
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-enabled metal-free pinacol coupling by hydrazine†
Zihang
Qiu‡
 ,
Hanh D. M.
Pham‡
,
Jianbin
Li
,
Chen-Chen
Li
,
Durbis J.
Castillo-Pazos
,
Rustam Z.
Khaliullin
* and
Chao-Jun
Li
*
,
Hanh D. M.
Pham‡
,
Jianbin
Li
,
Chen-Chen
Li
,
Durbis J.
Castillo-Pazos
,
Rustam Z.
Khaliullin
* and
Chao-Jun
Li
*
Department of Chemistry, FQRNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. W., Montreal, Quebec H3A 0B8, Canada. E-mail: rustam.khaliullin@mcgill.ca; cj.li@mcgill.ca
First published on 11th October 2019
Abstract
Efficient carbon–carbon bond formation is of great importance in modern organic synthetic chemistry. The pinacol coupling discovered over a century ago is still one of the most efficient coupling reactions to build the C–C bond in one step. However, traditional pinacol coupling often requires over-stoichiometric amounts of active metals as reductants, causing long-lasting metal waste issues and sustainability concerns. A great scientific challenge is to design a metal-free approach to the pinacol coupling reaction. Herein, we describe a light-driven pinacol coupling protocol without use of any metals, but with N2H4, used as a clean non-metallic hydrogen-atom-transfer (HAT) reductant. In this transformation, only traceless non-toxic N2 and H2 gases were produced as by-products with a relatively broad aromatic ketone scope and good functional group tolerance. A combined experimental and computational investigation of the mechanism suggests that this novel pinacol coupling reaction proceeds via a HAT process between photo-excited ketone and N2H4, instead of the common single-electron-transfer (SET) process for metal reductants.
1. Introduction
Development of economical and efficient methods for creating the carbon–carbon bond is of paramount importance in modern synthetic chemistry.1 In this regard, the pinacol coupling, though historically discovered over a century ago, is still one of the most efficient reactions to introduce the carbon–carbon bond in one step.2 Traditionally, more than the stoichiometric amount of metal reductants (Mg, Al, Zn, Mn, etc.) is required to realize the single-electron-transfer (SET) process from the metal to the carbonyl group and to generate the ketyl radical as the key intermediate to form the pinacol product with the help of various Brønsted or Lewis acid additives (H+, SiR3Cl, TiCl4, ZnCl2, AlCl3, etc.). The acids are vital to coordinate with the carbonyl group to lower the SET barrier and to increase the pinacol product selectivity over the simple alcohol product (the two-electron reduced form of ketone) (Scheme 1a). Although it is an apparently straight-forward process, carefully choosing the acid additives is always needed, otherwise it results in poor reactivity and selectivity towards pinacol.3 Besides, pre-activation of the excess amount of metals is often needed to remove the oxide from the metal surface and efficiently utilize them as reductants. Furthermore, the work-up processes of the excess metal are typically problematic, especially in large scale reactions, generating large amounts of metal waste and causing environmental concerns.4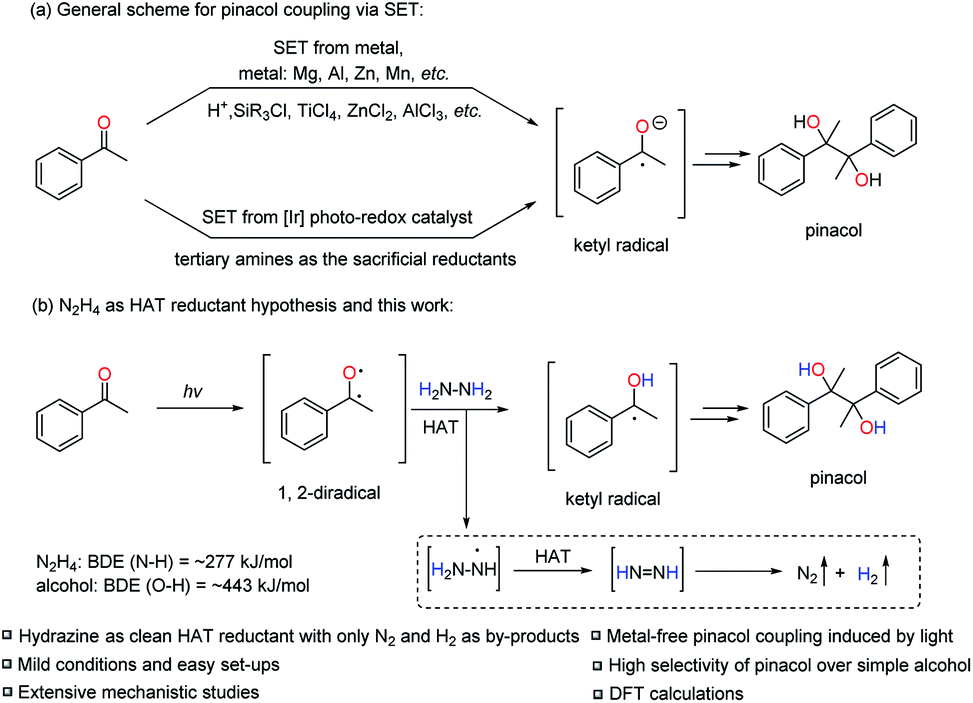 | ||
| Scheme 1 Previous work on pinacol coupling and this work. | ||
To partially solve the problems of excessive metal mentioned above, pioneering work by Rueping et al. applied [Ir] based photo-redox catalysts to generate the key ketyl radicals from ketones via SET with tertiary amines as sacrificial reductants instead of metals and to realize the pinacol coupling (Scheme 1a).5 Although successful, the method still required precious noble metal [Ir] based photo-redox catalysts. Therefore, we were pondering the possibility of designing a pinacol coupling without the use of any metal. Inspired by our previous work on [Ni]/N2H4 as the reductive system to realize diaryl reductive coupling reactions,6 we envisioned the feasibility of employing N2H4 as a clean reductant for the pinacol coupling, bearing in mind the innate reductive coupling nature in such a transformation. However, to use N2H4 as the reductant to directly reduce ketones to the ketyl radicals via the SET process could be highly challenging due to the unmatched redox potentials (e.g. acetophenone: Ered1/2 = −1.85 V vs. normal hydrogen electrode (NHE); N2H4: Ered1/2 = −1.49 V vs. NHE).5a,7
Faced with this dilemma and inspired by the recent work of Martin et al.,8 in which the aromatic ketone was identified as an efficient hydrogen-atom-transfer (HAT) reagent in its excited state, we anticipated the possibility of realizing the pinacol coupling reaction with N2H4via HAT instead of the well-known SET mechanism. In fact, there has been a long history of the use of aromatic ketones as HAT reagents in their excited states, pioneered by Norrish,9 Yang10 and others.11 The aromatic ketone can be excited under UV light irradiation to form the excited triplet state to act as a 1,2-diradical, which is very effective for the radical HAT process because of the electrophilic properties of the excited oxygen radical.8,11a,d On the other hand, N2H4 is potentially able to be used as a hydrogen atom donor in this HAT process due to the expected favorable thermodynamics: the relatively weak hydrazine N–H bond with a bond dissociation energy (BDE) of approximately 277 kJ mol−1 is replaced by a strong alcohol O–H bond with a BDE of ∼443 kJ mol−1.12
Based on the above mechanistic analysis, a working hypothesis is proposed in Scheme 1b. Initially, the 1,2-diradical can be formed when the aromatic ketone is excited by UV light. This is followed by hydrogen atom abstraction from N2H4 that generates the ketyl radical – the key precursor in the pathway to pinacol. Meanwhile, 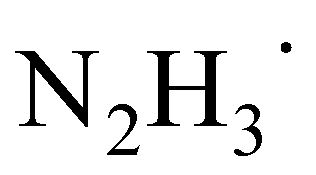 may be produced and then by another HAT, unstable diimide (N2H2) can be afforded, readily decomposing to N2 and H2 gases as by-products to be released out from the reaction system to further move forward this transformation.6,13 With this working hypothesis in mind, we report a light-driven metal-free pinacol coupling utilizing N2H4, for the first time, as a clean HAT reductant with traceless non-toxic N2 and H2 gases generated in situ as sole by-products.
may be produced and then by another HAT, unstable diimide (N2H2) can be afforded, readily decomposing to N2 and H2 gases as by-products to be released out from the reaction system to further move forward this transformation.6,13 With this working hypothesis in mind, we report a light-driven metal-free pinacol coupling utilizing N2H4, for the first time, as a clean HAT reductant with traceless non-toxic N2 and H2 gases generated in situ as sole by-products.
2. Results and discussion
To investigate the feasibility of our hypothesis, we initially examined the reaction by using acetophenone (0.2 mmol, 1 equiv.) as the model substrate with hydrazine monohydrate (0.1 mmol, 0.5 equiv.) as the reductant in 1 mL H2O at 25 °C with UV light irradiation (254 nm) for 12 h under an argon atmosphere (Table 1, entry 1). The desired corresponding pinacol product, 2,3-diphenylbutane-2,3-diol (3a), could be obtained in 12% NMR yield along with acetophenone hydrazone and azine as by-products that result from the condensation of acetophenone and hydrazine. To slow down the formation of hydrazone and azine, non-protic solvents were tested (entries 3–7) with CH3CN showing the best efficiency to produce 3a with 68% NMR yield (entry 7).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | N2H4 (x equiv.) | conv. (%) | Yield (%) | |
| 3a | 4a | ||||
| a General reaction conditions: acetophenone (0.2 mmol, 1 equiv.) and N2H4·H2O (x equiv.) in solvents (1 mL) were stirred under argon for 12 h with UV light (254 nm) irradiation at 25 °C; starting material conversion and NMR yields were given with 1,3,5-trimethoxybenzene as the internal standard; yields were calculated based on acetophenone. b Without UV light. c 24 h instead. d Under air instead of argon. e Isolated yield. f MeNH–NHMe (1 equiv.) was used instead of N2H4. g i PrOH (0.75 equiv.) was used instead of N2H4. For optimization of other reactants, please see the ESI. | |||||
| 1 | H2O | 0.5 | 90 | 12 | 2 |
| 2 | MeOH | 0.5 | 50 | 10 | 2 |
| 3 | EtOAc | 0.5 | 60 | 38 | 2 |
| 4 | PhCF3 | 0.5 | 50 | 42 | 2 |
| 5 | Benzene | 0.5 | 53 | 47 | 2 |
| 6 | 1,4-Dioxane | 0.5 | 73 | 62 | 2 |
| 7 | CH3CN | 0.5 | 77 | 68 | 2 |
| 8b | CH3CN | 0.5 | 14 | — | — |
| 9 | CH3CN | 0 | 12 | — | — |
| 10 | CH3CN | 0.75 | 84 | 72 | 2 |
| 11 | CH 3 CN | 0.75 | 98 | 80 (80) | 4 |
| 12c,d | CH3CN | 0.75 | 51 | 30 | 2 |
| 13c | CH3CN | 1.0 | 99 | 81 | 4 |
| 14c,f | CH3CN | 0.75 | 42 | 27 | 2 |
| 15c,g | CH3CN | 0.75 | 42 | 32 | 1 |
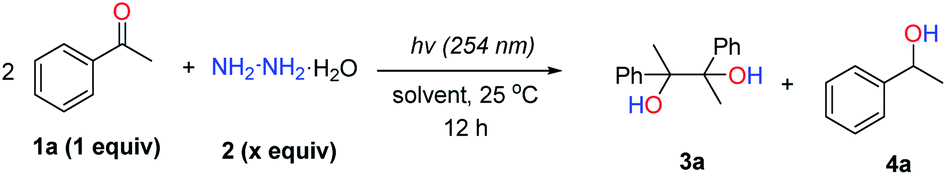
Control experiments (entries 8 and 9) revealed that both UV light and hydrazine were vital for this transformation to proceed. By increasing the hydrazine amount to 0.75 equiv. and prolonging the reaction time to 24 h (entries 10 and 11), delightfully, the desired pinacol product 3a could be obtained in 80% isolated yield (entry 11). The argon atmosphere was found to be necessary for this transformation, as decreased yield (30%) was observed when the reaction was run under air instead (entries 11 vs. 12). By further increasing the hydrazine amount to 1.0 equiv., the yield could not be increased accordingly. Other reductants, such as hydrazine derivatives (entry 14 and Table S1 in the ESI†) and iPrOH (entry 15), showed inferior reactivities compared to hydrazine.
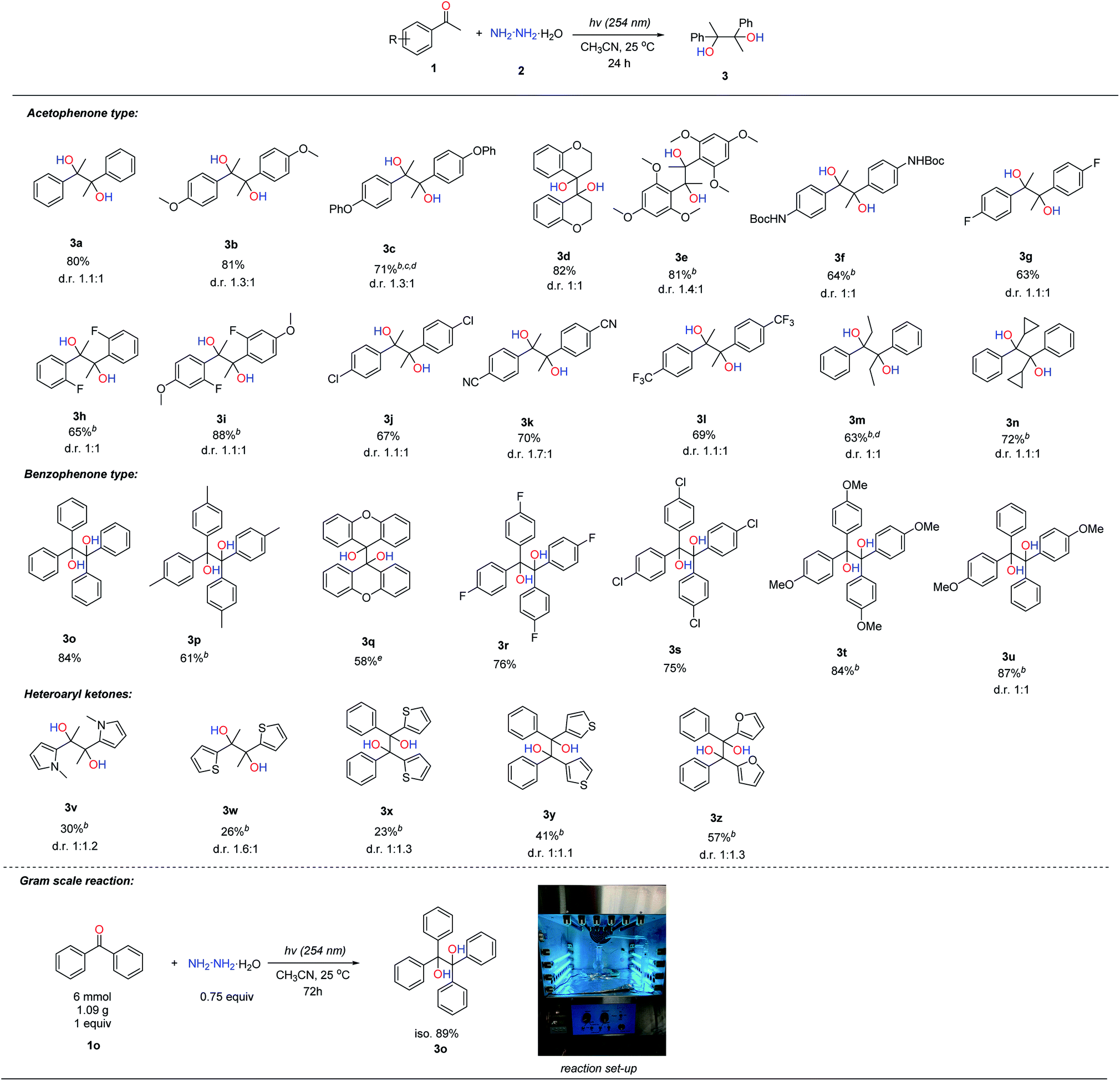
With the optimized reaction conditions in hand, we next examined the reaction scope of aromatic ketones as shown in Table 2. Acetophenones attached with electron-donating groups such as methoxy, phenoxy, and Boc-protected amine worked well in this transformation, affording the pinacol products (3b, 3c and 3f) in moderate to high yields (64–81%). It was noteworthy that cyclic aromatic ketone, e.g. 4-chromanone, was able to generate the pinacol product 3d in good yield (82%), and multiple electron-donating substituents such as 2,4,6-trimethoxy- acetophenone gave the product 3e in 81% yield. Acetophenones bearing electron-withdrawing substituents, such as –F, –Cl, –CN and –CF3 (3g, 3h, 3j, 3k and 3l), were also effective in this transformation, albeit giving slightly lower yields (63–70%) compared to electron-donating ones. Acetophenone bearing both electron-donating and electron-withdrawing substituents, such as 2-F-4-OMe-acetophenone, could afford the product 3i in 88% yield. However, 4-Br-acetophenone was an ineffective substrate, where the reductive photo-cleavage of the C–Br bond under UV irradiation was observed.14 Changing the methyl group in acetophenone to larger ethyl and cyclopropyl groups could also produce the corresponding products (3m and 3n) in moderate yields, 63% and 72% respectively. Benzophenone-type compounds reacted in a similar fashion to acetophenones, generating the corresponding pinacol products (3o–3u) in moderate to good yields. For the scope of heteroaryl ketones, we found that some heteroaryl ketones, containing pyrrole, thiophene and furan units (3v–3z), could afford the corresponding pinacol products in moderate yields (23% to 57%). To demonstrate the synthetic application of this protocol, a gram-scale reaction with benzophenone (1.09 g) as a model substrate was tested under standard conditions; and delightfully, 89% isolated yield could be obtained. Unfortunately, aldehydes, aliphatic ketones and aromatic imines were not compatible with this transformation probably due to their fast condensation with hydrazine to generate hydrazone and azine by-products.
| a General reaction conditions: ketone (0.2 mmol, 1 equiv.) and N2H4·H2O (0.75 equiv.) in CH3CN (1 mL) were stirred under argon for 24 h under UV light (254 nm) irradiation at 25 °C; isolated yields were given. b 1 equiv. of N2H4·H2O was used. c A xenon light was used. d Benzene was used as the solvent. e 2 equiv. of N2H4·H2O were used. |
|---|
|
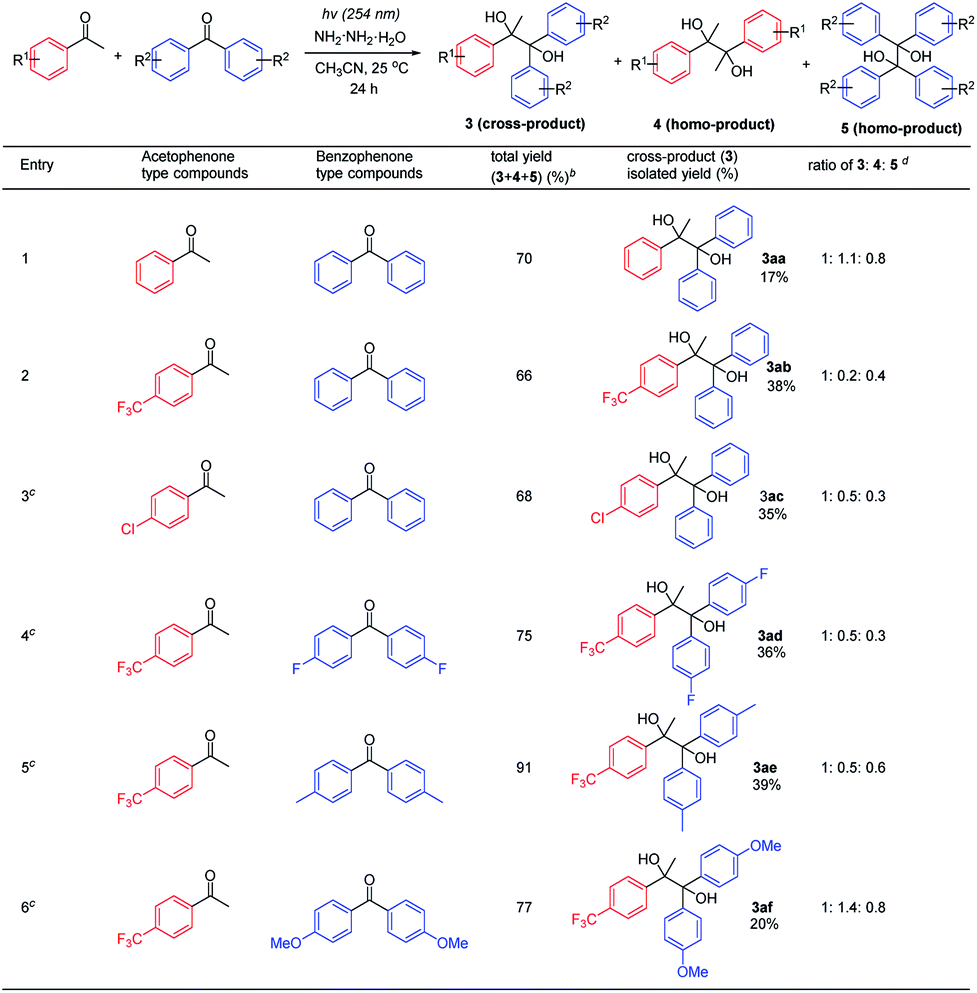
Next, the feasibility of realizing the cross-pinacol coupling was examined by mixing acetophenone and benzophenone-type compounds (Table 3 and see ESI† for details). It was found out that the acetophenones attached with electron-withdrawing groups such as 4-CF3 and 4-Cl gave better selectivity and yields of cross-pinacol products, compared to acetophenone (entries 1–3) and 4-OMe acetophenone (see ESI† for details). Benzophenones with –F and –CH3 substituents showed similar reactivities to simple benzophenone (entries 2, 4 and 5). Finally, benzophenone attached with –OMe could not further increase the cross-pinacol product selectivity and yield (entry 6).
| a General reaction conditions: acetophenone-type compounds (0.1 mmol, 1 equiv.), benzophenone-type compounds (0.1 mmol, 1 equiv.), and N2H4·H2O (0.15 mmol, 1.5 equiv.) in CH3CN (1 mL) were stirred under argon for 24 h under UV light (254 nm) irradiation at 25 °C. b NMR yields were given with mesitylene as the internal standard. c N2H4·H2O (0.2 mmol, 2 equiv.) was used instead. d Ratio was determined by analysis of crude 1H-NMR. |
|---|
|
3. Mechanistic studies
3.1 Identification of the ketyl radical formation process
The control experiments in Table 1 (entries 8 and 9) showed that without UV light or N2H4, the desired pinacol product 3a could not be generated, indicating that the ketyl radical could be formed from the HAT process between the photo-excited 1,2-diradical and N2H4. To further confirm this process, firstly, the origin of protons was studied by changing the solvent from CH3CN to CD3CN with benzophenone as the model substrate due to the ease of analyzing the crude NMR of the starting material and product (Fig. 1a). It was found that all the protons were derived from the N2H4 instead of the acetonitrile solvent, since no deuterium was incorporated into the benzopinacol (3o) product and the benzhydrol (4o) by-product as confirmed by deuterium NMR (2D-NMR) (Fig. 1a)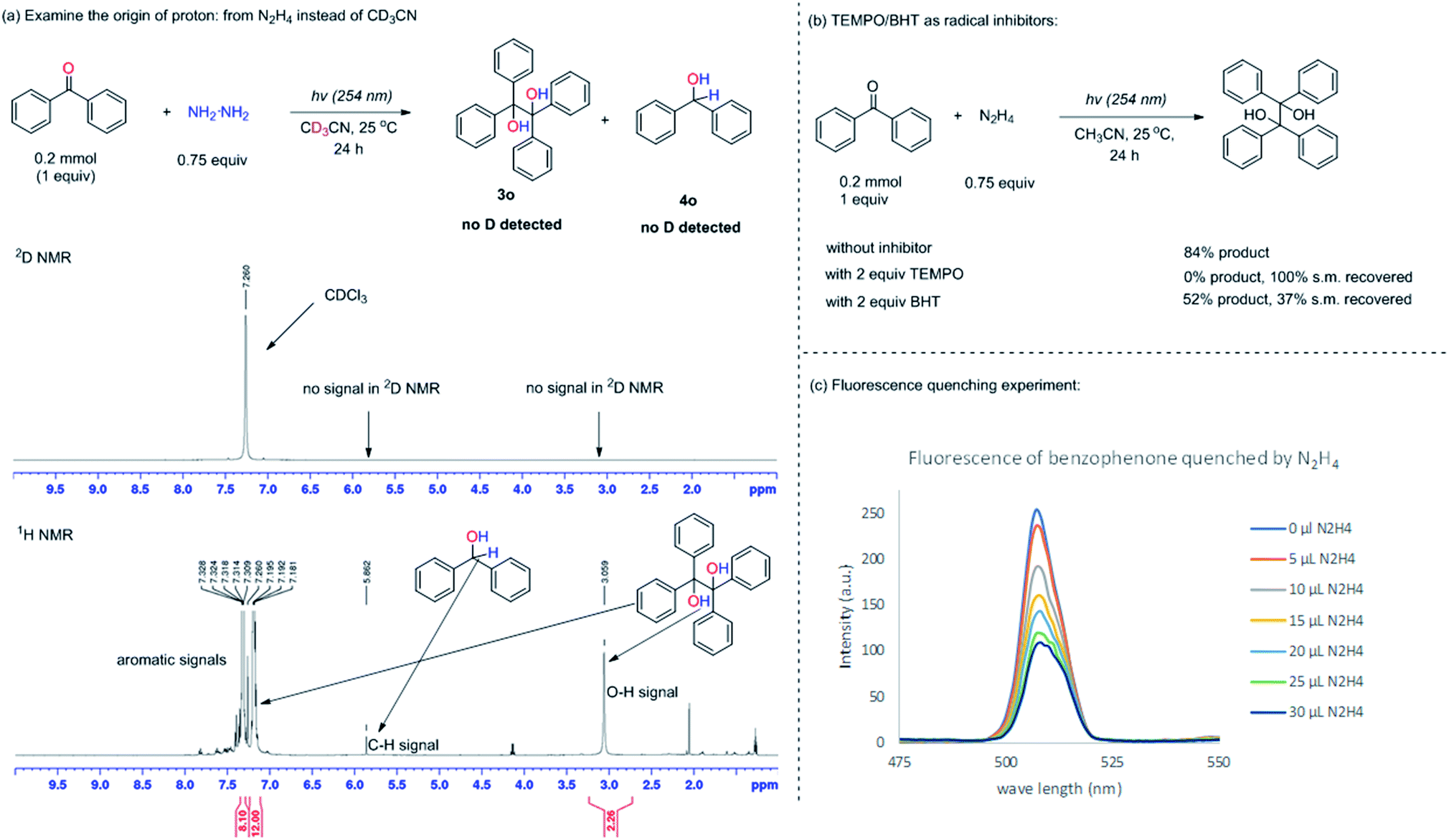 | ||
| Fig. 1 Examination of the mechanism of ketyl radical formation. | ||
Secondly, 2 equiv. of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) or butylated hydroxytoluene (BHT), as radical inhibitors, were added into the reaction system under the standard conditions. The reaction was totally suppressed when TEMPO was added, with the TEMPO being mainly reduced to 2,2,6,6-tetramethylpiperidine; whereas in the case of BHT, the reaction proceeded slower compared to the one without the inhibitor, demonstrating that the radical based mechanism was involved in this transformation (Fig. 1b).25 Thirdly, the fluorescence quenching experiment between benzophenone and N2H4 was performed (see ESI† for details). The fluorescence of benzophenone could be efficiently quenched by N2H4, suggesting the existence of a strong interaction between the excited benzophenone and N2H4 (Fig. 1c). From the three mechanistic studies shown above, it was reasonable to propose that the ketyl radical was formed by the HAT process between the 1,2-diradical, generated by the photoexcitation of the aromatic ketone and N2H4.
3.2 Identification of the HAT behavior of N2H4
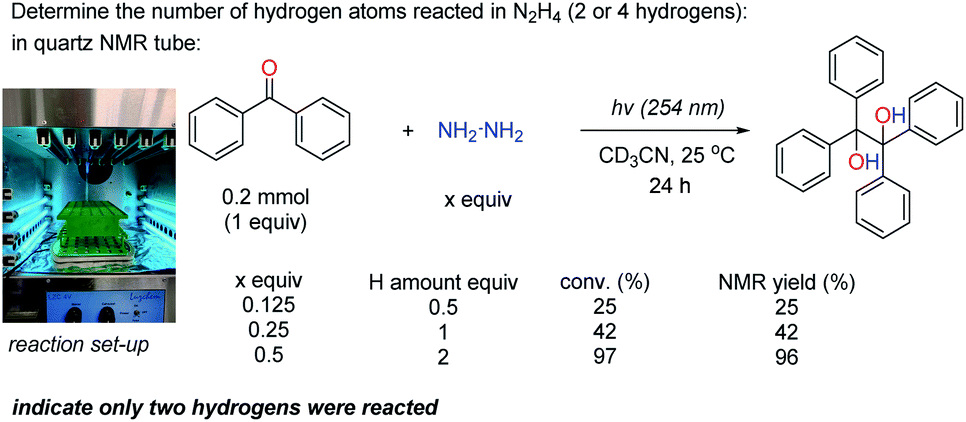 | ||
| Scheme 2 The number of hydrogens reacted in N2H4. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMe, the N2H2 analogue, was detected in the gas phase (see ESI† for details),17 further indicating the formation of N2H2 (Scheme 3b). Finally, analysis of the gas phase of the standard reaction conditions showed the presence of N2 but not NH3 (see ESI† for details). With all the above information shown, the reaction pathway B for N2H4 was more feasible.
NMe, the N2H2 analogue, was detected in the gas phase (see ESI† for details),17 further indicating the formation of N2H2 (Scheme 3b). Finally, analysis of the gas phase of the standard reaction conditions showed the presence of N2 but not NH3 (see ESI† for details). With all the above information shown, the reaction pathway B for N2H4 was more feasible.
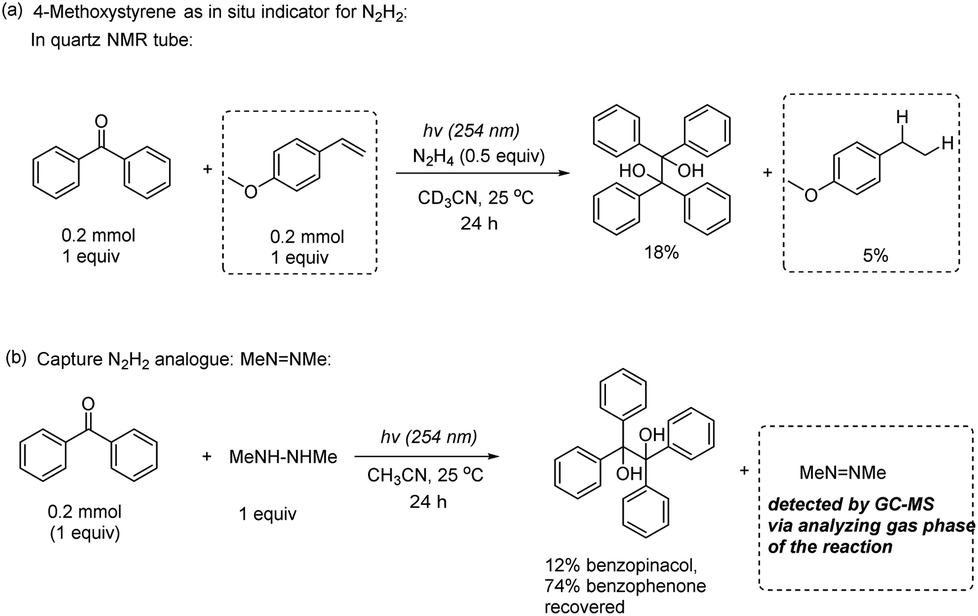 | ||
| Scheme 3 Evidence of N2H2. | ||
3.3 Mechanism of the pinacol C–C bond formation process
Historically, the commonly proposed mechanism for the UV-irradiation assisted benzopinacol type C–C bond formation was the homo-dimerization of two ketyl radicals (Scheme 4, left).11d,18 However, from modern perspectives, it is arguable that the C–C bond formation step arises from the homo-dimerization of two free radicals, simply because of the extremely low concentration and short life-times of the active radical species in solution,19 although it may represent the apparent results. Instead, another possible route to form the C–C bond is via the ketyl radical addition to the unsaturated CO (Scheme 4, right). Therefore, we were wondering whether the kinetics toolkit could be used to differentiate the two mechanisms to uncover this “historical mystery” of the C–C bond formation mechanism in this pinacol reaction.
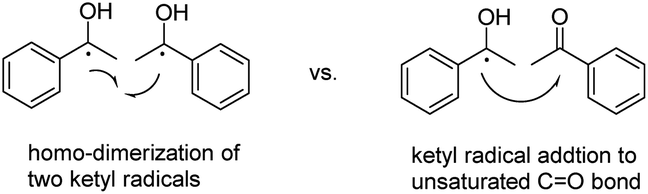 | ||
| Scheme 4 Two representative C–C formation mechanisms. | ||
Initially, the rate determining step (RDS) of this transformation was studied. Firstly, the kinetic isotope effect (KIE) was used to identify whether the HAT process was the RDS or not. The KIE (kH/kD) was around 1 when the N2D4 was used compared to the normal N2H4, indicating that the HAT process was not the RDS but a fast step (Scheme 5 and see ESI† for details). Since this reaction was initiated by UV irradiation, the photoexcitation might be the RDS. To examine this assumption, the reaction was run under UV irradiation with its light intensity reduced to 2/3 of its original one; and it was found that the reaction rate was reduced accordingly (vobs (2/3I0)/vobs (I0) ≈ 1.99/3 ≈ 2/3 (theoretical number), see ESI† for details), suggesting that the photoexcitation was the RDS.
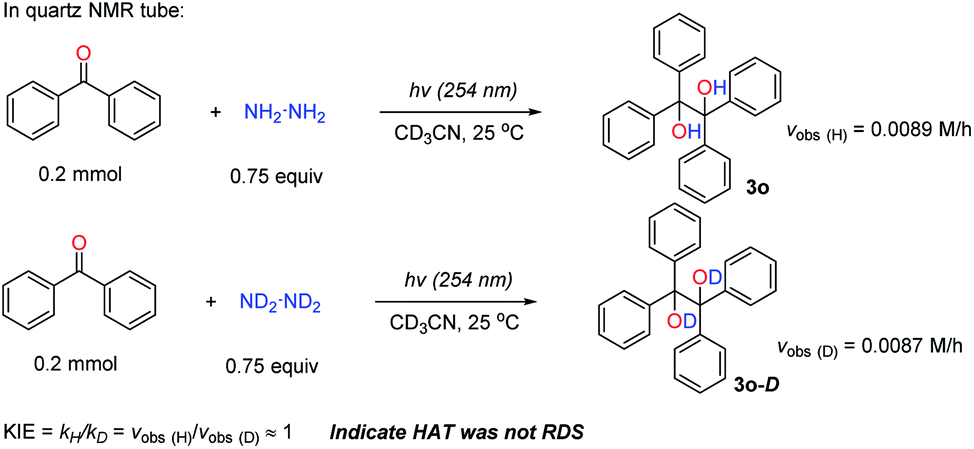 | ||
| Scheme 5 KIE studies. | ||
With the above kinetic information, if the mechanism for C–C bond formation was via ketyl radical homo-dimerization, the vobs (observed reaction rate) would be directly proportional to the I02 (the square of the light intensity). However, this assumption was not correct because the vobs was directly proportional to the I0, as stated before. On the other hand, if the mechanism was via ketyl radical addition to the unsaturated CO, the vobs would be directly proportional to the I0, which was very consistent with the experimental data. Thus, the latter one was the more likely mechanism (see ESI† for details).
3.4 DFT study of the mechanism
Density functional theory (DFT) calculations were carried out in order to verify the mechanism proposed based on the experimental study and to gain detailed insights into elementary steps and structural transformations along the pathway. Acetophenone (1a) reaction with hydrazine was chosen as a model of the pinacol coupling in calculations. All calculations were performed using the Gaussian software package (version 16, Revision B.01). The B3LYP approximation20 corrected to account for dispersion interactions21 was used as the exchange-correlation functional. The 6-31G(d,p) basis set was employed to represent molecular orbitals. Excited state calculations were performed using time-dependent DFT. Acetonitrile solvent was represented implicitly using the polarizable continuum model in the integral equation formalism.22 After structures of stable intermediates and transition states were optimized, frequency calculations were carried out and thermodynamic functions were calculated for the standard-state temperature 298.15 K and concentration 1 mol L−1 for all species in the ground electronic states.The stable intermediate structures and transition states are shown in Scheme 6 together with their energies (the free energy diagram is shown in the ESI†). For acetophenone, the intense UV-Vis absorption peak is calculated to lie at 239 nm (5.18 eV), in agreement with the experimentally observed transition (5.17 eV).23 Our excited state modeling also shows that a manifold of the excited triplet states of acetophenone lies between 3.31 and 3.35 eV above the ground state. This characterization is consistent with previous studies23 that show that an excited acetophenone undergoes vibrationally assisted intersystem crossing into a manifold of triplet states to generate the 1,2-diradical (Scheme 6). Our calculations indicate that the HAT from the triplet-state acetophenone to hydrazine is a barrierless process that leads to the formation of the ketyl radical bound to  .
.
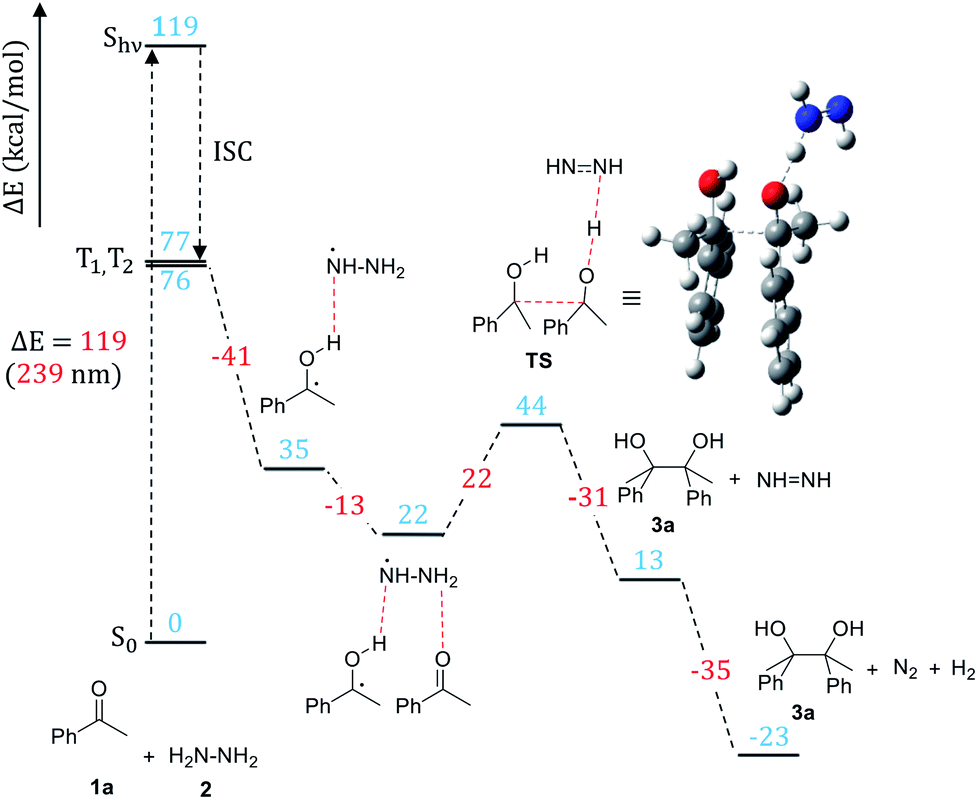 | ||
| Scheme 6 The B3LYP/6-31G(d,p) energy profile of the reaction. | ||
In the next major step of the reaction, the ketyl radical in its electronic ground state still bound to 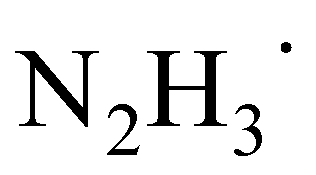 forms the carbon–carbon bond with another ground-state acetophenone molecule. Unsurprisingly, the carbon–carbon bond cannot be stabilized without another HAT from the nearby
forms the carbon–carbon bond with another ground-state acetophenone molecule. Unsurprisingly, the carbon–carbon bond cannot be stabilized without another HAT from the nearby 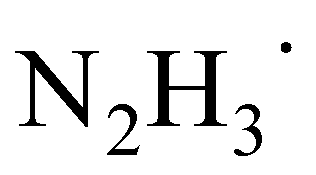 to the incoming acetophenone molecule. This is consistent with the observation of Glorius et al., who noted that the radical addition to CO is fully reversible without the assistance of hydrogen.24 Notably, it was found that the second HAT and C–C coupling can happen simultaneously via the transition state (TS) shown in Scheme 6. In the final step, the diimide N2H2 formed in the previous step decomposes into N2 and H2.
to the incoming acetophenone molecule. This is consistent with the observation of Glorius et al., who noted that the radical addition to CO is fully reversible without the assistance of hydrogen.24 Notably, it was found that the second HAT and C–C coupling can happen simultaneously via the transition state (TS) shown in Scheme 6. In the final step, the diimide N2H2 formed in the previous step decomposes into N2 and H2.
The results of computational modeling are fully consistent with the experimental observations and suggest that the energy the system acquires after the excitation is sufficient to overcome the transition state associated with the C–C bond formation.
4. Conclusions
We have developed a metal-free and clean pinacol coupling protocol enabled by light with N2H4, for the first time, as a HAT reductant under very mild conditions (room temperature). The reaction produces only traceless non-toxic N2 and H2 gases as by-products. The protocol solves the long-standing metal waste issues in the classical pinacol coupling reactions and has a relatively broad aromatic ketone scope and functional group tolerance. Potential synthetic applications are demonstrated by a gram-scale reaction. Furthermore, the fundamental steps regarding the mechanism such as the processes of ketyl radical formation, the HAT behavior of N2H4 and the C–C bond formation of the product were extensively studied experimentally and computationally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Canada Research Chair Foundation (to C.-J. Li), the CFI, FRQNT Center for Green Chemistry and Catalysis, NSERC, and McGill University for support of our research. R. Z. K. acknowledges funding by NSERC through the Discovery Grant (RGPIN-2016-05059). The authors are grateful to Compute Canada for computer time and thank Siting Ni for assistance with UV-visible and fluorescence spectra measurements. Z. Qiu is grateful to McGill's Chemistry Department for the Major Scholarship and Pall Dissertation Award. D. Castillo-Pazos thanks CONACYT Mexico (2018-000009-01EXTF-00413) for financial support.Notes and references
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- (a) T. Hirao, Catalytic Reductive Coupling of Carbonyl Compounds – The Pinacol Coupling Reaction in Metal Catalyzed Reductive C–C Bond Formation, ed. M. J. Krische, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, ch.120, pp. 53–75 Search PubMed; (b) T. Wirth, Angew. Chem., Int. Ed., 1996, 35, 61–63 CrossRef CAS.
- (a) B. K. Hazarika and D. K. Dutta, Synth. Commun., 2011, 41, 1088–1093 CrossRef CAS; (b) T. Mandal, S. Jana and J. Dash, Eur. J. Org. Chem., 2017, 4972–4983 CrossRef CAS; (c) G. M. Robertson, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, pp. 563–611, DOI:10.1016/b978-0-08-052349-1.00073-1.
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC; (b) C. J. Li, Green Chem., 2016, 18, 1836–1838 RSC.
- (a) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed; (b) Q. Xia, J. Dong, H. Song and Q. Wang, Chem.–Eur. J., 2019, 25, 2949–2961 CAS.
- L. Lv, Z. Qiu, J. Li, M. Liu and C. J. Li, Nat. Commun., 2018, 9, 4739 CrossRef PubMed.
- (a) T. Shimada, A. Tamaki, H. Nakai and T. Homma, Electrochemistry, 2007, 75, 45–49 CrossRef CAS; (b) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS; (c) V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed.
- R. G. W. Norrish and C. H. Bamford, Nature, 1937, 140, 195–196 CrossRef CAS.
- N. C. Yang and D. D. H. Yang, J. Am. Chem. Soc., 1958, 80, 2913–2914 CrossRef CAS.
- (a) C. Chen, Org. Biomol. Chem., 2016, 14, 8641–8647 RSC; (b) W. Liu, N. Chen, X. Yang, L. Li and C. J. Li, Chem. Commun., 2016, 52, 13120–13123 RSC; (c) W. B. Liu, X. B. Yang, Z. Z. Zhou and C. J. Li, Chem, 2017, 2, 688–702 CrossRef CAS; (d) S. G. Cohen, A. Parola and G. H. Parsons, Chem. Rev., 1973, 73, 141–161 CrossRef CAS.
- Y.-R. Luo, Comprehensive handbook of chemical bond energies, CRC press, 2007 Search PubMed.
- (a) K. D. Kim and J. H. Lee, Org. Lett., 2018, 20, 7712–7716 CrossRef CAS PubMed; (b) C. Smit, M. W. Fraaije and A. J. Minnaard, J. Org. Chem., 2008, 73, 9482–9485 CrossRef CAS PubMed; (c) D. Sellmann and A. Hennige, Angew. Chem., Int. Ed., 1997, 36, 276–278 CrossRef CAS.
- D. Cao, C. Yan, P. Zhou, H. Zeng and C. J. Li, Chem. Commun., 2019, 55, 767–770 RSC.
- (a) S. Hünig, H. R. Müller and W. Thier, Angew. Chem., Int. Ed., 1965, 4, 271–280 CrossRef; (b) D. Sellmann and A. Hennige, Angew. Chem., Int. Ed., 1997, 36, 276–278 CrossRef CAS.
- (a) D. Leow, Y. H. Chen, T. H. Hung, Y. Su and Y. Z. Lin, Eur. J. Org. Chem., 2014, 2014, 7347–7352 CrossRef CAS; (b) B. Pieber, S. T. Martinez, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2013, 52, 10241–10244 CrossRef CAS PubMed.
- B. N. Kenessov, J. A. Koziel, T. Grotenhuis and L. Carlsen, Anal. Chim. Acta, 2010, 674, 32–39 CrossRef CAS PubMed.
- (a) J. N. Pitts, R. L. Letsinger, R. P. Taylor, J. M. Patterson, G. Recktenwald and R. B. Martin, J. Am. Chem. Soc., 1959, 81, 1068–1077 CrossRef CAS; (b) S. G. Cohen and R. J. Baumgarten, J. Am. Chem. Soc., 1967, 89, 3471–3475 CrossRef CAS.
- (a) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS; (b) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201903726.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed; (c) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- (a) M. Huix-Rotllant and N. Ferré, J. Chem. Phys., 2014, 140, 134305 CrossRef PubMed; (b) M. Huix-Rotllant, I. Burghardt and N. Ferré, C. R. Chim., 2016, 19, 50–56 CrossRef CAS.
- (a) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652–13655 CrossRef CAS; (b) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 16219–16223 CrossRef CAS PubMed.
- The corresponding ketyl radical TEMPO and BHT adducts could be detected in trace amounts by ESI-HRMS. For TEMPO, HRMS (ESI): found for the adduct C22H29O2NNa ([M + Na]+) 362.2104, calculated 362.2091; for BHT, HRMS (ESI): found for the adduct C28H34O2Na ([M + Na]+) 425.2466, calculated 425.2451..
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03737c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |