
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubphthalocyanine–tetracyanobuta-1,3-diene–aniline conjugates: stereoisomerism and photophysical properties†‡
Kim A.
Winterfeld§
a,
Giulia
Lavarda§
b,
Julia
Guilleme§
b,
Dirk M.
Guldi
 *a,
Tomás
Torres
*bcd and
Giovanni
Bottari
*bcd
*a,
Tomás
Torres
*bcd and
Giovanni
Bottari
*bcd
aDepartment of Chemistry and Pharmacy, Interdisciplinary Center for Molecular Materials (ICMM), Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 3, 91058 Erlangen, Germany. E-mail: dirk.guldi@fau.de
bDepartamento de Química Orgánica, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: tomas.torres@uam.es; giovanni.bottari@uam.es
cIMDEA-Nanociencia, Campus de Cantoblanco, 28049 Madrid, Spain
dInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 19th September 2019
Abstract
Two subphthalocyanines (SubPcs) decorated at their peripheral (SubPc 1) or peripheral and axial (SubPc 2) positions with tetracyanobuta-1,3-diene (TCBD)–aniline moieties have been prepared as novel electron donor–acceptor (D–A) conjugates. In 1 and 2, the multiple functionalization of C3-symmetric SubPcs by TCBD moieties, each of them having a chiral axis, results in the formation of several stereoisomers. Variable temperature 1H-NMR studies in chlorinated solvents suggest that these latter species, which are detected at low temperatures, rapidly interconvert – on the NMR timescale – into each other at room temperature. Beside their unique structural and stereochemical features, 1 and 2 present interesting physicochemical properties. Steady-state absorption and fluorescence, as well as electrochemical studies on 1 and 2 clearly point to an important degree of electronic communication between the SubPc, the TCBD and the aniline subunits. Moreover, in both derivatives, photoexcitation of the SubPc moiety yields charge transfer products involving the electron-rich SubPc moiety and the electron-withdrawing TCBD fragment. Interestingly, such polarized excited state species evolve in 1 and 2 in different ways. While in the former compound, it directly decays to the ground state, the fourth axial TCBD moiety in 2 leads to the formation of an intermediate fully charge separated state prior to the ground state deactivation.
Introduction
Photoinduced electron transfer (PET) is a key step in the conversion of solar energy into chemical energy in many biological systems and a process with great relevance in research fields such as photocatalysis,1 photoconductivity,2 or molecular photovoltaics.3 In a general, yet simplistic scheme, it consists of two main events, namely, light absorption by a chromophore resulting in the formation of an excited state species, followed by a charge transfer (CT) between an electron donor (D) and an electron acceptor (A) leading to a charge separated (CS) D˙+–A˙− species.4 The optimization of these two processes, as well as the understanding of their mutual interplay, is of paramount importance for the development of efficient systems for solar-to-energy conversion schemes.Among the building blocks used for the construction of light-responsive D–A systems, subphthalocyanines (SubPcs) hold a privileged position. SubPcs are cone-shaped aromatic macrocycles that show intense absorptions in the 550–650 nm region, high fluorescence quantum yields, a rich redox chemistry, and a good photostability.5 Moreover, the rigid and nonplanar SubPc core affords small Stokes shifts and low reorganization energies, which are both important prerequisites for efficient intramolecular electron/energy transfer reactions. These unique physicochemical characteristics, which have prompted the use of SubPcs in diverse applications including, but not limited to, solar cells6 and photodynamic therapy,7 render these macrocycles promising light-harvesting synthons for their integration into D–A architectures. To date, several SubPc-based D–A arrays have been prepared, in which the macrocycle has been connected, either covalently or through supramolecular interactions, to electroactive moieties such as fullerenes,8 porphyrins,9 or phthalocyanines.10 In these conjugates, the SubPc acts as either D or A unit depending on the redox features of both the macrocycle – mainly dictated by the SubPc peripheral substituents – and the electroactive counterpart(s).
Recently, 1,1,4,4-tetracyanobuta-1,3-diene (TCBD) has been investigated as novel molecular partner for SubPcs.11–13 Up to now, few TCBD-based D–A arrays have been reported, in which the TCBD moiety, a strong electron-withdrawing group, has been linked to electroactive units such as anilines,14 porphyrins,15 corannulenes,16 or phthalocyanines.17 In most of these systems, photoinduced energy and/or electron transfer has been observed. Besides its excellent electronic properties, TCBD also presents some peculiar structural features resulting from the nonplanar arrangement of its two dicyanovinyl (DCV) halves14 and the restricted rotation around the C–C bond connecting them. As a consequence, atropisomers are formed and, in some experimental conditions, even isolated.11,18
Here, we report the synthesis and physicochemical properties of two novel electron D–A conjugates based on both SubPc and TCBD. In these systems, which have been characterized by a wide range of spectroscopic, spectrometric, and electrochemical techniques, the macrocycle has been decorated at its peripheral, or peripheral and axial positions with three (i.e., SubPc 1) or four (i.e., SubPc 2) TCBD–aniline moieties, respectively. In both arrays, the multiple functionalization of C3-symmetric SubPcs by TCBD moieties, each of them having a chiral axis, results in the formation of several stereoisomers. These species are detected in low temperature NMR experiments and rapidly interconvert – on the NMR timescale – into each other at room temperature. From the photophysical point of view, steady-state absorption, fluorescence, and electrochemical studies on 1 and 2 clearly point out to an important degree of electronic communication between the SubPc, the TCBD, and the aniline units. Moreover, in both derivatives, photoexcitation of the SubPc moiety yields CT products involving the electron-donating SubPc moiety and the electron-withdrawing TCBD fragment. Interestingly, the evolution of such polarized excited state species strongly depends on the functionalization position of the TCBD–aniline moiety on the SubPc (peripheral or peripheral/axial). While in 1, this state directly decays to the ground state, in 2 the formation of an intermediate fully CS product is observed prior to the ground state deactivation.
Results and discussion
Synthesis and stereoisomerism of (TCBD–aniline)-functionalized SubPcs 1 and 2
(TCBD–aniline)-functionalized SubPcs 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and 2 were prepared in a few-step synthesis starting from a common precursor, namely C3-symmetric triiodo-SubPc(Cl) 5 (Scheme 1).
19 and 2 were prepared in a few-step synthesis starting from a common precursor, namely C3-symmetric triiodo-SubPc(Cl) 5 (Scheme 1).
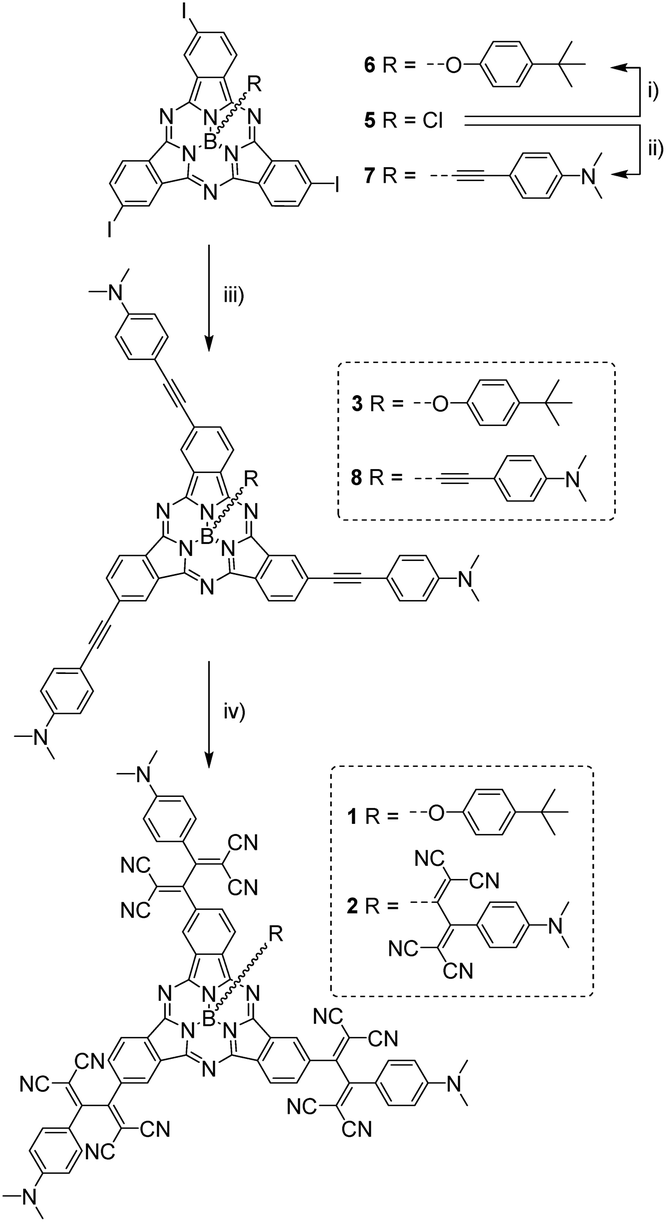 | ||
| Scheme 1 Synthesis of (TCBD–aniline)-functionalized SubPcs 1 and 2. Conditions: (i) 4-tert-butylphenol, toluene, reflux, 16 h; (ii) 4-ethynyl-MgBr-N,N′-dimethylamine, anhydrous THF, 60 °C, 4 h; (iii) PdCl2(PPh3)2, CuI, anhydrous THF/NEt3 (5:1), r.t., 16 h, and triiodo SubPcs 6 or 7 for the preparation of ethynyl-functionalized SubPcs 3 or 8, respectively; (iv) tetracyanoethylene, anhydrous THF, r.t., 1 h, and ethynyl-functionalized SubPcs 3 or 8 for the preparation of TCBD-functionalized SubPcs 1 or 2, respectively. | ||
For the synthesis of (TCBD–aniline)3-SubPc(tBuPhO) 1, SubPc 5 was initially transformed into its axially-substituted tert-butylphenoxy analog 6 and the latter was further reacted with 4-ethynyl-N,N′-dimethylaniline in a three-fold metal-catalyzed Sonogashira coupling reaction affording (ethynyl-aniline)3-SubPc(tBuPhO) 3 in 79% yield. 3 was then subjected to a three-fold cycloaddition–retroelectrocyclization (CA-REC) reaction20 with tetracyanoethylene obtaining conjugate 1 in 77% yield. For the synthesis of (TCBD–aniline)3-SubPc(TCBD–aniline) 2, the reaction between C3 triiodo-SubPc(Cl) 5 and the Grignard of 4-ethynyl-N,N′-dimethylaniline afforded triiodo-SubPc(ethynyl-aniline) 7 in 59% yield, a product which was further reacted in a three-fold Sonogashira reaction with 4-ethynyl-N,N′-dimethylaniline giving (ethynyl-aniline)3-SubPc(ethynyl-aniline) 8 in 87% yield. Finally, a fourfold CA-REC reaction between 8 and tetracyanoethylene led to (TCBD–aniline)3-SubPc(TCBD–aniline) 2 in 40% yield. Conjugates 1 and 2, as well as all SubPc precursors, were fully characterized by a wide range of spectroscopic, spectrometric, and electrochemical techniques (see ESI‡).
From the structural and stereochemical point of view, (TCBD–aniline)-functionalized SubPcs 1 and 2 present several interesting features.21 These latter SubPcs possess a C3-symmetric functionalization pattern resulting from the use of C3-symmetric triiodo-SubPc(Cl) 5 as common precursor for both syntheses. Moreover, the cone-shaped SubPc structure combined with the C3-symmetry of 5 gives rise to an additional topological feature in this SubPc, namely its isolation as a racemic mixture of two enantiomers, that is, M and P (Fig. 1a, b and S3.1‡).21 The same occurs for SubPcs 3 and 8, direct precursors of SubPcs 1 and 2, respectively. The chemical transformation of the three peripheral (in the case of 1) and four peripheral/axial (in the case of 2) ethynyl linkers into an equivalent number of TCBDs introduces, in conjugates 1 and 2, some new chiral elements. On one hand, TCBD possesses an axis of chirality arising from the restricted rotation around the TCBD C2–C3 carbon bond. As a consequence, TCBD has two atropisomers, which are also enantiomers, namely Ra and Sa (Fig. 1c, d and S3.2‡).21 Both enantiomers are observed in the crystal structures of TCBD-based derivatives, where the two DCV halves adopt a quasi-orthogonal arrangement between them.14 On the other hand, each of the three peripheral TCBDs in 1 and 2, beside adopting a Ra or Sa configuration, may present an additional conformational feature resulting from the nonplanar geometry of the SubPc.21 Taking all the above-mentioned structural and stereochemical features of (TCBD–aniline)-functionalized SubPcs 1 and 2 into account, at least 24 pairs of enantiomers are expected for (TCBD–aniline)3-SubPc(tBuPhO) 1 (Fig. S3.3‡).
