
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photochromic upconversion nanoarchitecture: towards activatable bioimaging and dual NIR light-programmed singlet oxygen generation†
Yongsheng
Mi‡
ab,
Hong-Bo
Cheng‡
ab,
Hongqian
Chu
a,
Jian
Zhao
a,
Mingming
Yu
 d,
Zhanjun
Gu
bc,
Yuliang
Zhao
*ab and
Lele
Li
*ab
d,
Zhanjun
Gu
bc,
Yuliang
Zhao
*ab and
Lele
Li
*ab
aCAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: zhaoyl@nanoctr.cn; lilele@nanoctr.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cCAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
dCollege of Chemistry, Zhengzhou University, Zhengzhou, 450001, China
First published on 18th October 2019
Abstract
The precise control of singlet oxygen (1O2) generation is in great demand for biological studies and precision medicine. Here, a nanoarchitecture is designed and synthesized for generating 1O2 in a dual NIR light-programmable manner, while shifting to the therapeutic window. The nanoarchitecture is constructed by controlled synthesis of mesoporous silica-coated upconversion nanoparticles (UCNPs), wherein the porphyrin photosensitizers (PSs) are covalently embedded inside the silica walls while NIR (808 nm)-responsive diarylethene (DAE) photochromic switches are loaded in the nanopores. Upon irradiation with 980 nm NIR light, the UCNP core absorbs low energy photons and transfers energy to the PSs in the silica wall, leading to efficient 1O2 generation. Furthermore, this 980 nm NIR light photosensitized activity can be remotely controlled by irradiation with a distinct NIR wavelength (808 nm). The 1O2 generation is inhibited when the DAE installed in the nanopores is in the closed form, whereas irradiation of the nanoconstruct with 808 NIR light leads to the transformation of DAE to the open form, and thus enabling full recovery of the 980 nm NIR light excited 1O2 generation capability. The NIR light-mediated on-demand “activation” of the nanoarchitecture for bioimaging and controllable photodynamic therapy is further demonstrated in vitro and in vivo.
Introduction
Light provides new opportunities as a noninvasive and controllable tool for biological studies because of its unparalleled precision both temporally and spatially.1,2 Optogenetics has revolutionized the field involving the use of light to regulate the neuronal activity in biological systems as complex as live animals.3–5 In particular, molecular photoswitches (e.g., diarylethene (DAE), azobenzene, and spiropyran), which undergo a reversible structure change upon light irradiation, have attracted great attention for biomedical applications, such as photocontrolled cell adhesion,6 bioimaging,7–12 and drug release.13–15 For example, photoswitchable fluorescence probes, whose fluorescence can be modulated from a bright-to-dark or two color state through light control, have been developed for bioimaging with improved resolution and specificity.7–12 More recently, the employment of molecular photoswitches for controlling the action of therapeutics has enabled the emerging field of photopharmacology.16–20 These switching units allow for remote control of the function of bioactive molecules with light, and thus could reduce off-target activity and achieve high precision in treatment.16–20 Photopharmacology has been explored for various therapeutic applications, including antimicrobials,21–23 cancer therapy,24 and regulation of neuronal networks.25,26 However, such applications are often limited to cultured cells because most molecular photoswitches require UV and visible light for deactivation/activation, and are not effective in vivo. Development of new photopharmacological systems with higher potency and better in vivo properties would significantly expand the toolbox of this technology.16,20In biomedical studies, singlet oxygen (1O2) is of great importance due to its crucial role in the cell-killing mechanism of photodynamic therapy (PDT), a valuable phototherapy modality for a variety of cancers.27–29 In PDT, 1O2 is the cytotoxic agent which is immediately produced upon excitation of a photosensitizer (PS), leading to direct damage of the targeted region.27 Nevertheless, 1O2 could cause direct and non-specific damage to the cells containing the photosensitizers under daylight exposure. Hence, on-demand control of 1O2 generation is highly necessary for biological studies and precision medicine.30 Recently, the employment of additional stimuli for “off–on” switching of light-excited 1O2 generation was proposed to minimize systemic toxicity and to enable enhanced safety of PDT.30–32 In particular, the principle of photopharmacology was introduced for light-mediated dynamic control of 1O2 generation based on intermolecular energy transfer between porphyrinic PSs and DAE photoswitches.11,33–36 Despite the progress made, one fundamental roadblock that hampers the use of these photoactivatable systems for PDT is that activation of the DAE derivatives and excitation of PSs require irradiation with two different wavelengths of visible light.11,33–36 Apparently, shifting such light sensitivities from the visible light range to the near-infrared (NIR) window would be more desirable due to its superior tissue penetration, which would allow for remote activation of PDT with relatively low interference and high precision.16
Thus motivated, here we report the development of a 980 nm NIR light photosensitized system, whose 1O2 generation activity can be remotely activated by 808 nm NIR light irradiation (Scheme 1). The nanoarchitecture was composed of a lanthanide-doped upconversion nanoparticle (UCNP) core and a mesoporous silica (MSN) shell, wherein the porphyrin PSs were covalently embedded inside the silica walls while the NIR-responsive DAE molecules were loaded in the mesopores in close proximity to the PSs. UCNPs, capable of upconverting NIR light into tunable shorter-wavelength UV and visible light via the multiphoton processes,37–39 have emerged as a promising platform to enable the delivery of light to deep tissues for various applications, including bioimaging,40–44 spatiotemporally regulated biosensing,45–47 optical memory,48 optogenetics,49 and controlled drug release.50–52 In particular, UCNPs have been widely used as light transducers to activate PSs with NIR light for improved PDT performance in deep tissues.53–55 Despite the progress made, to the best of our knowledge, building a smart “activatable” UCNP-PDT system with precisely controlled 1O2 generation has not yet been realized to date. In our nanoconstruct, the visible upconversion luminescence (UCL) of UCNPs (upon 980 nm light irradiation) overlays well with the broad absorption spectrum of PSs, and enables efficient energy transfer from the UCNP core to the PSs, leading to 980 nm light excited 1O2 production. Moreover, the NIR-responsive DAE molecules installed in the nanopores can noninvasively control this 1O2 generation activity. The 1O2 production was inhibited when the DAE is in the closed form (C-DAE) because of the energy transfer from PSs to C-DAE. Upon 808 nm light irradiation, the DAE unit is transformed to the colorless open form (O-DAE), resulting in recovery of the 980 nm light photosensitized 1O2 generation. Therefore, the nanoarchitecture enables dual NIR light programmed activatable 1O2 production. Furthermore, the system could be used for imaging-guided PDT in vitro and in vivo with superior controllability of 1O2 generation.
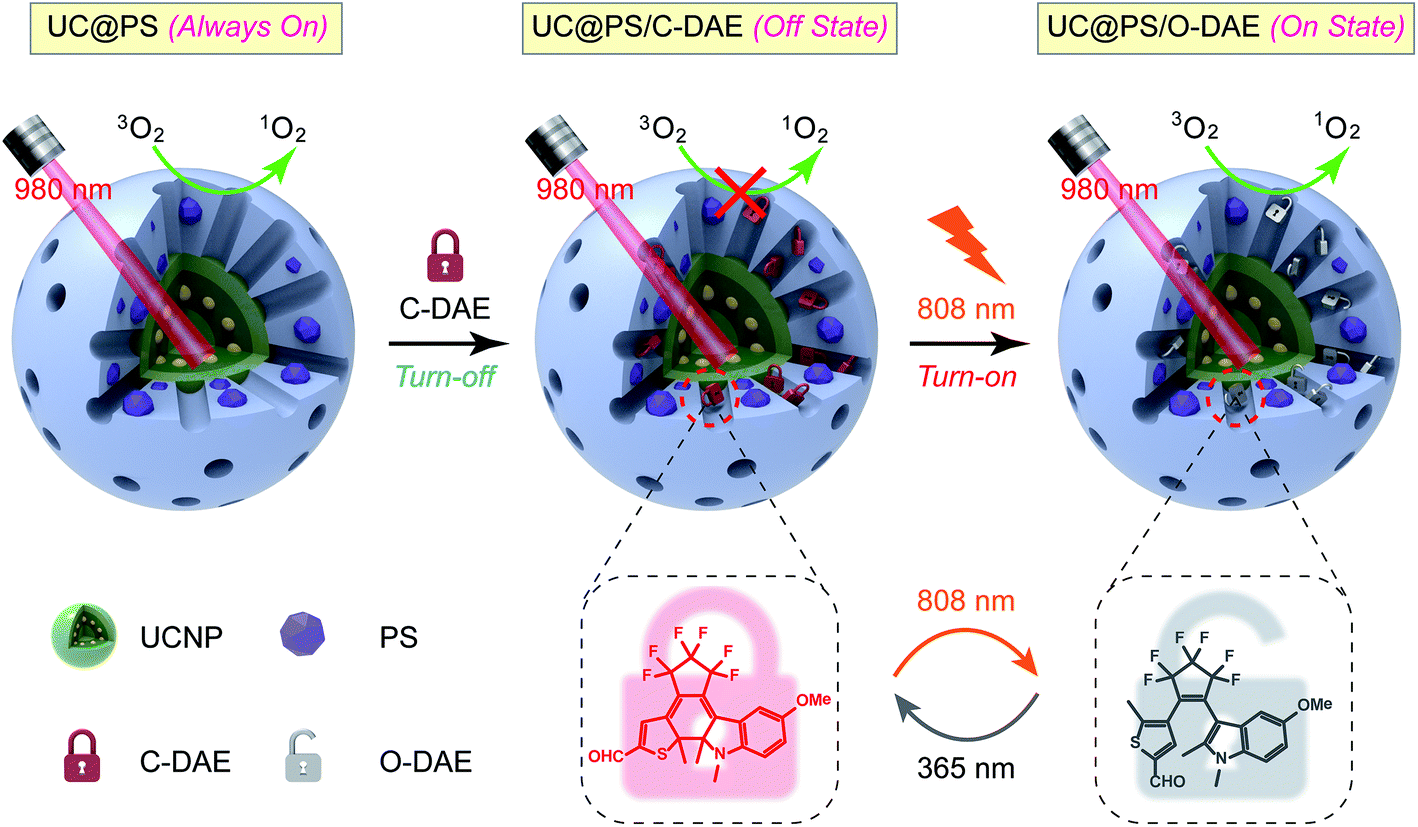 | ||
| Scheme 1 Schematic illustration of the 980 nm NIR light photosensitized UC@PS/C-DAE nanoconstruct, whose 1O2 generation capability can be remotely switched on by 808 nm NIR light irradiation. | ||
Results and discussion
Preparation and characterization of the nanoarchitecture
The UC@PS/C-DAE was prepared via a two-step process, as illustrated in Fig. 1a. Oleate-capped sandwich-structured NaGdF4@NaGdF4:Yb,Er@NaGdF4 UCNPs were first prepared through a seeded-growth method.56 Yb/Er ions were co-doped in the NPs to achieve visible UCL from Er3+ under 980 nm NIR light irradiation (Fig. S1†). Furthermore, the Er3+ ions are precisely doped in the inner shell near the surface of the NPs so that they can be close to external PSs for improving their energy transfer.55 The outer shell could efficiently enhance the upconversion efficiency via preventing the environmental quenching effect (Fig. S2†). Transmission electron microscopy (TEM) showed that the final UCNPs possess a mean size of ∼32 nm (Fig. 1b) and an outer shell thickness of ∼3 nm (Fig. S3†). Next, hydrolysis–condensation of tetra-substituted porphyrin silsesquioxane (TPS) and tetraethyl orthosilicate (TEOS) was performed on the surface of the UCNPs by using cetyltrimethyl ammonium bromide (CTAB) as the template. Tetrakis(4-carboxyphenyl)porphyrin (TCPP) is a widely used porphyrin PS, and its derivatives have been used as PDT agents in clinical applications.57,58 In our design, TCPP was modified with four co-condensable alkoxysilane arms to obtain the TPS (see the structure in Fig. S4†), which would enable covalent incorporation into the silica framework walls during the self-assembly process.59 This design also allows controllable PS loading and prevents leakage of PSs. Finally, CTAB molecules in the nanopores were removed, and the residual nanopores of the resulting UC@PS were ready to encapsulate DAE photoswitches through the impregnation process. The NIR-responsive DAE molecules were synthesized according to our previous reports.60 The loading content of DAE in the system was calculated to be 8 wt% (Fig. S5†). The core–shell mesostructure enabled close proximity of TCPP to both the UCNP core and DAE in the nanopores, which is indispensable for efficient energy transfer.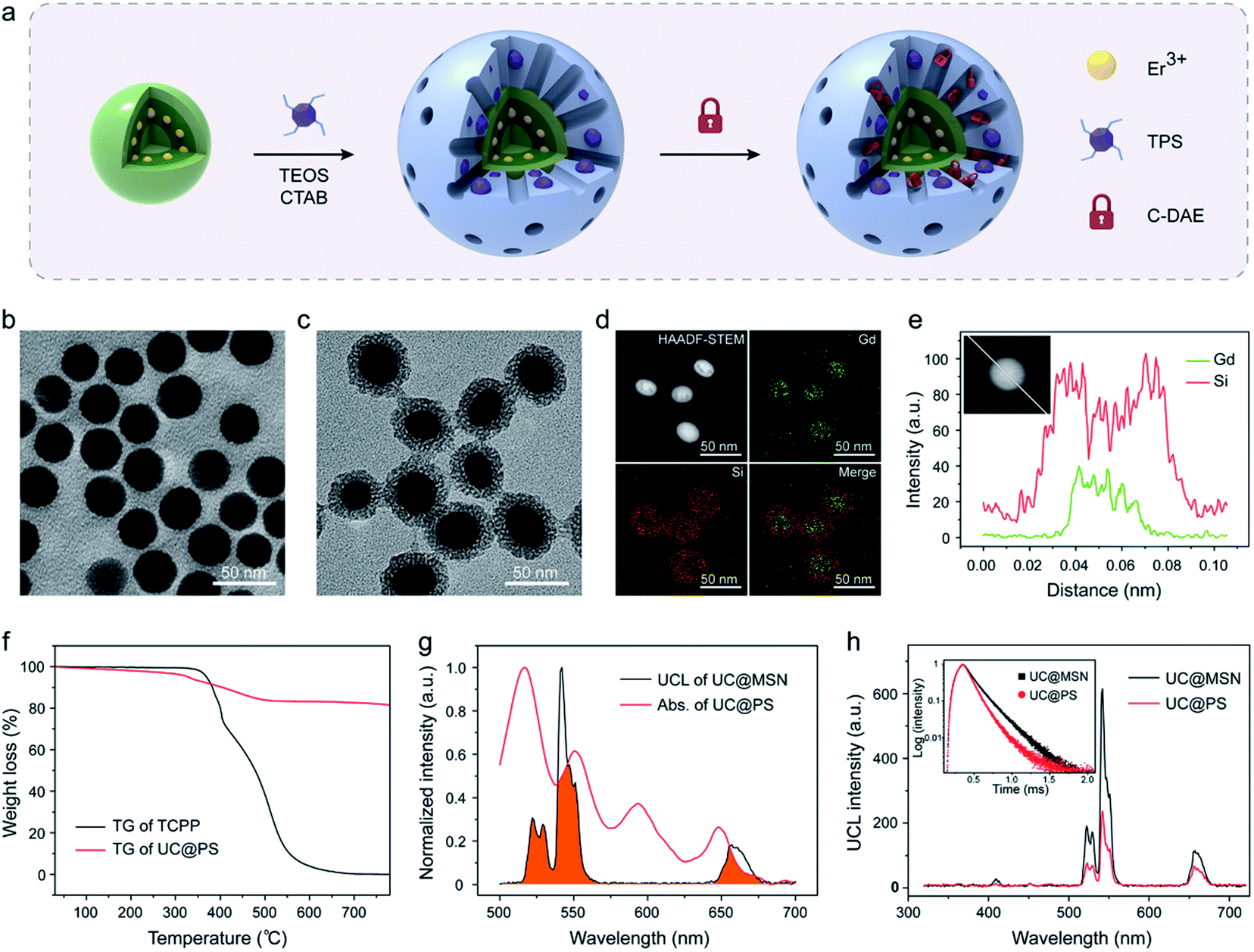 | ||
| Fig. 1 (a) Schematic showing the synthesis procedure of UC@PS/C-DAE. TEM image of (b) the sandwich-structured UCNPs and (c) UC@PS. (d) HAADF-STEM image and elemental mapping images of UC@PS. (e) EDS line scan profiles of UC@PS nanoparticles. (f) TG curves of free TCPP and UC@PS. (g) UCL spectrum of UC@MSN and absorption spectrum of UC@PS. The overlapping of spectra is indicated in orange. (h) UCL spectra of UC@MSN and UC@PS. Inset: UCL decay curves of UC@MSN and UC@PS (λex = 980 nm, λem = 542 nm). | ||
Fig. 1c shows the representative TEM images of UC@PS. The core–shell structure is clearly identified with characteristic worm-like mesopores in its silica shell. The silica shell is around 8 nm (Fig. S3†). Dynamic-light-scattering analysis indicated that UC@PS had an average hydrodynamic diameter of 182 ± 30 nm (Fig. S6†). The composition information of the nanoconstruct was evaluated in detail with high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). As shown in Fig. 1d, elemental mapping investigation of Gd and Si in the nanoconstruct confirms the core–shell structure in terms of the compositional distributions. This structure was also verified by HAADF-STEM-EDS line scanning analysis of single nanoparticles (Fig. 1e). The UCNP@PS showed a typical UV-vis absorbance and fluorescence spectrum of TCPP (Fig. S7†), indicating the successful encapsulation of TCPP PSs in the nanostructures. The content of TCPP units in the UCNP@PS was calculated to be around 18 wt% based on thermogravimetry and differential thermal analysis (TG-DTA) (Fig. 1f and S8†). The UCL spectra of the MSN-coated UCNPs with and without PSs embedded in the silica walls were subsequently measured. When the pure MSN coated UCNPs (UC@MSN) were illuminated with 980 nm NIR light, the Yb3+ ions absorb photons and transfer energy to Er3+ ions, resulting in typical UCL peaks at 522, 542 and 657 nm (Fig. 1g). For UC@PS, characteristic Q-bands of TCPP PSs at 517, 552, 593, and 648 nm were observed in the UV-vis absorption spectrum, which overlaps well with the UCL of UC@MSN (Fig. 1g). This spectral overlapping and confined location of the PSs on the surface of UCNPs could induce energy transfer from the UCNP core to PSs within the silica shell. The efficient energy transfer between them was confirmed by the strong decrease of the Er3+ UCL after incorporation of PSs (Fig. 1h). The ratio of green to red UCL (I542/I657 = 4.6) of the UC@PS is decreased in comparison with that of UC@MSN (I542/I657 = 7.6) due to the relatively more overlapping of the absorption spectrum of PSs with the green UCL of UCNPs. The energy transfer process was also confirmed by time-resolved photoluminescence measurements. The UCL decay curves of UC@MSN and UC@PS are shown in the inset of Fig. 1h, revealing an obvious decrease of the UCL lifetime at 542 nm (from 165 to 114 μs) upon the PS loading.
Dual NIR light-programmed singlet oxygen generation
Next, we investigated on-demand manipulation of the photochemical properties of UC@PS with the NIR-responsive DAE photochromic switches. As shown in Fig. 2a and S9†, under 808 nm light irradiation, the UV-vis-NIR absorption spectrum of the C-DAE showed that the peak in the vis-NIR range (the maximum absorbance at 712 nm) disappeared and the peak in the UV range below the isosbestic point of 378 nm gradually increased in intensity, indicating the transformation of DAE from C-DAE to O-DAE. Simultaneously, the DAE solution gradually changed from dark green to colorless (Fig. 2a, inset). Upon irradiation with UV light, the obtained colorless solution of O-DAE turned dark green again and a nearly complete recovery of the absorption spectrum of C-DAE was achieved (Fig. 2a and S10†), indicating the reversible transformation. Furthermore, the photo-switchable interconversion of DAE could be repeated at least ten times without any appreciable light fatigue phenomenon (Fig. 2b). The emission spectrum of UC@PS shows an emission maximum of TCPP PSs at 653 nm, which overlaps well with the absorption band of C-DAE in the vis-NIR range but not with that of O-DAE (Fig. 2c). Such a distinctive spectral overlap enables different energy transfer pathways for the PSs to regulate their fluorescence and 1O2 generation. Upon irradiation of UC@PS/C-DAE with 808 nm light for up to 30 s, the emission at 653 nm increased by around 6-fold (Fig. 2d and S11†), suggesting inhibitory energy transfer from PSs to the O-DAE. As expected, the fluorescence intensity decreased when the 808 nm light-irradiated solution was further treated with 365 nm irradiation because of the reverse photoisomerization from O-DAE to C-DAE (Fig. 2d and S12†). Significantly, this cycle of fluorescence switching on/off could be repeated at least ten times, indicating the good fatigue resistance of the dual-state nanoconstruct (Fig. 2e).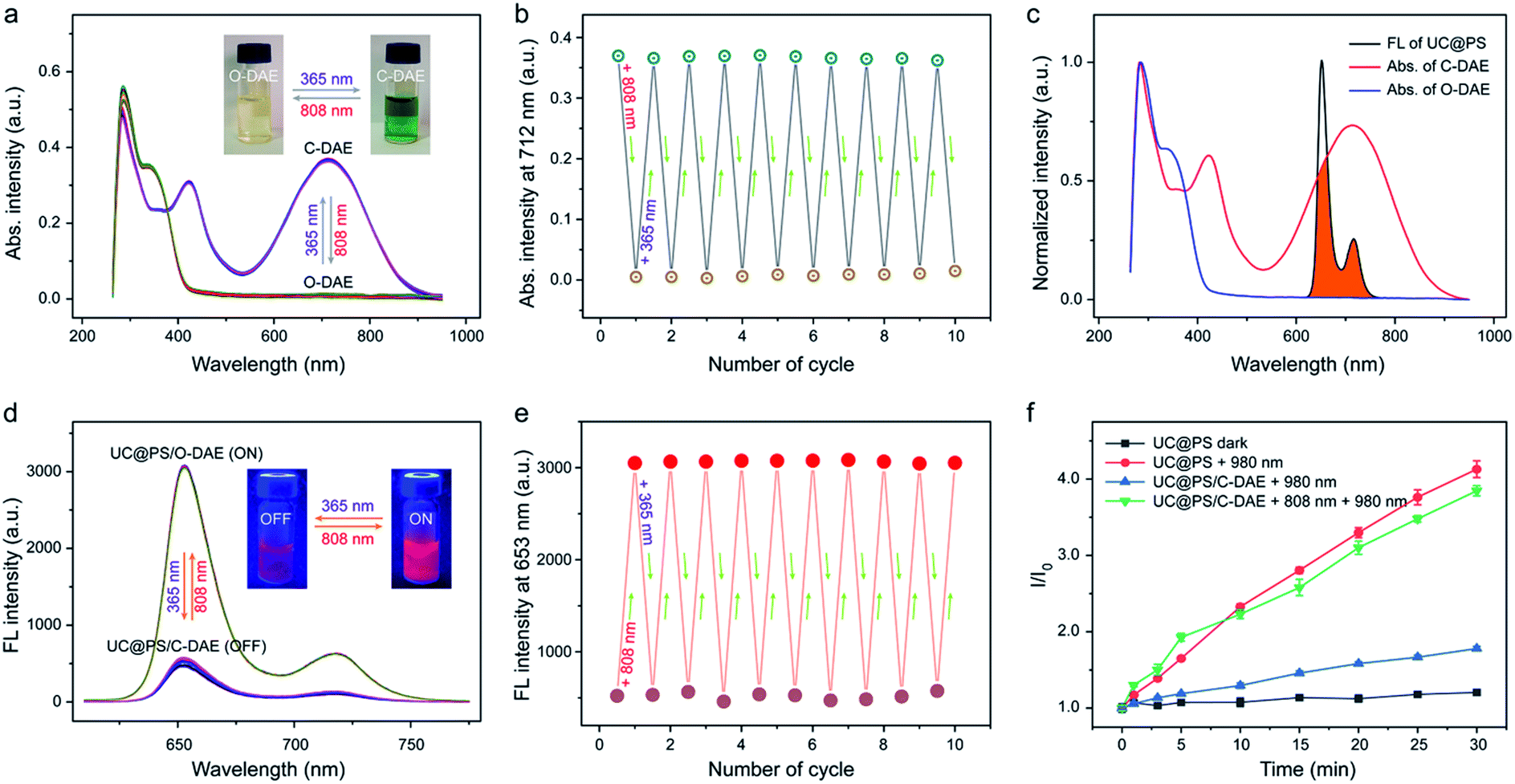 | ||
| Fig. 2 (a) UV-vis-NIR absorption spectra and (b) absorption intensity changes at 712 nm of the photochromic DAE upon alternating UV (365 nm, 5 mW cm−2, 1 min) and NIR (808 nm, 0.25 W cm−2, 30 s) light irradiation. The inset in (a) shows the changes of its photographic image. (c) UV-vis-NIR absorption (Abs.) spectra of two different forms of DAE and the fluorescence spectrum of UC@PS. The overlapping of spectra is indicated in orange. (d) Fluorescence spectra and (e) fluorescence intensity changes at λmax = 653 nm of UC@PS/C-DAE upon alternating UV (365 nm, 5 mW cm−2, 1 min) and NIR (808 nm, 0.25 W cm−2, 30 s) light irradiation (λex = 420 nm). The inset in (e) shows the changes of its photographic fluorescence image. (f) Singlet oxygen generation by UC@PS and UC@PS/DAE upon irradiation with light of different wavelengths in water (808 nm, 0.25 W cm−2, and 980 nm, 1.2 W cm−2), detected by SOSG assay. Data are means ± SD; N = 3. | ||
Then, activatable 1O2 generation of the nanoconstruct was investigated using a typical 1O2 indicator, namely SOSG due to its increased fluorescence in the presence of 1O2 (Fig. 2f). The SOSG fluorescence intensity in UC@PS solution was almost unchanged without irradiation, while irradiation with 980 nm laser light for 30 min led to a 4.1-fold fluorescence enhancement. As a control, the TPS molecule itself could not produce 1O2 upon the 980 nm laser irradiation (Fig. S13†). The results indicated that the UCNPs could efficiently transfer photon energy to the PSs in the silica walls, leading to 1O2 generation from surrounding oxygen molecules. In addition, the 1O2 produced by UC@PS showed a shorter lifetime (5.0 μs) compared with that of a free PS (TCPP) (6.8 μs), which could be attributed to the quenching effect of the silica microenvironment.61 In contrast, a slightly increased SOSG fluorescence was observed for UC@PS/C-DAE after 980 nm irradiation, indicating the suppression of the photosensitizing ability of UC@PS after loading with C-DAE in its nanopores. Upon irradiation of the UC@PS/C-DAE with 808 nm laser light for 30 s, the increased SOSG fluorescence intensity (3.8-fold) was comparable to that for UC@PS under 980 nm excitation, suggesting a recovery of the 1O2 generation capability. Taken together, the excited state of the PS can be quenched via the energy transfer from the PS to C-DAE, and thus the 980 nm light-excited 1O2 production is inhibited. Conversely, 1O2 can be efficiently generated when the energy transfer between the PS and O-DAE is inhibited (Fig. S14†).
In the reported systems for controlling 1O2 generation based on the combination of porphyrinic PSs with visible light-responsive DAE molecules, the excitation of PSs and activation of the DAE require visible light of two different wavelengths.11,33–36 In our design, UCNPs were employed as a transducer that can generate visible light in situ upon NIR light irradiation, allowing for 980 nm light excited 1O2 generation. Moreover, an 808 nm NIR light-responsive DAE molecule was synthesized and used as an additional tool for regulation of 1O2 generation. Therefore, the nanoconstruct enables shifting of both the light wavelengths for excitation of PSs and activation of DAE to the biological window, allowing dual NIR light-programmed activatable 1O2 production. We then compared the 1O2 generation efficacy of our system versus free PSs at the deep tissue level. As shown in Fig. S15†, free PSs excited by 640 nm visible light generated much more 1O2 compared to UC@PS/O-DAE excited by 980 nm NIR light. However, the 1O2 generation of TCPP significantly decreased by ∼70% and ∼99% when the 640 nm light was blocked by pork tissues of 3 mm and 7 mm, respectively. In contrast, 1O2 generated by UC@PS/O-DAE reduced by ∼8% and ∼50% when the 980 nm light was blocked by pork tissues with the same thicknesses, respectively. Notably, UC@PS/O-DAE produced more 1O2 than TCPP when using 7 mm thick pork tissues under different light sources. Therefore, this result validates the advantage of using UCNPs as transducers for NIR light triggered 1O2 production at the deep-tissue level.
Activatable bioimaging and dual NIR light-controlled PDT
Having confirmed dual NIR light controlled 1O2 generation in solution, we then evaluated the applicability of the nanoconstruct for activatable PDT in cancer cells. As shown in Fig. 3a, the 4T1 breast cancer cells treated with UC@PS displayed a strong red fluorescence from the PSs. In contrast, the cells treated with UC@PS/C-DAE showed much less fluorescence because of the proposed energy transfer between the PS and the C-DAE. Notably, upon irradiation of the resulting cells with 808 nm laser light (0.5 W cm−2, 3 min), the cells showed a significantly increased red fluorescence as intense as that of cells treated with UC@PS, suggesting activatable bioimaging in living cells. To validate these results by confocal microscopy, we quantified the cellular fluorescence signals using flow cytometry (Fig. 3b and c). The results showed that UC@PS/C-DAE displayed a 3.3-fold increase in cellular fluorescence intensity upon 808 nm light irradiation.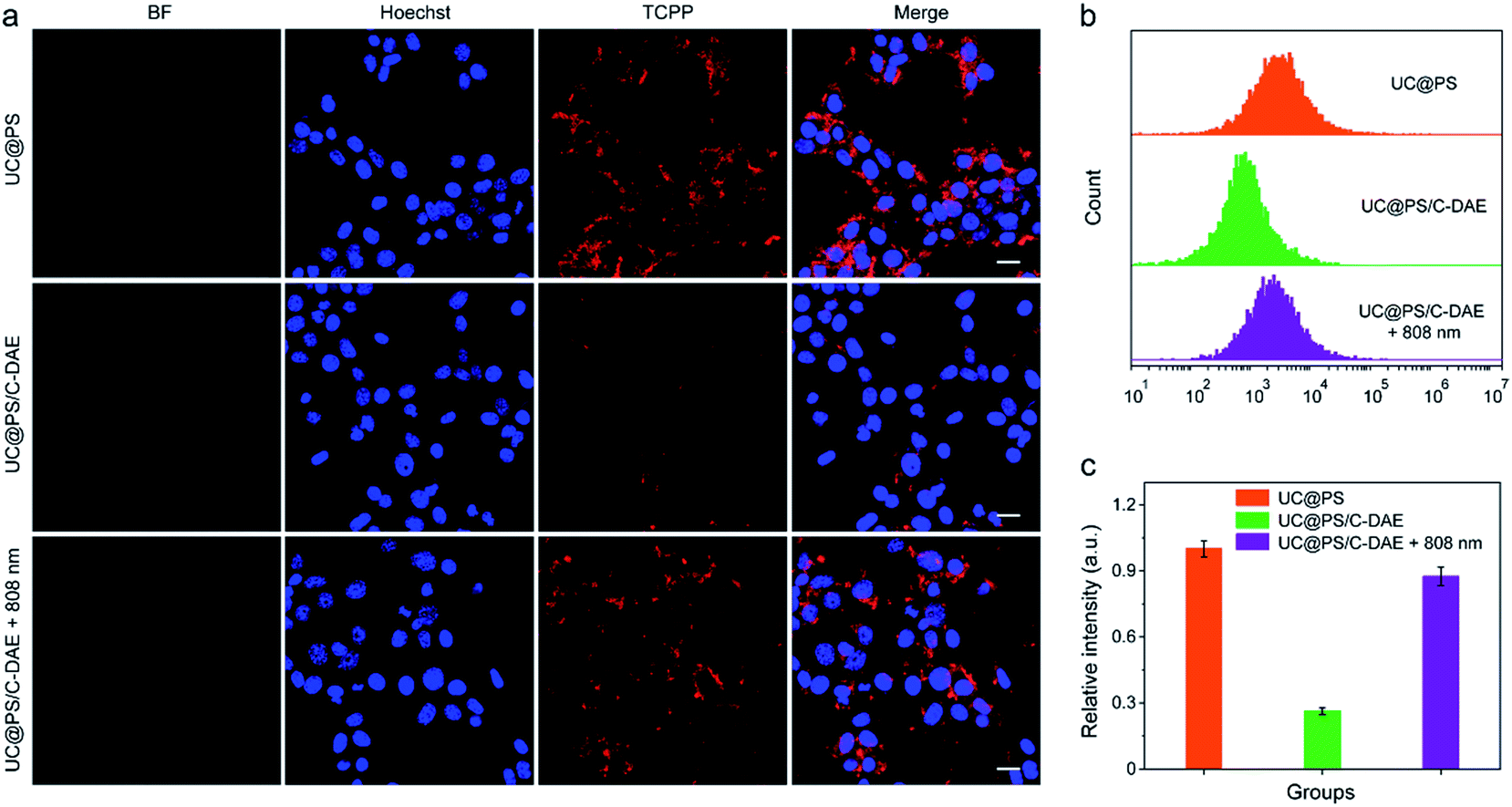 | ||
| Fig. 3 (a) CLSM images of 4T1 cells with different treatments. Nuclei stained with Hoechst 33342. For TCPP images, λex = 633 nm, λem = 675–725 nm. Scale bar: 20 μm. (b) Flow cytometry showing the fluorescence of 4T1 cells with different treatments. (c) Quantification of the flow cytometric data in (b). Data are represented as means ± SD (N = 3). | ||
We then investigated the 808 nm light-activated 1O2 generation by the nanoconstruct in cells using an intracellular 1O2 indicator (dichlorofluorescein diacetate (DCF-DA)) (Fig. 4a). Nonfluorescent DCF-DA can be oxidized to dichlorofluorescein (DCF) with increased fluorescence by 1O2. The 4T1 cells treated with UC@PS without 980 nm light irradiation showed negligible fluorescence of DCF (Fig. S16†). After 980 nm light irradiation (1.2 W cm−2, 20 min, 5 min break after 1.5 min irradiation to avoid the potential heating effect), the cells treated with UC@PS exhibited highly intense green fluorescence of DCF. In contrast, minimum fluorescence was seen in cells treated with UC@PS/C-DAE followed by 980 nm light irradiation (Fig. 4a), suggesting that the 1O2 generation capability of the nanoconstruct was inhibited. Upon activation of the nanoconstruct with 808 nm light, the 980 nm light-excited 1O2 generation recovered in cells, confirming the activatable behavior. These results were further validated using flow cytometry (Fig. 4b and c). For the UC@PS/C-DAE treated group, a 2.2-fold increase in fluorescence intensity of DCF was observed with 808 nm light activation.
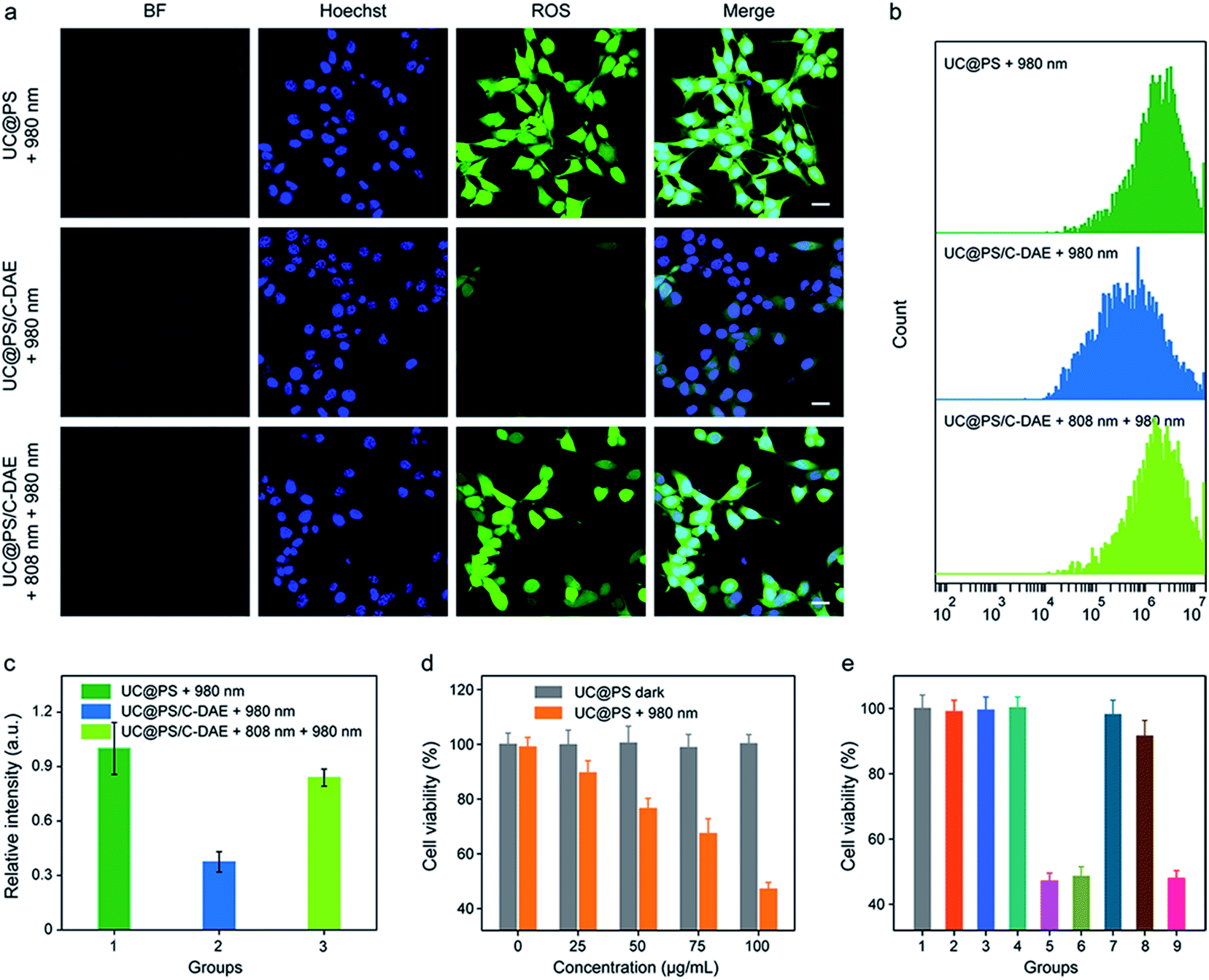 | ||
| Fig. 4 (a) CLSM images of 4T1 cells after different treatments. DCF-DA was used as an indicator of 1O2 in cells. Nuclei stained with Hoechst 33342. For DCF-DA images, λex = 488 nm, λem = 535–585 nm. Scale bar: 20 μm. (b) Flow cytometry showing the fluorescence of 4T1 cells with different treatments. (c) Quantification of the flow cytometric data in (b). Data are represented as means ± SD (N = 3). (d) Cytotoxicity assay of 4T1 cells incubated with different concentrations of UC@PS without or with 980 nm irradiation. (e) Cytotoxicity assay of the cells with different treatments. (1) Saline, (2) 980 nm, (3) 808 nm + 980 nm, (4) UC@PS, (5) UC@PS + 980 nm, (6) UC@PS + 808 nm + 980 nm, (7) UC@PS/C-DAE, (8) UC@PS@C-DAE + 980 nm, and (9) UC@PS/C-DAE + 808 nm + 980 nm. Data are represented as means ± SD (N = 4). For the irradiation, 808 nm (0.5 W cm−2, 3 min) and 980 nm (1.2 W cm−2, 20 min, 5 min break after 1.5 min irradiation) were applied. | ||
The in vitro PDT efficacy against 4T1 cells was then evaluated. UC@PS showed a dose-dependent PDT efficacy upon 980 nm light irradiation (Fig. 4d). The treatment with only NIR light, UC@PS or UC@PS/C-DAE led to a negligible change of the cell viability (Fig. 4e), suggesting the good biocompatibility of the nanoparticles. In particular, the cells treated with UC@PS/C-DAE and subsequent 980 nm light irradiation showed no apparent PDT effect, confirming our hypothesis that its 1O2 production could be inhibited in live cells. Furthermore, the PDT efficacy of UC@PS/C-DAE can be efficiently switched on by 808 nm NIR light irradiation, enabling obvious cytotoxicity to cancer cells.
Inspired by the good performance of the nanoconstruct in vitro, we further examined the activatable PDT in vivo. The 4T1 tumor-bearing mice were injected with UC@PS or UC@PS/C-DAE. For the groups treated with NIR irradiation, the tumor site was illuminated with an 808 nm laser or/and 980 nm laser 30 min postinjection. The mice treated with UC@PS/C-DAE and 808 nm light irradiation showed strong fluorescence of PSs at the tumor site, comparable to the UC@PS group, but much higher than that in the nonirradiated UC@PS/C-DAE group at each tested time point (e.g., ∼4-fold higher at 3 h post-injection) (Fig. 5a and b). The UC@PS group showed similar fluorescence intensity at the tumor site with and without 808 nm irradiation (Fig. S17†). These results indicated that an increased intratumoral fluorescence signal in the UC@PS/C-DAE + 808 nm group was indeed due to 808 nm light-triggered transformation of C-DAE to O-DAE.
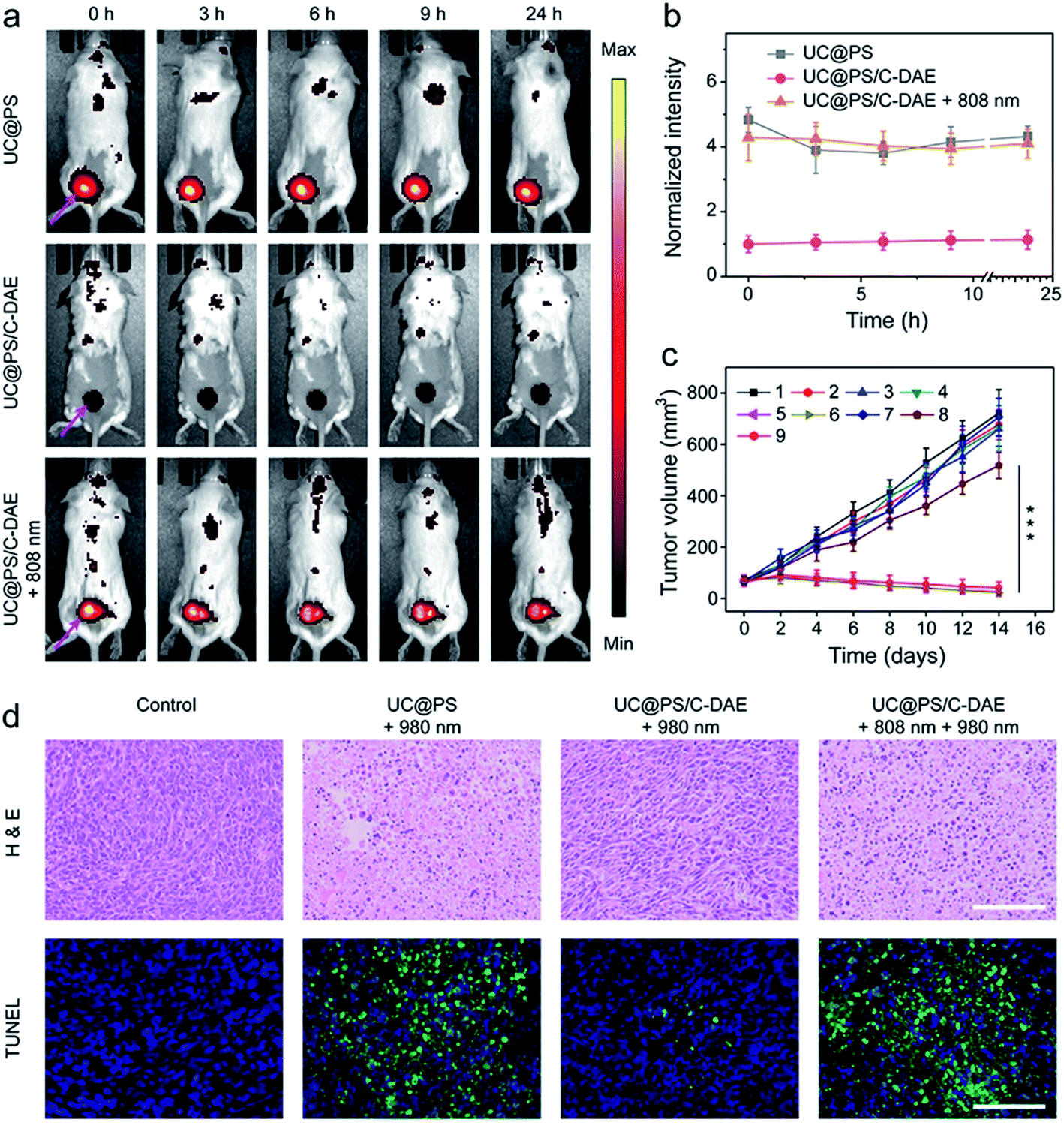 | ||
| Fig. 5 (a) Representative time-dependent in vivo fluorescence imaging of 4T1 tumor-bearing mice after exposure to different treatments. The arrows show the tumor sites. λex = 640 nm, λem = 700 nm. (b) Quantitative analysis of intratumoral fluorescence intensity over 24 h after different treatments, data normalized to the intensity at 0 h. Data are means ± SD; N = 4. (c) The 4T1 tumor growth curves after different treatments ((1) saline, (2) saline + 980 nm, (3) saline + 808 nm + 980 nm, (4) UC@PS, (5) UC@PS + 980 nm, (6) UC@PS + 808 nm + 980 nm, (7) UC@PS/C-DAE, (8) UC@PS@C-DAE + 980 nm, and (9) UC@PS/C-DAE + 808 nm + 980 nm). Data are means ± SD; N = 5. ***P < 0.001. For the irradiation, 808 nm (1.0 W cm−2, 10 min) and 980 nm (1.2 W cm−2, 20 min) lasers were applied. (d) Histological images of the tumor sections on day 14 post various treatments. The tissues were stained with H&E and TUNEL, respectively. Scale bar: 100 μm. | ||
Finally, NIR light activated PDT in vivo was evaluated. As shown in Fig. 5c, no obvious difference in the final size of tumor was observed in saline, saline + 808 nm, and saline + 808 nm + 980 nm treated groups, suggesting that NIR irradiation itself had no effect on tumor growth. The tumor growth of mice treated with UC@PS + 980 nm was efficiently delayed, while UC@PS without irradiation showed no impact on tumor progression, indicative of 980 nm light-excited PDT. It is noteworthy that treatment with UC@PS/C-DAE + 980 nm showed no obvious anti-tumor effect, while in the group treated with UC@PS/C-DAE + 808 nm + 980 nm, the tumor growth was completely suppressed, confirming the dual NIR light controlled therapeutic effect. The body weight of the mice showed no obvious change throughout the entire treatment (Fig. S18†). The histologic images obtained using hematoxylin and eosin (H&E) staining showed that the activation with 808 nm light induced much more tumor cell death than in the non-activated groups (Fig. 5d). Furthermore, in the images obtained with the TUNEL assay, a much higher level of cell apoptosis was observed in the harvested tumor from the mice treated with UC@PS/C-DAE and dual NIR light irradiation than that in control groups (Fig. 5d). Collectively, these results verified that the nanoplatform allows NIR-controlled activatable PDT in vivo.
Conclusions
In summary, we developed a 980 nm NIR light photosensitized nanoconstruct, whose activity can be remotely activated by irradiation with a distinct NIR wavelength (808 nm) for controllable 1O2 generation. Identification of a UCNP core with wavelength-converting ability and subsequent, controlled organization of porphyrin PSs and NIR-responsive photochromic switches on its surface led us to a nanoconstruct whose PDT capacity could be activated under 808 nm light irradiation. Importantly, the system enables shifting of the activating light from the visible range toward the NIR window. This nanoconstruct also reveals great advantages for efficient delivery of an integrated photosensitizing system in living cells, allowing activatable PDT in vitro and in vivo with efficient control of 1O2 release. This systemic platform provides a general solution to overcome the limitation of the photoactivatable PDT associated with the necessary use of visible light as an external regulatory tool.Ethical statement
All animal experiments were performed in strict accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the National Center for Nanoscience and Technology (NCNST). All animal protocols were approved by the IACUC of NCNST (Beijing, China).Conflicts of interest
All authors declare no conflict of interest.Acknowledgements
This work was supported financially by the NSFC (21822401, 21771044) and the Young Thousand Talented Program.Notes and references
- K. Deisseroth, Nat. Methods, 2011, 8, 26 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446 CrossRef CAS PubMed.
- R. L. Fork, Science, 1971, 171, 907 CrossRef CAS PubMed.
- S. Konermann, M. D. Brigham, A. E. Trevino, P. D. Hsu, M. Heidenreich, L. Cong, R. J. Platt, D. A. Scott, G. M. Church and F. Zhang, Nature, 2013, 500, 472 CrossRef CAS PubMed.
- M. Schönberger, M. Althaus, M. Fronius, W. Clauss and D. Trauner, Nat. Chem., 2014, 6, 712 CrossRef PubMed.
- J. Robertus, W. R. Browne and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 354 RSC.
- A. A. Beharry, L. Wong, V. Tropepe and G. A. Woolley, Angew. Chem., Int. Ed., 2011, 50, 1325 CrossRef CAS PubMed.
- K. Jeong, S. Park, Y.-D. Lee, C.-K. Lim, J. Kim, B. H. Chung, I. C. Kwon, C. R. Park and S. Kim, Adv. Mater., 2013, 25, 5574 CrossRef CAS PubMed.
- H. Wu, Y. Chen and Y. Liu, Adv. Mater., 2017, 29, 1605271 CrossRef PubMed.
- A. Parrot, A. Bernard, A. Jacquart, S. A. Serapian, C. Bo, E. Derat, O. Oms, A. Dolbecq, A. Proust, R. Métivier, P. Mialane and G. Izzet, Angew. Chem., Int. Ed., 2017, 56, 4872 CrossRef CAS PubMed.
- J. Qi, C. Chen, X. Zhang, X. Hu, S. Ji, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, Nat. Commun., 2018, 9, 1848 CrossRef PubMed.
- M. Li, J. Zhao, H. Chu, Y. Mi, Z. Zhou, Z. Di, M. Zhao and L. Li, Adv. Mater., 2018, 1804745 CrossRef PubMed.
- R. Tong, H. H. Chiang and D. S. Kohane, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19048 CrossRef CAS PubMed.
- A. M. Asadirad and N. R. Branda, J. Am. Chem. Soc., 2015, 137, 2824 CrossRef CAS PubMed.
- J. Liu, W. Bu, L. Pan and J. Shi, Angew. Chem., Int. Ed., 2013, 52, 4375 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178 CrossRef CAS PubMed.
- J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947 CrossRef CAS PubMed.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114 CrossRef PubMed.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 10978 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710 CrossRef PubMed.
- M. Wegener, M. J. Hansen, A. J. M. Driessen, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 17979 CrossRef CAS PubMed.
- W. A. Velema, J. P. van der Berg, M. J. Hansen, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924 CrossRef CAS PubMed.
- M. J. Hansen, J. I. C. Hille, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Chem, 2019, 5, 1293 CAS.
- M. Borowiak, W. Nahaboo, M. Reynders, K. Nekolla, P. Jalinot, J. Hasserodt, M. Rehberg, M. Delattre, S. Zahler, A. Vollmar, D. Trauner and O. Thorn-Seshold, Cell, 2015, 162, 403 CrossRef CAS PubMed.
- E. C. Carroll, S. Berlin, J. Levitz, M. A. Kienzler, Z. Yuan, D. Madsen, D. S. Larsen and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E776 CrossRef CAS PubMed.
- A. Reiner and E. Y. Isacoff, Nat. Chem. Biol., 2014, 10, 273 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38 RSC.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CrossRef CAS PubMed.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839 CrossRef CAS PubMed.
- Y. Ichikawa, M. Kamiya, F. Obata, M. Miura, T. Terai, T. Komatsu, T. Ueno, K. Hanaoka, T. Nagano and Y. Urano, Angew. Chem., Int. Ed., 2014, 53, 6772 CrossRef CAS PubMed.
- Y. Yuan, C.-J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780 CrossRef CAS PubMed.
- L. Hou, X. Zhang, T. C. Pijper, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 910 CrossRef CAS PubMed.
- J. Park, D. Feng, S. Yuan and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 430 CrossRef CAS PubMed.
- J. Park, Q. Jiang, D. Feng and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 7188 CrossRef CAS PubMed.
- Y. Qin, L.-J. Chen, F. Dong, S.-T. Jiang, G.-Q. Yin, X. Li, Y. Tian and H.-B. Yang, J. Am. Chem. Soc., 2019, 141, 8943 CrossRef CAS PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS PubMed.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061 CrossRef CAS PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808 CrossRef CAS PubMed.
- B. Zhou, B. Shi, D. Jin and X. Liu, Nat. Nanotechnol., 2015, 10, 924 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed.
- M. Zhao, R. Wang, B. Li, Y. Fan, Y. Wu, X. Zhu and F. Zhang, Angew. Chem., Int. Ed., 2019, 58, 2050 CrossRef CAS PubMed.
- Y. Liu, Y. Lu, X. Yang, X. Zheng, S. Wen, F. Wang, X. Vidal, J. Zhao, D. Liu, Z. Zhou, C. Ma, J. Zhou, J. A. Piper, P. Xi and D. Jin, Nature, 2017, 543, 229 CrossRef CAS PubMed.
- Y. Yang, Q. Shao, R. Deng, C. Wang, X. Teng, K. Cheng, Z. Cheng, L. Huang, Z. Liu, X. Liu and B. Xing, Angew. Chem., Int. Ed., 2012, 51, 3125 CrossRef CAS PubMed.
- J. Zhao, J. Gao, W. Xue, Z. Di, H. Xing, Y. Lu and L. Li, J. Am. Chem. Soc., 2018, 140, 578 CrossRef CAS PubMed.
- J. Zhao, H. Chu, Y. Zhao, Y. Lu and L. Li, J. Am. Chem. Soc., 2019, 141, 7056 CrossRef CAS PubMed.
- H. Chu, J. Zhao, Y. Mi, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2019, 58, 14877 CrossRef CAS PubMed.
- K. Zheng, S. Han, X. Zeng, Y. Wu, S. Song, H. Zhang and X. Liu, Adv. Mater., 2018, 30, 1801726 CrossRef PubMed.
- S. Chen, A. Z. Weitemier, X. Zeng, L. He, X. Wang, Y. Tao, A. J. Y. Huang, Y. Hashimotodani, M. Kano, H. Iwasaki, L. K. Parajuli, S. Okabe, D. B. L. Teh, A. H. All, I. Tsutsui-Kimura, K. F. Tanaka, X. Liu and T. J. McHugh, Science, 2018, 359, 679 CrossRef CAS PubMed.
- B. Yan, J.-C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558 CrossRef CAS PubMed.
- H. Chu, J. Zhao, Y. Mi, Z. Di and L. Li, Nat. Commun., 2019, 10, 2839 CrossRef PubMed.
- F. Hu, B. Liu, H. Chu, C. Liu, Z. Li, D. Chen and L. Li, Nanoscale, 2019, 11, 9201 RSC.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580 CrossRef CAS PubMed.
- H. Dong, S.-R. Du, X.-Y. Zheng, G.-M. Lyu, L.-D. Sun, L.-D. Li, P.-Z. Zhang, C. Zhang and C.-H. Yan, Chem. Rev., 2015, 115, 10725 CrossRef CAS PubMed.
- Y. Li, Z. Di, J. Gao, P. Cheng, C. Di, G. Zhang, B. Liu, X. Shi, L.-D. Sun, L. Li and C.-H. Yan, J. Am. Chem. Soc., 2017, 139, 13804 CrossRef CAS PubMed.
- H.-X. Mai, Y.-W. Zhang, R. Si, Z.-G. Yan, L.-D. Sun, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2006, 128, 6426 CrossRef CAS PubMed.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19 RSC.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340 RSC.
- K. Hayashi, M. Nakamura, H. Miki, S. Ozaki, M. Abe, T. Matsumoto and K. Ishimura, Adv. Funct. Mater., 2012, 22, 3539 CrossRef CAS.
- H.-B. Cheng, X. Tan and M.-L. Pang, Eur. J. Org. Chem., 2013, 7933 CrossRef CAS.
- B. Tambosco, K. Segura, C. Seyrig, D. Cabrera, M. Port, C. Ferroud and Z. Amara, ACS Catal., 2018, 8, 4383 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis, characterization and supplementary figures. See DOI: 10.1039/c9sc03524a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |