
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactions of thermally generated benzynes with six-membered N-heteroaromatics: pathway and product diversity†
Sahil
Arora
 ,
Juntian
Zhang
,
Vedamayee
Pogula
and
Thomas R.
Hoye
*
,
Juntian
Zhang
,
Vedamayee
Pogula
and
Thomas R.
Hoye
*
Department of Chemistry, University of Minnesota, 207 Pleasant St., SE, Minneapolis, MN 55455, USA. E-mail: hoye@umn.edu
First published on 14th August 2019
Abstract
We report here various pathways by which six-membered N-heteroaromatic compounds react with benzynes that are generated by the HDDA reaction. The initially formed 1,3-zwitterionic species (a) can collapse intramolecularly to give novel 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts of the heterocycle and benzyne; (b) can react with an externally added, electrophilic third-component to give functionalized heterocyclic products; or (c) can react with an external protic nucleophile to produce, following collapse of the ion pair resulting from protonation of the zwitterion, a variety of three-component assemblies. Mechanisms for formation of some of the 1:1 adducts are supported by DFT methods. The scope of the protic nucleophilic coupling was also expanded to a two-pot operation by using triflic acid as a protic “non-nucleophile”, followed by the addition of a suitably reactive nucleophile.
:1 adducts of the heterocycle and benzyne; (b) can react with an externally added, electrophilic third-component to give functionalized heterocyclic products; or (c) can react with an external protic nucleophile to produce, following collapse of the ion pair resulting from protonation of the zwitterion, a variety of three-component assemblies. Mechanisms for formation of some of the 1:1 adducts are supported by DFT methods. The scope of the protic nucleophilic coupling was also expanded to a two-pot operation by using triflic acid as a protic “non-nucleophile”, followed by the addition of a suitably reactive nucleophile.
Introduction
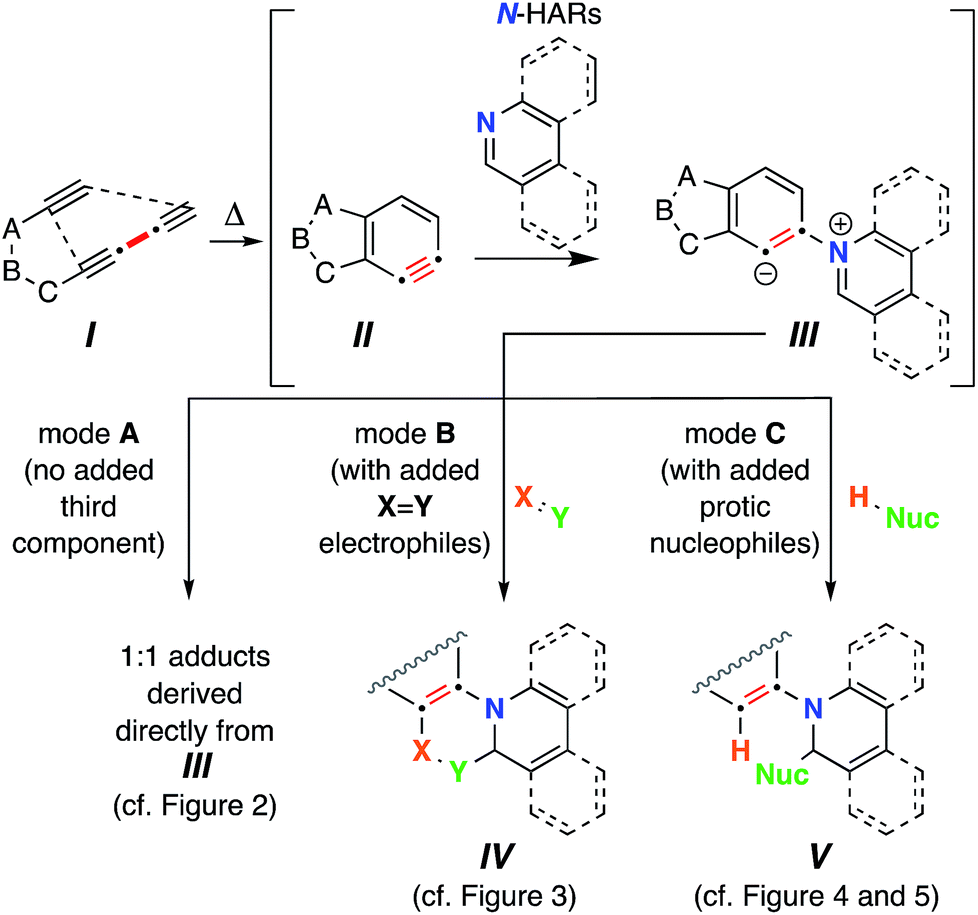
The thermal cycloisomerization of tethered triynes (I)‡ leads to formation of benzynes (II) under neutral conditions in a hexadehydro-Diels–Alder (HDDA) reaction.1 These short-lived, reactive intermediates then participate in myriad intermolecular trapping processes, including three-component reactions (TCRs).2 In earlier work, we have described a variety of modes of reactivity initiated by the reaction of these electrophilic benzynes with cyclic sulfides,3 tertiary amines,4 aliphatic cyclic amines,5 oxygen-containing heterocycles,6 and alkaloidal natural products.7In a number of instances, HDDA-generated benzynes have allowed the discovery of new modes of trapping reactions6,8 that are distinct from that seen for classically generated arynes (i.e., those formed by way of various ortho-elimination reactions of precursor arenes).9 This complementarity is one of the major, distinguishing features between classical and HDDA aryne chemistries. These differences in reactivity can be attributed to the fact that the trapping reactions of HDDA-generated benzynes are studied in a “thermal-only” and, thus, pristine environment, devoid of the reagents and byproducts necessary for nearly all classical benzyne-generating protocols.
Accordingly, HDDA-benzynes provide an opportunity to reveal new modes of reactivity for reagents already known to engage classical arynes. Although there are various examples in which six-membered N-heterocyclic arenes (prototypically, pyridine, quinoline, and isoquinoline) have engaged classically generated arynes, nearly all have been performed in the presence of an external, third component to give tractable outcomes.10 In those studies, aliphatic nitriles, terminal alkynes, and various carbonyl compounds have been used as third components. This reaction is presumed to be initiated by attack of the nucleophilic nitrogen atom in the heterocycle onto the electrophilic aryne, producing a 1,3-zwitterion (cf.III, Fig. 1‡). However, to our knowledge, formation of 1:1 adducts between benzynes and N-heterocyclic arene compounds (“N-hetaryls”, N-HARs) has never been definitively demonstrated. Fields and Meyerson (1966) speculated on the intermediacy of [2 + 2]-addition products from arynes and pyridine on the basis of gas chromatographic and mass spectrometric analyses of the complex product mixtures formed when, for example, phthalic anhydride, a benzyne precursor, was pyrolyzed in the presence of pyridine, but discrete products were not isolated.11
 | ||
| Fig. 1 Trapping of HDDA-generated benzynes II with N-hetaryls (N-HARs) of the pyridine family (pyridine, quinoline, isoquinoline, and phenanthridine, etc.) via diverse reaction pathways (Modes A–C). | ||
Two factors prompted the investigations reported here: (a) the possibility of identifying reactions leading to 1:1 adducts of N-HARs and benzynes; and (b) the opportunity to uncover new types of three-component reactions of benzynes with N-HARs. We herein report that upon formation of III, a variety of reaction pathways can ensue. The reactions that we have observed for zwitterions such as III can be categorized into three modes. Mode A leads to products that are 1:1 adducts between II and the N-HAR. In mode B, in situ bimolecular reaction of zwitterion III with an electrophile of type 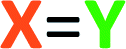 as a third component forms a new 6-membered ring in products IV. In mode C, protonation of III by a Brønsted acid [i.e., an in situ protic nucleophile (H-Nuc), the third component] and collapse of the nascent Nuc−/iminium+ ion pair produces adducts V. The full roster of reactants used in the studies we report here are compiled in Chart 1.
as a third component forms a new 6-membered ring in products IV. In mode C, protonation of III by a Brønsted acid [i.e., an in situ protic nucleophile (H-Nuc), the third component] and collapse of the nascent Nuc−/iminium+ ion pair produces adducts V. The full roster of reactants used in the studies we report here are compiled in Chart 1.
 | ||
| Chart 1 List of all reactants used in this study: (a) benzyne polyyne precursors; (b) N-heteroaromatics (N-HARs); (c) activated electrophiles; and (d) protic-nucleophiles. | ||
Results
Mode A (formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 adduct between an aryne and a N-HAR)
:1 adduct between an aryne and a N-HAR)
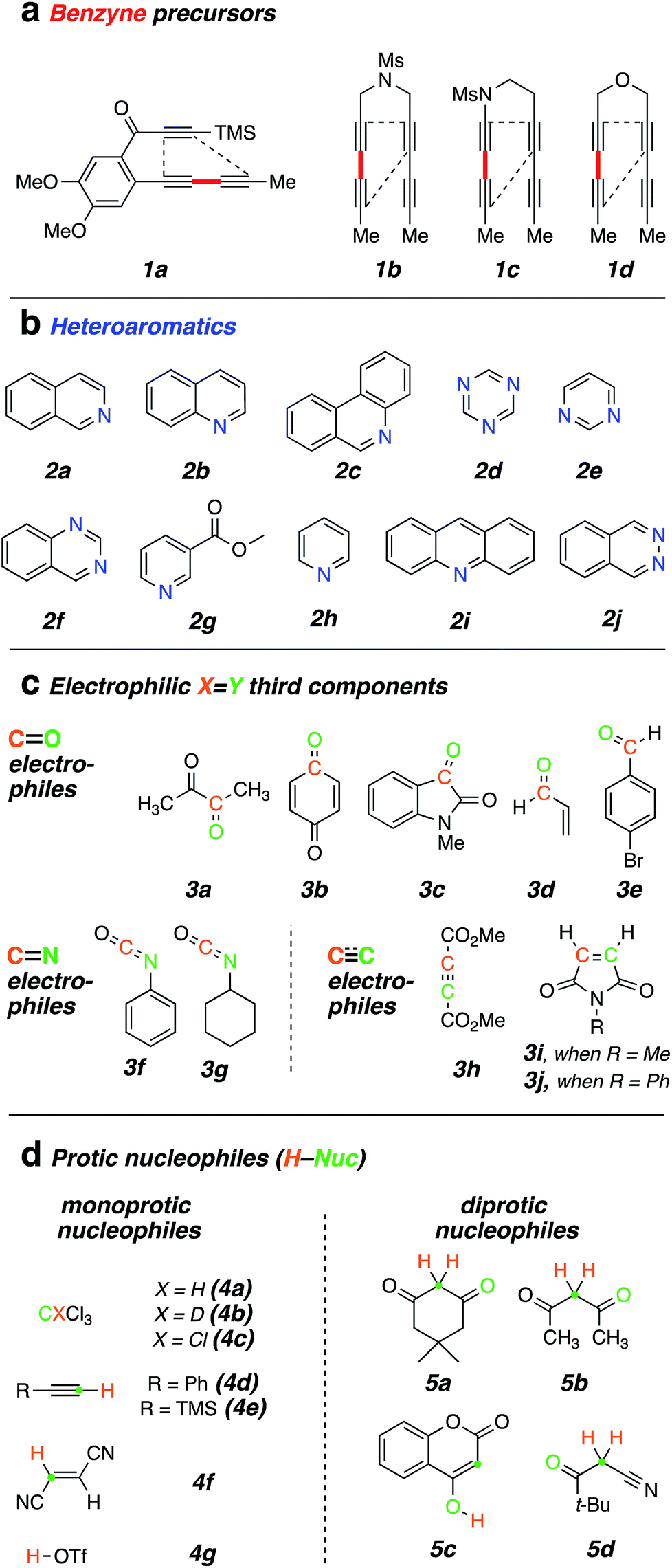
We first examined reactions of the triyne substrate 1a with several N-HARs in the absence of any added, potential third component. For example, when a benzene solution of 1a was heated to 85 °C in the presence of isoquinoline (2a), the four-membered benzoazetidine 6§ and 2-isoquinolone 7§ were formed (Fig. 2a) by in situ trapping of VI by 2a. Probably because of the oxidative lability of product 6, it was found to be unstable under ambient conditions and needed to be characterized immediately after isolation.
 | ||
| Fig. 2 (a) Trapping of the benzyne VI (from 1a, Chart 1) with N-hetaryls of the pyridine family (i.e., pyridine, quinoline, isoquinoline, phenanthridine, pyrimidine, 1,3,5-triazine, and quinazoline) follow different reaction pathways. (b) DFTi for VII + 2a showing competitive formation of IX and VIII and a high barrier for ring opening of IX to the azocine X. i[SMD(benzene)//M06-2X/6-311+G(d,p)]. | ||
The pathways leading to each of these products were then studied using DFT calculations of the reaction between a simplified aryne [3,6-dimethylbenzyne (VII)] and isoquinoline (2a) (Fig. 2b). The formation of 6 can be explained by a net [2 + 2]-addition of 2a to the benzyne, a process involving simple cyclization of the initial zwitterion (cf.VIII to IX). Formation of 7, on the other hand, can be rationalized by an intramolecular proton-transfer within the initially formed 1,3-zwitterion VIII and subsequent oxidation of the resulting carbene X by oxygen.12 This type of reactivity was earlier reported by Biju and coworkers.10c It is notable that the energies of the DFT transition structures for these two competing pathways (i.e., TSringclosingvs. TSprotonshift) are quite similar. The computations also suggest that electrocyclic ring-opening of the benzoazetidine IX to the 8-membered benzoazocine derivative XI is both endergonic as well as a high-barrier process, the latter reflecting, at least in part, the loss of benzenoid aromaticity that is fully revealed in structure XI.
Trapping of the benzyne VI with quinoline (2b) led to the isolation of only the four-membered species 8. This compound also was seen to degrade upon storage for extended periods of time. An analogous set of computations using quinoline 2b showed a lower barrier (by 2.3 kcal mol−1) favouring formation of azetidine relative to carbene (see Fig. S1†). Accordingly, a 2-quinolone product was not observed to accompany 8. When phenanthridine (2c) was used as the N-HAR, the yield of the analogous trapping product 9 was significantly higher (87%) than that of 6 or 8.
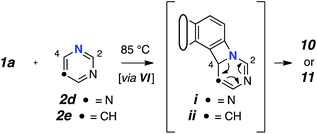
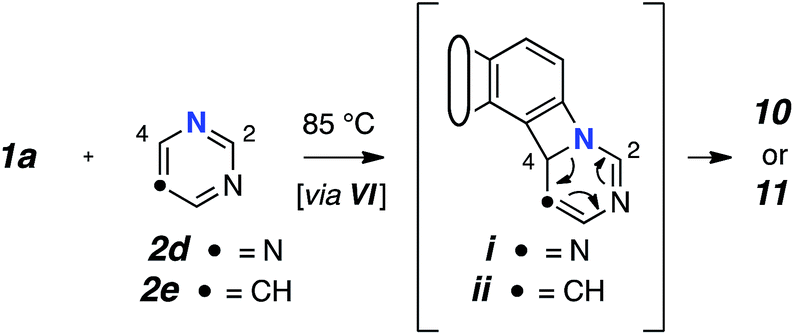
Reactions with monocyclic N-HARs containing two or more nitrogen atoms proved interesting. 1,3,5-Triazine (2d) gave rise to the 1,3,5-triazocine derivative 10;13 we can locate only a handful of examples of this (fully unsaturated) heterocycle, and they all bear an amino substituent on the carbon atom between two nitrogen atoms (i.e., guanidines).14 Compound 10 presumably arises from ring opening of an initially formed, four-membered, [2 + 2]-adduct analogous to IX (cf.i15). In this case, that strain-relieving event is not accompanied by loss of aromaticity, as would be the case for the opening of any of 6, 8, or 9. DFT calculations suggest that the electrocyclic ring-opening of i15 [see ESI Fig. S2†] has both an accessible activation barrier (ΔG‡ = 20.7 kcal mol−1) as well as a favourable exergonicity (ΔG° = −17.1 kcal mol−1), in contrast to the conversion of IX to XI (ΔG° = +9.8 kcal mol−1).
Pyrimidine (2e) reacted by a similar pathway to produce 11, preferring to close to C4 rather than C2 to give ii.15 The closure of ii at C4 maintains some of the N1–C2–N3 growing amidine character and stabilization throughout the reaction coordinate, which would be lost if cyclization were to take place at C2. Again, the diazocine 11 represents a relatively rare class of heterocycle, examples appearing in only four reports.14c,16
Finally, quinazoline (2f) gave rise to the unusual adduct 12, incorporating a molecule of adventitious water. We suggest that in this instance, ring-opening of the initial benzoazetidine XII is assisted by water, the C–N bond being weakened by virtue of the amidine character of XII that is absent in the benzoazetidines from quinoline (2b) or isoquinoline (2a, cf.IX). The structure assignment of 12 was secured by the clear HMBCs observed for both H6 (δ 6.18 ppm) to C15 (δ 66.8) and H15 (δ 6.67) to C6 (δ 82.1) as well as the chemical shifts of those four nuclei.
Three-component reactions
Modes B and C offer the potential to produce structurally complex, polycyclic structures in a single, one-pot, thermal operation. As mentioned, the former (mode B) is precedented10 for the case of certain carbonyl compounds but not for other types of electrophilic species. The latter (mode C) has been reported only for classical benzyne trapping with N-HARs in the presence of various terminal alkynes, carbonyl compounds and aliphatic nitriles, as the protic nucleophiles.10bMode B (TCR with N-HAR + X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) Y electrophile)
Y electrophile)
A successful mode B TCR (cf.Fig. 1) would likely need to meet the following criteria: (i) neither the N–HAR nor the  electrophile should react with the HDDA polyyne precursor faster than its rate of cyclization to the benzyne intermediate (the rate-limiting event); (ii) the N–HAR should engage the benzyne faster than it reacts with (at least irreversibly) the electrophile (
electrophile should react with the HDDA polyyne precursor faster than its rate of cyclization to the benzyne intermediate (the rate-limiting event); (ii) the N–HAR should engage the benzyne faster than it reacts with (at least irreversibly) the electrophile ( ); (iii) the electrophile should be sufficiently reactive to trap the 1,3-zwitterionic species (cf.III) to give products like IV before the former undergoes a ring-expansion process such as those discussed above in the mode A results.
); (iii) the electrophile should be sufficiently reactive to trap the 1,3-zwitterionic species (cf.III) to give products like IV before the former undergoes a ring-expansion process such as those discussed above in the mode A results.
We screened different electrophiles that might meet these requirements. Reactions were performed in benzene (or in a cosolvent of benzene and acetonitrile, depending on the solubility of the reactants) using an initial concentration of aryne precursor 1 of 0.02 M. Given the ready availability of all of the trapping components explored, we opted to use a stoichiometric ratio of the three reactants of, typically, 1:3:5 (benzyne precursor 1: N-HAR 2: electrophile 3); this choice was not further optimized.
Aliphatic aldehydes and ketones (e.g., butyraldehyde and methyl vinyl ketone) did not perform well as a TCR third component. These coupling partners may not be sufficiently electrophilic. Upon switching to biacetyl (3a), we observed efficient formation of 13 (Fig. 3a) as a mixture of diastereomers in 81% yield. In this reaction triyne 1a, quinoline (2b), and 3a were heated at 85 °C for 16 h. Other reactive carbonyl compounds could also serve as a third component, as demonstrated by products 14, 15, 16, and 17, which arise from incorporation of p-benzoquinone (3b), N-methylisatin (3c), acrolein (3d), or p-bromobenzaldehyde (3e),17 respectively. These results also show that both isoquinoline (2a) and quinoline (2b) can participate in these TCRs. We did not extensively investigate all combinations of the three reactive components. In some instances, both 2a and 2b reacted in qualitatively similar fashion to provide the analogous type of adduct; we believe that the reaction pathways we have identified would be generally applicable to many sets of three reaction partners.
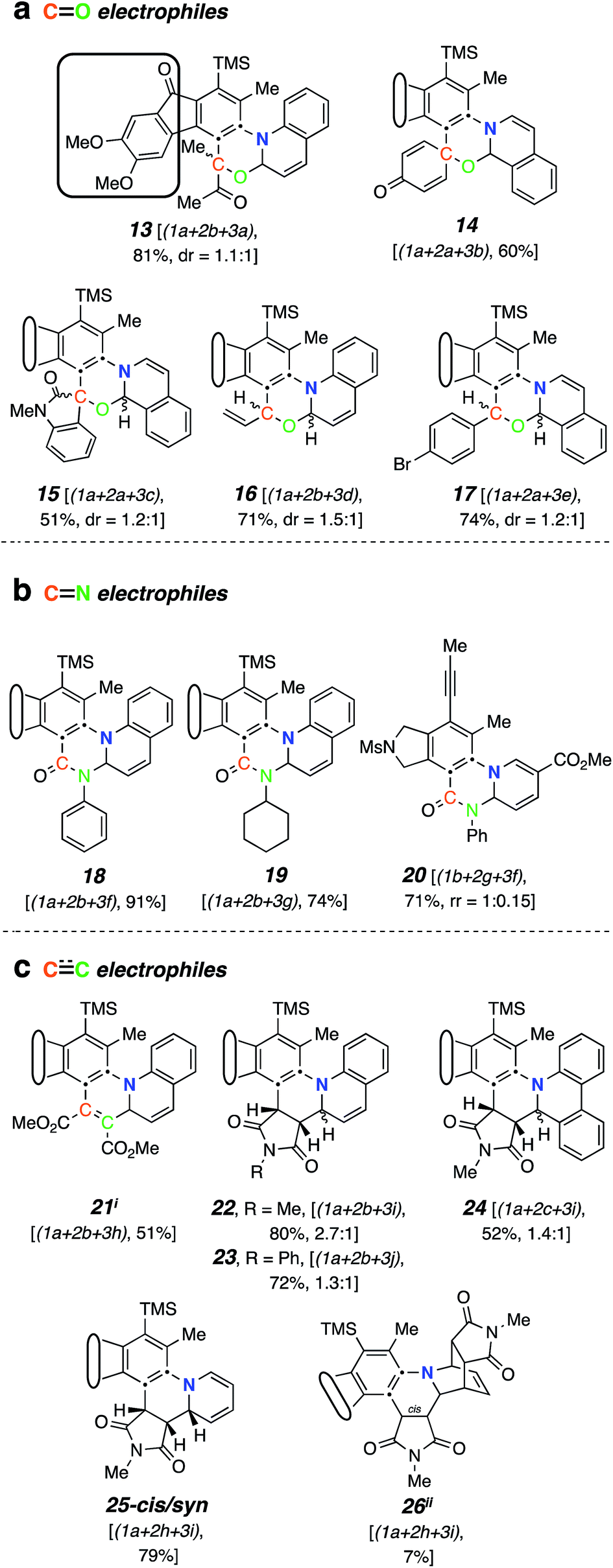 | ||
| Fig. 3 Products from mode B three-component reactions. Trapping of zwitterions (cf.III) by (a) carbonyl electrophiles, (b) isocyanates, and (c) electron-deficient alkenes or alkynes. iA byproduct comprising two molecules of dimethyl acetylenedicarboxylate and one of quinoline was isolated (in ca. equal mass to that of 21, see ESI†). iiRelative configuration not assigned. | ||
After exploring carbonyls as third components, we proceeded to identify other effective classes of  electrophiles. Phenyl and cyclohexyl isocyanate (
electrophiles. Phenyl and cyclohexyl isocyanate (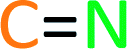 electrophiles 3f and 3g respectively, Chart 1c) participated in the coupling process to furnish the quinazolinone derivatives 18, 19 and 20 (Fig. 3b). The last involved the incorporation of a pyridine derivative, specifically methyl nicotinate (2g). When pyridine (2h) itself was used along with phenyl isocyanate (3f), the 1H NMR spectrum of the crude reaction product was fairly clean, suggesting an efficient initial transformation. However, the product proved difficult to isolate, perhaps a reflection of the lability of the 1,2-dihydropyridine that was initially formed. In contrast, the vinylogous carbamate character in 20 rendered that adduct readily tractable.
electrophiles 3f and 3g respectively, Chart 1c) participated in the coupling process to furnish the quinazolinone derivatives 18, 19 and 20 (Fig. 3b). The last involved the incorporation of a pyridine derivative, specifically methyl nicotinate (2g). When pyridine (2h) itself was used along with phenyl isocyanate (3f), the 1H NMR spectrum of the crude reaction product was fairly clean, suggesting an efficient initial transformation. However, the product proved difficult to isolate, perhaps a reflection of the lability of the 1,2-dihydropyridine that was initially formed. In contrast, the vinylogous carbamate character in 20 rendered that adduct readily tractable.
The final class of mode B TCRs involved trapping the initial 1,3-zwitterion with various electron-deficient alkenes or alkynes (Fig. 3c). Classic Diels–Alder dienophiles meet that definition. Dimethyl acetylenedicarboxylate (3h) as well as the N-substituted maleimides 3i and 3j all performed well, giving rise to adducts containing the new six-membered tetrahydropyridine ring present in each of 21–25 (Fig. 3c). In the case of formation of 24 (52%) from phenanthridine (2c) and N-methylmaleimide (3i), the 1:1 adduct 9 (cf.Fig. 2a) was formed competitively (34%). This suggests a shorter lifetime for the initially formed 1,3-zwitterion. This is expected because its cyclization to the azetidine 9 is not accompanied by as large of a loss in aromatic resonance stabilization as is the case for, say, quinoline or isoquinoline. Finally, when pyridine was the N-HAR component, 25 was produced efficiently (accompanied by a small amount of the cis-anti diastereomer, see ESI†). This product was also accompanied by one diastereomer of the Diels–Alder adduct 26, again reflecting a lability of the 1,2-dihydropyridine moiety.
Mode C (TCR with a monoprotic nucleophile)
We next studied the outcome of heterocyclic trapping of the benzynes in the presence of various protic nucleophiles (mode C, Fig. 1). We began by heating the triyne substrate 1a with quinoline (2b) in chloroform (4a, 0.02 M). The zwitterionic intermediate abstracted a proton from CHCl3, and the resulting ion pair collapsed to give the trichloromethylated adduct 27 (Fig. 4a). This molecule, as well as the other quinoline-derived biaryls, gave evidence of two atropisomers in its 1H and 13C NMR spectra. The analogous reaction with isoquinoline cleanly produced 28; the NMR spectra for this isomeric compound showed a single set, albeit of broadened, resonances, reflecting the expected lower barrier for biaryl rotation compared to that in 27.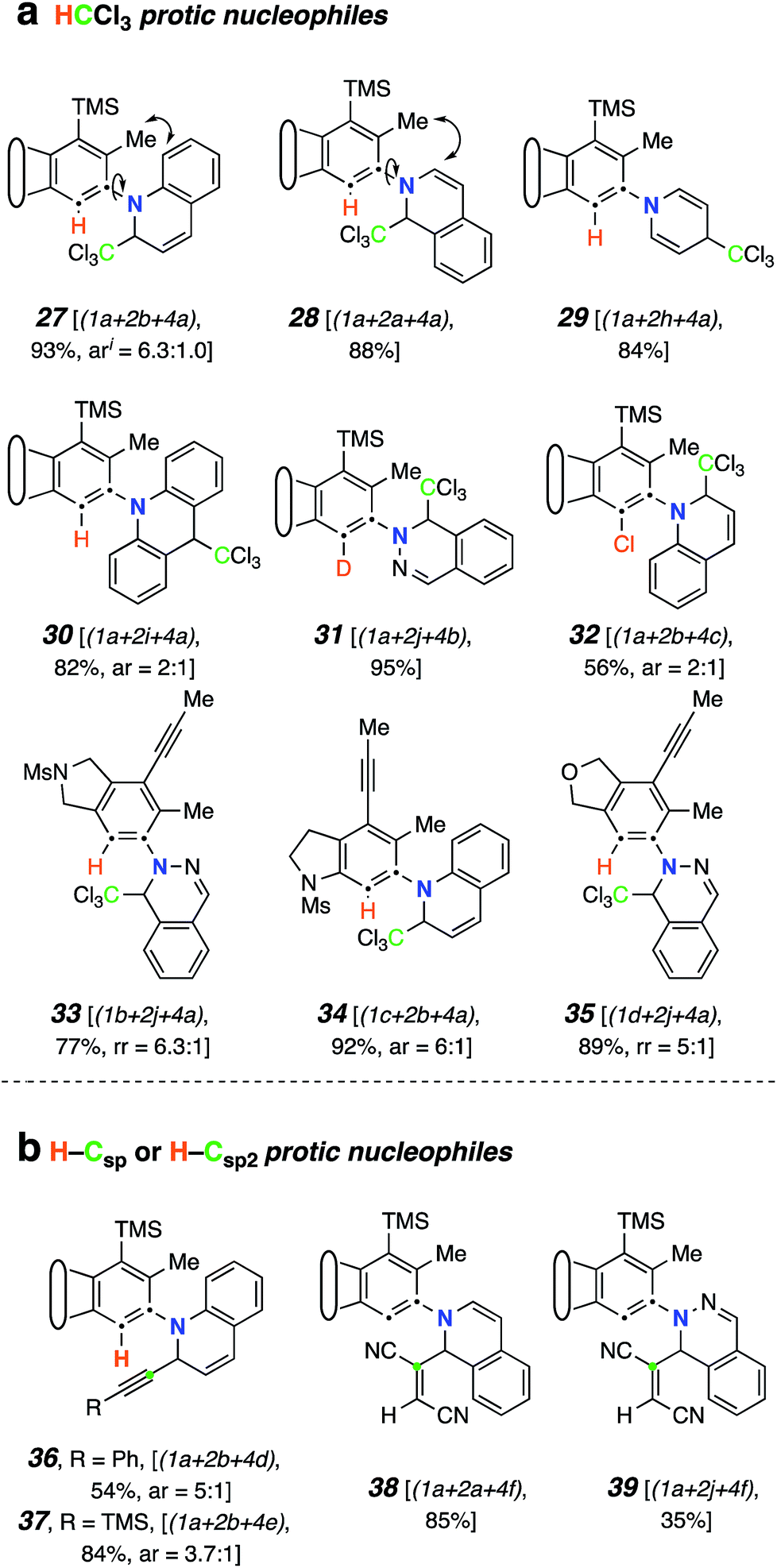 | ||
| Fig. 4 Products from mode C three-component reactions using monoprotic nucleophiles. Trapping of zwitterions (cf.III) by (a) CCl3-containing electrophiles and (b) carbon-acids (terminal alkynes or fumaronitrile). I ar = atropisomeric ratio (observed by 1H NMR spectroscopy, see ESI†). | ||
Because of the relatively high efficiency of the trapping by CHCl3, we examined a wider array of N-HARs as well as other benzyne precursors for this class of TCR (Fig. 4a). In the case where pyridine (2h) was used as the N-HAR, the 4-substituted product 29 was formed. Use of acridine (2i) also led to the formation of its para-functionalized product, 30, because the adduct arising from trapping by Cl3C− at an ortho-carbon would significantly interrupt aromaticity. The remote nature of the Cl3C-bearing carbon in 30 results in a only subtly different pair of atropisomers, a fact revealed in the NMR data for this adduct (see ESI†). The fact that chloroform was the source of the proton in these reactions was validated when CDCl3 was used as the third component; 31 was formed in 95% yield. This result also demonstrated that phthalazine (2j) was a participant in this TCR. We then examined several other aryne precursors for this transformation. Each of 33, 34, and 35 was produced in a very good yield, starting with 1b, 1c, and 1d, respectively. Finally, when carbon tetrachloride (4c) was used as solvent instead of CHCl3, chlorination of the 1,3-zwitterion from quinoline led to the formation of 32.18
TCRs using carbon acids were also shown to be effective. Specifically, we found that terminal alkynes (4d and 4e) were shown to have a sufficiently acidic proton to engage in a TCR to provide the alkynyl derivatives 36 and 37 (Fig. 4b). Surprisingly, fumaronitrile (4f), which we initially expected to capture the zwitterion in a net [4 + 2] manner (i.e., via mode B), had a different outcome. Protonation and capture by the conjugate base of 4f gave product 38 or 39 when isoquinoline (2a) or phthalazine (2j) was used as the N-HAR.
Mode C (TCR with a diprotic nucleophile)
We then studied the behavior of several β-dicarbonyl protic nucleophiles (5a–d, Chart 1d) and encountered a new type of net bicyclization reaction (Fig. 5a). For example, heating the substrate 1a with quinoline (2b) and dimedone (5a) resulted in the formation of the bridged benzoxazine 40 (77%) by sequential engagement of benzyne VI by 2b and, then, 5a. Thus, the third component here was serving as a “doubly protic” nucleophile. In this case the more crowded atropisomers interconverted sufficiently slowly that they now could be chromatographically separated. As the additional examples in Fig. 5a demonstrate, acetylacetone (5b), 4-hydroxycoumarin (5c), and α-cyanopinacolone (5d) all participated in this type of transformation. The structures of these novel products (40–44) were established on the basis of (i) the contiguous four-spin vicinal coupling array among the bridging methylene and bridgehead methine protons and, especially, (ii) the 13C NMR chemical shifts of the bridgehead aminal carbon (82–86 ppm). These spectroscopic signatures were consistently seen in all five of these adducts. HSQC and HMBC experiments were also consistent with these assignments.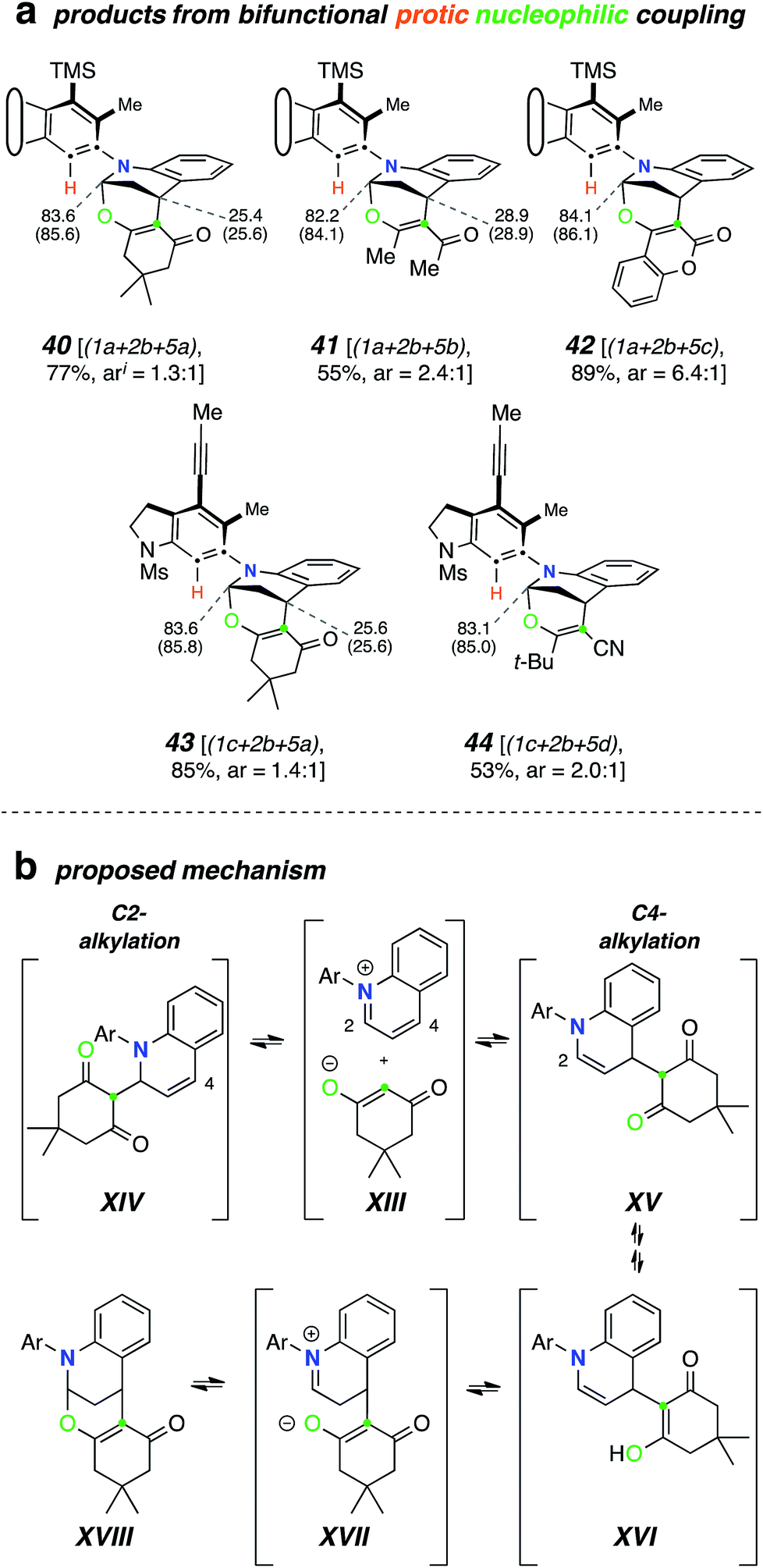 | ||
| Fig. 5 Products from mode C three-component reactions using diprotic nucleophiles. (a) Product structures [ratios of major and minor (separable) atropisomers]. (b) Mechanistic rationale consistent with the formation of the hemiaminal-containing skeletons in products 40–44. | ||
A mechanistic rationale for this interesting transformation is suggested in Fig. 5b. Nucleophilic addition of quinoline (2b) to the HDDA-benzyne and proton transfer from, for example, dimedone (5a) produces the enolate-quinolinium ion pair XIII. This can undergo collapse by attack at either C2 (to XIV) or C4 (to XV). Since C2-attack is the main event in the earlier TCRs, we suggest that XIII–XV are sufficiently close in energy to be in dynamic equilibrium with one another. Keto–enol tautomerization, while possible for either XIV or XV, can proceed further from the latter because of its embedded enamine moiety. Specifically, internal protonation within enol XVI gives zwitterion XVII, the collapse of which would generate XVIII (cf.40/43).
Finally, we devised a two-pot variant that allows for the use of a wider variety of nucleophiles in these types of reactions. Namely, generation of the benzyne from the polyyne precursor in the presence of the N-HAR and “non-nucleophilic” triflic acid (4g), typically in a molar ratio of 1:3:2 to ensure the presence of the free-base form of the N-HAR, gave rise to isolable salts XIX (Fig. 6a). Specifically, the N-arylated “inium” triflate salts 45–48 were efficiently generated. These reactions were seen to be most efficient using acetonitrile as the reaction solvent. Crude samples of the salts could be isolated by solvent removal and were characterized by 1H NMR and HRMS analyses. These samples still retained the excess N-HAR H+TfO− salts, but that did not seriously hamper their subsequent reactions with nucleophiles. These inium ions could then be redissolved (or suspended) in various solvents and further transformed to products that were either complementary to or not compatible with the one-pot TCRs. For example (Fig. 6b), the N-aryl pyridinium triflate 45 cleanly underwent hydrogenation in the presence of Adams catalyst, producing the piperidine derivative 49 in 71% yield at room temperature. In another reduction 46 was converted to the 1,2-dihydropyridine XX using sodium borohydride. As was the case with 1,2-dihydropyridines mentioned earlier, XX was also found to be unstable upon attempted silica gel chromatographic purification. However, the crude product could be further treated with N–methyl maleimide to furnish the Diels–Alder adduct 50 in 43% overall yield (from 1a). The triflate salts were also found to react readily with (excess) Grignard reagents. For example, the 1c-derived pyridinium salt 46 was reacted with MeMgBr in THF to produce XXI. This dihydropyridine was transformed directly into 51 using trifluoroacetic anhydride (58% overall yield from 1c). Grignard reagents were used to convert other heterocyclic derived salts, namely 47 and 48, into their corresponding alkylated products 52 and 53 in a clean manner.
 | ||
| Fig. 6 Use of the “non-nucleophile” triflic acid (4g) allows for the incorporation of a broader range of nucleophiles. (a) TCRs were performed using 3 equiv of the N-HAR and 2 equiv of triflic acid in acetonitrile (MeCN), which led to the formation of salts 45–48. (b) Examples demonstrating the subsequent diversification of some of these salts to give products 49–53. | ||
Conclusion
We have demonstrated that various modes (A–C) of reaction can be realized by reacting six-membered N-heteroaromatic compounds with benzynes generated by thermal HDDA cycloisomerization of triyne (or tetrayne) precursors. All reactions can be rationalized as passing through an initially formed 1,3-zwitterion arising from attack on the benzyne by the nitrogen atom of the heterocycle. In mode A and in unprecedented fashion, 1:1 adducts emanating from this zwitterion are produced. For the most part, these products belong to either of the benzoazetidine or benzoazocine family.
In mode B, the first of two types of three-component reaction, an intermediate electrophile is used to capture the 1,3-zwitterion. Mode B electrophiles can be suitably reactive carbonyl compounds, isocyanates, or electron-poor alkenes or alkynes. In mode C, the third component is a protic acid that quenches the anionic centre in the zwitterion to give an ion pair that collapses to product. Diprotic nucleophiles (e.g., β-dicarbonyl compounds) can be used to give novel bridged polycyclic products. Finally, the triflic acid produces isolable N-arylinium triflates that subsequently can be subjected to nucleophilic addition to further widen the strategic scope of the process.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institute of General Medical Sciences of the U.S. Department of Health and Human Services (R35 GM127097). Some of the NMR data were obtained with an instrument purchased with the support of the NIH Shared Instrumentation Grant program (S10OD011952). Computations were carried out using resources provided by the University of Minnesota Supercomputing Institute (MSI).Notes and references
- (a) O. J. Diamond and T. B. Marder, Org. Chem. Front., 2017, 4, 891–910 RSC; (b) A. Z. Bradley and R. P. Johnson, J. Am. Chem. Soc., 1997, 119, 9917–9918 CrossRef CAS; (c) K. Miyawaki, R. Suzuki, T. Kawano and I. Ueda, Tetrahedron, 1997, 38, 3943–3946 CrossRef CAS; (d) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208–212 CrossRef CAS PubMed.
- A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC.
- J. Chen, V. Palani and T. R. Hoye, J. Am. Chem. Soc., 2016, 138, 4318–4321 CrossRef CAS PubMed.
- (a) S. P. Ross, B. Baire and T. R. Hoye, Org. Lett., 2017, 19, 5705–5708 CrossRef CAS PubMed; (b) S. Arora, V. Palani and T. R. Hoye, Org. Lett., 2018, 20, 8082–8085 CrossRef CAS PubMed.
- S. P. Ross and T. R. Hoye, Org. Lett., 2018, 20, 100–103 CrossRef CAS PubMed.
- J. Zhang and T. R. Hoye, Org. Lett., 2019, 21, 2615–2619 CrossRef CAS PubMed.
- S. P. Ross and T. R. Hoye, Nat. Chem., 2017, 9, 523–530 CrossRef CAS PubMed.
- (a) D. Niu, P. H. Willoughby, B. Baire, B. P. Woods and T. R. Hoye, Nature, 2013, 501, 531–534 CrossRef CAS PubMed; (b) D. Niu and T. R. Hoye, Nat. Chem., 2014, 6, 34–40 CrossRef CAS PubMed; (c) J. Zhang, D. Niu, V. A. Brinker and T. R. Hoye, Org. Lett., 2016, 18, 5596–5599 CrossRef CAS PubMed.
- For reviews on aryne chemistry, see, e.g., (a) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef CAS PubMed; (b) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766–3778 CrossRef CAS PubMed; (c) T. Kitamura, Aust. J. Chem., 2010, 63, 987–1001 CrossRef CAS.
- (a) M. Jeganmohan and C. H. Cheng, Chem. Commun., 2006, 2454–2456 RSC; (b) M. Jeganmohan, S. Bhuvaneswari and C. H. Cheng, Chem. Asian J., 2010, 5, 153–159 CrossRef CAS PubMed; (c) A. Bhunia, T. Roy, P. Pachfule, P. R. Rajamohanan and A. T. Biju, Angew. Chem., Int. Ed., 2013, 52, 10040–10043 CrossRef CAS PubMed; (d) A. Bhunia, D. Porwal, R. G. Gonnade and A. T. Biju, Org. Lett., 2013, 15, 4620–4623 CrossRef CAS PubMed.
- E. K. Fields and S. Meyerson, J. Org. Chem., 1966, 31, 3307–3309 CrossRef CAS.
- (a) V. W. Kirmse, L. Horner and H. Hoffmann, Justus Liebigs Ann. Chem., 1958, 614, 19–30 CrossRef; (b) H. W. Wanzlick and E. Schikora, Chem. Ber., 1961, 94, 2389–2393 CrossRef CAS; (c) K. Ishiguro, T. Nojima and Y. Sawaki, J. Phys. Org. Chem., 1997, 10, 787–796 CrossRef CAS.
- The structure of each of these novel 8-membered heterocycles (10 and 11) were supported by two-dimensional NMR spectroscopic correlations. They were further validated by comparison of the experimental 1H and 13C chemical shifts with those computed (DFT) for several related model structures. [see ESI† for details].
- (a) M. Furukawa, Y. Kojima and S. Hayashi, Chem. Pharm. Bull., 1972, 20, 927–930 CrossRef CAS; (b) T. Saied, N. Jelaiel, M. L. Efrit, Y. Fort and C. Comoy, Tetrahedron, 2017, 73, 1489–1494 CrossRef CAS; (c) H. D. Perlmutter, Adv. Heterocycl. Chem., 1990, 50, 1–83 CrossRef CAS.
- The initial azetidine derivatives i and ii can arise from closure of an initially formed 1,3-zwitterion. Electrocyclic opening would then account for the formation of 10 and 11
 .
. - (a) R. N. Kumar, T. Suresh, T. Dhanabal and P. S. Mohan, Indian J. Chem., 2004, 43, 846–851 Search PubMed; (b) F. Bruni, A. Costanzo, S. Selleri, G. Guerrini, L. Giusti, C. Martini and A. Lucacchini, Farmaco, 1993, 48, 309–319 CAS; (c) A. Miyashita, N. Taido, S. Sato, K. Yamamoto, H. Ishida and T. Higashino, Chem. Pharm. Bull., 1991, 39, 282–287 CrossRef CAS; (d) V. V. Kaminski, R. N. Comber, A. J. Wexler and J. S. Swenton, J. Org. Chem., 1983, 48, 2337–2346 CrossRef CAS.
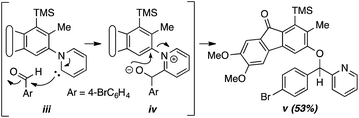
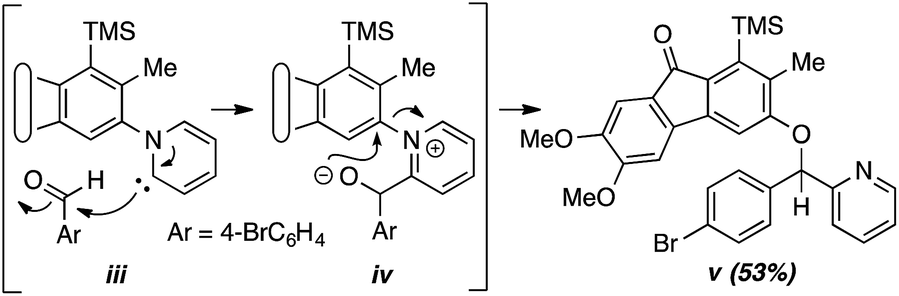
- In contrast the reaction of 1a in the presence of 4-bromobenzaldehyde (3e) with pyridine (2h) as the N-hetaryl instead of isoquinoline produced compound v (53%, see ESI†), presumably by way of intermediates like iii (cf. X, Fig. 2b) and iv, a pathway involving final ipso attack as observed and suggested for pyridine by Biju and coworkers.10c
 .
. - S. J. Li, Y. Wang, J. K. Xu, D. Xie, S. K. Tian and Z. X. Yu, Org. Lett., 2018, 20, 4545–4548 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03479j |
| ‡ Throughout the manuscript we have used Roman numerals to label generic or non-isolated structures and intermediates and Arabic numbering for structures of isolated compounds. |
| § The structures of isolated products bear a unique Arabic number. That number is followed (in brackets) by a listing of the reactants that have been incorporated into the compound (cf.Chart 1): polyyne 1 is the benzyne precursor, 2 the N-HAR, 3 an electrophilic third component, 4 a monoprotic-nucleophile, and/or 5 a diprotic nucleophile. |
| This journal is © The Royal Society of Chemistry 2019 |