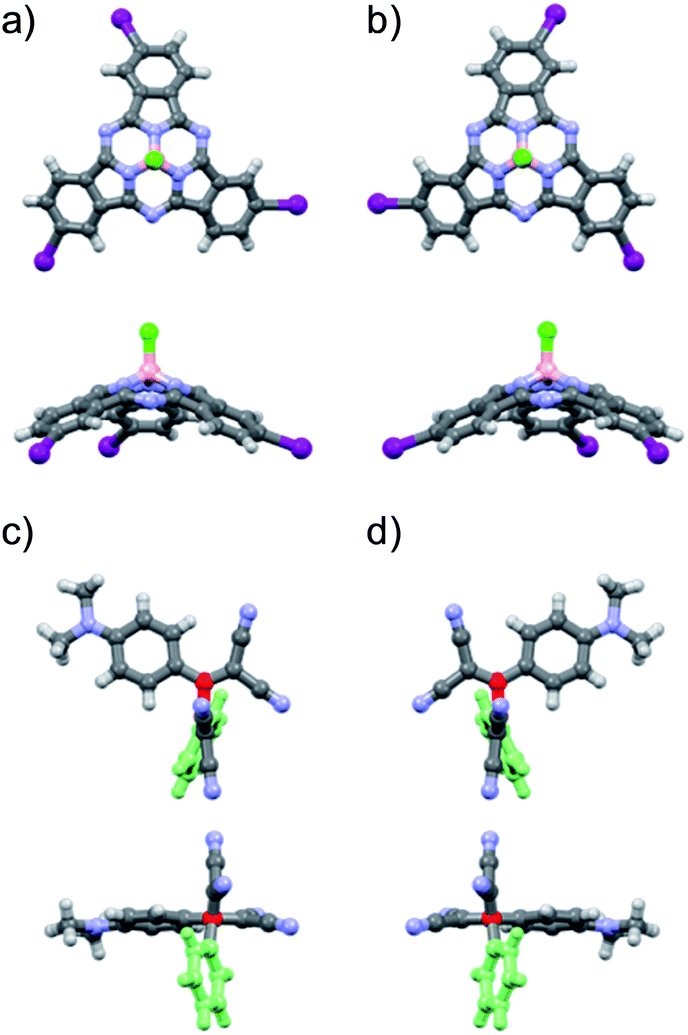 | ||
| Fig. 1 Top (top) and lateral (bottom) view (with respect to the SubPc B–Cl bond) of molecular modelling structures of (a) M and (b) P enantiomers of triiodo-SubPc(Cl) 5. Molecular modelling structures of the (c) Sa and (d) Ra atropisomers of TCBD–aniline derivative 4 (here chosen as example) visualized perpendicular to (top) and along the (bottom) butadiene C2–C3 chiral axis. Atoms' colour code: carbon in grey, hydrogen in white, nitrogen in light blue, iodine in violet, boron in light pink, and chlorine in green. For an easier visualization of the two atropisomers of 4, (i) the phenyl ring attached at the C2 butadiene moiety has been fully coloured in light green, and (ii) the butadiene C2 and C3 atoms as well as the bridging bond have been colored in red. | ||
This number doubles (i.e., 48 pairs of enantiomers) for (TCBD–aniline)3-SubPc(TCBD–aniline) 2 due to the introduction of an additional TCBD at the SubPc axial position.21 Nevertheless, despite the large number of possible stereoisomers of SubPcs 1 and 2, their 1H NMR spectra at room temperature in chlorinated solvents are quite sharp and present the multiplicity “theoretically” expected for achiral SubPcs with C3 symmetry (Fig. S2.5 and S2.17,‡ respectively). This result suggests that under these conditions, (i) the Ra/Sa interconversion of the TCBDs is fast on the NMR timescale, and/or (ii) the conformational flexibility around the C–C single bond connecting the SubPc to the peripheral TCBD is high. As a result, rapid interconversion occurs between the multiple stereoisomers. Low-temperature 1H-NMR experiments were also carried out leading to a rather different scenario, in which an increase in multiplicity, broadening, and shift of all the proton peaks was observed (Fig. 2 and S2.6‡). Such results are rationalized on the fact that the interconversion between the different stereoisomers of 1 and 2 is significantly slowed down upon lowering the temperature resulting in the coexistence, on the NMR timescale, of many magnetically distinct stereoisomers with C1 symmetry. A similar effect has been previously observed in variable temperature 1H-NMR experiments carried out on corannulene derivatives peripherally functionalized with two or five TCBDs.16
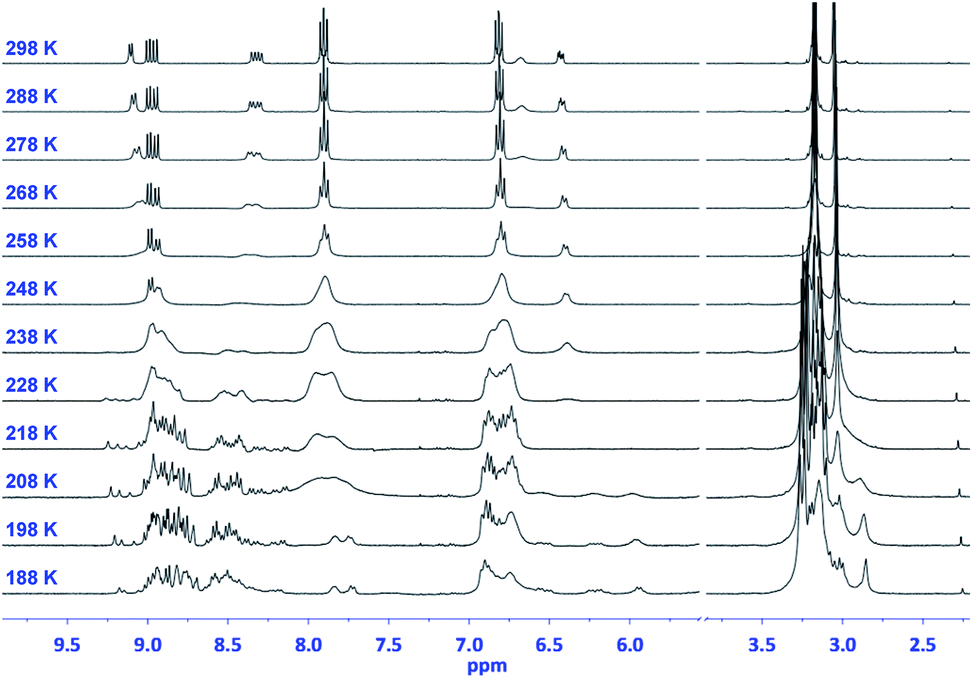 | ||
| Fig. 2 Variable-temperature 1H-NMR spectra (CD2Cl2) of (TCBD–aniline)3-SubPc(TCBD–aniline) 2. | ||
Photophysical and electrochemical studies
The absorption spectrum of (ethynyl-aniline)3-SubPc(tBuPhO) 3 in toluene is dominated by a sharp Q-band along with a less intense Soret-band maximizing at 601 and 329 nm, respectively (Fig. S5.1 and Table S5.1‡). In comparison to H12SubPc(Cl), which lacks the three peripheral ethynyl-aniline moieties of 3, significant differences are discernible (Fig. S5.1‡). On one hand, the Soret-band absorption in 3 is slightly broadened and more intense than in H12SubPc(Cl), probably due to the contribution of the aniline absorption in the UV region of the solar spectrum. On the other hand, the Q-band absorption in 3 is bathochromically shifted (i.e., 36 nm) and broadened with respect to H12SubPc(Cl), probably due to an extension of the SubPc π-conjugation by the three peripheral electron-donating ethynyl-aniline groups.5,23,24 Additional transition bands are observed for 3 at 394 and 439 nm that are attributable to n → π* transitions stemming from the peripheral nitrogen lone pairs.5,10a,24 Interestingly, the 439 nm transition presents a bathochromic shift upon increasing the solvent polarity (i.e., 16 nm moving from toluene to benzonitrile) (Fig. S5.2‡). A likely rationale for such positive solvatochromism is the stabilization of the “polar” excited state populated via the n → π* transition upon increasing the solvent polarity.
Next, the absorption features of (TCBD–aniline)-functionalized SubPcs 1 and 2 were investigated. Both compounds showed significant differences with respect to (ethynyl-aniline)-functionalized SubPc 3 and H12SubPc(Cl) (Fig. 3 and S5.3, and Table S5.1‡). For 1, characteristic Soret- and Q-band absorptions are observable at 310 and 622 nm, respectively (Fig. 3). The latter band is broadened and bathochromically shifted with respect to the Q-band of 3 and H12SubPc(Cl) (i.e., 21 and 57 nm, respectively). Beside the aforementioned Soret- and Q-band absorptions in 1 in toluene, an additional feature is present at 462 nm. Valuable insights into the nature of such transition came from the UV-vis absorption analysis of phenyl-TCBD–aniline 4, which presents a broad absorption maximum at 463 nm and a shoulder at 410 nm (Fig. 3). Such transitions in 4 are attributable to a push–pull CT absorption resulting from ground state interactions between the electron-rich aniline and the electron-withdrawing TCBD units. Moreover, a positive solvatochromism of this transition has been observed.17
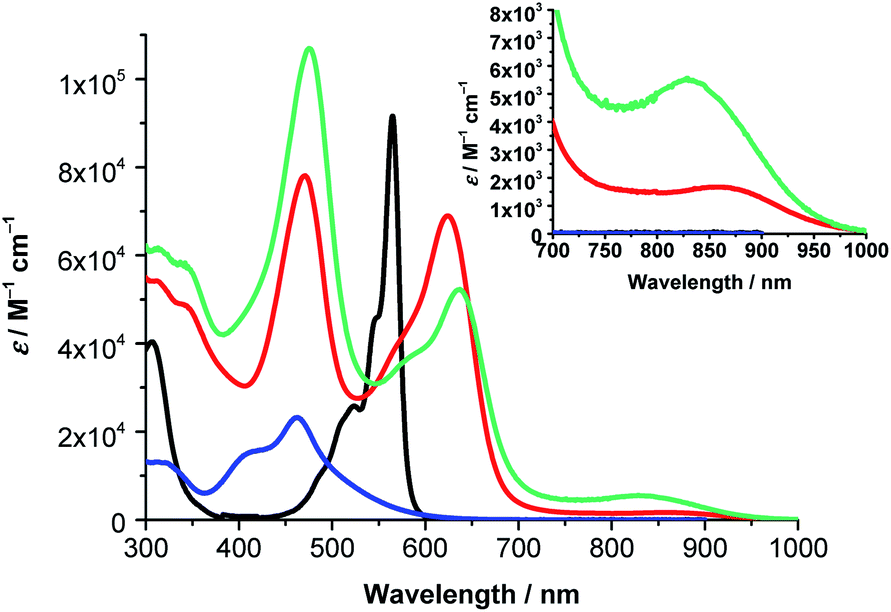 | ||
| Fig. 3 Steady-state absorption spectra of (TCBD–aniline)3-SubPc(TCBD–aniline) 2 (green) in anisole, (TCBD–aniline)3-SubPc(tBuPhO) 1 (red) in anisole, phenyl-TCBD–aniline 4 (blue) in toluene, and H12SubPc(Cl) (black) in toluene. Inset: zoom of the steady-state absorption spectra in the region between 700 and 1000 nm. | ||
Similar to 4, the absorption of 1 at 462 nm in toluene is subject to a bathochromic shift upon increasing the solvent polarity (i.e., up to 473 nm in benzonitrile) (Fig. S5.4 and Table S5.1‡). Interesting is the threefold increase in the extinction coefficient of the push–pull CT band of 1 with respect to 4, reflecting the presence of three TCBD–aniline moieties in the former conjugate as opposed to one in the latter. A closer look at the absorption spectra of 1 reveals an additional broad absorption around 860 nm, which is more prominent in the more polar solvents, i.e. chlorobenzene, anisole, and benzonitrile (Fig. S5.4‡). Furthermore, a slight broadening of the Q-band absorption is observable upon increasing the solvent polarity. A possible rationale for these phenomena includes the formation of a CT state between the electron-rich SubPc macrocycle and the covalently linked electron-withdrawing TCBDs (vide infra).
Turning to (TCBD–aniline)3-SubPc(TCBD–aniline) 2, its absorption spectrum in anisole exhibits Soret- and Q-band transitions at 313 and 636 nm, respectively, and a TCBD–aniline CT absorption band at 475 nm (Fig. 3).25 Moreover, a low-energy band was also observed at 832 nm. In contrast to 1, 2 lacks any appreciable bathochromic shifts of the TCBD–aniline CT absorption band as the solvent polarity is varied (Fig. S5.5 and Table S5.1‡). However, a noticeable broadening of the low-energy band is observed together with its red-shift moving from anisole and chlorobenzene (i.e., 832 and 838 nm, respectively) to benzonitrile (i.e., 860 nm). Considering the positive solvatochromism of this latter transition and the solvent-independent nature of the TCBD–aniline CT absorption in 2, we postulate that the axial TCBD enhances the electron-withdrawing effect, which is exerted onto the macrocycle, as compared to 1. Therefore, the polarized CT state, which is populated upon excitation in the low energy regime, is more susceptible to external influences, such as solvent polarity, and, hence, subject to polarizability and energy stabilization to a larger extent with increasing solvent polarity.
Turning to (TCBD–aniline)3-SubPc(tBuPhO) 1, its fluorescence spectrum in toluene is a mirror image of the corresponding absorption, dominated by a maximum at 650 nm and a Stokes shift of 28 nm (Fig. S5.7‡). In comparison to precursor 3, the fluorescence of 1 is broader and further shifted to the red. Upon testing more polar solvents, the broadening augments and the Stokes shift increases from 28 nm in toluene to 33 nm in benzonitrile. Moreover, ΦF of this conjugate is very low in all investigated solvents, ranging between 1 × 10−4 (benzonitrile) and 5 × 10−4 (anisole), which corresponds to a fluorescence quenching of more than 99.9% (Table S5.2‡).
In the case of (TCBD–aniline)3-SubPc(TCBD–aniline) 2, a further fluorescence quenching with respect to the TCBD-functionalized SubPc 1 was observed with ΦF lower than 1 × 10−5, which corresponds to a fluorescence quenching of more than 99.99% (Fig. S5.8 and Table S5.2‡). The Stokes shift ranges from 26 nm in benzonitrile to 32 nm in anisole and resembles that of 1. The strongly quenched fluorescence of 1 and 2 is indicative of a deactivation pathway possibly involving an intramolecular CT process from the photoexcited SubPc to the electron-accepting TCBD moiety. Moreover, the stronger quenching observed in the case of (TCBD–aniline)3-SubPc(TCBD–aniline) 2 with respect to 1 could result from the presence of an additional TCBD unit at its axial position.
With these results in hand, for conjugate 1 the process at −0.79 V is attributable to the reduction of the DCV adjacent to the SubPc, whereas the one at −1.11 V is located at the DCV connected to the aniline.11,17 With regard to SubPc, the SubPc-centered reduction at −1.81 V is cathodically shifted by about 0.41 V relative to SubPc precursor 3, most probably as a result of the repulsive negative charges at the TCBD units in 1 covalently linked to the macrocycle. As in the case of the reductive processes, multiple GAUSS peak fittings were necessary in order to clearly identify the oxidation potentials of 1. In this case, one one-electron oxidation peak at +0.85 V and one three-electron oxidation peak at +0.91 V were obtained. The latter potential, which nicely matches that of the oxidation of the aniline moiety in reference TCBD–aniline 4, is attributable to the three aniline units in 1, whereas the oxidation at +0.85 is SubPc-centered (Table 1).
Finally, the redox features of (TCBD–aniline)3-SubPc(TCBD–aniline) 2 were investigated. Similar to TCBD-functionalized SubPc 1, all redox processes in 2 are subject to a strong coalescence, which had to be unraveled by means of multiple GAUSS peak fits (Fig. S4.3‡). With regard to the reduction, five processes were identified at −0.71, −0.81, −1.14, −1.31, and −1.45 V (Tables 1 and S4.1‡). On one hand, the second and third reduction at −0.81 and −1.14 V, respectively, are three-electron reduction processes with potentials very similar to the first two reductions of 1 at −0.79 and −1.11 V. Therefore, we assign these reductions to the three peripheral DCVs adjacent to the SubPc and the three peripheral DCVs closer to aniline, respectively. On the other hand, the one-electron reductions at −0.71 and −1.31 V are attributable to processes involving the axial DCVs adjacent to SubPc and aniline, respectively. The fifth and final reduction at −1.45 V is SubPc-centered. In the oxidative voltammograms of (TCBD–aniline)3-SubPc(TCBD–aniline) 2, two oxidations, namely one one-electron oxidation at +0.88 V and one three-electron oxidation at +0.93 V are observed, which are SubPc- and aniline-centered, respectively. The SubPc-centered oxidation is anodically shifted by 0.03 V relative to 1, probably due to the slightly higher electron-deficiency exerted by the additional axial TCBD unit.
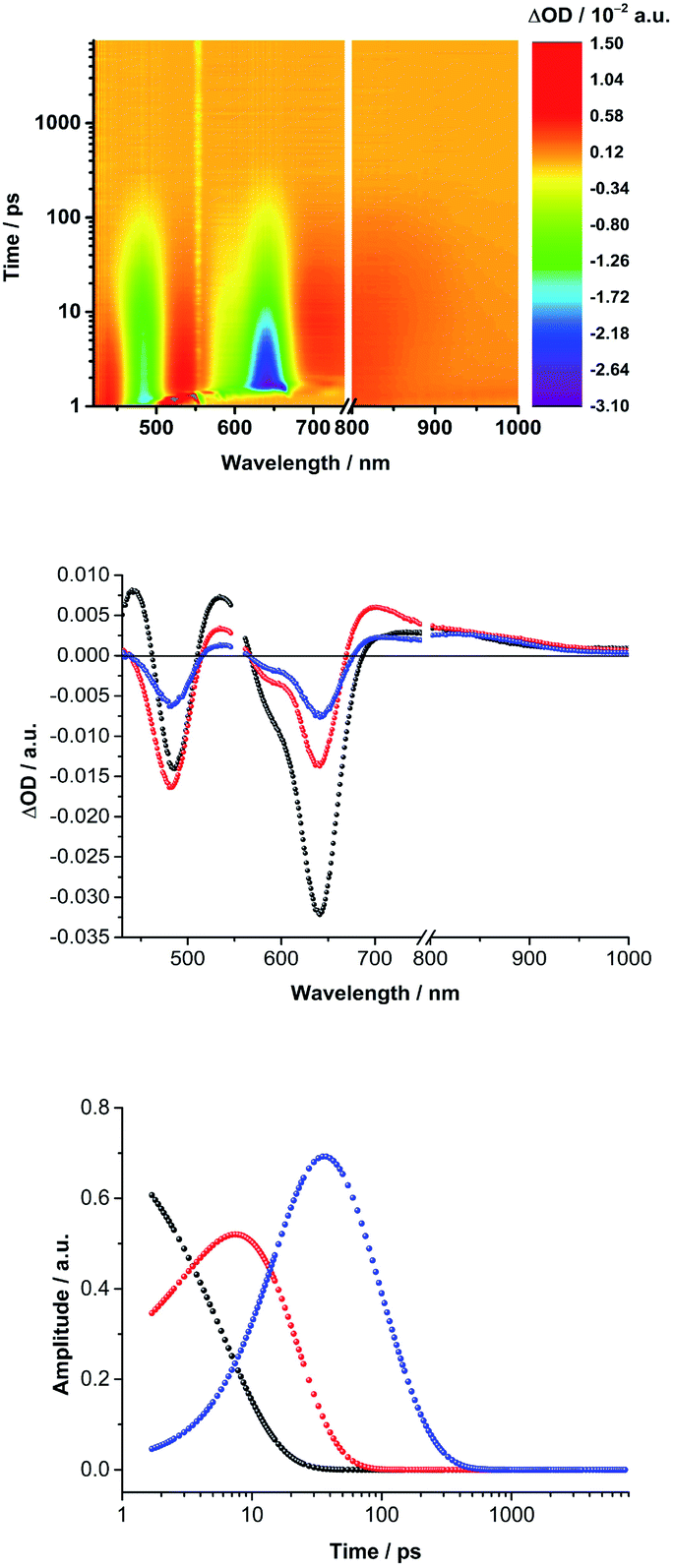
Turning to (TCBD–aniline)3-SubPc(tBuPhO) 1, following 550 nm excitation in anisole, newly developing differential absorption changes in the form of transient minima at 474 and 626 nm are a good reflection of the ground state absorption (Fig. 4). These features are complemented by maxima at 520 and 687 nm, as well as a broad absorption reaching far into the NIR. As time progresses, several features evolve. Firstly, the broad NIR broad absorption and the ground state bleaching of the SubPc Q-band decrease, whereas the ground state bleaching of the TCBD–aniline CT band at 474 nm intensifies and experiences a slight hypsochromic shift to 470 nm. Secondly, a new signature maximizing at 674 nm grows-in with monoexponential kinetics. These findings point out to the formation of the one-electron reduced form of TCBD similar to what was observed in recent spectroelectrochemical investigations.11 Based on these results, we assume a CT process from the SubPc to one of the adjacent TCBDs leading to the formation of a highly polarized *(SubPcδ+-[(TCBD–aniline)3]δ−) species (Fig. 5a). The same transient absorption features were observed in toluene, chlorobenzene, and benzonitrile (Fig. S5.10a–c‡). Multiwavelength and global analyses afforded ultrafast formation/decay kinetics, namely, 3.3/13 ps in toluene, 4.2/15 ps in chlorobenzene, 3.6/14 ps in anisole, and 2.0/15 ps in benzonitrile. However, the formation of a fully CS state is not discernible for 1 in any of the investigated solvents.
 | ||
| Fig. 4 (Top) Differential absorption spectra obtained by femtosecond transient absorption pump–probe experiments (550 nm, 400 nJ) of (TCBD–aniline)3-SubPc(tBuPhO) 1 (2 × 10−5 M) in argon-saturated anisole with several time delays between 1 to 7500 ps. (Middle) Deconvoluted evolution associated spectra and (bottom) associated time-dependent amplitudes obtained via global analysis in GloTarAn monitoring formation and decay kinetics. | ||
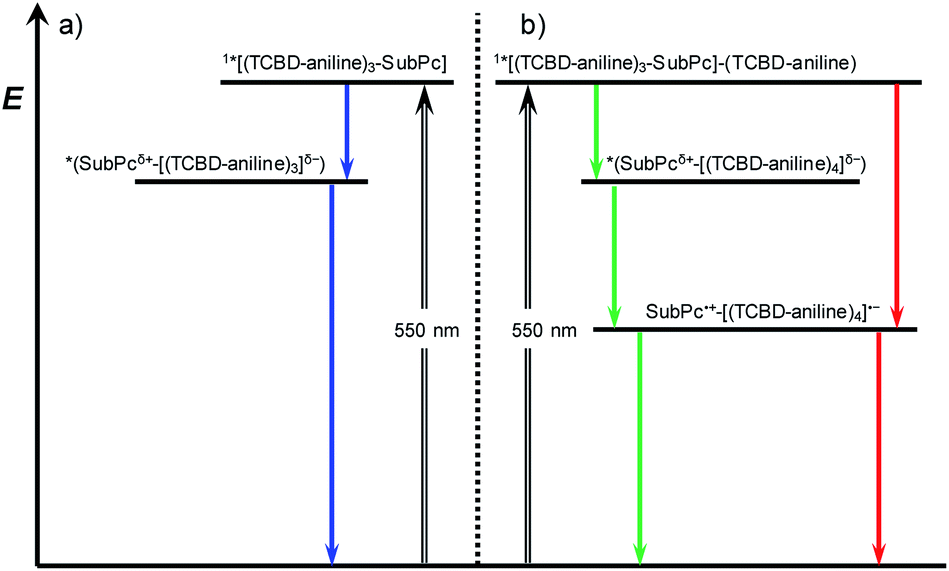 | ||
| Fig. 5 Energy level diagram of (a) (TCBD–aniline)3-SubPc(tBuPhO) 1 in toluene, chlorobenzene, anisole, and benzonitrile (blue arrows) and (b) (TCBD–aniline)3-SubPc(TCBD–aniline) 2 in chlorobenzene and anisole (green arrows), and benzonitrile (red arrows) reflecting the energetic pathways after excitation of SubPc (λex = 550 nm, black arrow). Blue, green, and red arrows refer to nonradiative processes. | ||
Next, (TCBD–aniline)3-SubPc(TCBD–aniline) conjugate 2 in chlorobenzene, anisole, and benzonitrile was probed (Fig. 4 and S5.11‡). Laser excitation of 2 at 550 nm in argon-saturated anisole results in the instantaneous formation of sharp maxima at 441 and 534 nm, a broad maximum at 805 nm, as well as minima at 484 and 641 nm (Fig. 6). All these fingerprints are assigned to the SubPc singlet excited state formation. Notably, the NIR absorption is more prominent than in 1, which reflects a stronger electronic coupling between the SubPc and the TCBD units. As time progresses, a slight hypsochromic shift and an increase of the CT ground state bleaching at 484 to 482 nm, as well as a significant decay of the SubPc ground state bleaching are seen. In parallel, a maximum at 701 nm raises with monoexponential kinetics. All these features resemble those of the highly polarized excited CT state seen for 1. From this, we conclude that the SubPc singlet excited state transforms into *(SubPcδ+-[(TCBD–aniline)4]δ−) (Fig. 5b). This happens with 3.6 ps in chlorobenzene and 4.2 ps in anisole. Global analysis reveals the presence of yet another component, whose characteristics, that is, minima at 484 and 642 nm, are fingerprints of the TCBD–aniline CT and the SubPc Q-band transition, respectively. These features are complemented by transient maxima at 539 and 703 nm, as well as a broad absorption with a maximum at 836 nm. We assign the latter to a significant electronic interaction between the SubPc and the TCBD units as already indicated in the context of steady-state absorption and fluorescence spectroscopy (vide supra). Together with the 703 nm maximum and the 484 nm minimum as markers of the one-electron reduced form of TCBD as well as the 642 nm minimum of the one-electron oxidized form of SubPc, we postulate the formation of a SubPc˙+-[(TCBD–aniline)4]˙− CS state (Fig. 5b).11 Global analysis yielded charge separation and charge recombination kinetics of 20/120 ps in chlorobenzene and 17/89 ps in anisole, respectively. Interestingly, in benzonitrile, global analysis was based on a two-species kinetic model (Fig. S5.11b‡). The first species is assigned to the SubPc singlet excited state, which transforms ultrafast within 1.8 ps into a second species, whose differential absorption spectra are dominated by a distinct maximum at 700 nm and a broad maximum around 825 nm. These latter spectral features resemble those attributed to the radical cation and anion species formed in chlorobenzene and anisole. Basically, in a polar solvent such as benzonitrile, the singlet excited state transforms directly into the highly stabilized SubPc˙+-[(TCBD–aniline)4]˙− CS state species, which finally deactivates to the ground state within 29 ps. The observed PET process in 2 is in line with the dramatic decrease of ΦF and the higher intensity of the CT absorption band in the NIR region in comparison to 1.
 | ||
| Fig. 6 (Top) Differential absorption spectra obtained by femtosecond transient absorption pump–probe experiments (550 nm, 400 nJ) of (TCBD–aniline)3-SubPc(TCBD–aniline) 2 (3 × 10−5 M) in argon-saturated anisole with several time delays between 1 to 7500 ps. (Middle) Deconvoluted evolution associated spectra and (bottom) associated time-dependent amplitudes obtained via global analysis in GloTarAn monitoring formation and decay kinetics. | ||
It is important to notice that in case of 2, CS is seemingly possible as opposed to 1 due to the presence of an electron-accepting TCBD unit in the SubPc's axial position. In the former conjugate, the axial TCBD–aniline is decoupled from the SubPc through the nodal plane at the boron atom, which allows for a significant stabilization of the electron transfer product. In contrast, the peripheral TCBDs are part of the SubPc's π-conjugated system. This, in turn, enhances the electronic communication, but, at the same time, destabilizes the electron transfer product. Therefore, the peripheral TCBDs and the SubPc in 1 are subject to CT interactions rather than undergoing electron transfer.
Conclusions
In this work, we have reported two novel D–A conjugates comprising a SubPc functionalized at its peripheral, or peripheral and axial positions with three (1) or four (2) TCBD–aniline moieties, respectively. In 1 and 2, the combination of multiple axially chiral TCBD units, the C3 symmetry of the SubPcs' peripheral functionalization pattern, and the cone-shaped structure of the macrocycle result in the formation of several stereoisomers. Variable temperature 1H-NMR experiments in chlorinated solvents suggest that manifold stereoisomers are observed at low temperatures, whereas at room temperature a rapid interconversion – on the NMR timescale – takes place. In depth electrochemical and photophysical characterization of 1 and 2 was also carried out. These studies suggest that the covalent functionalization of the macrocycle by several TCBD–aniline moieties has dramatic consequences on the physicochemical properties. In detail, conjugates 1 and 2 present a red-shift of the SubPc Q-band absorption (∼20–30 nm) and a strong fluorescence quenching (>99.9%) with respect to their (ethynyl-aniline)-functionalized SubPc precursors 3 and 8, respectively. On the same line, the electrochemical analysis of both 1 and 2 revealed that the chemical transformation of the multiple ethynyl linkers in 3 and 8 into an equivalent number of TCBD units significantly perturbs the redox features of the SubPc and aniline moieties in 1 and 2. These studies point to a significant degree of electronic communication between SubPc, TCBD, and aniline units in 1 and 2. Corroboration came from steady-state absorption studies, which revealed the occurrence of new transitions in the NIR regime, which are based on ground state CT interactions between the SubPc and the electron-withdrawing TCBD units. In this connection, the introduction of an axial TCBD unit in 2 as compared to 1 remarkably enhances this transition. As a matter of fact, this NIR absorption is, to the best of our knowledge, unprecedented in the context of SubPc based chemistry. Additional proofs of electronic communication in these conjugates come from transient absorption studies. In 1, a CT from the photoexcited SubPc to one of the three peripheral TCBD moieties occurs in toluene, chlorobenzene, anisole, and benzonitrile. As a result, a polarized transient species *(SubPcδ+-[(TCBD–aniline)3]δ−) is formed, which directly deactivates to the ground state. In turn, the introduction of an additional TCBD–aniline group at the axial position of the SubPc in conjugate 2 significantly changes the conjugate's photophysics relative to 1. For the former derivative, similarly to 1, the deactivation of the photoexcited SubPc yields a polarized transient species *(SubPcδ+-[(TCBD–aniline)4]δ−) in solvents of low or medium polarity (i.e., chlorobenzene and anisole). However, in 2, this latter polarized transient species further transforms into a fully CS species (SubPc˙+-[(TCBD–aniline)4]˙−) before deactivating to the ground state. Moving to a solvent with a higher dielectric constant such as benzonitrile, the “intermediate” polarized excited state was not detected and the direct conversion of the photoexcited SubPc into a fully CS state was observed. The different behavior of 1 and 2 in their excited-state deactivation suggests that the introduction of an axial TCBD unit has a dramatic impact on the excited state interactions between SubPc and TCBD.In summary, we have prepared two novel SubPc-based D–A conjugates, which present fluxional stereoisomerism and strongly coupled electron D (i.e., SubPc and aniline) and A (i.e., TCBD) units. Physicochemical studies revealed (i) a broad and panchromatic absorption in the UV-vis region of the solar spectrum, (ii) intense ground state CT interactions between SubPc and TCBD, and (iii) photoinduced charge and/or electron transfer, which strongly depends on the SubPc's functionalization pattern (i.e., peripheral or peripheral/axial). All these phenomena render these systems promising materials for applications in the field of molecular photovoltaics.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the “Solar Energy goes Hybrid” Initiative of the Bavarian Ministry for Science, Culture and Education (SolTech) and Spanish MICINN (CTQ2017-85393-P) is acknowledged. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686).Notes and references
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- L. Zang, Acc. Chem. Res., 2015, 48, 2705 CrossRef CAS PubMed.
- D. M. Stoltzfus, J. E. Donaghey, A. Armin, P. E. Shaw, P. L. Burn and P. Meredith, Chem. Rev., 2016, 116, 12920 CrossRef CAS PubMed.
- (a) M. Gilbert and B. Albinsson, Chem. Soc. Rev., 2015, 44, 845 RSC; (b) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS PubMed.
- C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192 CrossRef CAS PubMed.
- (a) T. M. Grant, D. S. Josey, K. L. Sampson, T. Mudigonda, T. P. Bender and B. H. Lessard, Chem. Rec., 2019, 19, 1093 CrossRef CAS PubMed; (b) G. de la Torre, G. Bottari and T. Torres, Adv. Energy Mater., 2017, 1601700 CrossRef.
- E. van de Winckel, M. Mascaraque, A. Zamarron, A. Juarranz de la Fuente, T. Torres and A. de la Escosura, Adv. Funct. Mater., 2018, 1705938 CrossRef.
- (a) D. Gonzalez-Rodriguez, E. Carbonell, D. M. Guldi and T. Torres, Angew. Chem., Int. Ed., 2009, 48, 8032 CrossRef CAS PubMed; (b) I. Sanchez-Molina, C. G. Claessens, B. Grimm, D. M. Guldi and T. Torres, Chem. Sci., 2013, 4, 1338 RSC; (c) I. Sánchez-Molina, B. Grimm, R. M. Krick Calderon, C. G. Claessens, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2013, 135, 10503 CrossRef PubMed.
- (a) M. E. El-Khouly, D. K. Ju, K.-Y. Kay, F. D'Souza and S. Fukuzumi, Chem.–Eur. J., 2010, 16, 6193 CrossRef CAS PubMed; (b) R. Menting, J. T. F. Lau, H. Xu, D. K. P. Ng, B. Roder and E. A. Ermilov, Chem. Commun., 2012, 48, 4597 RSC; (c) M. Managa, J. Mack, D. Gonzalez-Lucasb, S. Remiro-Buenamañana, C. Tshangana, A. N. Cammidge and T. Nyokong, J. Porphyrins Phthalocyanines, 2016, 20, 1 CrossRef.
- (a) D. Gonzalez-Rodriguez, C. G. Claessens, T. Torres, S. G. Liu, L. Echegoyen, N. Vila and S. Nonell, Chem.–Eur. J., 2005, 11, 3881 CrossRef CAS PubMed; (b) Z. Zhao, A. N. Cammidge and M. J. Cook, Chem. Commun., 2009, 7530 RSC.
- K. A. Winterfeld, G. Lavarda, J. Guilleme, M. Sekita, D. M. Guldi, T. Torres and G. Bottari, J. Am. Chem. Soc., 2017, 139, 5520 CrossRef CAS PubMed.
- H. Gotfredsen, T. Neumann, F. E. Storm, A. V. Muñoz, M. Jevric, O. Hammerich, K. V. Mikkelsen, M. Freitag, G. Boschloo and M. B. Nielsen, ChemPhotoChem, 2018, 2, 976 CrossRef CAS.
- A. V. Muñoz, H. Gotfredsen, M. Jevric, A. Kadziola, O. Hammerich and M. B. Nielsen, J. Org. Chem., 2018, 83, 2227 CrossRef PubMed.
- T. Michinobu, C. Boudon, J.-P. Gisselbrecht, P. Seiler, B. Frank, N. N. P. Moonen, M. Gross and F. Diederich, Chem.–Eur. J., 2006, 12, 1889 CrossRef CAS PubMed.
- (a) F. Tancini, F. Monti, K. Howes, A. Belbakra, A. Listorti, W. B. Schweizer, P. Reutenauer, J.-L. Alonso-Gómez, C. Chiorboli, L. M. Urner, J.-P. Gisselbrecht, C. Boudon, N. Armaroli and F. Diederich, Chem.–Eur. J., 2014, 20, 202 CrossRef CAS PubMed; (b) D. Koszelewski, A. Nowak-Król and D. T. Gryko, Chem.–Asian J., 2012, 7, 1887 CrossRef CAS PubMed.
- Y.-L. Wu, M. C. Stuparu, C. Boudon, J.-P. Gisselbrecht, W. B. Schweizer, K. K. Baldridge, J. S. Siegel and F. Diederich, J. Org. Chem., 2012, 77, 11014 CrossRef CAS PubMed.
- M. Sekita, B. Ballesteros, F. Diederich, D. M. Guldi, G. Bottari and T. Torres, Angew. Chem., Int. Ed., 2016, 55, 5560 CrossRef CAS PubMed.
- M. Yamada, P. Rivera-Fuentes, W. B. Schweizer and F. Diederich, Angew. Chem., Int. Ed., 2010, 49, 3532 CrossRef CAS PubMed.
- The synthesis and steady-state absorption features of a C1-symmetric analog of C3-symmetric (TCBD–aniline)3-SubPc(tBuPhO) 1 have been recently reported in ref. 12. However, no additional studies such as variable temperature 1H-NMR, electrochemistry, and steady-state fluorescence and time-resolved absorption spectroscopy have been realized on that conjugate.
- T. Michinobu and F. Diederich, Angew. Chem., Int. Ed., 2018, 57, 3552 CrossRef CAS PubMed.
- For a detailed account of the chiral elements in (TCBD–aniline)-functionalized SubPcs 1 and 2, and possible stereoisomers, see Section 3 in the ESI.‡.
- It is important to notice here that (ethynyl-aniline)3-SubPc(tBuPhO) 3 and (ethynyl-aniline)3-SubPc(ethynyl-aniline) 8, immediate precursors of (TCBD–aniline)-functionalized SubPcs 1 and 2, respectively, present substantially identical optical features (i.e., in SubPcs, the absorption and fluorescence features are mainly dictated by the nature of the peripheral substituents and are only marginally influenced by changes in the nature of the axial ligand). For this reason, SubPc 3 has been used as the only (ethynyl-aniline)-functionalized SubPc for comparative studies with SubPcs 1 and 2.
- D. Gonzalez-Rodriguez, T. Torres, D. M. Guldi, J. Rivera, M. A. Herranz and L. Echegoyen, J. Am. Chem. Soc., 2004, 126, 6301 CrossRef CAS PubMed.
- G.-R. David and T. Tomás, Eur. J. Org. Chem., 2009, 2009, 1871 Search PubMed.
- The absorption spectrum of 2 in toluene could not be recorded due to solubility issues.
- Singlet excited state energies of SubPc were calculated as an average value by means of the following equation:
λmax(abs.) = absorption maximum at the longest wavelength, λmax(em.) = fluorescence maximum at the shortest wavelength. - K. Kimura, H. Tsubomura and S. Nagakura, Bull. Chem. Soc. Jpn., 1964, 37, 1336 CrossRef CAS.
- Since the oxidation peaks of 3 coalesce, they were deconvoluted by multiple GAUSS peak fittings.
- L. Feng, M. Rudolf, O. Trukhina, Z. Slanina, F. Uhlik, X. Lu, T. Torres, D. M. Guldi and T. Akasaka, Chem. Commun., 2015, 51, 330 RSC.
- M. Rudolf, O. Trukhina, J. Perles, L. Feng, T. Akasaka, T. Torres and D. M. Guldi, Chem. Sci., 2015, 6, 4141 RSC.
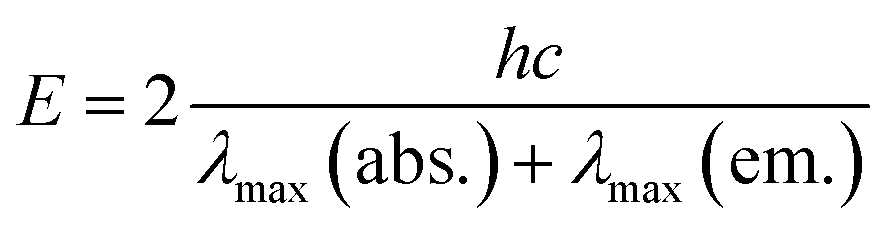
Footnotes |
| † This article is dedicated to Prof. Michael R. Wasielewski on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Procedures for synthesis, characterization data, and supplementary figures. See DOI: 10.1039/c9sc03970h |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |