
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving MOF stability: approaches and applications
Meili
Ding
a,
Xuechao
Cai
 ab and
Hai-Long
Jiang
*a
ab and
Hai-Long
Jiang
*a
aHefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Soft Matter Chemistry, Collaborative Innovation Center of Suzhou Nano Science and Technology, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: jianglab@ustc.edu.cn
bCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China
First published on 2nd October 2019
Abstract
Metal–organic frameworks (MOFs) have been recognized as one of the most important classes of porous materials due to their unique attributes and chemical versatility. Unfortunately, some MOFs suffer from the drawback of relatively poor stability, which would limit their practical applications. In the recent past, great efforts have been invested in developing strategies to improve the stability of MOFs. In general, stable MOFs possess potential toward a broader range of applications. In this review, we summarize recent advances in the design and synthesis of stable MOFs and MOF-based materials via de novo synthesis and/or post-synthetic structural processing. Also, the relationships between the stability and functional applications of MOFs are highlighted, and finally, the subsisting challenges and the directions that future research in this field may take have been indicated.
 Meili Ding | Meili Ding received her BS degree (2014) in chemistry from Anhui Normal University. She is currently a PhD student under the guidance of Prof. Hai-Long Jiang at the University of Science and Technology of China (USTC). Her research focuses on the preparation of stable metal–organic framework-based materials. |
 Hai-Long Jiang | Hai-Long Jiang earned his PhD (2008) from Fujian Institute of Research on the Structure of Matter, CAS. He subsequently worked at the National Institute of Advanced Industrial Science and Technology (AIST, Japan), first as an AIST Fellow and later as a JSPS Fellow during 2008–2011. After a postdoctoral stint at Texas A&M University (USA), he was appointed a full professor at USTC in 2013. He is a Fellow of the Royal Society of Chemistry (FRSC) and was recognized as a highly cited researcher (2017 & 2018 & 2019) in chemistry by Clarivate Analytics. His main research interest is the development of crystalline porous and nanostructured materials for catalysis. |
1. Introduction
Over the past two decades, the emergence of metal–organic frameworks (MOFs) which are constructed by metal ions/clusters and multidentate organic linkers via coordination bonds (reticular synthesis) has significantly enriched the domain of porous materials.1 Based on the flexibility of the constituents' geometry, size, and functionality, MOFs with a crystalline nature generally possess extremely high surface areas (typically ranging from 1000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 g−1) and tunable pore sizes/characteristics.1 These aspects endow MOFs with fantastic functionalities/properties for a variety of applications, for example, gas adsorption and separation, catalysis, sensors, drug delivery, proton conduction, etc.1–18 For various applications of MOFs, stability, mainly including chemical, thermal and mechanical stability, is one of the major concerns. The chemical stability of MOFs, specifically, refers to their resistance to the effects of exposure to various chemicals in their environment, e.g. moisture, solvents, acids, bases, and aqueous solutions containing coordinating anions. The thermal stability and mechanical stability of MOFs usually correlate with the ability of MOFs to preserve their structural integrity upon exposure to heat, vacuum or pressure treatment. However, most of the reported MOFs have low endurance in the above-mentioned operating environments. This drawback considerably hampers MOFs' practical application. Therefore, the preparation and application of stable MOFs have attracted immense attention owing to both importance and urgency.
000 m2 g−1) and tunable pore sizes/characteristics.1 These aspects endow MOFs with fantastic functionalities/properties for a variety of applications, for example, gas adsorption and separation, catalysis, sensors, drug delivery, proton conduction, etc.1–18 For various applications of MOFs, stability, mainly including chemical, thermal and mechanical stability, is one of the major concerns. The chemical stability of MOFs, specifically, refers to their resistance to the effects of exposure to various chemicals in their environment, e.g. moisture, solvents, acids, bases, and aqueous solutions containing coordinating anions. The thermal stability and mechanical stability of MOFs usually correlate with the ability of MOFs to preserve their structural integrity upon exposure to heat, vacuum or pressure treatment. However, most of the reported MOFs have low endurance in the above-mentioned operating environments. This drawback considerably hampers MOFs' practical application. Therefore, the preparation and application of stable MOFs have attracted immense attention owing to both importance and urgency.
Under normal conditions, the stability of MOFs arises from a variety of factors such as thermodynamic factors, kinetic factors and other factors in the operating environment.3,19 Thermodynamic factors are mainly related to the metal–ligand coordination bond strength.3,19 The coordination bond strength might be predicted by Pearson's hard soft acid base (HSAB) theory. In the de novo synthesis of MOFs, ligands with relatively high pKa (e.g. azoles) easily produce robust frameworks with low-valent metal ions, while linkers with relatively low pKa (e.g. carboxylic acids) tend to bind with high-valent metal ions to give stable structures.3,19,20 The inertness of metal clusters generally endows MOFs with excellent chemical stability, as confirmed in many MOFs, such as MIL-101(Cr).21 However, even with the same metal cluster and framework topology, the chemical stability of some MOFs decreases with the lengthening of the linker and the enlargement of pore size (as observed in UiO-66 series, SUMOF-7 series, etc.).22,23 This is mostly caused by kinetic factors, which are mainly related to the rigidity of the linker, coordination number, surface hydrophobicity, framework interpenetration, etc.3,19 In general, dense and rigid frameworks constructed by rigid and highly connected building blocks (metal ions/clusters and ligands) usually demonstrate high stability due to their high tolerance toward partial lattice collapse.19,24,25 Surface hydrophobicity prevents the adsorption of water into pores and/or the condensation of water around the metal clusters, which enhances the MOF stability in the presence of water.3 On the other hand, improved stability from framework interpenetration is likely to be caused by increased steric hindrances to ligand displacement.3 Further, the influence of reduced systematic energy of the interpenetrated structure cannot be ignored.26 Beyond these, the operating environment as an external factor also plays an important role in MOF stability. For instance, moisture-labile MOF-5 can retain its crystallinity upon heating at 300 °C in air for 24 h.27 Hence, the assessment of MOF stability for identification of suitable MOFs should be varied to meet the specific conditions in an operating environment.
The development of stable MOFs can be divided into several important stages and that began with the advent of the first porous MOF (MOF-5).27 In the early stage, the study of MOF stability was mainly focused on their thermal and mechanical stability.21,28–35 With the in-depth understanding of the framework collapse of MOFs in air,36 the development of stable MOFs entered the second stage. Numerous water-stable MOFs were reported based on their structural designability and chemical tunability. In the third stage, different techniques of crystal growth and crystal structure determination gave rise to a large number of extraordinarily stable MOFs, which effectively widened their application ranges and benefited their commercialization.
Over the past two decades, various promising methods have been developed to tune MOF stability, especially chemical stability, which might be the most important and significant prerequisite of MOFs for their diverse applications.19,37–39 Therefore, we believe that it is a good time for us to provide a systematic review of recent advances in the preparation of stable MOFs and their applications (Scheme 1). Our discussions begin with a summary of strategies that have been developed for improving the chemical stability of MOFs while emphasizing the key factors in the synthesis of stable MOFs and MOF-based materials. Next, the unique attributes of stable MOFs and their extensive applications, including adsorption and separation, heterogeneous catalysis, fluorescence sensing, biological and medical application, and proton conductivity, are introduced. At the end of the review, a summary and proposed future developments in this research field will be presented. Hopefully, this review will inspire the enthusiasm of scientists in chemistry and materials science and interdisciplinary researchers toward this field.
 | ||
| Scheme 1 Schematic illustration showing stable MOFs composed of metal clusters (blue or orange balls), organic linkers (grey lines) and dangling functional groups (green or pink segments) for diverse applications. | ||
2. Chemical stability
Although many MOFs are vulnerable to structural destruction even in an ambient atmospheric environment due to the lability of coordination bonds between metal ions and ligands, a growing number of MOFs with exceptional chemical stability have been reported in recent years. In general, two main types of approaches have been adopted to improve the chemical stability of MOFs. One is the de novo synthesis of stable unknown MOFs and the other is to improve the stability of existing MOFs. The chemical stabilities of some representative MOFs are summarized in Table 1.| MOFs | Metal | Ligands | Testing conditiona | Time | Ref. |
|---|---|---|---|---|---|
| a RT: room temperature. RH: relative humidity. Conc.: concentrated. | |||||
| MAF-X27-Cl | Co(II) | bbta | 0.001 M HCl, 1.0 M KOH | 1 week | 88 |
| H3[(Cu4Cl)3-(BTTri)8] | Cu(II) | BTTri | Boiling water | 3 days | 87 |
| pH = 3 | 1 day | ||||
| FJI-H14 | BTTA | pH = 2–12 (373 K) | 24 h | 95 | |
| BUT-155 | tdhb | pH = 4–10, boiling water | 24 h | 97 | |
| USTC-6 | 4,4′-(Hexafluoroisopropylidene)diphthalicanhydride | pH = 2–10 | 7 days | 109 | |
| Water | 1 month | ||||
| Ni3(BTP)2 | Ni(II) | BTP | Boiling solutions of pH = 2–14 | 14 days | 83 |
| PCN-601 | TPP | Saturated NaOH (100 °C) | 24 h | 25 | |
| 0.1 mM HCl (RT) | |||||
| PCN-602(Ni) | TPPP | 1 M Na2CO3, K3PO4, KF, pH = 4–14 | 24 h | 84 | |
| ZIF-8 | Zn(II) | MeIM | Boiling benzene, methanol, and water | 7 days | 79 |
| 8 M NaOH (100 °C) | 24 h | ||||
| USTC-7 | TZBPDC | Boiling organic solvents, pH = 2–12 (RT) | 12 h | 94 | |
| Water (RT) | 4 months | ||||
| oCB-MOF-1 | bdc, oCB-L | pH = 2–12 (RT) | 15 h | 108 | |
| MIL-53(Al) | Al(III) | BDC | pH = 4–12 | 3 days | 44 |
| NH2-MIL-53(Al) | NH2-BDC | 2 months | |||
| Al-TCPP | TCPP | pH = 5–8 (RT) | 1 week | 49 | |
| PCN-333(Al) | TATB | pH = 3–9 | Overnight | 50 | |
| 467-MOF | BTTB | pH = 0–13 (RT) | 36 h | 74 | |
| Water (RT) | 30 days | ||||
| Boiling water | Over 1 week | ||||
| AlTCS-1 | TCS | pH = 1–11, aqua regia solution | 24 h | 75 | |
| MIL-53(Cr) | Cr(III) | BDC | Solution of 7.0 × 10−2 M NaOH | 6 h | 21 |
| Solution of 7.0 × 10−2 M HCl | 48 h | ||||
| MIL-101(Cr) | pH = 0–12 | 2 months | 44 | ||
| MIL-100(Cr) | BTC | Water | 12 months | 64 | |
| PCN-333(Fe) | Fe(III) | TATB | pH = 3–9 | Overnight | 50 |
| PCN-600(Fe) | TCPP-Fe | pH = 2–11 | 24 h | 61 | |
| MIL-100(Fe) | BTC | pH = 1–10 | 7 days | 65 | |
| PCN-250(Fe2Co) | Fe(II,III), Co(II) | BTB, 4,4′-(diazene-1,2-diyl)dibenzoate | Water | 6 months | 60 |
| pH = 1–12 | 24 h | ||||
| [(CH3)2NH2]2[Eu6(μ3-OH)8(NDC)6(H2O)6] | Eu(III) | NDC | pH = 3.5–10 (373 K) | 24 h | 106 |
| [La(TTCA) (H2O)]·DMF·H2O | La(III) | TTCA | Boiling water, pH = 1–13 | 7 days | 52 |
| [La(BTB)H2O] | BTB | pH = 2–14 (100 °C) | 3 days | 54 | |
| USTC-8(In) | In(III) | TCPP | pH = 0–11 | 12 h | 76 |
| MIL-177-LT | Ti(IV) | mdip | Water (RT) | 30 days | 66 |
| Boiling water | 24 h | ||||
| Conc. HCl, HNO3, H2SO4 and HPO4, aqua regia (RT) | 7 days | ||||
| UiO-66 | Zr(IV) | BDC | pH = 0–12 | 2 months | 44 |
| PCN-777 | TATB | pH = 3–11 | 12 h | 51 | |
| DUT-98 | CPCDC | Boiling water or solutions of 2–6 M HCl (298 K) | 24 h | 53 | |
| PCN-222(Fe) | TCPP-Fe | Boiling water, conc. HCl | 24 h | 55 | |
| PCN-223 | TCPP | pH = 0–10 | 24 h | 56 | |
| PCN-224(Ni) | TCPP-Ni | pH = 0–11 | 24 h | 57 | |
| PCN-225 | TCPP | pH = 1–11 | 12 h | 58 | |
| PCN-228 | TCP-1 | pH = 0–12 | 24 h | 59 | |
| PCN-229 | TCP-2 | ||||
| PCN-230 | TCP-3 | ||||
| BUT-12 | CTTA | Conc. HCl, pH = 10 | 24 h | 63 | |
| BUT-13 | TTNA | ||||
| NUPF-1 | 4,4′,4′′,4′′′-((4,4′,4′′,4′′′-(Porphyrin-5,10,15,20-tetrayl)tetrakis(benzoyl))tetrakis(azanediyl))tetrabenzoic acid | 12 M HCl, pH = 9 | 3 days | 67 | |
| MOF-808 | BTC | Boiling water, conc. HCl, pH = 10 | 24 h | 68 | |
| MIP-202(Zr) | Aspartic acid | Boiling water, pH = 0–12 | — | 73 | |
| Zr(H4L1) | Tetraphenylsilane tetrakis-4-phosphonic acid (H8L1) | pH = 12 (100 °C), conc. HNO3 (100 °C) | 7 days | 77 | |
| ZrPP-1 | THPP | 0.1 M HCl to 20 M NaOH | 7 days | 78 | |
| UiO-67-o-2CF3 | BPDC-o-2CF3 | Boiling water, 8 M HCl, 50 ppm NaF aqueous solution | 24 h | 100 | |
2.1 Governing MOFs towards high stability via de novo synthesis
It is widely believed that the degradation of MOFs generally has two aspects, namely, the metal–ligand bond breaking, and the formation of products that are more stable as compared with pristine MOFs.40,41 Hence, the chemical stability of MOFs depends strongly on the intrinsic structures of MOFs (internal factors), including the charge density of metal ions, connection numbers of metal ions/clusters, basicity and configuration as well as hydrophobicity of ligands, etc. Following these considerations, judicious selection of metal nodes and design of linkers enable the construction of MOFs with sound stability.42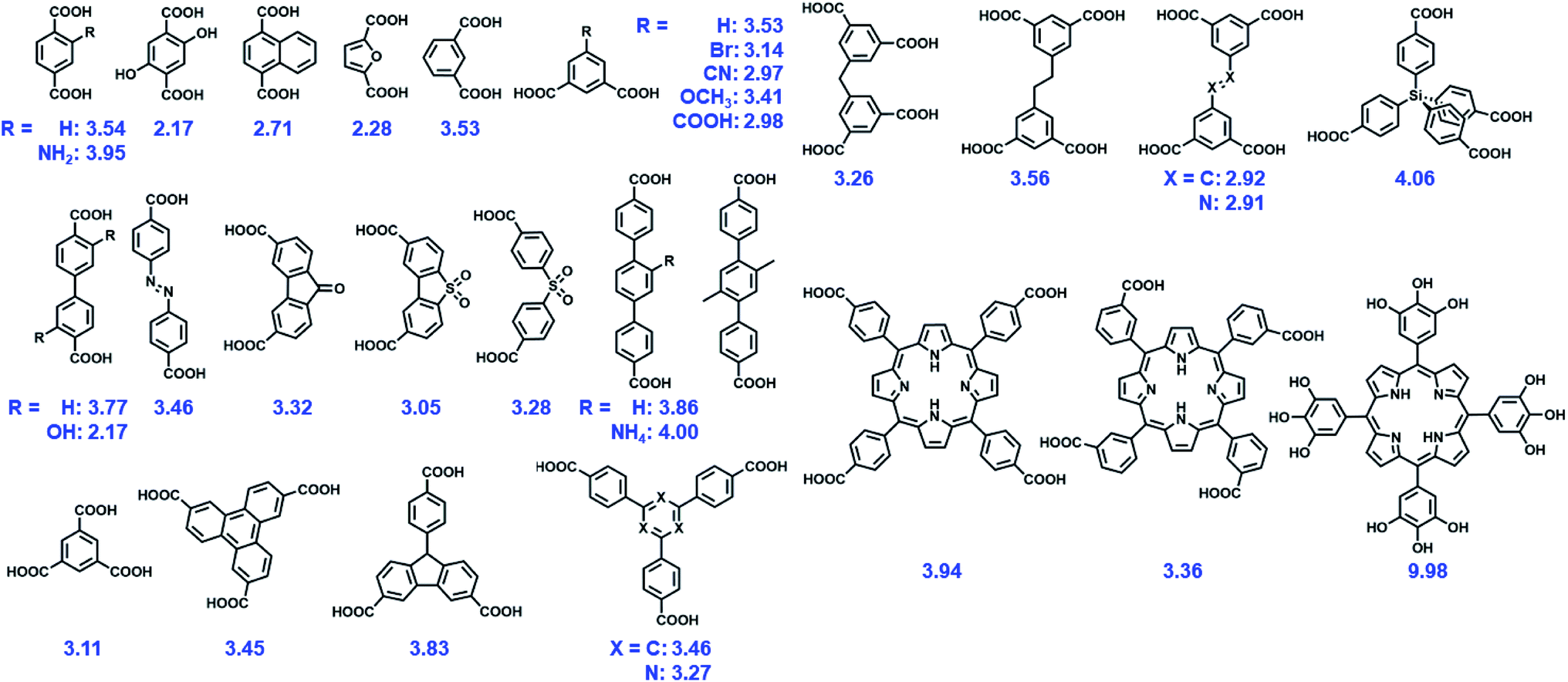 | ||
| Fig. 1 Some representative O donor ligands, with their pKas referred in this text. | ||
As an early example, Férey and co-workers fabricated a Cr(III)-based MOF, MIL-101(Cr), with very large pore sizes (2.9 and 3.4 nm) and Brunauer–Emmett–Teller (BET) surface area (∼4000 m2 g−1) via the combination of targeted chemistry and computational design.43 By virtue of the inertness of Cr–carboxylate oxygen bonds, MIL-101(Cr) is highly resistant to the attack of acid and alkali, making it stable in aqueous solutions of pH = 0–12 for 2 months.44 Using a similar strategy, a variety of high-valent metal-containing MOFs with high chemical stability have been reported, including MIL-100(Cr), MIL-53(Cr), UiO series, MIL-125, Al-TCPP, PCN series, etc.22,45–68 However, the strong M–O bonds often lead to the poor crystallinity and the small sizes of resultant MOF particles. This increases the difficulty of structural characterization with single-crystal X-ray diffraction. For this reason, most of the crystal structures of the above-mentioned MOFs are determined by using powder X-ray diffraction.
To address this issue, Behrens's group developed a modulated synthesis strategy to regulate the size of Zr-MOFs from nanosized crystallites to large single crystals, and reported the first Zr-MOF single crystal that was sufficiently large for single-crystal X-ray diffraction.69 By adjusting the concentration of the modulator (i.e. monocarboxylic acid) in the synthetic system, the nucleation rate of MOFs can be controlled, which ultimately leads to the formation of highly crystalline products or even large single crystals.69,70 With this in mind, Zhou's group employed Fe-tetrakis(4-carboxyphenyl)porphyrin, Zr4+ and benzoic acid separately as the ligand, metal ion and modulator to yield needle-shaped single crystals, PCN-222(Fe), also called MOF-545 and MMPF-6.55,71,72 The stability experiments showed that this MOF was able to survive even in concentrated aqueous HCl solution for 24 h due to the strong Zr–O bonds in the 8-connected Zr-clusters and the chelating effect between Fe(III) and porphyrin. A similar phenomenon was observed in other PCN-series reported by the same team.50,56–62 In fact, monocarboxylic acids can also work as solvents in the preparation of stable Zr(IV)-based MOFs. The common solvent in MOF synthesis, dimethylformamide (DMF), was replaced by a mixture of monocarboxylic acid (formic acid) and acetic anhydride to afford a large-pore MOF, MIP-200, based on Zr4+ and 3,3′,5,5′-tetracarboxydiphenylmethane (H4mdip).73 Remarkably, MIP-200 exhibited excellent stability under extremely harsh conditions, including concentrated strong acids (HCl and HNO3), highly concentrated H3PO4 and H2SO4, and NH4OH vapor. The modulated synthesis strategy applies equally to trivalent metal-based MOFs. Using formic acid as the modulator, Du's group successfully fabricated an Al-based MOF (467-MOF) based on a flexible ligand (4,4′,4′′-[benzene-1,3,5-triyl-tris(oxy)]tribenzoic acid, H3BTTB).74 Chemical stability investigation for the 467-MOF indicated that it remained stable in aqueous solutions of HCl or NaOH of a broad pH range from 1 to 11. Similarly, with the aid of hydrofluoric acid, large and good-quality single crystals of Al-MOF, AlTCS-1, were obtained based on tetrakis(4-oxycarbonylphenyl)silane.75 Strikingly, AlTCS-1 could be stable in both aqueous solutions of pH 1 to 11 and aqua regia solution for at least 24 h. Other M3+ ions like In3+ can also form stable MOFs with O donor linkers. Recently, Jiang's group synthesized an unusual out-of-plane (OOP) porphyrin-based MOF, USTC-8(In), by employing In(OH)3 and HNO3 separately as the metal precursor and modulator.76 Due to the strong coordination bonds between In3+ ions and porphyrin carboxylate ligands, USTC-8(In) could remain intact in aqueous solutions with pH values ranging from 2 to 11 for 12 h.
Note that high-valent metal-containing MOFs with remarkable chemical stability include not only carboxylate-based MOFs, but also those with phosphonate and phenolate-based frameworks. Sun's group selected tetraphenylsilane tetrakis-4-phosphonic acid as the ligand and constructed a zirconium-phosphonate network that displayed distinctive tolerance to aqueous solutions with a wide range of pH values (1–12) and concentrated acids.77 As for phenolate-based MOFs, the relatively high pKa of the phenolate ligands can afford stronger M–O bonds with Zr4+ ions in comparison to carboxylate linkers. Lin's group demonstrated the fabrication of a series of zirconium polyphenolate-(metallo)porphyrin MOFs, in which the highly stable 5-membered rings generated by the chelating effect of polyphenols greatly improved the stability of resultant MOFs (Fig. 2a).78 Remarkably, the prototypic ZrPP-1 not only remained intact in strong acid (HCl, pH = 1) but also presented high robustness in saturated NaOH solution for 168 h (Fig. 2b), offering an ideal platform for diverse applications.
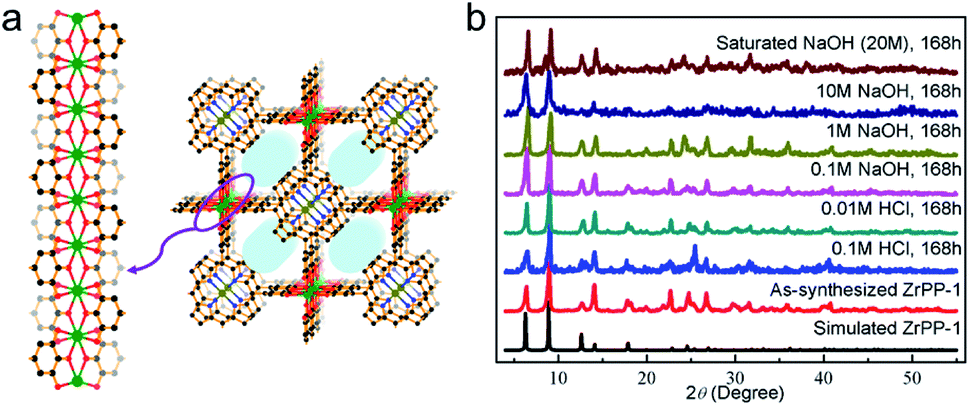 | ||
| Fig. 2 (a) The structure of ZrPP-1 constructed from Zr4+ and 5,10,15,20-tetrakis(3,4,5-trihydroxyphenyl)porphyrin with the view of the ZrIV-pyrogallate (=1,2,3-trioxobenzene) coordination chain. Color code: C, black; O, red; N, blue; Zr, green; and metal (in the porphyrin core), olive green. H atoms are omitted for clarity. (b) Powder X-ray diffraction (PXRD) patterns of ZrPP-1 under different conditions. Adapted from ref. 78 with permission from Wiley-VCH, copyright 2018. | ||
To summarize, the basic principle for the fabrication of stable high-valent metal-containing MOFs is the strong coordination bond, high coordination number of metal ions, inertness of metal and others, which enhance the MOF tolerance of hydrolysis before framework collapse occurs.
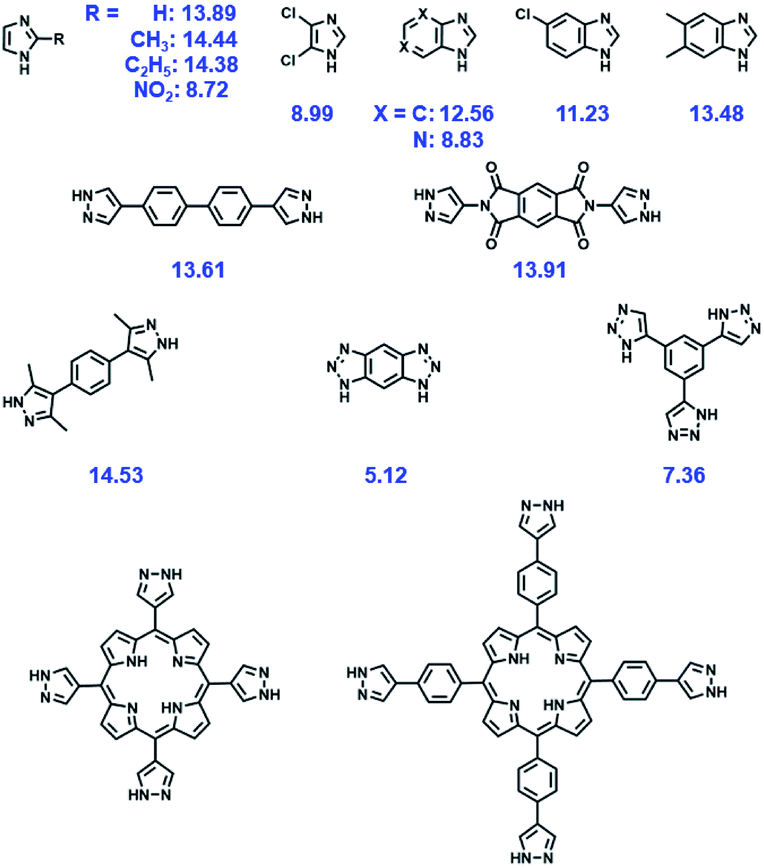 | ||
| Fig. 3 Some representative N donor ligands, with their pKas referred in this text. | ||
As a representative example, zeolitic imidazolate framework (ZIF)-8 (also called MAF-4) which possesses a zeolite-type topology composed of four-coordinated Zn2+ ions linked by imidazolate linkers is very stable in an aqueous environment.79,80 In particular, the structure of ZIF-8 can be well maintained even in 8 M aqueous NaOH for 24 h at 100 °C, indicating the exceptional stability of this framework under alkaline conditions. A similar stability can be observed also in other Zn-based ZIF series like ZIF-68, -69, and -70.81 Not limited to imidazoles, reacting the pyrazole-derived ligand with suitable low-valent metal salts can also lead to the formation of MOFs with high chemical stability. Volkmer's group deliberately selected 1,4-bis[(3,5-dimethyl)-pyrazol-4-yl]benzene (H2bdpb) and Co(II) salts to fabricate a hydrolytically stable MOF (MFU-1), which is isostructural with MOF-5.82 The relatively strong bonds in the {CoON3} coordination units of MFU-1 give rise to the decent thermodynamical stability of the framework under ambient conditions for over 6 months. Similarly, using the reaction between the pyrazol-type ligand (1,3,5-tris(1H-pyrazol-4-yl)benzene, H3BTP) and Ni2+ ions, an ultrastable framework, Ni3(BTP)2, was obtained and the chemical resistance experiments indicated that it was stable in boiling aqueous solutions of pH 2 to 14 for 2 weeks.83 Recently, Zhou, Li and coworkers utilized a top-down strategy to rationally synthesize an alkali stable MOF, PCN-601, based on a pyrazolate-based porphyrinic ligand (Fig. 4a).25 All the strong coordination bonds, relatively short ligand length, and high connectivities of the porphyrinic ligand and [Ni8] cluster ([Ni8(OH)4(H2O)2Pz12], Pz = pyrazolate) guaranteed the high chemical resistance of PCN-601 to saturated NaOH solution (∼20 M) at 100 °C. In view of the small window size of PCN-601 (∼2.1 × 8.0 Å), the groups then fabricated PCN-602(Ni) which is isostructural with PCN-601 using the reticular synthesis strategy (Fig. 4a).84 Apart from the high resistance to alkaline solution (pH = 14), PCN-602(Ni) also displayed reasonable tolerance to aqueous solutions with coordinating anions (e.g. CO32−, PO43−, F−) (Fig. 4b and c), which was very rare in other high-valent metal-based MOFs like PCN-224(Co), PCN-222(Fe), and PCN-600(Mn). Actually, the investigation of [Ni8] cluster-based MOFs can be traced back to the year 2010.85,86 However, unlike carboxylate-based MOFs and the above-mentioned PCN-601 and PCN-602(Ni), [Ni8] cluster-based MOFs fabricated with 4,4′-bis(1H-pyrazol-4-yl)biphenyl and 2,6-bis(1H-pyrazol-4-yl)pyrrolo[3,4-f]isoindole-1,3,5,7(2H,6H)-tetrone are polycrystalline products.85
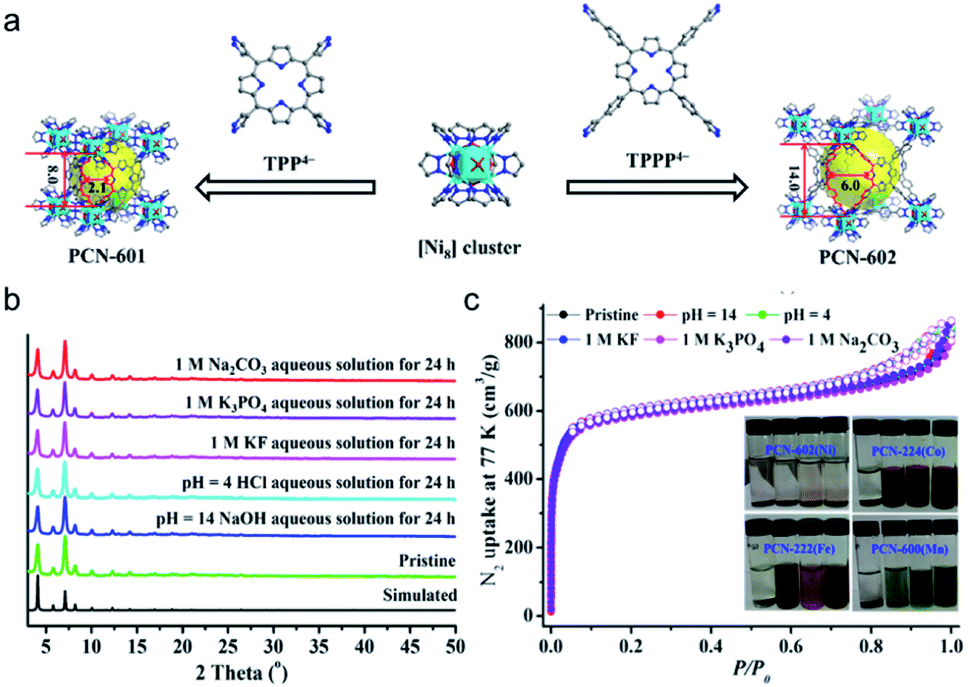 | ||
| Fig. 4 (a) Reticular design and construction of PCN-601 and PCN-602(Ni) from TPP4− and TPPP4− ligands, respectively. Color code: C, gray; O, red; N, blue; and Ni, cyan. H atoms are omitted for clarity. (b) PXRD patterns of PCN-602(Ni) under different conditions. (c) N2 sorption isotherms of the pristine PCN-602(Ni) sample and of those after soaking in different aqueous solutions (inset: photographs of PCN-602(Ni), PCN-224(Co), PCN-222(Fe), and PCN-600(Mn) soaked in different solutions for 24 h: deionized water, 1 M KF, 1 M Na2CO3, and 1 M K3PO4 aqueous solutions, from left to right). Adapted from ref. 84 with permission from the American Chemical Society, copyright 2017. | ||
The self-assembly from low-valent metal ions and triazole-based linkers can also afford highly stable MOFs. Long's group reported a triazolate-bridged framework, Cu-BTTri, which displayed high tolerance even after soaking in boiling water and in a solution of HCl (pH = 3) for 3 days and 1 day, respectively.87 Lu et al. discovered that MAF-X27-Cl as a triazole-based framework was able to retain its structural integrity not only in acidic solution (0.001 M HCl) but also in strong alkaline solution (1.0 M KOH) for over 1 week.88 It should be noted that, when tetrazole-based linkers are utilized as ligands, the resultant low-valent metal containing MOFs often present weak stability on account of their relatively low basicity and weak coordination ability.19,89
In short, low-valent metal ions afford hydrostable skeletons with N donor linkers on account of the relatively high basicity (high pKa) of these linkers. However, these resultant frameworks usually show poor resistance to acidic solutions due to the fairly strong affinity between protons and azolate groups.
For instance, to improve the hydrostability of MOF-5, Zn2+ ions were partially replaced with Ni2+ ions to give the Ni-doped MOF-5.90 In particular, when the Ni content reached 22%, the resultant Ni-doped MOF-5, due to the stable Nix(Zn)4−xO6+ cluster secondary building units (SBUs), was able to retain its integrity when exposed to static air conditions (25 °C, 30–37% RH) for one week. Furthermore, Ni-doping increased the Langmuir specific surface area and pore size of the resultant MOF on account of the prevention of framework interpenetration. Employing a similar strategy, a few metal ions, including Cu2+, Cd2+, and Fe2+, were separately doped into a gyroidal MOF, STU-1.91 Unlike the poor hydrostability of pure STU-1, it was found that all metal doped STU-1s retained their morphology and crystallinity even after soaking in boiling water for 7 days. Beyond that, after doping with metal ions, the significantly increased surface hydrophobicity of STU-1 was proved by water vapor adsorption isotherm curves. The authors speculated that the enhanced hydrophobicity could be ascribed to the perturbation on the MOF surface by doping with metal ions, which impeded the formation of water clusters on the pore surface and improved the water stability of the resultant MOFs.
Indeed, this strategy powerfully enhances the chemical stability of MOFs. However, this advantage is offset by the fact that many metal ions are difficult to dope into the framework by this process. Additionally, the sites of doped metal ions are usually indistinct even when observed by means of the single-crystal X-ray diffraction technology. These factors pose challenges for the mechanism study.
By substituting the carboxylate ligand (BDC) with an azole-containing carboxylic acid ligand (3,5-dimethyl-4-carboxypyrazole, H2dmcapz), Montoro et al. successfully obtained a MOF-5 type framework with Zn2+ ions.92 Because of more robust Zn–N bonds, this MOF was highly stable in water and boiling organic solvents for 24 h as compared with the moisture-sensitive MOF-5. In addition, the methyl groups of the linker improved the hydrophobicity of the MOF, which provided protection to the weak Zn–O bonds against hydrolysis. Even when 4-(3,5-dimethyl-1H-pyrazol-4-yl)benzoic acid (HMe2pzC6H4CO2H) was employed as a linker, the resultant MOF-5 analogs could withstand prolonged contact with the water/DMF mixture.93 Using a π-conjugated ligand, 4′-(1H-tetrazol-5-yl)-[1,10-biphenyl]-3,5-dicarboxylic acid (H3TZBPDC), involving both soft- and hard-base coordination sites, a zinc-based MOF, USTC-7, with large single-crystal sizes, was successfully synthesized by Jiang's group.94 As expected, USTC-7 demonstrated its outstanding chemical stability by preserving its framework integrity not only in various boiling solvents but also in pH = 2–12 aqueous solutions for 12 h. Besides zinc azolate-carboxylate frameworks, recently, Liang et al. reported a highly stable MOF, FJI-H14, synthesized by a self-assembly process from Cu2+ ions and 2,5-di(1H-1,2,4-triazol-1-yl)terephthalic acid (H2BTTA).95 Both the penta-coordinated Cu(II) ion subunit formed by Cu–N bonds and Cu–O bonds, and the abundant free N atoms effectively improved the stability of FJI-H14 under acidic and alkaline conditions.
To illustrate the effectiveness of hydrophobic ligands, a methyl group modified MOF-5 was synthesized.96 The H2 adsorption isotherm revealed that the H2 uptake capacity of 2,5-dimethyl-modified MOF-5 was preserved even after a four-day exposure to ambient air. Chen et al. designed and synthesized an octatopic carboxylic ligand, 3,3′,5,5′-tetrakis(3,5-dicarboxyphenyl)-2,2′,4,4′,6,6′-hexamethylbiphenyl, to prepare a copper(II)-paddlewheel MOF (BUT-155).97 Compared with the common copper(II)-paddlewheel MOFs, the high connectivity and abundance of hydrophobic methyl groups endowed BUT-155 with exceptional chemical stability in aqueous solutions of a wide pH range (4–10) and boiling water for 24 h.
To investigate the influence of side-chain length, a series of isostructural NbO-type MOFs were tested.98 Upon extending the length of dialkoxy-substituents (from C1 to C6) on the organic linker, the moisture stability of the resultant Cu(II)-MOF improved noticeably while the thermal stability decreased. Furthermore, an inevitable problem was the gradual drop in porosity of the resultant MOFs after the incorporation of these hydrophobic functional groups. In addition to the length of the side chain, the position of the hydrophobic groups relative to metal clusters also plays a critical role in improving MOF chemical stability.99,100 A convincing comparison was made between MOF-508 and its two analogs (SCUTC-18 and SCUTC-19).99 Compared with bipyridine of MOF-508, there were two methyl-substituents, respectively, located at the ortho-positions and meta-positions of the coordinating N atoms of bipyridine in SCUTC-18 and SCUTC-19. Upon exposure to humid air for 30 days, only the porous structure of SCUTC-18 was maintained due to the introduction of methyl-substituents at sites more adjacent to the metal clusters.
Apart from alkyl chains,96–101 introduction of fluorinated groups (e.g. –F, –CF3, etc.) or other hydrophobic substituent groups into organic linkers is also an efficient means for enhancing MOFs' moisture/water resistance.102–114 Jiang's group reported a copper-based MOF, USTC-6, based on 4,4′-(perfluoropropane-2,2-diyl)diphthalic acid.109 Although most Cu–O coordination bonds in MOFs are vulnerable to hydrolysis, USTC-6 exhibits exceptional tolerance to water and aqueous solutions in the pH range 2 to 10 on account of the hydrophobicity of the corrugated –CF3 surface. Cohen's group developed a bridging co-ligand strategy to construct a series of polymer-metal-organic frameworks (polyMOFs) (Fig. 5).107 Remarkably, all polyMOFs inherited the hydrophobicity from the substituted H2BDC derivatives with water contact angles (WCA) of 110–120°. As expected, several polyMOFs displayed outstanding stability even after exposure to 90% RH at 25 °C for 7 days.
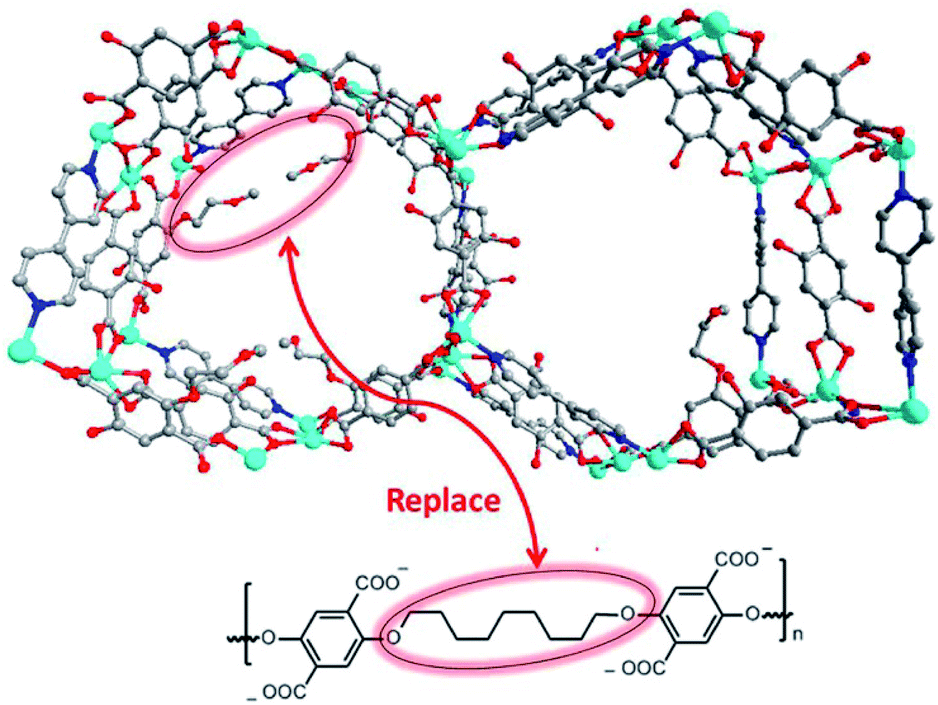 | ||
| Fig. 5 (top) Packing diagram of representative [Zn2(BME-bdc)2(bpy)]]n (BME-bdc = 2,5-bis(2-methoxyethoxy)-1,4-benzenedicarboxylate; bpy = 4,4′-bipyridine) along the c-axis direction; (bottom) design concept for producing a polyMOF analogue of [Zn2(BME-bdc)2(bpy)]]nvia replacing dangling groups by polymer chains. Color code: C, gray; O, red; N, blue; and Zn, cyan. H atoms are omitted for clarity. Reproduced from ref. 107 with permission from the American Chemical Society, copyright 2016. | ||
By adding bpy into the synthetic system of [Cu2(obb)2(DMF)2]·2DMF (H2obb = 4,4′-oxybis(benzoic acid)), the coordinated DMF molecules were substituted with bridging bpy ligands, which generated [Cu2(obb)2(bpy)0.5(DMF)]·2DMF.116 Unlike the negligible gas uptake capacities of the pristine MOF, [Cu2(obb)2(bpy)0.5(DMF)]·2DMF exhibited significantly increased adsorptive properties for N2, CO2 and H2 on account of its enhanced stability and rigid framework. Using a similar strategy, bpy was employed as size-matching ligand braces to bridge two metal clusters in [Co3(μ3-O)(adc)3(DMA)3]2(C2H6NH2) (H2adc = 9,10-anthracenedicarboxylic acid, DMA = N,N′-dimethylacetamide), affording a robust [Co3(μ3-O)(adc)3(bpy)(DMA)]2(C2H6NH2) with permanent porosity and preferential adsorption of CO2 and O2 over N2 due to the increase in host–guest interactive sites.117
In the case of MIL-88 type MOFs, organic linkers with C3 symmetry are the ideal size-matching MBBs. For example, both [Co2(ina)3(H2O)2]+ (ina = isonicotinate) and 2,4,6-tri(4-pyridyl)-1,3,5-triazine (tpt) were inserted into the open channels during the MOF formation (Fig. 6).118 The grand canonical Monte Carlo (GCMC) simulations and density functional theory (DFT) calculations revealed that the framework energy was drastically reduced upon the introduction of these two MBBs, especially for tpt. The stability tests then proved that the tpt substituted MOF showed greater stability in an aqueous environment as compared to the [Co2(ina)3(H2O)2]+ substituted counterpart and the experimental results agreed closely with the results obtained by computation. The MBBs could also be generated during the MOF synthesis.119,120 Chen et al. utilized the solvothermal reaction of CoCl2·6H2O and H2TZB (TZB = 4-(1H-tetrazol-5-yl)benzoate) to fabricate a MIL-88 type MOF.119 By adding isonicotinic acid into the synthetic system, [Co2(INT)3(H2O)2] (INT = isonicotinate) MBBs were formed in situ and incorporated into the resultant framework. As tridentate brackets, [Co2(INT)3(H2O)2] MBBs effectively improved the rigidity of the MOF, avoiding the framework collapse during solvent exchange. A similar phenomenon was also observed in NENU-401.120
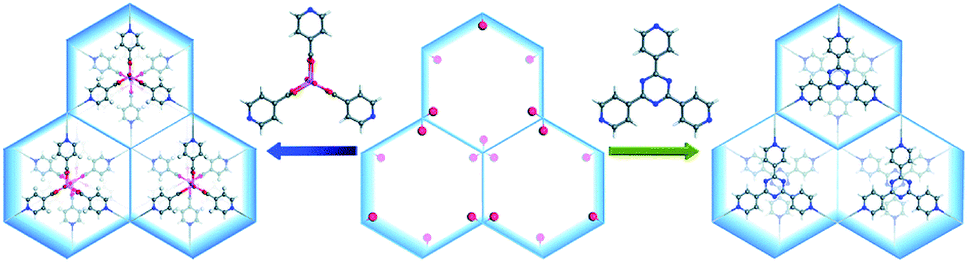 | ||
| Fig. 6 Structural comparison of two analogous MOFs in which the channels are inserted with [Co2(ina)3(H2O)2]+ (left) and 2,4,6-tri(4-pyridyl)-1,3,5-triazine brackets (right), respectively. Color code: C, gray; O, red; N, blue; H, white; and Co, pink. Reproduced from ref. 118 with permission from The Royal Society of Chemistry, copyright 2017. | ||
For example, the stabilities of two isostructural pillared MOFs, MOF-508 and DMOF, upon exposure to water were compared by Walton's group.121 Upon exposure to RH = 90%, the surface area of non-interpenetrated DMOF dramatically declined (from 1980 to 7 m2 g−1). In contrast, the two-fold interpenetrated MOF-508 showed no significant change in the surface area after the same treatment. This clearly demonstrated the enhanced framework stability resulting from catenation. A similar phenomenon was also observed in two-fold catenated Co-BTTB-DMBPY and Zn-BTTB-DMBPY.122 Actually, the influence of the interpenetration direction on framework stability is also very apparent. Using the solvothermal reaction of Zn(NO3)2 and 4-(3,5-dimethyl-1H-pyrazol-4-yl)benzoic acid (H2mpba), three isomeric MOFs with different polar nets were reported.123 Interestingly, although all these three MOFs present 4-fold interpenetrating structures, the different interpenetration directions endow them with entirely different levels of porosity and stability. Molecular mechanics (MM) calculations revealed that the framework energy of the nonporous isomer was the lowest. As expected, after being immersed in water, the other two nanoporous isomers transformed into the most stable framework (nonporous isomer).
Recently, Bu's group demonstrated that a self-penetrated network was more stable than an interpenetrated framework (Fig. 7).124 By employing 5,5′-((thiophene-2,5-dicarbonyl)bis(azanediyl))diisophthalic acid and 5,5′-([2,2′-bipyridine]-5,5′-dicarbonyl)bis(azanediyl)diisophthalic acid separately as the organic linkers, NKU-112 with a two-fold interpenetrated framework and NKU-113 with a self-interpenetrated framework were prepared. In NKU-113, the coordinated DMF molecules of metal clusters were substituted by chelating the bipyridine moiety of 5,5′-([2,2′-bipyridine]-5,5′-dicarbonyl)bis(azanediyl)diisophthalic acid. This greatly increased the framework rigidity of NKU-113 as compared to NKU-112. After solvent exchange and supercritical drying, NKU-113 retained its framework and displayed a moderate BET surface area of 1486 m2 g−1, while only a negligible amount of N2 was adsorbed in NKU-112 due to its collapsed structure.
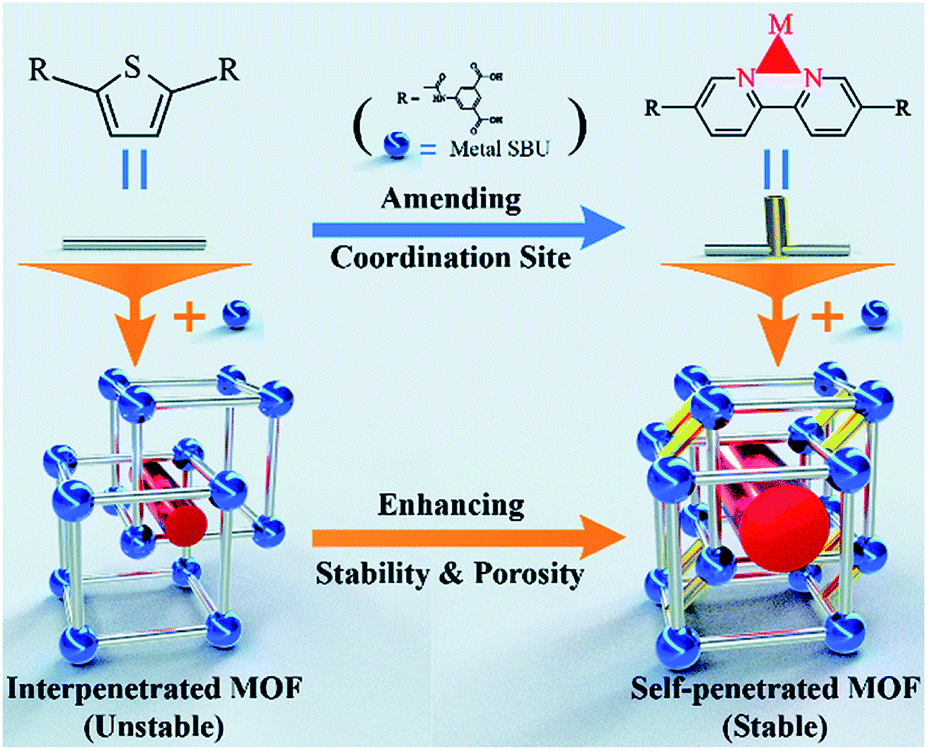 | ||
| Fig. 7 Schematic diagram showing the framework and the modification strategy. The blue-colored balls, silver-colored sticks, yellow-colored sticks, and red-colored cylinders represent metal SBUs, ligands, coordinate bonds and pores, respectively. Reproduced from ref. 124 with permission from The Royal Society of Chemistry, copyright 2018. | ||
2.2 Improving the chemical stability of existing MOFs
Given that the rational strategy for preparing stable MOFs via de novo syntheses remains limited, a large number of strategies for enhancing the stability of existing MOFs have been developed. In this section, four major approaches, including post-synthetic exchange, post-synthetic modification, hydrophobic surface treatment, and composite fabrication, will be discussed.As regards metal-ion metathesis, an early example was found in SDU-1.125 By soaking into a Cu2+ containing solution, the Zn2+ ions of SDU-1 were exchanged by Cu2+ ions in a single-crystal-to-single-crystal (SC–SC) fashion, giving a stable Cu-SDU-1. After the removal of the guest solvent, the N2 adsorption capacity of Cu-SDU-1 was ∼3 times higher than that of SDU-1, demonstrating the increase of framework stability upon PSE. In 2014, Zhou's group developed a postsynthetic metathesis and oxidation strategy to transform the labile PCN-426-Mg into two robust MOFs, PCN-426-Fe(III) and PCN-426-Cr(III).126 Although PCN-426-Mg was labile in water, PCN-426-Fe(III) could remain intact even after immersion in water for one day. When it came to PCN-426-Cr(III), essentially unchanged crystallinity was found in solutions in the range from pH = 12 to extremely acidic conditions for over 12 h. Inspired by the high stability of Cr(III)-based MOFs, PCN-333(Cr) was obtained by the reduction of PCN-333(Fe) with Cr(II) ions and the subsequent reductive labilization process.127 PCN-333(Fe) could only maintain its structural integrity in aqueous solutions with a pH range from 3 to 9, while PCN-333(Cr) was stable at pH ranging from 0 to 11. Wang et al. demonstrated that exchange solvent played a vital role in the PSE process of MOFs.128 Acetone was proven to be a promising solvent for a postsynthetic metal exchange due to the direct participation of the carbonyl group via coordination interaction with metal ions. With the aid of acetone, several Fe/Cr-exchanged MOFs were obtained from the Fe(III)-based counterparts under very mild conditions and all these modified MOFs presented enhanced chemical stability.
Compared with investigations into postsynthetic metal exchange, there are fewer investigations into postsynthetic linker exchange and counterion exchange for improving the stability of MOFs. Utilizing DUT-67 as the template, the polarity of the inner surface was tuned through the postsynthetic exchange of the modulator by fluorinated monocarboxylates, such as trifluoroacetic acid, pentafluorobenzoic acid, perfluorooctanoic acid, etc.129 The incorporation of fluorinated linkers rendered the resultant MOF hydrophobic, greatly increasing their tolerance to water. Upon water adsorption at 298 K, only 0.3% porosity loss was observed in the perfluorooctanoic acid-modified DUT-67, while the same treatment caused a porosity loss of 34% in the parent DUT-67. As for postsynthetic counterion exchange, a series of alkylammonium cations were employed to adjust the surface wettability of bio-MOF-1.130 Water stability tests demonstrated that the large-sized tetraethylammonium (TEA) and tetrabutylammonium (TBA) cations with hydrophobic nature effectively protected bio-MOF-1 from the attack of water molecules. In contrast, significant deformation took place in the parent bio-MOF-1 and the small-sized tetramethylammonium (TMA) or hydrophilic triethylhydroxylmethylammonium cation modified frameworks after exposure to water vapor.
As a very early example, IRMOF-3 was modified with various alkyl anhydrides to give moisture-resistant IRMOF-3-AMX (X = 1–6, 15, iPr, iBu).132 The longer the chain of alkyl anhydride was, the more hydrophobic the resultant modified MOF became. In sharp contrast to the significantly reduced crystallinity of IR-MOF-3 (WCA: ∼0°), no evident change was found in the crystallinity of IRMOF-3-AM15 (WCA: 123 ± 5°) upon exposure to ambient air for 4 days, revealing the protective effects from the modified alkyl chains on MOFs. The same approach was also applied to NH2-MIL-53(Al) and Cu3(NH2BTC)2, greatly increasing their stability.132,133 Using a diazotization reaction between amidogen and 1-methylindole, NH2-UiO-66 was successfully transformed into UiO-66-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-ind.134 By comparison, the resistance to the acidic and alkaline environment of UiO-NN-ind was expanded from the pH range of 1–9 (NH2-UiO-66) to 1–12. However, the action was not limited to the aforementioned functional groups. Hydrophobic phenylisocyanate could also be modified on the framework to protect the MOF from the attack of water molecules.135
N-ind.134 By comparison, the resistance to the acidic and alkaline environment of UiO-NN-ind was expanded from the pH range of 1–9 (NH2-UiO-66) to 1–12. However, the action was not limited to the aforementioned functional groups. Hydrophobic phenylisocyanate could also be modified on the framework to protect the MOF from the attack of water molecules.135
As for the post-synthetic modification of metal clusters, hydrophobic groups were grafted at the Zr6-oxo nodes of NU-1000 via the solvent-assisted ligand incorporation (SALI) process.136 The dangling organic carboxylates could effectively increase the tolerance of Zr6-oxo nodes to water, albeit with the reduction of pore volume. Specifically, perfluorodecanoic-acid-functionalized NU-1000 could maintain its crystallinity and porosity even after 20 cycles of water vapor adsorption and desorption, proving its strong hydrostability. Moreover, when monotopic carboxylate was replaced with the phosphonate ligand, the resultant MOF exhibited higher resistance to hydroxide as a result of stronger bonds between the phosphonate ligand and Zr6-oxo nodes.137 The same effect was also found in the PCN-222 system by employing the SALI strategy with diphenylphosphinic acid,138 demonstrating the universal applicability of this method. Recently, Kim's group obtained a super-hydrophobic MOF (WCA: 161°), NH2-UiO-66-shp, by modifying the Zr6-oxo clusters of NH2-UiO-66 (WCA: 0°) with phenylsilane via pore-surface engineering (Fig. 8).139 The stability tests demonstrated that NH2-UiO-66-shp not only possessed the same acidic stability as NH2-UiO-66 but also showed excellent tolerance to 0.1 M NaOH solution for 5 h, surpassing the alkali labile NH2-UiO-66.
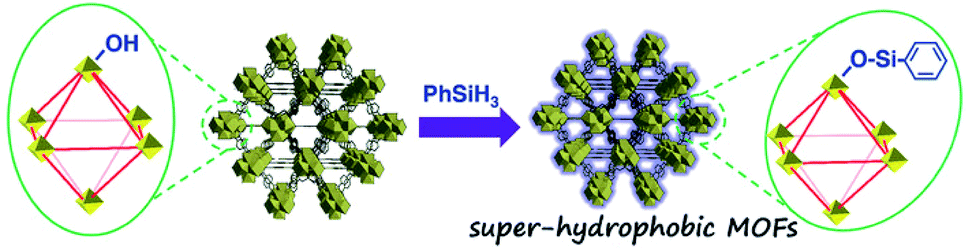 | ||
| Fig. 8 Preparation diagram of super-hydrophobic NH2-UiO-66-shp. Reproduced from ref. 139 with permission from Wiley-VCH, copyright 2019. | ||
Surface hydrophobic coating is a very simple and direct yet effective approach to improve MOF stability with retained porosity. In 2014, Zhang et al. developed a facile and general polydimethylsiloxane (PDMS)-coating strategy to enhance the stability of MOFs in the presence of moisture or water (Fig. 9a).140 The hydrophobic PDMS coating layer yielded impressive results. The hydrostability of three randomly selected and structurally different MOFs, namely, MOF-5, HKUST-1, and ZnBT, was significantly enhanced. The coating layer prevented the attack from water. It is worth noting that the BET surface area and gas uptake performance of the PDMS-coated MOFs are almost maintained, demonstrating the good permeability of the coating layer. Furthermore, the PDMS coating layer on the surface of MOFs did not block the catalytically active sites from coming into contact with the substrates during the catalytic reaction. When the PDMS-coated MOF and the pristine MOF are subjected to the same water vapor treatment, the PDMS-coated MOF is observed to retain its catalytic activity better than the pristine MOF. Since this work, a number of similar approaches have been developed and applied to improve MOF stability.141–144 Qian et al. successfully deposited a hydrophobic organosilicone layer on the exterior surface of MOFs via a facile solution-immersion process without any heat treatment.141 Upon exposure to water for 5 days, it was found that the porosity of the MOFs HKUST-1, MIL-125, and ZIF-67 was retained to a great extent due to the enhanced hydrophobicity. Hou et al. developed a post-synthetic polymerization method to form polymer brushes on the external surfaces of MOFs.142 Interestingly, upon irradiation with UV-light, polymerization could be initialized by active hydrogen species (surface radicals) on the MOFs to generate the polymer brushes on the MOFs to obtain MOF-based materials with enhanced chemical stabilities. Recently, Queen's group successfully developed a two-step post-synthetic polymerization method to improve the stability of seven different types of MOFs.143 First, polydopamine (PDA) as an adhesive was firstly coated on the surface of these MOFs under an oxygen atmosphere. Then, based on a Michael addition, the hydrophobic 1H,1H,2H,2H-perfluorodecanethiol was covalently grafted on the surface of the MOF@PDA that resulted from the previous step. As expected, all the highly hydrophobic MOF composites displayed superior chemical stability under acidic or alkaline conditions compared with the pristine MOFs. Thermal polymerization of protective layers on the MOF surfaces can also strengthen their chemical stability. Using direct polymerization of fluorinated 4-undecylcatechol (fdcat), HKUST-1 was coated with hydrophobic layers under mild reaction conditions.144 Compared with unmodified HKUST-1, after soaking in water for a week, the permeable coatings protected HKUST-1 crystals against water molecules while retaining 92% of the porosity.
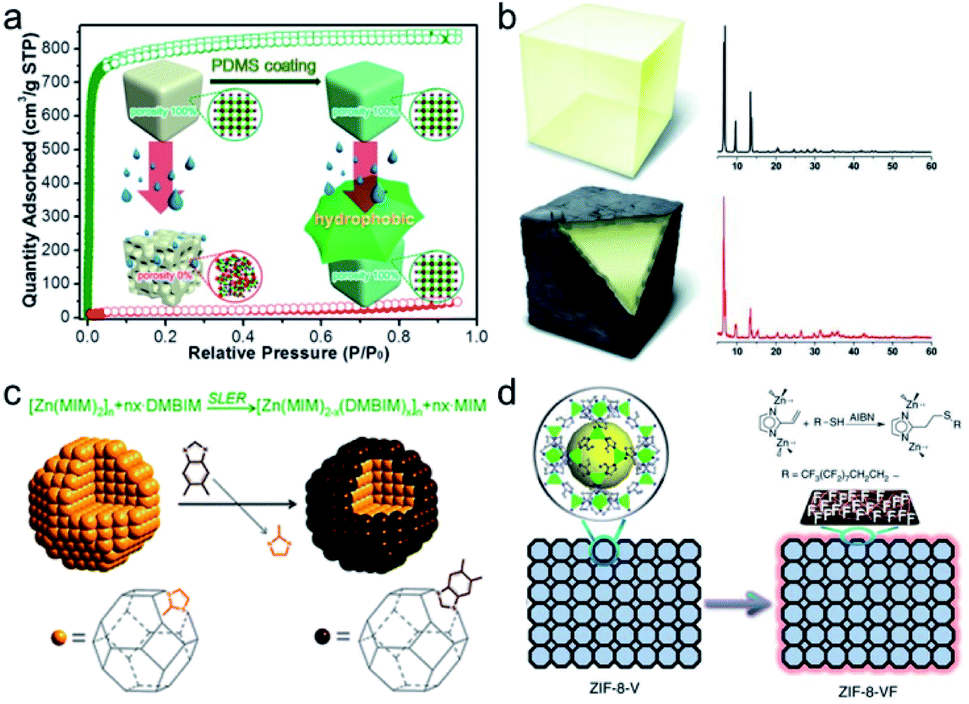 | ||
| Fig. 9 Schematic representation of (a) PDMS-coating on the MOF surface for improved moisture/water resistance, (b) showing MOF-5 (top) and MOF-5 after thermal modification to produce amorphous carbon-coated MOFs (bottom) and related powder XRD patterns of corresponding samples (right), (c) shell-ligand-exchange-reaction process of ZIF-8, and (d) imparting amphiphobicity on ZIF-8-V via grafting perfluoroalkyl groups on the exterior surface. (a) Reproduced from ref. 140 with permission from the American Chemical Society, copyright 2014. (b) Reproduced from ref. 145 with permission from Wiley-VCH, copyright 2012. (c) Reproduced from ref. 147 with permission from The Royal Society of Chemistry, copyright 2013. (d) Reproduced from ref. 148 with permission from Nature Publishing Group, copyright 2016. | ||
Inspired by the high hydrostability of “carbonaceous grease”, Park’s group prepared highly moisture-resistant black-colored MOF-5 through a simple heat treatment at a specific temperature followed by immediate cooling to room temperature (Fig. 9b).145 When the pyrolysis temperature was raised to 530 °C, the BET surface area of the resultant material (1740 m2 g−1) was half of that (3450 m2 g−1) of pristine MOF-5. Nevertheless, thermally modified MOF-5 retained its crystallinity and porosity to a great extent upon exposure to ambient air for at least two weeks. This indicated its high hydrostability. Gadipelli et al. investigated the framework decomposition of MOF-5 crystals by varying the annealing temperature.146 After annealing MOF-5 at 400 °C (that is, below the onset decomposition temperature of 500 °C) for 3 h, the resultant sample could retain certain porosity even after exposure to air for 15 days.
As an example of post-synthetic surface modification, 2-methylimidazolate located on the exterior surface of ZIF-8 particles was substituted with 5,6-dimethylbenzimidazole (DMBIM) via a shell-ligand-exchange-reaction (SLER) to improve the hydrothermal stability (Fig. 9c).147 After four cycles of isobutanol sorption, the uptake amount of ZIF-8 fell to ∼0 caused by the collapse of the structure. In contrast, no obvious change in adsorption capacity was found in the DMBIM modified ZIF-8 thanks to the maintenance of the framework. By deliberately utilizing 2-vinyl-imidazole as the organic linker, Sun et al. successfully fabricated ZIF-8-V, a ZIF-8 type framework and hydrophobically treated its external surface with perfluoroalkyl groups based on the thiol–ene click reaction (Fig. 9d).148 The resultant ZIF-8-VF was an amphiphobic material and was able to remain intact upon exposure to 100% RH under a CO2 atmosphere at 45 °C for over 720 h, while almost all ZIF-8 structures degraded under the above conditions in 240 h. The method was also applied to MOF-5, producing a similar effect. Analogously, alkylphosphonic acids with long alkyl chains were modified on the Zr6-nodes on the exterior surface of MOFs, namely, UiO-66, UiO-66-SO3H, and PCN-222, without any disturbance to the internal pores.149 By virtue of the external superhydrophobic modification, the tolerance of the resultant MOFs toward different harsh environments was appreciably enhanced.
In 2009, Park's group successfully incorporated acid-treated multiwalled carbon nanotubes (MWCNTs) into MOF-5 to give a hybrid composite (MOFMC).150 This composite growth helped with removing the guest molecules during activation which enhanced the Langmuir specific surface area. Moreover, the rigid MWCNTs could protect the moisture-sensitive MOF-5, making MOFMC stable even after one week in air. The same effects can also be achieved when MWCNTs are replaced by graphite oxide or carboxyl-functionalized attapulgite.151,152 Matzger's group produced MOF-5-polystyrene composites using the polymerization of styrene in the presence of MOF-5.153 Polystyrene was uniformly distributed throughout the MOF-5 particles. The resultant composites were endowed with much better moisture stability at RH of 53% as compared with the pristine MOF-5. Apart from polystyrene, polynaphthylene (PN) could also be formed in the channels of MOF-5 as partitions via a radical reaction, affording a hydrophobic PN@MOF-5 composite with significantly improved hydrostability.154 In fact, so long as the hydrophobic molecules are introduced into pores, MOFs would display improved water stability. Decoste's group prepared hydrophobic HKUST-1 by treating the parent MOF with plasma-enhanced chemical vapor deposition (PECVD) of perfluorohexane (PFH).155 With the protection from adsorbed PFH, hydrophobic HKUST-1 could remain resistant to the attack by water for at least 24 h.
As an alternative approach, MOFs can be encapsulated into other materials to prevent the hydrolysis/collapse.156–160 For example, MOF-5 crystals were formed inside the channels of SBA-15 to give MOF-5@SBA-15 composites.156 Due to the support from rigid SBA-15, the crystallinity of composites remained essentially unchanged even after exposure to air for one week (∼50% RH and 25 °C). However, the decomposition of MOF-5 occurred under the same conditions within 36 hours, proving the great effect of SBA-15 on the hydrostability of the composite. Maspoch's group developed a one-step, rapid, and scalable spray-drying synthesis method to encapsulate HKUST-1 into polystyrene, which yielded microscale HKUST-1@polystyrene spheres.157 In contrast to the pristine HKUST-1, the hydrophobic composite retained its water uptake capacity even after three consecutive cycles, demonstrating its strong hydrostability. Recently, a water sensitive MOF, DUT-5, was imbedded within polymer monoliths using microwave-assisted polymerization.158 As expected, the DUT-5-polymer hybrid composite could retain its integrity even after 24 h immersion in 50 mM phosphoric acid solution. Besides these composites, MOF-based membranes involving polymer or other materials, even the MOF@MOF composites, have shown enhanced chemical stability.161–163 The Rosi group prepared a stable core–shell MOF material by growing a bio-MOF-14 shell on the bio-MOF-11/14 mixed core.164 In contrast to the water-sensitive core, the resultant core–shell structure remained resistant to water for one day due to the protection afforded by the more stable bio-MOF-14 shell.
3. Properties and applications
3.1 Adsorption and separation
The tunable porous structures and large surface areas of MOFs make them ideal candidates for molecular sorption and separation. Most of the sorption and separation studies for MOFs are based on energy-related gases such as hydrogen, carbon dioxide, methane, etc.7,18 Specifically, water or moisture loaded environments and many other harsh environments are unavoidable in most of the practical sorption/separation processes, e.g. in the CO2 separation from flue gas generated by coal-fired power plants, which is composed of 15–16% CO2, 73–77% N2, 5–7% H2O, 3–4% O2 and traces of SOx, NOx, etc. For such applications, MOFs with decent chemical stability are required.95 By employing two distinct imidazolates as linkers, three hydrophobic ZIFs with chabazite topology were fabricated with Zn2+ ions.105 All three ZIFs displayed nearly equal CO2 uptake capacity under both dry and humid (80% RH) conditions. More importantly, these ZIFs could be completely regenerated within 15 min by a low energy consumption and highly time-efficient process of subjecting them to pure N2 flow at ambient temperature within 15 min, demonstrating the low energy consumption and high time efficiency of the regeneration process. A hydrostable and highly connected cadmium(II)-based MOF was produced using a triaminepentacarboxylic acid ligand.165 Flue gas simulation (3.9% H2O:14.4% CO2:81.7% N2) experiments showed that this MOF preserved its CO2 adsorption performance even after three consecutive cycles, indicating its outstanding water stability. Stable MOF-based composites showed similar results.154 Given the significance of the removal of trace CO2 from air for the environment and health, a made-to-order MOF, SIFSIX-3-Cu was utilized to selectively adsorb trace CO2 at 74% RH.166 It was remarkable that high humidity did not affect the apparent selectivity of SIFSIX-3-Cu at 1000 ppm CO2. The MOF, thus, displayed excellent stability and strong CO2 affinity.In view of the good hydrostability of water-resistant MOFs, they were also used in other applications that involved water adsorption.3,111,167–170 The water adsorption performance of 23 types of porous materials, including 20 different Zr-based MOFs, was systematically investigated.169 Amongst them, MOF-801-P (P = microcrystalline powder form) and MOF-841 exhibited the best water uptake properties (450–640 cm3 g−1 at P/P0 = 0.9) with outstanding recyclability through five adsorption/desorption cycles (Fig. 10). Taking advantage of the outstanding water adsorption performance of MOF-801, Kim et al. firstly designed and fabricated a water harvesting device.170 The amount of water collected by the device reached 2.8 liters per kilogram of MOF at 20% RH upon exposure to natural daylight for 1 day. The hydrolytically stable MOF materials can also be used in drying natural gas as even a trace amount of water in natural gas can cause catastrophic blockage during the methane ice formation. Cadiau et al. demonstrated energy-efficient dehydration by using a stable fluorinated MOF, AlFFIVE-1-Ni, with a one-dimensional channel structure as a water adsorbent.111 The performance of AlFFIVE-1-Ni at selectively adsorbing water vapor from a gas mixture containing CO2, N2, and CH4 at 75% RH was impressive. Also, the MOF maintained its adsorption performance through several cycles of the adsorption column breakthrough test.
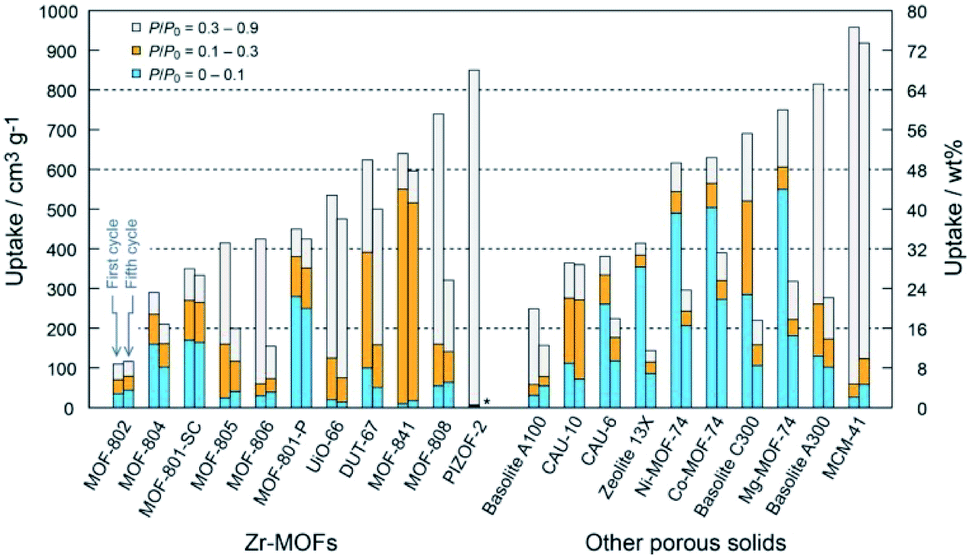 | ||
| Fig. 10 Water uptake capacity of Zr-based MOFs (left) and other representative porous solids (right) in different pressure ranges (asterisk (*) indicates no data). Left and right bars represent the first and fifth cycles, respectively. For MOF-801-SC (SC = single crystal form), uptake capacities of the first and second cycles were demonstrated. Reproduced from ref. 169 with permission from the American Chemical Society, copyright 2014. | ||
In addition, stable and/or hydrophobic MOF-based materials can be employed as powerful adsorbents for the capture of corrosive gases, the removal of target compounds in aqueous solution, and oil/water separation.109,161,171–176 In order to adsorb toxic and corrosive ammonia (NH3), the Morris group synthesized a Cu(II)-based MOF, STAM-17-OEt, based on the 5-ethoxy isophthalate linker.172 Interestingly, upon exposure to a humid environment, the weak Cu–O bonds located on the paddlewheel cluster of the dehydrated MOF would transform into its original hydrated form. This conversion prevents the breakup of the Cu–O bonds in the paddlewheel clusters which hold the framework together, which improves the stability of the framework. STAM-17-OEt was then employed as an adsorbent for NH3 and infrared spectroscopy demonstrated that the MOF structure was retained even after 5 days of the ammonia breakthrough experiment (90% RH). DeCoste et al. prepared a set of chemically stable mixed-matrix membranes (MMMs) based on HKUST-1 and polyvinylidene difluoride (PVDF).161 By virtue of the enhanced hydrophobicity and unsaturated Cu2+ sites, the HKUST-1 MMMs exhibited outstanding NH3 uptake performance and a large proportion of NH3 adsorption capacity survived at 90% RH at 25 °C for 28 days. Given the ultra-efficient metal ion permeation, excellent stability and processability of the MOF membrane, Liu et al. incorporated these two MOFs into various polymer matrixes to create a sequence of UiO-66 and NH2-UiO-66 membranes. They employed the membranes as adsorbents in the recovery of palladium Pd(II) and platinum Pt(IV) ions from strongly acidic solutions (pH = 1.0).176 By virtue of the high affinity to Pt/Pd and the striking stability caused by the strong π–π interactions between the polymer and the MOFs, the resultant membranes showed amazing adsorption capacities for the above ions in strongly acidic solutions, and also excellent recycle performance. Recently, a hydrophobic porous coordination polymer, USTC-6, was uniformly grown throughout a graphene oxide-modified sponge to yield a macroscopic USTC-6@GO@sponge adsorbent with excellent chemical stability for highly efficient oil/water separation.109 Remarkably, USTC-6@GO@sponge could be combined with tubes and a self-priming pump to fabricate a model apparatus for continuous oil recovery from water, showing a promising future for stable MOFs for oil/water separation.
Undoubtedly, highly stable MOFs have exhibited their unique advantages in sorption and separation. The immediate challenges lie in the design of MOF-based adsorbents such as MOF membranes with low (production and regeneration) cost, desirable working capacity and selectivity, and long-term stability according to the demands of practical use. Beyond these, more efforts are needed in mechanistic studies of the co-adsorption behaviour of some adsorbates in MOFs.
3.2 Heterogeneous catalysis
The Lewis acid/basic sites, Brønsted acid sites, and redox active sites on the metal clusters and/or the organic linkers make stable MOFs efficient heterogeneous catalysts for various reactions.12,15,17,177 Since many of these reactions take place in water or even in acidic or basic solutions, MOF-based catalysts should be stable to tolerate such conditions.By employing MIL-101-SO3H as the acid catalyst, reasonable hydrolysis efficiency of cellulose was achieved in aqueous solution systems.178 To beneficially utilise the outstanding chemical stability of PCN-602(Ni), Lv et al. employed PCN-602 with Mn3+–porphyrin centers (denoted as PCN-602(Mn)) for the C–H bond halogenation under harsh conditions (in aqueous solutions and dichloromethane mixtures of 0.165 M NaClO).84 Thanks to the presence of the Mn–porphyrin centre and rigid framework structure, a high yield of chlorocyclohexane (92%) was achieved on PCN-602(Mn) after 5 h reaction, much higher than that (8%) achieved by the homogeneous catalyst, Mn(TPP)Cl. Apart from organocatalysis, many electrocatalytic and photocatalytic properties of MOFs have also been studied in aqueous or even in acidic or basic solutions. For instance, the stable MIL-101(Fe) was chosen as an effective catalyst for visible-light-driven water oxidation.179 With the aid of [Ru(bpy)3]2+, MIL-101(Fe) presented a decent initial turnover frequency (0.10 s−1) with a high oxygen yield of 36.5%. Furthermore, the recycling tests and X-ray photoelectron spectra (XPS) results showed that MIL-101(Fe) was stable after water oxidation. Lu et al. obtained an alkaline-stable MOF, MAFX27-OH, through the post-synthetic ion exchange of MAF-X27-Cl during a linear sweep voltammetric procedure.88 In 1.0 M KOH aqueous solution, MAFX27-OH with both OMSs and hydroxides offered an overpotential of 292 mV at 10.0 mA cm−2 with remarkable durability in 20 h of electrocatalytic oxygen evolution reaction (OER) processes. Shen et al. developed a modular synthesis strategy to stabilize the Co2(RCOO)4(H2O)2 (R = substituent group) cluster into MCF-37 by exchanging the original paddle-wheel type Fe-carboxylate core, affording a modified alkaline-stable MOF, MCF-49 (Fig. 11).180 By virtue of the high catalytic activity of Co2-(RCOO)2(LT)2 (LT = terminal ligand) towards electrocatalytic OER and the high alkaline stability of the framework, MCF-49 showed a very low overpotential of 225 mV at 10 mA cm−2 with nearly 100% Faraday efficiency in aqueous solution (pH = 13). In terms of hydrogen evolution reaction (HER), Qin et al. rationally designed two highly stable polyoxometalate (POM)-based MOFs, NENU-500 and NENU-501, and employed them as HER electrocatalysts in 0.5 M H2SO4 aqueous solution.181 NENU-500 gave a noticeably low onset overpotential of 180 mV with a Tafel slope of 96 mV dec−1. In addition, the two MOFs demonstrated their ultrahigh stability by continuing their activities even after 2000 cycles. Based on the outstanding chemical stability and unique photocatalytic properties of porphyrins, Al-TCPP was utilized as a photocatalyst for hydrogen generation from water.49 With the aid of methyl viologen and ethylenediaminetetraacetic acid, Al-TCPP gave an outstanding H2 generation performance of 600 μmol g−1 after light irradiation for 3 h. When the TCPP linker was substituted by TCPP-Co, the resultant Al-TCPP-Co could be an ideal platform for the aqueous electrochemical reduction of CO2.182 Specifically, Al-TCPP-Co demonstrated a selectivity of >76% and per-site TON of 1400 along with high stability that exceeded 7 h. It is noteworthy that the post-synthetic metalation of stable MOFs such as MOF-525 also achieved a similar effect.183
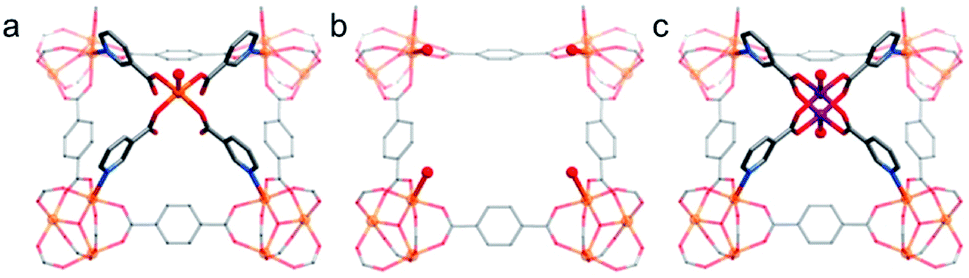 | ||
| Fig. 11 Key local structures of (a) MCF-37, (b) the [Fe3(μ3-O) (BDC)3(LT)3] scaffold, (c) MCF-49. Color code: C, gray; O, red; N, blue; Fe, orange; and Co, purple. H atoms are omitted for clarity. Adapted from ref. 180 with permission from the American Chemical Society, copyright 2017. | ||
Interestingly, MOF stability can be adjusted by altering their pore environments, which further improves the catalytic performance. Inspired by the mechanism of pH buffer solutions, 2,4-bis(3,5-dicarboxyphenylamino)-6-ol triazine (H4BDPO) was designed and utilised as an organic linker to synthesize a Cu-based MOF, JUC-1000.184 The –O−/–OH, –NH–/–NH+–, and –N/–NH+ pairs acted as buffer pairs that provided stability to JUC-1000 in aqueous solutions in a broad pH range from 1.5 to 12.5. Moreover, the synergistic effect among Lewis acid sites, Lewis basic sites and Brønsted acid sites from metal clusters and organic linkers endowed JUC-1000 with striking catalytic activities for the cycloaddition reaction of CO2 and epoxides. In view of the outstanding stability of MOFs coated with PDMS reported by the Jiang group,140 the same group wrapped Pd/UiO-66 with PDMS layers using the chemical vapor deposition (CVD) process.185 Since hydrophobic substrates were enriched around the active sites by taking advantage of the hydrophobic PDMS layer, the resultant Pd/UiO-66@PDMS showed superior catalytic performances and stability for the reduction of nitrobenzene compared to Pd/UiO-66.
As stated above, enhanced stability has widened the scope for the use of MOFs and MOF-based materials as catalysts. In future, it can be expected that the use of operando spectroscopy for gaining a better understanding of the catalytic mechanism, precise control of active sites, and multifunction synergy will be of great importance for heterogeneous catalysis using stable MOFs. In addition, to address the challenges involved in MOF catalysis such as mass transfer, conductivity, and high cost, is also the important consideration.
3.3 Fluorescence sensing
The tunable structures and fluorescence of MOFs make them a great platform for fluorescence sensing.10,186–188 By altering the metal ions and organic linkers, the fluorescence properties of the resulting MOFs can be regulated to a large extent. Particularly, composition, structure, functional groups, interaction sites, etc. on the MOFs might be responsive to external stimuli, such as temperature, pressure, ions, solvents, and so on.10,186–188 Given that MOF fluorescence is sensitive to the external stimuli, MOFs can be used as probes or sensors. To this end, the prerequisite is that the frameworks of MOF sensors should be maintained during the fluorescence change under different testing conditions.For example, ITQMOF-3-Eu was constructed with Eu3+ and 1,10-phenanthroline-2,9-dicarboxylic acid and subsequently joined to an optical fiber to fabricate a miniaturized linear sensor.189 By means of the fine balance among absorption, emission rate, and energy transfer, ITQMOF-3-Eu could be applied as a pH sensor in the pH range of 5–7.5 without any additional external calibration. Jiang et al. reported a Zr-based MOF, PCN-225, which was stable in aqueous solutions of pH range 1–11 for 12 h.58 Utilising the high stability and pH-dependent fluorescence of PCN-225, this MOF could recognize aqueous solutions with different pHs ranging from 0 to 10.2. Deibert et al. developed a reversible colorimetric fluorescence pH sensor based on highly stable PCN-222.190 Upon exposure to aqueous solution with pH = 0, the color of PCN-222 (purple) turned green and this colorimetric response was reversible and reproducible. In order to detect and remove organic pollutants in wastewater such as antibiotics and organic explosives, a stable Zr-based MOF, BUT-13, was fabricated via a topological design approach.63 Due to the combined effect of electron and energy transfer, this MOF displayed outstanding ability to detect nitrofurazone (NZF), nitrofurantoin (NFT), 2,4,6-trinitrophenol (TNP), and 4-nitrophenol (4-NP) in aqueous solutions with a low limit of detection (LOD). More recently, Ebrahim et al. presented a luminescent lanthanide-based MOF, SION-105, which proved to be highly sensitive to fluoride ions in aqueous solution due to the strong interaction between three-coordinate boron atoms of the linker and the F− ions.191 This MOF, due to its good stability, showed excellent regenerability even after ten cycles. Unlike these chemical sensors, a Cu(I) triazolate framework, MAF-2, was designed as a gaseous oxygen sensor based on its high moisture stability, fast uptake and sorption kinetics for oxygen and other optical properties.192 Additionally, upon combining MAF-2 with silicone rubbers, the resultant MAF-2 thin film exhibited extraordinary sensitivity to oxygen and superior stability even in an acidic atmosphere due to the protection provided by the silicone rubbers.
Apart from the de novo processes, stable MOFs with remarkable sensing properties can also be obtained via post-synthetic modification.134,193,194 For instance, the aforementioned UiO-66-NN-ind obtained by a post-synthetic modification diazotisation strategy showed enhanced chemical stability and also broadened the sensing range (pH = 1–12), compared with that of the pristine UiO-66 (pH = 1–9).134 Farha's group presented a halochromic derivative of NU-1000, NU-1000-CF, through a SALI process with 5(6)-carboxynaphthofluorescein (CNF).193 On the basis of the wonderful chemical stability of NU-1000 and the unique optical properties of CNF, NU-1000-CF revealed visual detection of pH over the range from pH 2 to 9. ZIF-90 was functionalized by converting aldehyde groups on the linkers into dicyanovinyl (DCV) groups with a simple Knoevenagel condensation reaction.194 The DCV groups could act as the specific recognition sites for CN− ions and then endowed the modified ZIF-90 with remarkable aqueous-phase sensing ability for CN−.
In conclusion, stable MOFs have shown great potential for sensing various analytes. However, there are many improvements necessary and several issues to be solved before practical implementation. First, more effort should be devoted to improve sensitivity, selectivity, response time, detection limit and long-term stability of fluorescent MOF materials based on the requirements of practical use. Second, further investigation into MOF-based turn-on sensors and their turn-on mechanism will prove valuable. Last but not least, the integration of MOFs into sensing devices is still at a very early stage. Hence, more endeavour is necessary in this direction.
3.4 Biological and medical applications
For their tunable pore sizes, high surface area, and versatile physical and chemical properties, MOFs are extensively used in biological and medical applications such as biosensing, drug release, biomimetic catalysis, etc.5,195–201 For these applications, MOFs with excellent chemical stability are a must. They should possess strong resistance to hydrolysis/collapse in physiological environments in which they would be expected to function, e.g. stomach acidity, intestinal alkalinity and the peristalsis in the esophagus, stomach, and intestines.As for biosensing, a representative example is Cu-TCA constructed with Cu2(O2CR)4 paddlewheel units and tricarboxytriphenyl amine (TCA) linkers.202 Upon the addition of NO, the coordination interaction between NO and Cu(II) complexes reduced Cu2+ to Cu+ ions and caused the recovery of luminescence from Cu-TCA in aqueous solutions. The brightness of emission of Cu-TCA made it an ideal candidate for biological imaging of NO in living cells. Wu et al. utilized mixed ligands to fabricate a lanthanide-based MOF (Ln-MOF) that shows high 48 h stability in water.203 The competition for the absorption of irradiated light between biomarkers serotonin (5-hydroxytryptamine, HT) and 5-hydroxyindole-3-acetic acid (HIAA)) and the organic linkers, and the dynamic quenching process caused fluorescence quenching, thus endowing the Ln-MOF with remarkable sensitivity for selective detection of the two biomarkers in aqueous solution. On the basis of the intrinsic peroxidase-like catalytic activity, two Fe-based MOFs, MIL-68 and MIL-100, were found to behave as colorimetric biosensors to detect H2O2.204 The high chemical stability of MIL-68 and MIL-100(Fe) allowed them to be used to catalyze the oxidation of different peroxidase substrates with H2O2 in acetate buffer with pH = 4.0. Moorthy's group obtained a luminescent and stable homochiral MOF, Zn-PLA, based on a concave shaped pyrene-tetralactic acid.205 Interestingly, histidine was found to cause more significant fluorescence quenching in the aqueous dispersion of Zn-PLA than many other amino acids. This led to the use of Zn-PLA, an enantiodifferentiating sensor of histidine. The authors speculated that this fluorescence quenching was due to the exchange of dimethylammonium cations in Zn-PLA by deprotonated histidine in water.
As an early attempt at the use of MOFs for drug release, two flexible frameworks, MIL-53(Cr) and MIL-53(Fe), were selected as carrier systems for delivering ibuprofen.206 Due to the unique flexibility, a very slow and complete release of ibuprofen over a period of three weeks was accomplished with both MIL-53(Cr) and MIL-53(Fe). Deng's group revealed that the guest release rate was strongly correlated with the type and proportion of functional groups in MOFs.207 By constructing different multivariate MOFs from MIL-101(Fe), the maximum release amount of doxorubicin (DOX) could be shifted from the 17th to the 29th day within a 40 day release period. In order to improve the biocompatibility of carriers, adenine was employed as a biomolecular ligand to fabricate an ionic framework, bio-MOF-1, which could maintain the structural integrity in biological buffers for weeks.208 Unlike common MOFs, the release of procainamide from bio-MOF-1 could be triggered by the ionic interactions between the drug and framework. Recently, Farha's group demonstrated the immobilization of insulin in an acid-stable MOF, NU-1000.209 By virtue of the protective effect from NU-1000, insulin was stable even upon exposure to stomach acid (pH = 1.5–3.5) and pepsin (Fig. 12). Moreover, ∼40 wt% of insulin could be released from the host framework under simulated physiological conditions (pH = 7.0) (Fig. 12).
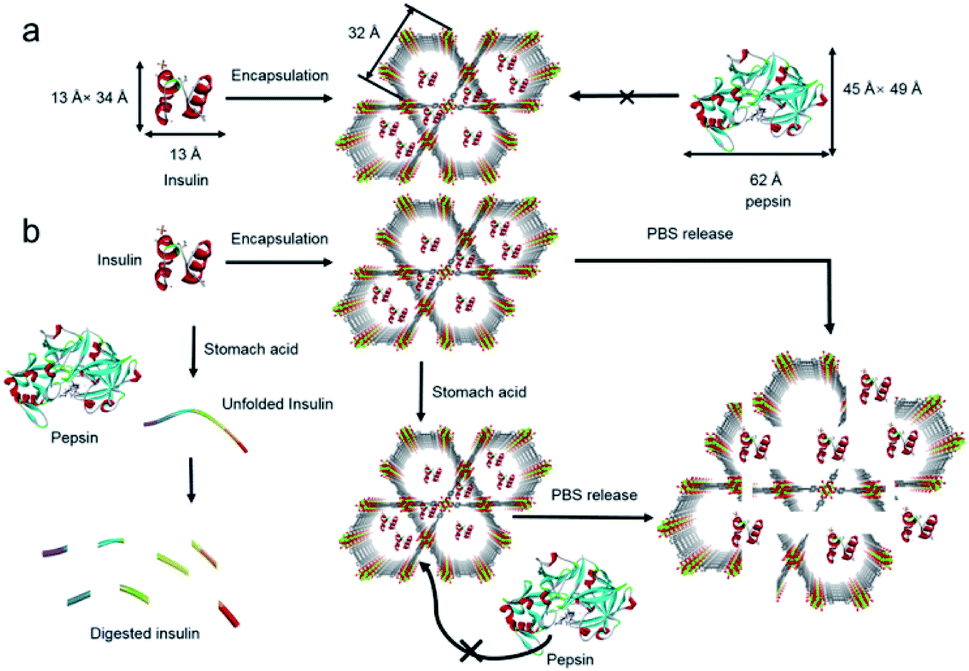 | ||
| Fig. 12 Schematic illustration of (a) encapsulation of insulin in the mesopores (32 Å) of NU-1000 and exclusion of pepsin from the MOF framework and (b) exposure of insulin and insulin@NU-1000 to stomach acid. Free insulin denatures in stomach acid and is digested by pepsin. The release of insulin from NU-1000 happens when insulin@NU-1000 is exposed to a PBS solution. Reproduced from ref. 209 with permission from the American Chemical Society, copyright 2018. | ||
As for the biocatalysis, Reynolds's group utilized Cu-BTTri with high stability in fresh citrated whole-blood (30 min, pH 7.4, 37 °C) to produce polymeric medical devices with biomedical grade polyurethane.210 Fortunately, the catalytic activity from the exposed Cu2+ active sites of Cu-BTTri can be retained, and the medical devices realized the surface-localized generation of NO from endogenous sources even in fresh citrated whole blood. Inspired by the bimetallic Zn–OH–Zn active site in phosphotriesterase enzymes, UiO-66 with similar Zr–OH–Zr bonds in [Zr6O4(OH)4] clusters was utilized as a biomimetic catalyst for the hydrolysis of methyl paraoxon in an aqueous solution containing 0.45 M N-ethylmorpholine (pH = 10).211 Besides directly behaving as biocatalysts, stable MOFs can also act as catalyst supports for immobilizing enzymes for biocatalysis.50,212 Three enzymes of different sizes, namely, horseradish peroxidase (HRP), cytochrome c (Cyt c) and microperoxidase-11 (MP-11), were separately encapsulated in PCN-333(Al).50 Compared with the same enzymes immobilized in SBA-15, the enzymes encapsulated in PCN-333(Al) presented much superior catalytic stability through five cycles of o-phenylenediamine oxidation due to the outstanding chemical stability of PCN-333(Al) and strong interaction between the enzymes and frameworks. Using PCN-888 with three different sized cavities as the carrier, Lian et al. fabricated a tandem nanoreactor via a unique stepwise encapsulation of two different enzymes (glucose oxidase (GOx) and horseradish peroxidase (HRP)).212 It was remarkable that strong interactions between immobilized moieties and cages of PCN-888 not only prevented the trypsin digestion of encapsulated enzymes but also ensured the maintenance of catalytic activity through four catalytic cycles.
Nevertheless, the study of MOFs and MOF-based materials for biological and medical applications is still in its infancy. One remaining critical issue is the fabrication of nontoxic MOFs with outstanding chemical stability, excellent biocompatibility, and appropriate pore size and pore volume. In addition, control of MOF particle size is also very important in view of the endocytosis by the living cells and systemic circulation in blood. As for the drug delivery, the degradation mechanism of MOFs should be studied closely before the in vivo investigations and clinical applications.
3.5 Proton conductivity
Over the past decades, MOFs have attracted rapidly increased attention in the field of proton conductivity on account of their tailorable structures.213–217 Different from other porous materials, proton conduction of MOFs can be easily controlled by tuning the hydrophilicity and acidity of the surface structure of MOFs. To date, many proton conduction studies on MOFs have been reported, though only a limited number of MOFs displayed both high conductivity and good stability.218 In principle, the stability of MOFs as proton conductors mainly refers to long-term stability under different temperature and/or humidity conditions. It is important to note here that proton transport relies mainly on the generation of hydrogen-bonded water networks in the cavities of MOFs according to the Grotthuss or proton-hopping mechanism.213,219,220 Therefore, design, synthesis and modification of MOFs with high chemical stability will provide an excellent platform for investigating their proton-conducting behaviors.In light of the easy proton release of strong acids such as H2SO4, UiO-66(SH)2 was oxidized by H2O2 to afford UiO-66(SO3H)2 with SO3H groups covalently linked to the framework, which greatly favored the proton conductivity.221 At 80 °C and 90% RH, UiO-66(SO3H)2 displayed an extraordinary protonic conductivity of 8.4 × 10−2 S cm−1 with long-term stability over 96 h, demonstrating its high stability. Ponomareva et al. developed a facile method of incorporating nonvolatile H2SO4 and H3PO4 within the pores of MIL-101(Cr) to give H2SO4@MIL-101 and H3PO4@MIL-101 for proton conductivity.222 Specifically, upon confining ∼70% H2SO4 (∼10 M) or ∼80% H3PO4 (∼14 M) in MIL-101, the proton conductivity of the resultant composites reached up to 4.0 × 10−2 and 2.5 × 10−2 S cm−1, respectively, at low temperature and 20% RH. Not limited to acids, NH3 and amino were also modified within MOFs via the de novo process or post-synthetic strategy, improving their proton-conducting ability.223,224 Wang et al. reported a highly stable MOF, MIP-202(Zr), assembled by L-aspartate and Zr4+ and investigated its proton-conduction properties at 90 °C and 95% RH.224 The interaction between NH3+ groups (proton source) and water molecules in the cavities generated an extended hydrogen-bonded network in MIP-202(Zr), endowing it with a desirable and steady proton conductivity of 0.011 S cm−1. Hong and co-workers utilized a microwave-assisted solvothermal reaction to obtain Ni-MOF-74, which remained stable even at a pH value as low as 1.8.225 When Ni-MOF-74 was immersed in sulfuric acid solutions with different pH values, H+@Ni-MOF-74 was generated with different proton-conducting properties. Particularly, at pH 1.8, the resultant H+@Ni-MOF-74 presented a remarkable proton conductivity of 2.2 × 10−2 S cm−1 under the conditions of 95% RH and 80 °C. In order to prepare a stable MOF-based electrolyte material with good conductivity at low RH, a highly stable and flexible MOF, BUT-8(Cr)A, was constructed from naphthalene-2,6-dicarboxylate, 4,8-disulfonaphthalene-2,6-dicarboxylate and Cr3O(OH) (CO2)6 SBUs.226 In contrast to other rigid MOFs, BUT-8(Cr)A presented a decent proton conductivity of 1.27 × 10−1 S cm−1 at 80 °C and 100% RH due to the chemically stable structure with high-density –SO3H sites. Its high conductivity could be maintained across a wide range of RH. The authors speculated that this unique feature was ascribable to the water-content-dependent structural transformation in BUT-8(Cr)A (i.e. framework ‘self-adaption’) (Fig. 13).
 | ||
| Fig. 13 Illustration of the proposed self-adaption mechanism in the flexible MOF (top) and rigid MOF (bottom) with a high density of sulfonic acid sites for proton conduction. Reproduced from ref. 226 with permission from Nature Publishing Group, copyright 2017. | ||
Although the proton conductivity of certain MOFs has exceeded that of the commercially available Nafion, there still exist many challenging problems to be solved. The key task is to gain a deep understanding of the proton conduction mechanism of MOF materials. Besides, from a practical perspective, more attention should be paid to the commercial development of MOF-based electrochemical devices with desirable mechanical stability.
4. Conclusions and perspectives
Over the past two decades, much effort has been dedicated to the de novo synthesis of novel MOFs with high stability via different strategies, including preparation of high-valent metal-carboxylate or low-valent metal-azolate MOFs, use of azole-containing carboxylate linkers, mixed metal ions, and hydrophobic ligands, insertion of building blocks, framework interpenetration, etc. Beyond these, post-synthetic structural processing and composite material engineering have also been employed to improve the stability of existing MOFs. Amongst MOF chemical stability investigations, water or moisture stability is the most preliminary and important, as handling MOF samples in air (containing moisture) is hardly avoidable for almost all MOF applications. Fortunately, a very large number of MOF-based materials that are stable in air and water have been developed. Moreover, more and more MOFs resistant to harsh chemical environments (acidic/alkaline media) have also been reported. As extreme examples, AlTCS-1 shows strong chemical resistance in aqua regia solution over 24 h;75 the structure of PCN-601 is not disturbed during alkaline treatment in saturated NaOH solution at 100 °C;25etc. These important breakthroughs together with many other advances give the MOF community great encouragement and confidence. The stability (especially chemical stability) of MOFs, which was once regarded as a critical bottleneck for their real-world applications, may no longer be a severe difficulty and should be addressed in the near future with the continuous efforts of scientists. The preparation of stable MOFs and the strategy of improving MOF stability would become more rational along with powerful prediction of computational chemistry. Accordingly, the applications of MOFs might become widespread and more practicable. As a step further, on the basis of the intriguing properties of stable MOFs, the scope of MOF applications has significantly expanded. In this review, only a minority of MOF applications have been covered. Undoubtedly, the development of MOF stability and its interaction with other research fields will bring more promising and innovative applications.Although great progress has been made in this field, many challenges remain. Due to the particular requirements of each strategy, it is rather difficult to find a general approach that is applicable to different MOF systems. Also, a number of pre- or post-synthetic modification methods usually cause the change of pore features, which poses challenges for their subsequent applications. To address these issues, some targeted approaches such as hydrophobic surface treatment might be worthy of further investigation. Furthermore, experimental trials can be rationally combined with computational design to afford new framework materials with high stability and particular properties. In addition, so far, there appear to be no systematic investigations on the mechanical stability of MOFs, which is a crucial factor for their industrialization. Maybe, certain specific techniques such as single crystal X-ray diffraction analysis and theoretical calculation will prove helpful in the characterisation of mechanical stability of MOFs. Moreover, it is necessary to establish a unified evaluation index system for assessing the mechanical properties of MOFs. With regard to the experimental stability testing, the combination of multiple means of detection is more reliable to qualify the maintenance of intrinsic properties of MOFs.227 Particularly, compared with static stability analysis, the investigation of the dynamic change course of MOFs under harsh conditions may be more meaningful.
In the future, the fabrication of very stable MOFs is likely to remain a dream and that would pave the way to their applications to diverse ends. For example, stable and well-designed multivariate MOFs or MOF-based materials, which possess the features of each individual part, can provide extraordinary performance for targeted application. In addition, the precise location control of customisable functional groups within stable MOFs through stepwise synthetic techniques will produce great potential or optimal performance for diverse applications. Moving forward, mechanism analysis related to applications and device-engineering are also the attractive goals for stable MOFs. We also hope that such a Minireview can stimulate the interest of scientists who engage in the interdisciplinary area to explore the value of MOFs from an academic and/or industrial perspective.
In conclusion, MOFs have entered a new period of rapid development and are no longer a category of moisture-sensitive frameworks. The endeavor and progress on enhanced MOF (chemical) stability do greatly promote the development and applications of MOFs. We believe in a bright future for MOF chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the editors for kind invitation and the reviewers for insightful suggestions. This work was supported by the NSFC (21725101, 21673213, 21871244, and 21521001) and Fujian Institute of Innovation, CAS.References
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- Z.-G. Gu, C. Zhan, J. Zhang and X. Bu, Chem. Soc. Rev., 2016, 45, 3122–3144 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- Y. Cui, J. Zhang, H. He and G. Qian, Chem. Soc. Rev., 2018, 47, 5740–5785 RSC.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- L. Jiao, Y. Wang, H.-L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- G. Li, S. Zhao, Y. Zhang and Z. Tang, Adv. Mater., 2018, 30, 1800702 CrossRef PubMed.
- C. Wang, B. An and W. Lin, ACS Catal., 2019, 9, 130–146 CrossRef CAS.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed.
- L. Jiao, J. Y. R. Seow, W. S. Skinner, Z. U. Wang and H.-L. Jiang, Mater. Today, 2019, 27, 43–68 CrossRef CAS.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H.-C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- T. Devic and C. Serre, Chem. Soc. Rev., 2014, 43, 6097–6115 RSC.
- I. J. Kang, N. A. Khan, E. Haque and S. H. Jhung, Chem.–Eur. J., 2011, 17, 6437–6442 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- Q. Yao, A. B. Gómez, J. Su, V. Pascanu, Y. Yun, H. Zheng, H. Chen, L. Liu, H. N. Abdelhamid, B. Martín-Matute and X. Zou, Chem. Mater., 2015, 27, 5332–5339 CrossRef CAS.
- X.-L. Lv, S. Yuan, L.-H. Xie, H. F. Darke, Y. Chen, T. He, C. Dong, B. Wang, Y.-Z. Zhang, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2019, 141, 10283–10293 CrossRef CAS PubMed.
- K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef CAS PubMed.
- H.-L. Jiang, T. A. Makal and H.-C. Zhou, Coord. Chem. Rev., 2013, 257, 2232–2249 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- T. Loiseau, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- S. Ma, X.-S. Wang, D. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4130–4133 CrossRef CAS PubMed.
- D. T. de Lill and C. L. Cahill, Chem. Commun., 2006, 4946–4948 RSC.
- H. Wu, T. Yildirim and W. Zhou, J. Phys. Chem. Lett., 2013, 4, 925–930 CrossRef CAS PubMed.
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2009, 131, 17546–17547 CrossRef CAS PubMed.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed.
- W. Li, A. Thirumurugan, P. T. Barton, Z. Lin, S. Henke, H. H.-M. Yeung, M. T. Wharmby, E. G. Bithell, C. J. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2014, 136, 7801–7804 CrossRef CAS PubMed.
- Z. Li and H. C. Zeng, J. Am. Chem. Soc., 2014, 136, 5631–5639 CrossRef CAS PubMed.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- N. u. Qadir, S. A. M. Said and H. M. Bahaidarah, Microporous Mesoporous Mater., 2015, 201, 61–90 CrossRef CAS.
- N. Li, J. Xu, R. Feng, T.-L. Hu and X.-H. Bu, Chem. Commun., 2016, 52, 8501–8513 RSC.
- M. Shengqian and P.A. Jason, Elaboration And Applications of Metal–Organic Frameworks, World Scientific Publishing Company, 2018 Search PubMed.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed.
- R. Antwi-Baah and H. Liu, Materials, 2018, 11, 2250 CrossRef PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. G. III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- K. Leus, T. Bogaerts, J. D. Decker, H. Depauw, K. Hendrickx, H. Vrielinck, V. V. Speybroeck and P. V. D. Voort, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Rémy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326–6327 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- A. Fateeva, P. A. Chater, C. P. Ireland, A. A. Tahir, Y. Z. Khimyak, P. V. Wiper, J. R. Darwent and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2012, 51, 7440–7444 CrossRef CAS PubMed.
- D. Feng, T.-F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y.-P. Chen, X. Wang, K. Wang, X. Lian, Z.-Y. Gu, J. Park, X. Zou and H.-C. Zhou, Nat. Commun., 2015, 6, 5979 CrossRef PubMed.
- D. Feng, K. Wang, J. Su, T.-F. Liu, J. Park, Z. Wei, M. Bosch, A. Yakovenko, X. Zou and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 149–154 CrossRef CAS PubMed.
- Y.-N. Gong, T. Ouyang, C.-T. He and T.-B. Lu, Chem. Sci., 2016, 7, 1070–1075 RSC.
- S. Krause, V. Bon, U. Stoeck, I. Senkovska, D. M. Tçbbens, D. Wallacher and S. Kaskel, Angew. Chem., Int. Ed., 2017, 56, 10676–10680 CrossRef CAS PubMed.
- J. Duan, M. Higuchi, S. Horike, M. L. Foo, K. P. Rao, Y. Inubushi, T. Fukushima and S. Kitagawa, Adv. Funct. Mater., 2013, 23, 3525–3530 CrossRef CAS.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, Y.-P. Chen, J. Park, Z. Wei, Y. Sun, M. Bosch, S. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 17714–17717 CrossRef CAS PubMed.
- D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS PubMed.
- H.-L. Jiang, D. Feng, K. Wang, Z.-Y. Gu, Z. Wei, Y.-P. Chen and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 13934–13938 CrossRef CAS PubMed.
- T.-F. Liu, D. Feng, Y.-P. Chen, L. Zou, M. Bosch, S. Yuan, Z. Wei, S. Fordham, K. Wang and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 413–419 CrossRef CAS PubMed.
- D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed.
- K. Wang, D. Feng, T.-F. Liu, J. Su, S. Yuan, Y.-P. Chen, M. Bosch, X. Zou and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 13983–13986 CrossRef CAS PubMed.
- S. Yuan, J.-S. Qin, L. Zou, Y.-P. Chen, X. Wang, Q. Zhang and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6636–6642 CrossRef CAS PubMed.
- B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed.
- K. A. Cychosz and A. J. Matzger, Langmuir, 2010, 26, 17198–17202 CrossRef CAS PubMed.
- Y.-Y. Fu, C.-X. Yang and X.-P. Yan, J. Chromatogr. A, 2013, 1274, 137–144 CrossRef CAS PubMed.
- S. Wang, T. Kitao, N. Guillou, M. Wahiduzzaman, C. Martineau-Corcos, F. Nouar, A. Tissot, L. Binet, N. Ramsahye, S. Devautour-Vinot, S. Kitagawa, S. Seki, Y. Tsutsui, V. Briois, N. Steunou, G. Maurin, T. Uemura and C. Serre, Nat. Commun., 2018, 9, 1660 CrossRef PubMed.
- L. Xu, Y.-P. Luo, L. Sun, Y. Xu, Z.-S. Cai, M. Fang, R.-X. Yuan and H.-B. Du, Chem.–Eur. J., 2016, 22, 6268–6276 CrossRef CAS PubMed.
- W. Liang, H. Chevreau, F. Ragon, P. D. Southon, V. K. Peterson and D. M. D'Alessandro, CrystEngComm, 2014, 16, 6530–6533 RSC.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- S. Diring, S. Furukawa, Y. Takashima, T. Tsuruoka and S. Kitagawa, Chem. Mater., 2010, 22, 4531–4538 CrossRef CAS.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed.
- Y. Chen, T. Hoang and S. Ma, Inorg. Chem., 2012, 51, 12600–12602 CrossRef CAS PubMed.
- S. Wang, J. S. Lee, M. Wahiduzzaman, J. Park, M. Muschi, C. Martineau-Corcos, A. Tissot, K. H. Cho, J. Marrot, W. Shepard, G. Maurin, J.-S. Chang and C. Serre, Nat. Energy, 2018, 3, 985–993 CrossRef CAS.
- Z.-W. Wang, M. Chen, C.-S. Liu, X. Wang, H. Zhao and M. Du, Chem.–Eur. J., 2015, 21, 17215–17219 CrossRef CAS PubMed.
- Y. Guo, J. Zhang, L.-Z. Dong, Y. Xu, W. Han, M. Fang, H.-K. Liu, Y. Wu and Y.-Q. Lan, Chem.–Eur. J., 2017, 23, 15518–15528 CrossRef CAS PubMed.
- F. Leng, H. Liu, M. Ding, Q. Lin and H.-L. Jiang, ACS Catal., 2018, 8, 4583–4590 CrossRef CAS.
- C.-Y. Gao, J. Ai, H.-R. Tian, D. Wu and Z.-M. Sun, Chem. Commun., 2017, 53, 1293–1296 RSC.
- E.-X. Chen, M. Qiu, Y.-F. Zhang, Y.-S. Zhu, L.-Y. Liu, Y.-Y. Sun, X. Bu, J. Zhang and Q. Lin, Adv. Mater., 2018, 30, 1704388 CrossRef PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- M. Tonigold, Y. Lu, B. Bredenkotter, B. Rieger, S. Bahnmuller, J. Hitzbleck, G. Langstein and D. Volkmer, Angew. Chem., Int. Ed., 2009, 48, 7546–7550 CrossRef CAS PubMed.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC.
- X.-L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 211–217 CrossRef CAS PubMed.
- N. Masciocchi, S. Galli, V. Colombo, A. Maspero, G. Palmisano, B. Seyyedi, C. Lamberti and S. Bordiga, J. Am. Chem. Soc., 2010, 132, 7902–7904 CrossRef CAS PubMed.
- C. Pettinari, A. Tăbăcaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–31 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS PubMed.
- X. F. Lu, P. Q. Liao, J. W. Wang, J. X. Wu, X. W. Chen, C. T. He, J. P. Zhang, G. R. Li and X. M. Chen, J. Am. Chem. Soc., 2016, 138, 8336–8339 CrossRef CAS PubMed.
- A. Tăbăcaru, C. Pettinari and S. Galli, Coord. Chem. Rev., 2018, 372, 1–30 CrossRef.
- H. Li, W. Shi, K. Zhao, H. Li, Y. Bing and P. Cheng, Inorg. Chem., 2012, 51, 9200–9207 CrossRef CAS PubMed.
- X.-W. Zhu, X.-P. Zhou and D. Li, Chem. Commun., 2016, 52, 6513–6516 RSC.
- C. Montoro, F. Linares, E. Q. Procopio, I. Senkovska, S. Kaskel, S. Galli, N. Masciocchi, E. Barea and J. A. R. Navarro, J. Am. Chem. Soc., 2011, 133, 11888–11891 CrossRef CAS PubMed.
- C. Heering, I. Boldog, V. Vasylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757–9768 RSC.
- Y. Hu, M. Ding, X.-Q. Liu, L.-B. Sun and H.-L. Jiang, Chem. Commun., 2016, 52, 5734–5737 RSC.
- L. Liang, C. Liu, F. Jiang, Q. Chen, L. Zhang, H. Xue, H.-L. Jiang, J. Qian, D. Yuan and M. Hong, Nat. Commun., 2017, 8, 1233 CrossRef PubMed.
- J. Yang, A. Grzech, F. M. Mulder and T. J. Dingemans, Chem. Commun., 2011, 47, 5244–5246 RSC.
- Y. Chen, B. Wang, X. Wang, L.-H. Xie, J. Li, Y. Xie and J.-R. Li, ACS Appl. Mater. Interfaces, 2017, 9, 27027–27035 CrossRef CAS PubMed.
- T. A. Makal, X. Wang and H.-C. Zhou, Cryst. Growth Des., 2013, 13, 4760–4768 CrossRef CAS.
- D. Ma, Y. Li and Z. Li, Chem. Commun., 2011, 47, 7377–7379 RSC.
- K. Wang, H. Huang, X. Zhou, Q. Wang, G. Li, H. Shen, Y. She and C. Zhong, Inorg. Chem., 2019, 58, 5725–5732 CrossRef CAS PubMed.
- L. Liu and S. G. Telfer, J. Am. Chem. Soc., 2015, 137, 3901–3909 CrossRef CAS PubMed.
- J. He, K.-K. Yee, Z. Xu, M. Zeller, A. D. Hunter, S. S.-Y. Chui and C.-M. Che, Chem. Mater., 2011, 23, 2940–2947 CrossRef CAS.
- C. Yang, U. Kaipa, Q. Z. Mather, X. Wang, V. Nesterov, A. F. Venero and M. A. Omary, J. Am. Chem. Soc., 2011, 133, 18094–18097 CrossRef CAS PubMed.
- J. M. Taylor, R. Vaidhyanathan, S. S. Iremonger and G. K. H. Shimizu, J. Am. Chem. Soc., 2012, 134, 14338–14340 CrossRef CAS PubMed.
- N. T. T. Nguyen, H. Furukawa, F. Gándara, H. T. Nguyen, K. E. Cordova and O. M. Yaghi, Angew. Chem., Int. Ed., 2014, 53, 10645–10648 CrossRef CAS PubMed.
- D.-X. Xue, Y. Belmabkhout, O. Shekhah, H. Jiang, K. Adil, A. J. Cairns and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 5034–5040 CrossRef CAS PubMed.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920–925 CrossRef CAS PubMed.
- S. Rodríguez-Hermida, M. Y. Tsang, C. Vignatti, K. C. Stylianou, V. Guillerm, J. Pérez-Carvajal, F. Teixidor, C. Viñas, D. Choquesillo-Lazarte, C. Verdugo-Escamilla, I. Peral, J. Juanhuix, A. Verdaguer, I. Imaz, D. Maspoch and J. G. Planas, Angew. Chem., Int. Ed., 2016, 55, 16049–16053 CrossRef PubMed.
- Z.-R. Jiang, J. Ge, Y.-X. Zhou, Z. U. Wang, D. Chen, S.-H. Yu and H.-L. Jiang, NPG Asia Mater., 2016, 8, e253 CrossRef CAS.
- M. Joharian, A. Morsali, A. A. Tehrani, L. Carlucci and D. M. Proserpio, Green Chem., 2018, 20, 5336–5345 RSC.
- A. Cadiau, Y. Belmabkhout, K. Adil, P. M. Bhatt, R. S. Pillai, A. Shkurenko, C. Martineau-Corcos, G. Maurin and M. Eddaoudi, Science, 2017, 356, 731–735 CrossRef CAS PubMed.
- C. Serre, Angew. Chem., Int. Ed., 2012, 51, 6048–6050 CrossRef CAS PubMed.
- Z. Zhang, H. T. H. Nguyen, S. A. Miller and S. M. Cohen, Angew. Chem., Int. Ed., 2015, 54, 6152–6157 CrossRef CAS PubMed.
- N. M. Padial, E. Q. Procopio, C. Montoro, E. López, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290–8294 CrossRef CAS PubMed.
- Q.-G. Zhai, X. Bu, X. Zhao, D.-S. Li and P. Feng, Acc. Chem. Res., 2017, 50, 407–417 CrossRef CAS PubMed.
- Y.-X. Tan, Y.-P. He and J. Zhang, Inorg. Chem., 2012, 51, 9649–9654 CrossRef CAS PubMed.
- X. Wang, W.-Y. Gao, J. Luan, L. Wojtas and S. Ma, Chem. Commun., 2016, 52, 1971–1974 RSC.
- D.-M. Chen, N.-N. Zhang, J.-Y. Tian, C.-S. Liu and M. Du, J. Mater. Chem. A, 2017, 5, 4861–4867 RSC.
- D.-M. Chen, J.-Y. Tian, C.-S. Liu and M. Du, Chem. Commun., 2016, 52, 8413–8416 RSC.
- S.-Y. Jiang, W.-W. He, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Inorg. Chem., 2018, 57, 6118–6123 CrossRef CAS PubMed.
- H. Jasuja and K. S. Walton, Dalton Trans., 2013, 42, 15421–15426 RSC.
- H. Jasuja, Y. Jiao, N. C. Burtch, Y.-g. Huang and K. S. Walton, Langmuir, 2014, 30, 14300–14307 CrossRef CAS PubMed.
- C.-T. He, P.-Q. Liao, D.-D. Zhou, B.-Y. Wang, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2014, 5, 4755–4762 RSC.
- R. Feng, Y.-Y. Jia, Z.-Y. Li, Z. Chang and X.-H. Bu, Chem. Sci., 2018, 9, 950–955 RSC.
- J. Yang, X. Wang, F. Dai, L. Zhang, R. Wang and D. Sun, Inorg. Chem., 2014, 53, 10649–10653 CrossRef CAS PubMed.
- T.-F. Liu, L. Zou, D. Feng, Y.-P. Chen, S. Fordham, X. Wang, Y. Liu and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 7813–7816 CrossRef CAS PubMed.
- X. Lian, D. Feng, Y.-P. Chen, T.-F. Liu, X. Wang and H.-C. Zhou, Chem. Sci., 2015, 6, 7044–7048 RSC.
- J.-H. Wang, Y. Zhang, M. Li, S. Yan, D. Li and X.-M. Zhang, Angew. Chem., Int. Ed., 2017, 56, 6478–6482 CrossRef CAS PubMed.
- F. Drache, V. Bon, I. Senkovska, C. Marschelke, A. Synytska and S. Kaskel, Inorg. Chem., 2016, 55, 7206–7213 CrossRef CAS PubMed.
- A. B. Spore and N. L. Rosi, CrystEngComm, 2017, 19, 5417–5421 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed.
- J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560–4561 CrossRef CAS PubMed.
- H. N. Rubin and M. M. Reynolds, Inorg. Chem., 2017, 56, 5266–5274 CrossRef CAS PubMed.
- J. Aguilera-Sigalat and D. Bradshaw, Chem. Commun., 2014, 50, 4711–4713 RSC.
- T. Wittmann, R. Siegel, N. Reimer, W. Milius, N. Stock and J. Senker, Chem.–Eur. J., 2015, 21, 314–323 CrossRef CAS PubMed.
- P. Deria, Y. G. Chung, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 5172–5176 RSC.
- P. Deria, W. Bury, I. Hod, C.-W. Kung, O. Karagiaridi, J. T. Hupp and O. K. Farha, Inorg. Chem., 2015, 54, 2185–2192 CrossRef CAS PubMed.
- J. Hynek, S. Ondrušová, D. Bůžek, P. Kovář, J. Rathouský and J. Demel, Chem. Commun., 2017, 53, 8557–8560 RSC.
- D. Sun, P. R. Adiyala, S.-J. Yim and D.-P. Kim, Angew. Chem., Int. Ed., 2019, 58, 7405–7409 CrossRef CAS PubMed.
- W. Zhang, Y. Hu, J. Ge, H.-L. Jiang and S.-H. Yu, J. Am. Chem. Soc., 2014, 136, 16978–16981 CrossRef CAS PubMed.
- X. Qian, F. Sun, J. Sun, H. Wu, F. Xiao, X. Wu and G. Zhu, Nanoscale, 2017, 9, 2003–2008 RSC.
- L. Hou, L. Wang, N. Zhang, Z. Xie and D. Dong, Polym. Chem., 2016, 7, 5828–5834 RSC.
- S. Yang, L. Peng, D. T. Sun, M. Asgari, E. Oveisi, O. Trukhina, S. Bulut, A. Jamali and W. L. Queen, Chem. Sci., 2019, 10, 4542–4549 RSC.
- J. Castells-Gil, F. Novio, N. M. Padial, S. Tatay, D. Ruíz-Molina and C. Martí-Gastaldo, ACS Appl. Mater. Interfaces, 2017, 9, 44641–44648 CrossRef CAS PubMed.
- S. J. Yang and C. R. Park, Adv. Mater., 2012, 24, 4010–4013 CrossRef CAS PubMed.
- S. Gadipelli and Z. Guo, Chem. Mater., 2014, 26, 6333–6338 CrossRef CAS.
- X. Liu, Y. Li, Y. Ban, Y. Peng, H. Jin, H. Bux, L. Xu, J. Caro and W. Yang, Chem. Commun., 2013, 49, 9140–9142 RSC.
- Q. Sun, H. He, W.-Y. Gao, B. Aguila, L. Wojtas, Z. Dai, J. Li, Y.-S. Chen, F.-S. Xiao and S. Ma, Nat. Commun., 2016, 7, 13300 CrossRef CAS PubMed.
- Y. Sun, Q. Sun, H. Huang, B. Aguila, Z. Niu, J. A. Perman and S. Ma, J. Mater. Chem. A, 2017, 5, 18770–18776 RSC.
- S. J. Yang, J. Y. Choi, H. K. Chae, J. H. Cho, K. S. Nahm and C. R. Park, Chem. Mater., 2009, 21, 1893–1897 CrossRef CAS.
- D.-D. Zu, L. Lu, X.-Q. Liu, D.-Y. Zhang and L.-B. Sun, J. Phys. Chem. C, 2014, 118, 19910–19917 CrossRef CAS.
- B. Yuan, X.-Q. Yin, X.-Q. Liu, X.-Y. Li and L.-B. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 16457–16464 CrossRef CAS PubMed.
- N.-D. H. Gamage, K. A. McDonald and A. J. Matzger, Angew. Chem., Int. Ed., 2016, 55, 12099–12103 CrossRef CAS PubMed.
- N. Ding, H. Li, X. Feng, Q. Wang, S. Wang, L. Ma, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 10100–10103 CrossRef CAS PubMed.
- J. B. Decoste, G. W. Peterson, M. W. Smith, C. A. Stone and C. R. Willis, J. Am. Chem. Soc., 2012, 134, 1486–1489 CrossRef CAS PubMed.
- C.-M. Wu, M. Rathi, S. P. Ahrenkiel, R. T. Koodali and Z. Wang, Chem. Commun., 2013, 49, 1223–1225 RSC.
- A. Carné-Sánchez, K. C. Stylianou, C. Carbonell, M. Naderi, I. Imaz and D. Maspoch, Adv. Mater., 2015, 27, 869–873 CrossRef PubMed.
- Y.-H. Shih, Y.-C. Kuo, S. Lirio, K.-Y. Wang, C.-H. Lin and H.-Y. Huang, Chem.–Eur. J., 2017, 23, 42–46 CrossRef CAS PubMed.
- J. Kim, S. Yeo, J.-D. Jeon and S.-Y. Kwak, Microporous Mesoporous Mater., 2015, 202, 8–15 CrossRef CAS.
- M. Armstrong, P. Sirous, B. Shan, R. Wang, C. Zhong, J. Liu and B. Mu, Microporous Mesoporous Mater., 2018, 270, 34–39 CrossRef CAS.
- J. B. DeCoste, M. S. Denny Jr, G. W. Peterson, J. J. Mahle and S. M. Cohen, Chem. Sci., 2016, 7, 2711–2716 RSC.
- Y. Yoo, V. Varela-Guerrero and H.-K. Jeong, Langmuir, 2011, 27, 2652–2657 CrossRef CAS PubMed.
- B. L. Ouay, S. Kitagawa and T. Uemura, J. Am. Chem. Soc., 2017, 139, 7886–7892 CrossRef PubMed.
- T. Li, J. E. Sullivan and N. L. Rosi, J. Am. Chem. Soc., 2013, 135, 9984–9987 CrossRef CAS PubMed.
- A. J. Emerson, C. S. Hawes, M. Marshall, G. P. Knowles, A. L. Chaffee, S. R. Batten and D. R. Turner, Chem. Mater., 2018, 30, 6614–6618 CrossRef CAS.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- J. Canivet, A. Fateeva, Y. Guo, B. Coasne and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef PubMed.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- H. Kim, S. Yang, S. R. Rao, S. Narayanan, E. A. Kapustin, H. Furukawa, A. S. Umans, O. M. Yaghi and E. N. Wang, Science, 2017, 356, 430–434 CrossRef CAS PubMed.
- A. J. Rieth and M. Dincă, J. Am. Chem. Soc., 2018, 140, 3461–3466 CrossRef CAS PubMed.
- L. N. McHugh, M. J. McPherson, L. J. McCormick, S. A. Morris, P. S. Wheatley, S. J. Teat, D. McKay, D. M. Dawson, C. E. F. Sansome, S. E. Ashbrook, C. A. Stone, M. W. Smith and R. E. Morris, Nat. Chem., 2018, 10, 1096–1102 CrossRef PubMed.
- H. Wang, W. P. Lustig and J. Li, Chem. Soc. Rev., 2018, 47, 4729–4756 RSC.
- J.-H. Wang, M. Li and D. Li, Chem.–Eur. J., 2014, 20, 12004–12008 CrossRef CAS PubMed.
- A. V. Desai, B. Manna, A. Karmakar, A. Sahu and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 7811–7815 CrossRef CAS PubMed.
- Y. Liu, S. Lin, Y. Liu, A. K. Sarkar, J. K. Bediako, H. Y. Kim and Y.-S. Yun, Small, 2019, 1805242 CrossRef PubMed.
- S. Xu, S. Chansai, C. Stere, B. Inceesungvorn, A. Goguet, K. Wangkawong, S. F. R. Taylor, N. Al-Janabi, C. Hardacre, P. A. Martin and X. Fan, Nat. Catal., 2019, 2, 142–148 CrossRef CAS.
- G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294–3297 CrossRef CAS PubMed.
- L. Chi, Q. Xu, X. Liang, J. Wang and X. Su, Small, 2016, 12, 1351–1358 CrossRef CAS PubMed.
- J.-Q. Shen, P.-Q. Liao, D.-D. Zhou, C.-T. He, J.-X. Wu, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2017, 139, 1778–1781 CrossRef CAS PubMed.
- J.-S. Qin, D.-Y. Du, W. Guan, X.-J. Bo, Y.-F. Li, L.-P. Guo, Z.-M. Su, Y.-Y. Wang, Y.-Q. Lan and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 7169–7177 CrossRef CAS PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- H. He, Q. Sun, W. Gao, J. A. Perman, F. Sun, G. Zhu, B. Aguila, K. Forrest, B. Space and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 4657–4662 CrossRef CAS PubMed.
- G. Huang, Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef CAS PubMed.
- Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS.
- J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- B. V. Harbuzaru, A. Corma, F. Rey, J. L. Jordá, D. Ananias, L. D. Carlos and J. Rocha, Angew. Chem., Int. Ed., 2009, 48, 6476–6479 CrossRef CAS PubMed.
- B. J. Deibert and J. Li, Chem. Commun., 2014, 50, 9636–9639 RSC.
- F. M. Ebrahim, T. N. Nguyen, S. Shyshkanov, A. Gładysiak, P. Favre, A. Zacharia, G. Itskos, P. J. Dyson and K. C. Stylianou, J. Am. Chem. Soc., 2019, 141, 3052–3058 CrossRef CAS PubMed.
- S.-Y. Liu, X.-L. Qi, R.-B. Lin, X.-N. Cheng, P.-Q. Liao, J.-P. Zhang and X.-M. Chen, Adv. Funct. Mater., 2014, 24, 5866–5872 CrossRef CAS.
- S.-Y. Moon, A. J. Howarth, T. Wang, N. A. Vermeulen, J. T. Hupp and O. K. Farha, Chem. Commun., 2016, 52, 3438–3441 RSC.
- A. Karmakar, N. Kumar, P. Samanta, A. V. Desai and S. K. Ghosh, Chem.–Eur. J., 2016, 22, 864–868 CrossRef CAS PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed.
- H.-S. Wang, Coord. Chem. Rev., 2017, 349, 139–155 CrossRef CAS.
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS PubMed.
- M.-X. Wu and Y.-W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- J. D. Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef PubMed.
- E. Gkaniatsou, C. Sicard, R. Ricoux, J.-P. Mahy, N. Steunou and C. Serre, Mater. Horiz., 2017, 4, 55–63 RSC.
- J.-S. Qin, S. Yuan, C. Lollar, J. Pang, A. Alsalme and H.-C. Zhou, Chem. Commun., 2018, 54, 4231–4249 RSC.
- P. Wu, J. Wang, C. He, X. Zhang, Y. Wang, T. Liu and C. Duan, Adv. Funct. Mater., 2012, 22, 1698–1703 CrossRef CAS.
- S. Wu, Y. Lin, J. Liu, W. Shi, G. Yang and P. Cheng, Adv. Funct. Mater., 2018, 28, 1707169 CrossRef.
- J.-W. Zhang, H.-T. Zhang, Z.-Y. Du, X. Wang, S.-H. Yu and H.-L. Jiang, Chem. Commun., 2014, 50, 1092–1094 RSC.
- P. Chandrasekhar, A. Mukhopadhyay, G. Savitha and J. N. Moorthy, Chem. Sci., 2016, 7, 3085–3091 RSC.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- Z. Dong, Y. Sun, J. Chu, X. Zhang and H. Deng, J. Am. Chem. Soc., 2017, 139, 14209–14216 CrossRef CAS PubMed.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed.
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS PubMed.
- J. L. Harding, J. M. Metz and M. M. Reynolds, Adv. Funct. Mater., 2014, 24, 7503–7509 CrossRef CAS.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497–501 CrossRef CAS PubMed.
- X. Lian, Y.-P. Chen, T.-F. Liu and H.-C. Zhou, Chem. Sci., 2016, 7, 6969–6973 RSC.
- X. Meng, H.-N. Wang, S.-Y. Song and H.-J. Zhang, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
- A.-L. Li, Q. Gao, J. Xu and X.-H. Bu, Coord. Chem. Rev., 2017, 344, 54–82 CrossRef CAS.
- M. Yoon, K. Suh, S. Natarajan and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 2688–2700 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- S. C. Sahoo, T. Kundu and R. Banerjee, J. Am. Chem. Soc., 2011, 133, 17950–17958 CrossRef CAS PubMed.
- H. Wu, F. Yang, X.-L. Lv, B. Wang, Y.-Z. Zhang, M.-J. Zhao and J.-R. Li, J. Mater. Chem. A, 2017, 5, 14525–14529 RSC.
- D. D. Borges, S. Devautour-Vinot, H. Jobic, J. Ollivier, F. Nouar, R. Semino, T. Devic, C. Serre, F. Paesani and G. Maurin, Angew. Chem., Int. Ed., 2016, 55, 3919–3924 CrossRef CAS PubMed.
- M. Sadakiyo, T. Yamada, K. Honda, H. Matsui and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7701–7707 CrossRef CAS PubMed.
- W. J. Phang, H. Jo, W. R. Lee, J. H. Song, K. Yoo, B. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2015, 54, 5142–5146 CrossRef CAS PubMed.
- V. G. Ponomareva, K. A. Kovalenko, A. P. Chupakhin, D. N. Dybtsev, E. S. Shutova and V. P. Fedin, J. Am. Chem. Soc., 2012, 134, 15640–15643 CrossRef CAS PubMed.
- M. Bazaga-García, R. M. P. Colodrero, M. Papadaki, P. Garczarek, J. Zoń, P. Olivera-Pastor, E. R. Losilla, L. León-Reina, M. A. G. Aranda, D. Choquesillo-Lazarte, K. D. Demadis and A. Cabeza, J. Am. Chem. Soc., 2014, 136, 5731–5739 CrossRef PubMed.
- S. Wang, M. Wahiduzzaman, L. Davis, A. Tissot, W. Shepard, J. Marrot, C. Martineau-Corcos, D. Hamdane, G. Maurin, S. Devautour-Vinot and C. Serre, Nat. Commun., 2018, 9, 4937 CrossRef PubMed.
- W. J. Phang, W. R. Lee, K. Yoo, D. W. Ryu, B. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2014, 53, 8383–8387 CrossRef CAS PubMed.
- F. Yang, G. Xu, Y. Dou, B. Wang, H. Zhang, H. Wu, W. Zhou, J.-R. Li and B. Chen, Nat. Energy, 2017, 2, 877–883 CrossRef CAS.
- R. A. Dodson, A. G. Wong-Foy and A. J. Matzger, Chem. Mater., 2018, 30, 6559–6565 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |