
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical ring expansion reactions: synthesis of tetrahydrofuran derivatives and mechanism studies†
Sripati
Jana
 a,
Zhen
Yang
a,
Chao
Pei
b,
Xinfang
Xu
*bc and
Rene M.
Koenigs
*a
a,
Zhen
Yang
a,
Chao
Pei
b,
Xinfang
Xu
*bc and
Rene M.
Koenigs
*a
aRWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
bKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Suzhou 215123, China
cGuangdong Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: xuxinfang@mail.sysu.edu.cn
First published on 6th September 2019
Abstract
The reaction mechanism of oxygen and sulfur ylide mediated rearrangements is even today a matter of debate. In this report, we describe ring expansion reactions of oxetane and thietane heterocycles that allow probing the underlying reaction mechanism under metal-free, photochemical conditions. This ring expansion proves highly efficient and allows the synthesis of tetrahydrofuran and thiolane heterocycles under mild and operationally simple reaction conditions. These studies reveal marked differences in the stereoselectivity of the ring expansion of oxygen or sulfur ylides, which were further investigated computationally. DFT calculations show that carbenes react under ylide formation and that the corresponding ring expansion reactions proceed via a diradical pathway. The different bond lengths in free oxygen or sulfur ylide intermediates cause the distinctive stereochemical outcome.
Introduction
Ylides are important reaction intermediates in sigmatropic rearrangement reactions and serve as highly versatile synthons for the construction of complex molecular scaffolds.1 Despite significant advances over the past decades,2,3 the basic understanding of the rearrangement process of oxygen or sulfur ylides is still limited and currently available synthesis methods underpin a substantial difference of the reaction mechanism of sulfur vs. oxygen ylides.3,4 Both are typically accessed from unsymmetrically substituted (thio)ethers in the presence of a metal catalyst. The influence of the ether substitution pattern and the catalyst environment on the stereochemical outcome of the rearrangement step can thus not be completely ruled out and both metal-bound or free ylide reaction mechanisms are currently under discussion.5 With the re-emergence of photochemical carbene transfer reactions in the past years,6–8 free ylides can now be efficiently accessed in a metal-free approach. However, the reaction types with these in situ generated free carbene intermediates are limited to X–H insertion, cyclopropanation, and few others.7,8 Substantially, the exploration and the development of stereoselective versions are challenging and highly desirable with this reactive species.We envisioned that the formation of ylides starting from 4-membered ring heterocycles should provide achiral free ylide intermediates, or ylide intermediates bearing a stereochemical information in the proximity of the ylidic bond and may thus serve as a tool to study differences in the reactivity of oxygen vs. sulfur ylides and to obtain evidence on the underlying reaction mechanism. Against this background, the rearrangement reaction of 4-membered ring heterocycles9 under photochemical, metal-free conditions is highly desirable as it provides an experimental probe to elucidate the reaction mechanism of rearrangement reactions. Combining these experimental findings with DFT calculations on this transformation would provide important insight into the reaction mechanism of photochemical Stevens rearrangements and allow the identification of differences between oxygen and sulfur ylides. Moreover, it opens up a pathway to selectively yield 5-membered, saturated heterocycles in an expeditious fashion without overreaction to larger ring sizes.9–11 This approach would streamline currently available multi-step protocols for tetrahydrofuran synthesis following a de novo 2-step photochemical synthetic strategy via Paternò Büchi and consecutive ring expansion reaction (Scheme 1).
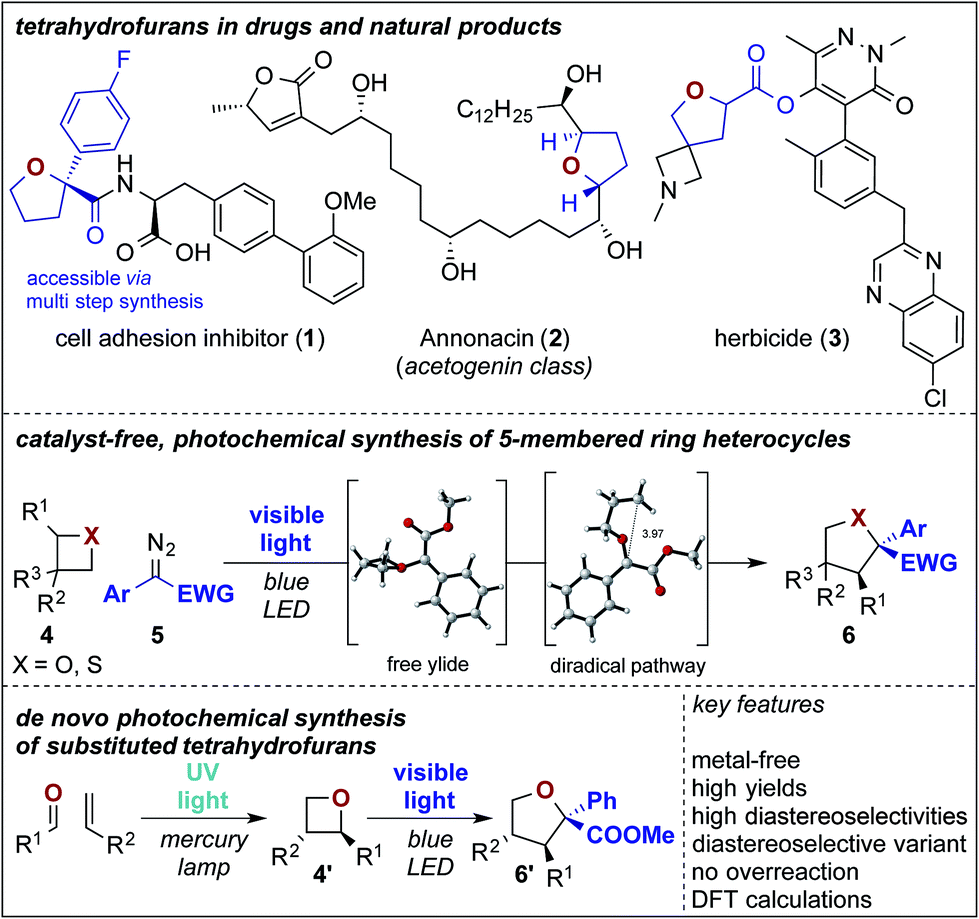 | ||
| Scheme 1 Photochemical ring expansion reactions. | ||
Results and discussion
We thus set out our investigations by studying the reaction of 3,3-dimethyl oxetane (4a) with methyl phenyldiazoacetate 5a under metal-free photochemical conditions and after a short optimization the tetrahydrofuran product 6a was obtained in excellent isolated yield; the best yield was obtained when using chloroform as solvent (Table 1, entry 2 and Table S1 in ESI†).12 Not even trace amounts of further ring-expanded products, such as pyran or oxepane were observed. Importantly, when carrying out the reaction in the absence of light, no reaction was observed (Table 1, entry 3). When studying the parent oxetane and thietane heterocycles, the respective ring expanded products were obtained in good yields. Importantly, for the parent unsubstituted heterocycles the crude reaction mixture was treated with KMnO4 for oxetane or Br2 for thietane to remove alkene by-products resulting from ring opening reactions of the heterocycle.12,13 The importance of light mediated carbene transfer reactions is highlighted in studies using Rh2(OAc)4 as catalyst, which provided the ring-expansion products 6a and 8a in decreased yield, while for 7a a similar yield was observed.|
|
||||
|---|---|---|---|---|
| Entrya | Reaction conditions | Solvent | X, R | Yieldb (%) |
| a Reaction conditions: 4a–c (0.4 mmol, 2.0 eq.) was dissolved in 1.0 mL of solvent and 5a (0.2 mmol, 1.0 eq.) was added by syringe pump over a period of 2 h and then stirred for another hour while irradiating with blue LEDs (3 W, 470 nm). b Yields refer to isolated products. c Reaction in the dark. d Reaction with 1.0 eq. 4a and 2.0 eq. 5a. e The reaction mixture was treated with aq. KMnO4 after completion of the reaction. f The reaction mixture was treated with Br2 after completion of the reaction. | ||||
| 1 | 470 nm | DCM | O, Me (4a) | 71 (6a) |
| 2 | 470 nm | CHCl3 | O, Me (4a) | 93 (6a) |
| 3c | Dark reaction | CHCl3 | O, Me (4a) | No reaction |
| 4d | 470 nm | CHCl3 | O, Me (4a) | 68 (6a) |
| 5e | 470 nm | CHCl3 | O, H (4b) | 70 (7a) |
| 6f | 470 nm | CHCl3 | S, H (4c) | 73 (8a) |
| 7 | Rh2(OAc)4 (1 mol%) | CHCl3 | O, Me (4a) | 43 (6a) |
| 8e | Rh2(OAc)4 (1 mol%) | CHCl3 | O, H (4b) | 61 (7a) |
| 9f | Rh2(OAc)4 (1 mol%) | CHCl3 | S, H (4c) | 55 (8a) |
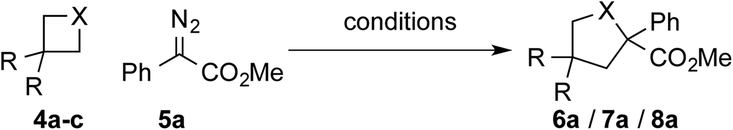
In further studies, we examined the general applicability of this transformation and studied different 4-membered ring heterocycles as well as different diazoesters. In general, the substitution pattern of the aromatic ring of the diazoester had only little influence on the ring expansion reaction of 3,3-dimethyloxetane, oxetane and thietane and in all cases the desired products (Scheme 2, 6–18) were isolated in high yields. Investigations on different acceptor groups of the diazoalkane reaction partner revealed that the nitrile group was well tolerated (6e), while the corresponding trifluoromethyl substituted diazoalkane only underwent a decomposition reaction.
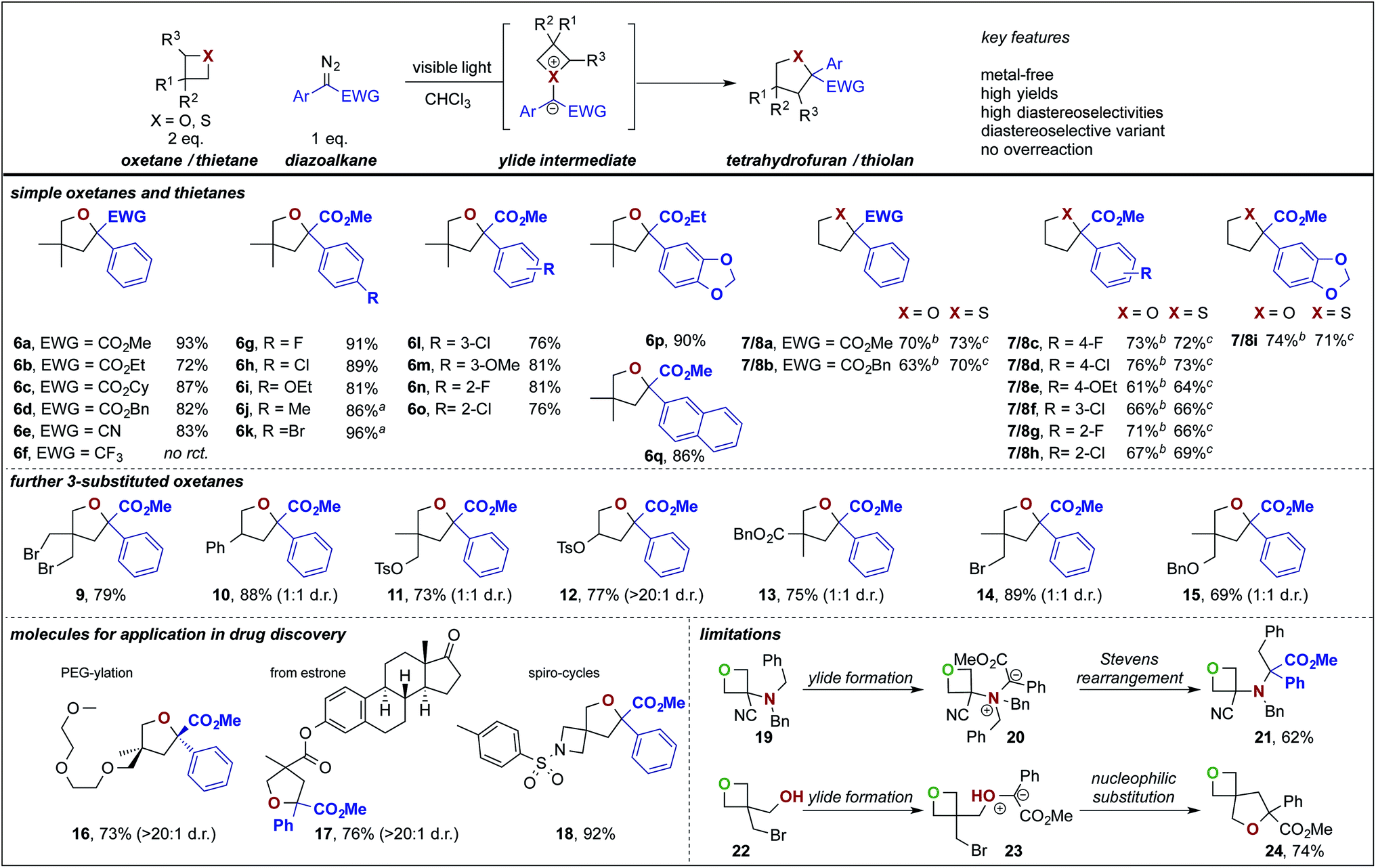 | ||
| Scheme 2 Substrate scope, applications for drug discovery and limitations of the photochemical ring expansion reaction. aEthyl ester instead of methyl ester. Reaction conditions: oxetane/thietane (0.4 mmol, 2.0 eq.) was dissolved in 1.0 mL of solvent and diazoalkane (0.2 mmol, 1.0 eq.) was added by syringe pump over a period of 2 h and then stirred for another hour while irradiating with blue LEDs (3 W, 470 nm). Yields refer to isolated products. bTreatment with aq. KMnO4 after completion of the reaction. cTreatment with Br2 after completion of the reaction. | ||
Notably, unsymmetrically substituted oxetanes gave the ring expansion product in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of both diastereoisomers (10–15). This 1:1 mixture can be explained by an unselective ylide formation due to missing side-differentiation by the remote substituents leading to two diastereomeric ylides and thus the reaction product is generated as a 1:1 mixture of both diastereoisomers. To showcase the potential in functionalization of drugs or drug-like molecules, we investigated more complex oxetane heterocycles. The estrone derived furan 17 was obtained in good yield as a single diastereoisomer. Despite multiple ether functional groups, the PEG-ylated furan 16 was obtained in a diastereoselective ring expansion in good yield. This PEG-ylation is a common strategy to prevent drugs from entering the central nervous system and thus prevent centrally mediated side effects.14 We also investigated the ring expansion of a spirocyclic oxetane that allows a high-yielding, unprecedented, access to rare 6-oxa-2-azaspiro[3.4]octane building blocks (18) that find applications as herbicides15a or S1P modulating agents.15b Limitations of the present methodology lie within amines (19) and free alcohols (22) that smoothly react under ylide formation and subsequent Stevens rearrangement (21) or cyclization (24).
:1 mixture of both diastereoisomers (10–15). This 1:1 mixture can be explained by an unselective ylide formation due to missing side-differentiation by the remote substituents leading to two diastereomeric ylides and thus the reaction product is generated as a 1:1 mixture of both diastereoisomers. To showcase the potential in functionalization of drugs or drug-like molecules, we investigated more complex oxetane heterocycles. The estrone derived furan 17 was obtained in good yield as a single diastereoisomer. Despite multiple ether functional groups, the PEG-ylated furan 16 was obtained in a diastereoselective ring expansion in good yield. This PEG-ylation is a common strategy to prevent drugs from entering the central nervous system and thus prevent centrally mediated side effects.14 We also investigated the ring expansion of a spirocyclic oxetane that allows a high-yielding, unprecedented, access to rare 6-oxa-2-azaspiro[3.4]octane building blocks (18) that find applications as herbicides15a or S1P modulating agents.15b Limitations of the present methodology lie within amines (19) and free alcohols (22) that smoothly react under ylide formation and subsequent Stevens rearrangement (21) or cyclization (24).
Next, we studied the photochemical ring expansion reaction of 2-phenyl oxetane with aryl diazoesters. This reaction proceeded in good to very good yields and gave exclusive formation of the cis-2,2,3-trisubstituted furan heterocycle as the major product (d.r. > 20:1, Scheme 3a, 25a–i). The high diastereoselectivity of this rearrangement can be rationalized by diastereoselective ylide formation followed by a stereospecific rearrangement reaction that leads to the cis-substituted 5-membered ring heterocycle. In this context, we also studied different 2-phenyl substituted oxygen containing heterocycles, yet no ring expansion was observed when using an epoxide, tetrahydrofuran or pyran heterocycle.
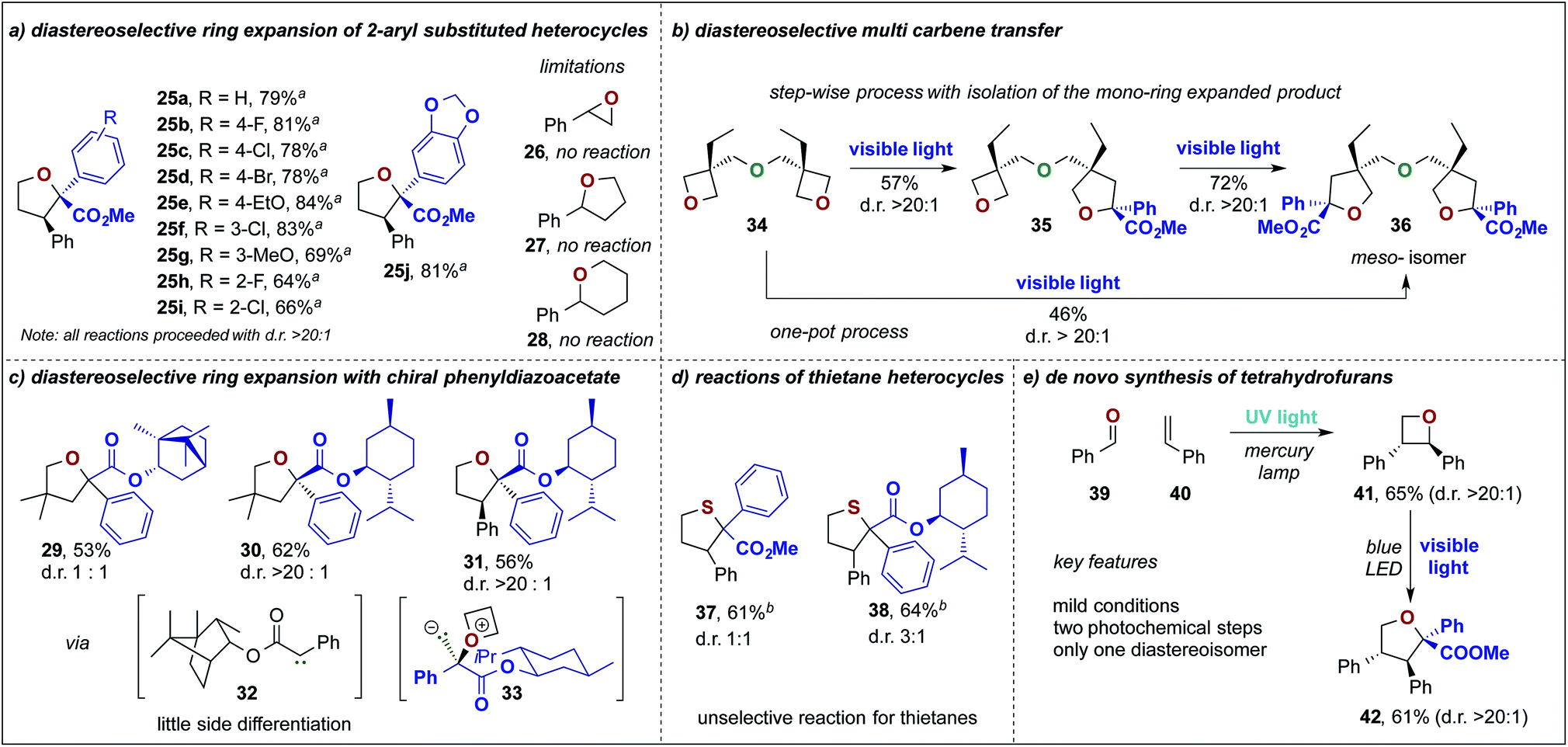 | ||
| Scheme 3 Diastereoselective ring expansion of 2-phenyl oxetane and 2-phenyl thietane and investigations on other oxygen-containing heterocycles: (a) substrate scope of aryldiazoacetates; (b) multi-carbene transfer reactions for the synthesis of bis-tetrahydrofurans; (c) ring expansion reactions with chiral diazoesters; (d) reactions of thietanes; (e) de novo photochemical synthesis of tetrahydrofurans. aTreatment with aq. KMnO4 after completion of the reaction. bTreatment with Br2 after completion of the reaction. | ||
Encouraged by the above observation, we set out to study the reaction of chiral phenyldiazoacetates to render this process diastereoselective and to enable an auxiliary-mediated stereoselective formation of the 5-membered heterocycles. In this context, we investigated chiral phenyldiazoacetates, based on (−)-borneol and (−)-menthol in the reaction with 3,3-dimethyloxetane. While only little selectivity was observed using the borneol derived phenyldiazoester (29), almost exclusive formation of furan 30 (d.r. > 20:1) was observed when using the (−)-menthol derived diazoester. This outcome can be rationalized by a highly diastereoselective ylide formation (33), which is a result of steric shielding of one face of the carbene by the iso-propyl group of the chiral auxiliary. Similarly, 2-phenyl oxetane underwent a highly diastereoselective ring expansion reaction with (−)-menthyl phenyldiazoacetate to yield 31 essentially as a single isomer via32 (Scheme 3c). In contrast to oxetane heterocycles, thietanes did not undergo diastereoselective ring expansion reactions. 2-Phenyl thietane underwent a chemoselective ring expansion reaction to yield the 2,2,3-trisubstituted thiolane, yet only little diastereoselectivity was observed (37, d.r. 1:1). Notably, in the case of 2-phenyl oxetane and 2-phenyl thietane oxidative workup procedures had to be performed to remove ring-opened by-products.
Similarly, only a minor chiral induction was detected when using (−)-menthyl phenyldiazoacetate (Scheme 3d, 38). Intrigued by the high efficiency of this ring expansion reaction, we looked into reactions involving multiple carbene transfer events, which find only rare examples in the literature.16 For this purpose, we studied the reaction of methyl phenyldiazoacetate 5a with bis-oxetane 34 in a consecutive two-step protocol and a one-pot protocol. Indeed, we could obtain the product of a single carbene transfer (35, 57%) when subjecting 5a with 2 eq. of 34 without formation of the double ring expanded product. In a second, consecutive ring expansion the bis-tetrahydrofuran 36 was obtained in good yield (72%). When subjecting 34 with an excess of diazoalkane (4 eq.) we could obtain exclusively the double ring expanded product 36 in 46% yield; in both cases only the meso-isomer was obtained in a highly stereoselective manner (Scheme 3b).
The synthesis of oxetane heterocycles might pose a challenge, for broader applications of this ring expansion reaction, due to its lengthy and tedious synthesis.17 To overcome this challenge, we studied a two-step approach towards furan heterocycles via UV-light mediated Paternò–Büchi reaction of benzaldehyde and styrene to yield in a first photochemical reaction the oxetane heterocycle 41 in moderate yield. When subjecting 41 to the ring expansion reaction, we obtained the tetra-substituted tetrahydrofuran 42 as a single diastereoisomer and could thus showcase a convenient de novo synthesis of substituted tetrahydrofuran heterocycles from cheap commodity chemicals (Scheme 3e).
For a better understanding of the underlying reaction mechanism, we explored different mechanistic pathways of this ring expansion reaction by DFT calculations at the SMD(chloroform)/(U)B3LYP/6-311+G(d,p)//SMD(chloroform)/(U)B3LYP/6-31G(d) level. With regards to the blue-light-induced carbene transfer reactions, a free singlet carbene should be the core intermediate that participates in downstream transformations. Besides, for simplification of the calculations, we began our calculations with the free singlet carbene 43 and oxetane as the model substrates.8a,18 As shown in Scheme 4, the initial step involves the facile formation of an ylide intermediate INT1via transition states TS1 with an activation free energy of 15.5 kcal mol−1. Then we have located three distinct pathways to account for the formation of the product. Firstly, we have considered a concerted [1,2]-sigmatropic rearrangement, which seems as an unfavorable pathway with a high energy barrier of 34.0 kcal mol−1via transition state TScon2 (Scheme 4a, red). Secondly, we considered a heterolytic cleavage of the C–O bond, which would result in a stepwise, ionic process to give the final product. Yet, all attempts in locating a suitable transition state led to other unfavorable transition states, which do not account for product formation (for details, see Fig. S4†).12
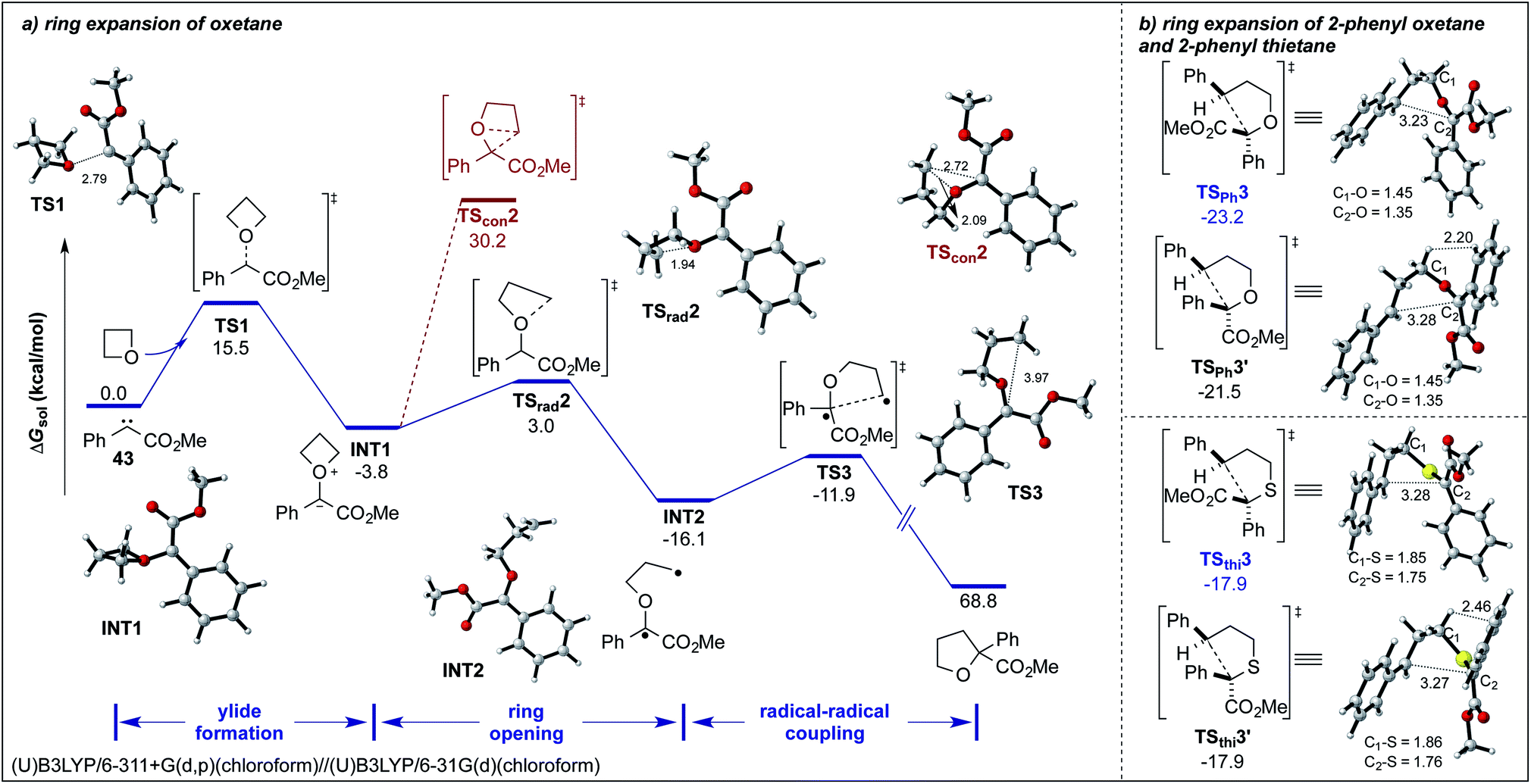 | ||
| Scheme 4 (a) Potential energy surface and 3D structures of key species of this ring expansion reaction. The bond lengths in the structures at the bottom are given in Å. (b) DFT calculated 3D structures of intramolecular coupling transition states of this ring expansion reaction. The bond lengths in the structures at the bottom are given in Å. | ||
Finally, we have located the transition state TSrad2 of the homolytic dissociation of the C–O bond, which requires an activation free energy of only 6.8 kcal mol−1. The generated intermediate INT2 is a diradical species, which undergoes an intramolecular radical–radical coupling and leads to the exergonic formation of the final tetrahydrofuran derivative via an early transition state TS3 with an activation energy of 4.2 kcal mol−1 (Scheme 4a, black).5
Furthermore, we have considered the selectivity of the Stevens rearrangement of methyl phenyldiazoacetate 5a with 2-phenyloxetane and 2-phenylthietane.12 The detailed potential energy surfaces are given in the ESI.† For the final radical–radical coupling step, we were able to locate two facile transition states TSPh3 and TSPh3′ in the case of the reaction of 5a with 2-phenyloxetane (Scheme 4). The calculated results suggest that TSPh3 (the two phenyl groups in trans position) is favored by 1.7 kcal mol−1 compared to TSPh3′ (the two phenyl groups in cis position), and implies that the predicted diastereomeric ratio of this process is about 18:1 at room temperature, which is consistent with the experimental findings (20:1). However, in the reaction of 5a with 2-phenyl thietane, transition states TSthi3 and TSthi3′ are very close in energy, which indicates cis-37 and trans-37 should have similar reaction yields, which is in agreement with the experimental results (d.r. = 1:1). This selectivity difference results from the transition state TSPh3′ that suffers steric repulsion between internal hydrogen atoms (the corresponding H⋯H distance is 2.20 Å, within the sum of van der Waals radii).19 In contrast, the transition state TSthi3′ reduces the steric repulsion due to the longer length of C–S bonds (1.86 Å and 1.76 Å) compared to C–O bonds (1.45 Å and 1.35 Å) in TSPh3′.
Conclusions
In summary, we herein report on a simple photochemical carbene transfer protocol that allows the diastereoselective synthesis of valuable tetrahydrofuran and thiolane heterocycles via ylide mediated ring expansion reactions of oxetane heterocycles, including multi-carbene transfer reactions for the synthesis of bis-oxetanes (61 examples, up to 96% yield, up to >20:1 d.r.). We have demonstrated a two-step, photochemical approach via Paternò–Büchi reaction and subsequent ring expansion that now enables a simple, photochemical de novo access of tetrahydrofuran heterocycles. Moreover, mechanistic studies based on DFT calculations revealed that the reaction mechanism of both oxonium and sulfur ylide mediated ring expansion reactions proceed via a diradical pathway. Based on this strategy more applications towards the synthesis of 5-membered rings, multi-carbene transfer reactions and telescoped synthetic methodology are expected.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project no. 408033718. Funded by the NSF of China (Grant No. 21971262).References
- (a) A. C. Jones, J. A. May, R. Sarpong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 2556 CrossRef CAS PubMed; (b) Y. Zhang and J. Wang, Coord. Chem. Rev., 2010, 254, 941 CrossRef CAS; (c) J. D. Neuhaus, R. Oost, J. Merad and N. Maulide, Top. Curr. Chem., 2018, 376, 15 CrossRef PubMed.
- Z. Sheng, Z. Zhang, C. Chu, Y. Zhang and J. Wang, Tetrahedron, 2017, 73, 4011 CrossRef CAS.
- Z. Zhang, Z. Sheng, W. Yu, G. Wu, R. Zhang, W.-D. Chu, Y. Zhang and J. Wang, Nat. Chem., 2017, 9, 970 CrossRef CAS PubMed.
- K. J. Hock and R. M. Koenigs, Angew. Chem., Int. Ed., 2017, 56, 13566 CrossRef CAS PubMed.
- (a) D. M. Hodgson, F. Y. T. M. Pierard and P. A. Stupple, Chem. Soc. Rev., 2001, 30, 50 RSC; (b) G. Ghigo, S. Cagnina, A. Maranzana and G. Tonachini, J. Org. Chem., 2010, 75, 3608 CrossRef CAS PubMed; (c) Y. Zheng, R. Bian, X. Zhang, R. Yao, L. Qiu, X. Bao and X. Xu, Eur. J. Org. Chem., 2016, 3872 CrossRef CAS.
- (a) L. W. Ciszewski, K. Rybicka-Jasinska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432 RSC; (b) N. R. Candeias and C. A. M. Afonso, Curr. Org. Chem., 2009, 13, 763 CrossRef CAS; (c) O. S. Galkina and L. L. Rodina, Russ. Chem. Rev., 2016, 85, 537 CrossRef CAS; (d) C. Empel and R. M. Koenigs, Synlett, 2019 DOI:10.1055/s-0037-1611874.
- (a) I. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112 RSC; (b) J. Yang, J. Wang, H. Huang, G. Qin, Y. Jiang and T. Xiao, Org. Lett., 2019, 21, 2654 CrossRef CAS PubMed; (c) T. Xiao, M. Mei, Y. He and L. Zhou, Chem. Commun., 2018, 54, 8865 RSC; (d) Z. Zhang, D. Yadagiri and V. Gevorgyan, Chem. Sci., 2019 10.1039/c9sc02448d.
- (a) R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203 CrossRef CAS PubMed; (b) S. Jana and R. M. Koenigs, Asian J. Org. Chem., 2019, 8, 683 CrossRef CAS; (c) Z. Yang, Y. Guo and R. M. Koenigs, Chem.–Eur. J., 2019, 25, 6703 CrossRef CAS PubMed; (d) F. He and R. M. Koenigs, Chem. Commun., 2019, 55, 4881 RSC.
- (a) H. Nozaki, H. Takaya and R. Noyori, Tetrahedron, 1966, 22, 3393 CrossRef CAS; (b) K. Friedrich, U. Jansen and W. Kirmse, Tetrahedron Lett., 1985, 26, 193 CrossRef CAS; (c) W. Kirmse, R. Lelgemann and K. Friedrich, Chem. Ber., 1991, 124, 1853 CrossRef CAS; (d) K. Ito and T. Katskui, Chem. Lett., 1994, 1857 CrossRef CAS; (e) K. Ito, M. Yoshitake and T. Katsuki, Heterocycles, 1996, 42, 305 CrossRef CAS; (f) V. Nair, S. M. Nair, S. Mathai, J. Liebscher, B. Ziemer and K. Narsimuli, Tetrahedron Lett., 2004, 45, 5759 CrossRef CAS; (g) G. K. Murphy and F. G. West, Org. Lett., 2005, 7, 1801 CrossRef CAS PubMed.
- (a) D. M. Jaber, R. N. Burgin, M. Helper, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2012, 14, 1676 CrossRef CAS PubMed; (b) D. M. Jaber, R. N. Burgin, M. Helper, P. Zavalij and M. P. Doyle, Chem. Commun., 2011, 47, 7623 RSC; (c) E. J. Roskamp and C. R. Johnson, J. Am. Chem. Soc., 1986, 108, 6062 CrossRef CAS PubMed; (d) E. D. Butova, A. V. Barabash, A. A. Petrova, C. M. Kleiner, P. R. Schreiner and A. A. Fokin, J. Org. Chem., 2010, 75, 6229 CrossRef CAS PubMed.
- (a) A. Boto and L. Alvarez, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, p. 97 Search PubMed; (b) K. C. Majumdar and S. Mondal, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, p. 377 CrossRef; (c) C. M. White, M. A. McGowan, Y. Han and N. Sciammetta, WO2019074748, 2019; (d) M. T. Rudd, Z. Meng, J. Wai, D. J. Bennett, E. J. Brnardic, N. J. Liverton, S. J. Stachel, Y. Han, P. Tempest, J. Zhu, X. Xu and B. Zhu, WO2018068295, 2018; (e) V. J. Colandrea, W. K. Hagmann, J. J. Hale and I. E. Whitney, WO2007136607, 2007; (f) G. A. Doherty, G. X. Yang, E. Borges, L. L. Chang, M. MacCoss, S. Tong, U. Kidambi, L. A. Egger, E. McCauley, G. Van Riper, R. A. Mumford, J. A. Schmidt and W. K. Hagmann, Org. Biomol. Chem., 2002, 12, 1501 CAS; (g) W. K. Hagmann, S. E. Delaszlo, G. Doherty, L. L. Chang and G. X. Yang, WO2001012183, 2001.
- For details, please see the ESI.†.
- Oxetane and thietane heterocycles bearing a proton in the 3-position might undergo ring opening side reactions to give minor amounts of ring-opened by-products under both photochemical and metal-catalyzed conditions. To remove the alkene by-products and for ease of purification, oxidative workup with KMnO4 or Br2 was performed. This ring opening reaction did not occur in case of 3,3-disubstituted oxetane heterocycles
 .
. - J. Riggs-Sauthier, B.-L. Deng and T. A. Riley, US Pat., 20100048602, 2010.
- (a) T. Ueki, R. Yamada, T. Okita, S. Takami, M. Nakamura and H. Tanaka, WO2019065283, 2019; (b) K. Guckian, G. Kumaravel, B. Ma, S. Mi, H. Peng, Z. Shao, L. Sun, A. Taveras, Z. Xin, L. Zhang, WO2014018891, 2014.
- (a) U. P. N. Tran, R. Hommelsheim, Z. Yang, C. Empel, K. J. Hock, T. V. Nguyen and R. M. Koenigs, ChemRxiv, DOI:10.26434/chemrxiv.7545623.v1; (b) T. Miura, T. Nakamuro, S. G. Stewart, Y. Nagata and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 3334 CrossRef CAS PubMed.
- Selected references: (a) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150 CrossRef CAS PubMed; (b) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed.
- Y. Zhang, J. Kubicki and M. S. Platz, J. Am. Chem. Soc., 2009, 131, 13602 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04069b |
| This journal is © The Royal Society of Chemistry 2019 |