425–21433 RSC.
- S. S. Andersen, A. W. Saad, R. Kristensen, T. S. Pedersen, L. J. O'Driscoll, A. H. Flood and J. O. Jeppesen, Org. Biomol. Chem., 2019, 17, 2432–2441 RSC.
- M. C. Lipke, Y. Wu, I. Roy, Y. Wang, M. R. Wasielewski and J. F. Stoddart, ACS Cent. Sci., 2018, 4, 362–371 CrossRef CAS PubMed.
- M. C. Lipke, T. Cheng, Y. Wu, H. Arslan, H. Xiao, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 3986–3998 CrossRef CAS PubMed.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- K. M. Tomczyk, M. Woźny, S. Domagała, A. Wiȩckowska, J. Pawłowska, K. Woźniak and B. Korybut-Daszkiewicz, New J. Chem., 2017, 41, 6004–6013 RSC.
- M. Woźny, K. M. Tomczyk, A. Więckowska, S. Sutuła, D. Trzybiński, K. Woźniak and B. Korybut-Daszkiewicz, Dalton Trans., 2019, 48, 6546–6557 RSC.
- X. Ma, J. Zhang, J. Cao, X. Yao, T. Cao, Y. Gong, C. Zhao and H. Tian, Chem. Sci., 2016, 7, 4582–4588 RSC.
- M. Steffenhagen, A. Latus, T. M. N. Trinh, I. Nierengarten, I. T. Lucas, S. Joiret, J. Landoulsi, B. Delavaux-Nicot, J. F. Nierengarten and E. Maisonhaute, Chem.–Eur. J., 2018, 24, 1701–1708 CrossRef CAS PubMed.
- G. Yu, Y. Suzaki and K. Osakada, RSC Adv., 2016, 6, 41369–41375 RSC.
- T. Hosomi, R. Harada, H. Masai, T. Fujihara, Y. Tsuji and J. Terao, Chem. Commun., 2018, 54, 2487–2490 RSC.
- R. Harada, T. Hosomi, H. Masai and J. Terao, Tetrahedron Lett., 2018, 59, 2930–2933 CrossRef CAS.
- M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879–889 CrossRef CAS PubMed.
- Y. Trolez, A. D. Finke, F. Silvestri, F. Monti, B. Ventura, C. Boudon, J. P. Gisselbrecht, W. B. Schweizer, J. P. Sauvage, N. Armaroli and F. Diederich, Chem.–Eur. J., 2018, 24, 10422–10433 CrossRef CAS PubMed.
- C. R. Benson, A. I. Share, M. G. Marzo and A. H. Flood, Inorg. Chem., 2016, 55, 3767–3776 CrossRef CAS PubMed.
- C. R. Benson, C. Maffeo, E. M. Fatila, Y. Liu, E. G. Sheetz, A. Aksimentiev, A. Singharoy and A. H. Flood, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9391–9396 CrossRef CAS PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- A. C. Fahrenbach, S. C. Warren, J. T. Incorvati, A. J. Avestro, J. C. Barnes, J. F. Stoddart and B. A. Grzybowski, Adv. Mater., 2013, 25, 331–348 CrossRef CAS PubMed.
- Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Nielsen, E. DeIonno, G. Ho, J. Perkins, H.-R. Tseng, T. Yamamoto, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2002, 3, 519–525 CrossRef CAS PubMed.
- J. R. Heath and M. A. Ratner, Phys. Today, 2003, 56, 43–49 CrossRef CAS.
- J. E. Green, J. W. Choi, A. Boukai, Y. Bunimovich, E. Johnston-Halperin, E. DeIonno, Y. Luo, B. A. Sheriff, K. Xu, Y. S. Shin, H. R. Tseng, J. F. Stoddart and J. R. Heath, Nature, 2007, 445, 414–417 CrossRef CAS PubMed.
- T. J. Huang, B. Brough, C.-M. Ho, Y. Liu, A. H. Flood, P. A. Bonvallet, H.-R. Tseng, J. F. Stoddart, M. Baller and S. Magonov, Appl. Phys. Lett., 2004, 85, 5391–5393 CrossRef CAS.
- E. Katz, O. Lioubashevsky and I. Willner, J. Am. Chem. Soc., 2004, 126, 15520–15532 CrossRef CAS PubMed.
- A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- P. R. McGonigal, P. Deria, I. Hod, P. Z. Moghadam, A. J. Avestro, N. E. Horwitz, I. C. Gibbs-Hall, A. K. Blackburn, D. Chen, Y. Y. Botros, M. R. Wasielewski, R. Q. Snurr, J. T. Hupp, O. K. Farha and J. F. Stoddart, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11161–11168 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Eur. J. Org. Chem., 2019, 2019, 3433–3441 CrossRef CAS.
- T. A. Barendt, I. Rasovic, M. A. Lebedeva, G. A. Farrow, A. Auty, D. Chekulaev, I. V. Sazanovich, J. A. Weinstein, K. Porfyrakis and P. D. Beer, J. Am. Chem. Soc., 2018, 140, 1924–1936 CrossRef CAS PubMed.
- N. Mihara, Y. Yamada, H. Takaya, Y. Kitagawa, S. Aoyama, K. Igawa, K. Tomooka and K. Tanaka, Chem.–Eur. J., 2017, 23, 7508–7514 CrossRef CAS PubMed.
- Y. Yamada, M. Okamoto, K. Furukawa, T. Kato and K. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 709–713 CrossRef CAS.
- D. C. Milan, M. Krempe, A. K. Ismael, L. D. Movsisyan, M. Franz, I. Grace, R. J. Brooke, W. Schwarzacher, S. J. Higgins, H. L. Anderson, C. J. Lambert, R. R. Tykwinski and R. J. Nichols, Nanoscale, 2017, 9, 355–361 RSC.
- M. Mohankumar, M. Holler, E. Meichsner, J. F. Nierengarten, F. Niess, J. P. Sauvage, B. Delavaux-Nicot, E. Leoni, F. Monti, J. M. Malicka, M. Cocchi, E. Bandini and N. Armaroli, J. Am. Chem. Soc., 2018, 140, 2336–2347 CrossRef CAS PubMed.
- C. Schäfer, G. Ragazzon, B. Colasson, M. La Rosa, S. Silvi and A. Credi, ChemistryOpen, 2016, 5, 120–124 CrossRef PubMed.
- I. Prigogine, Introduction to Thermodynamics of Irreversible Processes, Wiley, New York, USA, 3rd edn, 1968 Search PubMed.
- C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46, 5491–5507 RSC.
- M. R. Wilson, J. Sola, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235–240 CrossRef CAS PubMed.
- S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340–343 CrossRef CAS PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10, 70–75 CrossRef CAS PubMed.
- H. Logtenberg, J. Areephong, J. Bauer, A. Meetsma, B. L. Feringa and W. R. Browne, ChemPhysChem, 2016, 17, 1895–1901 CrossRef CAS PubMed.
- B. S. L. Collins, J. C. M. Kistemaker, E. Otten and B. L. Feringa, Nat. Chem., 2016, 8, 860–866 CrossRef CAS.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- C. Pezzato, M. T. Nguyen, C. Cheng, D. J. Kim, M. T. Otley and J. F. Stoddart, Tetrahedron, 2017, 73, 4849–4857 CrossRef CAS.
- C. Pezzato, M. T. Nguyen, D. J. Kim, O. Anamimoghadam, L. Mosca and J. F. Stoddart, Angew. Chem., Int. Ed., 2018, 57, 9325–9329 CrossRef CAS.
- A. J. Avestro, D. M. Gardner, N. A. Vermeulen, E. A. Wilson, S. T. Schneebeli, A. C. Whalley, M. E. Belowich, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 4442–4449 CrossRef CAS PubMed.
- Y. Wang, M. Frasconi and J. F. Stoddart, ACS Cent. Sci., 2017, 3, 927–935 CrossRef CAS PubMed.
- H. V. Schröder and C. A. Schalley, Beilstein J. Org. Chem., 2018, 14, 2163–2185 CrossRef PubMed.
- J. L. Zhang, J. Q. Zhong, J. D. Lin, W. P. Hu, K. Wu, G. Q. Xu, A. T. Wee and W. Chen, Chem. Soc. Rev., 2015, 44, 2998–3022 RSC.
- W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373–3382 RSC.
Footnote |
| † Present address: Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, USA. |
| This journal is © The Royal Society of Chemistry 2019 |