
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-permeable polyamide membranes harvested by covalent organic framework nanofiber scaffolds: a two-in-one strategy†
Zhe
Zhang
,
Xiansong
Shi
,
Rui
Wang
,
Ankang
Xiao
and
Yong
Wang
 *
*
State Key Laboratory of Materials-Oriented Chemical Engineering, Jiangsu National Synergetic Innovation Center for Advanced Materials, College of Chemical Engineering, Nanjing Tech University, Nanjing 210009, P. R. China. E-mail: yongwang@njtech.edu.cn
First published on 13th August 2019
Abstract
Polyamide-based thin-film composite nanofiltration membranes are widely used for potable water decontamination, brackish water desalination and wastewater reutilization. However, enhancing the water permeance of the polyamide layer within the thin-film composite nanofiltration membrane still remains a great challenge, because of the relatively large thickness and high transport resistance of the polyamide layer produced by intractable interfacial polymerization. Herein, we reported a two-in-one strategy to prepare ultra-permeable nanofiltration membranes via covalent organic framework (COF) nanofiber scaffold mediated interfacial polymerization. The highly porous and hydrophilic COF nanofiber scaffolds enhanced the controlled release of amine monomers, leading to ultrathin polyamide layers. Also, the relatively smooth COF nanofiber scaffolds can be spontaneously evolved into rugged and uneven architectures during interfacial polymerization, providing rough substrates for enlarging the actual areas of polyamide layers. Therefore, the increased areas of polyamide layers were employed as additional water permeable domains. Arising from the synergetic effect of the ultrathin and increased water permeation domains, the produced membranes exhibit exceptional nanofiltration performance with a water permeance of up to 31.1 L m−2 h−1 bar−1 and a Na2SO4 rejection rate of about 95%, outperforming most other nanofiltration membranes. This highly accessible technique opens a new avenue for the design and engineering of ultra-permeable thin-film composite nanofiltration membranes, highlighting its great potential in providing clean water.
Introduction
The growing requirements for clean water have become global issues, on account of rapid population growth and environmental contamination. Nanofiltration (NF) as a burgeoning membrane separation technology enables clean water supply by brackish water desalination and wastewater reclamation. Thin-film composite (TFC) membranes, composed of polyamide (PA) separation layers and porous support layers, are highly demanded for water NF owing to their high structural stability, effective separation capability and up-scalable processability.1,2 However, these TFC NF membranes suffer from their relatively low water permeance because the PA layers are derived from rapid and intractable reaction–diffusion driven interfacial polymerization (IP), thus leading to rather thick PA layers.3–5 Hence, reducing the thickness of PA layers is a straightforward way for improving the water permeance.6,7Substrates (support layers) as the platform for implementing the IP process play a vital role in the formation of the PA layers, including the thickness of the PA layers. The pore channels inside the substrate not only store the aqueous solutions of amine monomers, but also diffuse them into the substrate surface to accomplish the IP. Due to the heterogeneous pore structures of widely used substrates for the IP process, such as commercial microfiltration membranes or ultrafiltration membranes, the release and diffusion extents of amine monomers within the pore channels are difficult to control. Therefore, reconstruction of conventional substrates via mediating the physicochemical properties of substrates, including surface porosity,8 pore structure,8 surface hydrophilicity,9 and roughness of the substrate,10 would improve the controlled release of amine monomers. Thus, it could create the reduced thickness of the PA layer through the controlled IP process. For instance, Livingston and co-workers reported an interesting approach for producing ultrathin polyamide nanofilms on substrates decorated with cadmium hydroxide nanostrands.11 By virtue of their favourable hydrophilicity and uniform porosity, the nanostrand layers promote the uptake and controlled release of monomers during the IP process, leading to sub-10 nm thick PA layers. The ultrathin nanofilms show greatly enhanced organic solvent permeance compared with those commercial NF membranes with similar solute rejection rates. This unique method provides a promising way for improving the permeance of PA layers through substrate reconstruction. Similarly, various materials have been utilized for substrate reconstruction to reduce the thickness of the PA layers, such as carbon nanotubes,12–15 copper hydroxide nanostrands,16 cellulose nanocrystals,17 polydopamine18,19 and polyphenol.20,21 It is noteworthy that, for the state-of-the-art NF membranes, the thickness of the PA layers has been successfully reduced to ∼10 nm.6,12,15,22 Therefore, it is necessary to find other methods for improving the water permeance of PA layers, because it would be very challenging to further reduce the thickness of PA layers to smaller than 10 nm. Otherwise, the integrity of PA layers would be deeply compromised and fail to give satisfactory solute rejection rates.
It is generally accepted that increasing the permeable area for water transport per projected area on the substrate can enhance water permeance. Recently, a breakthrough progress was made by Zhang and co-workers, who varied the morphologies of PA layers from dense to void structures (Turing structures).23 They manipulated the diffusion rates of monomers during the IP process through hydrogen bond interactions between piperazines and introduced water-soluble macromolecules, for creating the Turing structures. Because of the existence of spatially distributed effective water permeable sites in those structures, the membranes exhibited superior water–salt separation properties excelling the upper-bound line of conventional PA membranes. Alternatively, Jin and co-workers pioneered a nanoparticle-template technique for generating ultrathin PA layers with abundant crumpled nanoscale structures.22 In their work, such a morphology was obtained through interlayer mediation and template dissolution on nanoparticle pre-loaded carbon nanotube network layers. Benefitting from the increased water permeable areas of the crumpled ultrathin PA layers, the membranes showed unprecedented water permeation. Moreover, other research also verified the contribution of increased permeable areas within membrane separation layers to the permeance enhancement.11,22,24,25 Thinning the PA layers and increasing the permeable areas are both beneficial for elevating water permeance. Integrating them into one approach thus would be a better way to further improve water permeance. Although several studies have already been reported to integrate both methods,11,22 most of the other studies are separately focused on thinning the PA layers or increasing the permeable areas.
Covalent organic frameworks (COFs), as emerging crystalline porous materials, are tailored from pure organic building blocks. These features endow COFs with valuable application prospects in various fields.26–28 Their highly porous backbones and strong compatibility with polymers make COFs an appealing candidate for substrate reconstruction to meet the requirements for fabricating ultra-permeable TFC NF membranes. Herein, we proposed a two-in-one strategy for rationally designing ultra-permeable TFC NF membranes via simultaneously reducing the thickness of PA layers and increasing water permeation areas (domains). We constructed a distinctive porous scaffold on a macroporous polyethersulfone (PES) substrate with fibrous cationic COFs via a simple vacuum filtration method. In the IP process, the cationic COF scaffolds act as a controlled release platform and a rough structure generator. The highly porous structure, extremely low density and cationic feature endow the COF scaffolds with excellent water wettability for enhancing the uptake and controlled release of piperazine monomers to produce thinner PA layers. Furthermore, such advantages also facilitate the formation of extensive nanofiber aggregates on the COF scaffolds during the IP process, thus generating the rugged and fluctuant morphology of PA layers for the improvement of water permeance (see Scheme 1). The prepared TFC NF membranes demonstrated a superhigh water permeance of 31.1 L m−2 h−1 bar−1 and a Na2SO4 rejection rate of about 95%, outperforming most reported NF membranes. This work enables a promising opportunity for the fabrication of ultra-permeable TFC NF membranes through a simple and effective substrate reconstruction strategy.
 | ||
| Scheme 1 Schematic illustration of the preparation process of TFC NF membranes. | ||
Results and discussion
Synthesis and characterization of TpTGCl nanofibers
The fibrous COF TpTGCl was synthesized via the Schiff base condensation reaction between aldehyde monomer Tp and amine monomer TGCl (Fig. 1a). The chemical composition of TpTGCl was confirmed by the FTIR spectra (Fig. 1b). The carbonyl stretching vibration peak (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 1643 cm−1) and C–H stretching vibration peak (2893 cm−1) of aldehyde groups in the Tp monomer and the N–H stretching vibration peak (3322 cm−1) of primary amines in the TGCl monomer were absent in the resultant product, indicating the complete conversion from the original monomers to the final product. Moreover, after the condensation reaction, two characteristic peaks of CC stretching vibration (1613 cm−1) and C–N stretching vibration (1291 cm−1) in TpTGCl appeared. This evidence verified the successful synthesis of cationic COF TpTGCl. The XRD pattern (Fig. 1c) of TpTGCl shows a moderate crystallinity, which is due to the insertion of chloride ions into the interspaces between two adjacent monomer units, disturbing their structural regularity.29 The diffraction peaks at ∼11° and ∼27° correspond to the intrinsic (100) and (001) crystallographic planes of TpTGCl, respectively. The crystalline characteristic of TpTGCl endows it with high porosity and structural stability for further utilization.
O, 1643 cm−1) and C–H stretching vibration peak (2893 cm−1) of aldehyde groups in the Tp monomer and the N–H stretching vibration peak (3322 cm−1) of primary amines in the TGCl monomer were absent in the resultant product, indicating the complete conversion from the original monomers to the final product. Moreover, after the condensation reaction, two characteristic peaks of CC stretching vibration (1613 cm−1) and C–N stretching vibration (1291 cm−1) in TpTGCl appeared. This evidence verified the successful synthesis of cationic COF TpTGCl. The XRD pattern (Fig. 1c) of TpTGCl shows a moderate crystallinity, which is due to the insertion of chloride ions into the interspaces between two adjacent monomer units, disturbing their structural regularity.29 The diffraction peaks at ∼11° and ∼27° correspond to the intrinsic (100) and (001) crystallographic planes of TpTGCl, respectively. The crystalline characteristic of TpTGCl endows it with high porosity and structural stability for further utilization.
 | ||
| Fig. 1 Characterization of TpTGCl nanofibers. (a) Synthetic scheme of TpTGCl. (b) FTIR spectra of TGCl, Tp and TpTGCl. (c) XRD pattern of TpTGCl (inset shows the structural mode of TpTGCl). (d) SEM image of TpTGCl nanofibers (inset shows the diameter distribution of TpTGCl nanofibers). (e) TEM image of TpTGCl nanofibers. | ||
The morphology of synthesized TpTGCl was examined by SEM and TEM (Fig. 1d and e). COF TpTGCl exhibits fibrous structures loosely stacked together. The COF nanofibers possess a low aspect ratio with a diameter of about 36 nm and a length of about several hundred nanometers. It is notable that, however, the morphology of the obtained COF TpTGCl is quite different from that reported by Banerjee and co-workers.29 The possible reason for the formation of the fibrous morphology may be ascribed to a specific dissolution–recrystallization process in the presence of excess polar solvents,30 such as water. This phenomenon has also been observed in other studies.31,32 Furthermore, because of the presence of excessive inherent positively charged guanidinium units (Fig. 1a), the synthesized COF TpTGCl nanofibers can be easily dispersed in water as a result of strong electrostatic repulsion, forming a highly stable dispersion without any precipitate within 3 months (Fig. S2†). Moreover, the structures of the COF nanofibers can be retained after water treatment, further implying their structural stability (Fig. S2†).
Surface properties of the membranes
The TFC NF membranes were fabricated via vacuum filtration of water-dispersed TpTGCl nanofibers onto macroporous PES substrates followed by IP. The chemical compositions of the PES substrate, the COF-deposited PES substrate and the PA composite membrane were revealed by the ATR-FTIR spectra (Fig. 2a). Two characteristic peaks of CO stretching vibration (1626 cm−1 and 1443 cm−1) of amide groups in the PA composite membrane appeared after IP, evidencing the formation of the PA layer. It is noteworthy that a new characteristic peak of CO (1615 cm−1) stretching vibration was shown in the spectrum of the PA composite membrane, which was attributed to amide groups formed between the TMC monomers and the secondary amine groups in TpTGCl backbones (Fig. S3†). To further verify this speculation, we soaked the COF-deposited PES substrate in a TMC/n-hexane organic solution which was used in the IP process. As we expected, a characteristic peak of CO (1617 cm−1) stretching vibration arises, evidencing the formation of covalent linkages between the TpTGCl backbones and PA networks (Fig. S4†). Therefore, the interfacial compatibility of TpTGCl and the PA layer would be enhanced, which is beneficial for the stability of the TFC NF membranes.
 | ||
| Fig. 2 Surface properties of different membranes. (a) ATR-FTIR spectra of the PES substrate, the COF-deposited PES substrate and the PA composite membrane. (b) Zeta potentials of the PES substrate, the COF-deposited PES substrate and the PA composite membrane. (c) Digital photos of water droplets penetrating through different membranes with the same duration (on the left the optical images of the corresponding membranes are shown). | ||
We analyzed the surface charging properties of different membranes by a zeta potential test (Fig. 2b). Under a neutral pH environment, all the membranes were negatively charged. For the COF-deposited PES substrate, although the TpTGCl nanofibers are intrinsically positively charged, the surface of the deposited membrane still exhibits negatively charged properties. The highly porous and negatively charged COF layer may facilitate the capture and binding of positively charged piperazine molecules,33 for further mediating the vulnerable rapid-reacting IP. The surface of the PA composite membrane shows strong negatively charged properties compared with the charge properties of the COF-deposited PES substrate. This could be attributed to the formation of carboxyl groups via hydrolysis of residual acyl chloride groups within the PA networks. The highly negatively charged PA layer will enhance the rejection of divalent anions by the Donnan exclusion effect, which will be discussed later.
The hydrophilicity of different membranes was revealed by the water contact angles (Fig. 2c). The macroporous PES substrate shows excellent water wettability and the water droplet penetrates into the membrane within ∼2 seconds. This fast penetration indicates the low mass transfer resistance and hydrophilic surface of the PES substrate, which is favorable for fabricating high-flux TFC NF membranes. Likewise, such fast water penetration also occurred on the surface of the COF-deposited PES substrate. However, due to the presence of the COF layer, the penetration on the surface of the COF-deposited PES substrate was slowed down to ∼10 seconds, which is slower than that on the PES substrate. The excellent surface hydrophilicity and wettability enable high piperazine solution storage capability of the COF-deposited PES substrate. This would strengthen the controllability of the IP reaction through the homogeneous release and diffusion of piperazine molecules toward the organic phase, leading to thinner PA layers.17 The PA composite membrane also exhibits a high surface hydrophilicity, resulting from the presence of abundant carboxyl groups within the PA layer as mentioned above. Moreover, there is no obvious penetration of water droplets through the PA composite membrane in 10 seconds, implying the formation of a continuous and dense PA layer on the top of the COF layer.
Microstructures of the membranes
To understand the formation and evolution process of the PA layer, we firstly conducted SEM examination on the surface and cross section of different membranes (Fig. 3a–e). A surface view of the COF-deposited PES substrate (Fig. 3a) exhibits that the COF nanofibers are randomly stacked together, forming a continuous COF layer on the macroporous PES substrate (pore size, 0.1 μm). The macropores of the PES substrate are covered and substituted by the smaller stacking pores of COF nanofibers (Fig. S6†), further making the reaction–diffusion process during IP more controllable.34 The cross-sectional view of the COF-deposited PES substrate (Fig. 3b) shows a continuous and uneven COF nanofiber scaffold layer with an average thickness of about 650 nm. After the IP process, a continuous and defect-free PA layer was formed on the COF nanofiber scaffold layer (Fig. 3c). The PA layer shows a rugged and fluctuant morphology, and the underneath COF layer with a similar rough morphology is visible, suggesting the ultrathin thickness of the PA layer. We also used SEM to examine the cross-sectional morphology of the PA composite membrane (Fig. 3d and e). Likewise, the cross section of the PA composite membrane exhibits the same rugged and fluctuant morphology as observed on its top surface. In the high magnification image (Fig. 3e), an ultrathin PA layer with a thickness of about 20 nm was revealed. Therefore, we can conclude that a continuous, defect-free and ultrathin PA layer with a rugged and fluctuant morphology on the COF layer was formed after the IP process. | ||
| Fig. 3 SEM and AFM characterization of different membranes. SEM images of the COF-deposited PES substrate: (a) surface and (b) cross section. SEM images of the PA composite membrane: (c) surface, (d) cross section at low magnification and (e) cross section at high magnification. (f) Schematic structure of the PA composite membrane. AFM topography images of (g) the COF-deposited PES substrate and (h) the PA composite membrane (scanning area was 5 μm × 5 μm). (i) The root mean square roughness and the surface area increase of the different membranes (the results were obtained from AFM measurements). | ||
To further understand the formation and evolution mechanism of such a PA layer, we subsequently performed AFM measurements on these membranes (Fig. 3g–i). The topography of the COF-deposited PES substrate shows a relatively flat surface (Fig. 3g) with a low roughness (Fig. 3i, ∼17.6 nm). However, no obvious fibrous morphology can be seen on the COF-deposited PES substrate surface. This is mainly because the COF nanofibers with a low aspect ratio were randomly stacked, thus exhibiting an anisotropic orientation on the top surface of the PES substrate. In contrast, the PA composite membrane shows a high surface roughness (Fig. 3i, ∼100.9 nm) with massive aggregated hill-like structures on its surface (Fig. 3h). This morphology is consistent with SEM observations. Furthermore, we compared the surface area increase (surface area ratios) of different membranes (Fig. 3i), which were directly acquired using software equipped on the AFM. It is calculated as follows:
| Surface area increase (%) = (Sactual area/Sprojected area − 1) × 100% | (1) |
According to the SEM and the AFM analyses, we understand the possible formation and evolution mechanism of this unique PA layer. The COF scaffold with cationic nature, low density, high porosity and intrinsic pores enables the aqueous solution to fully wet and infiltrate it. Because the COF nanofibers with a low aspect ratio (short fibers) are difficult to intertwine with each other to form smooth and thin network layers, the COF scaffold constructed from randomly stacked low aspect ratio COF nanofibers would become much looser and more disordered, leading to the formation of large amounts of hill-like nanofiber aggregates on the outer surface of the COF scaffold. After adding a TMC-containing organic solution, IP occurred on the top of such nanofiber aggregates, forming the PA layer with a rugged and uneven morphology. The COF layer not only created a ∼20 nm thick PA layer via controlled released of piperazine monomers, but also formed a continuous and defect-free nanofilm tightly attached on the top of the uneven COF layer. Ultimately, the resultant PA composite membrane consists of a rough COF layer closely capsulated by the PA nanofilm, which bonded on the macroporous PES substrate. In the present work, the reduction of the thickness and increase of the permeable area of the PA layer for water permeation are achieved through a one-step substrate reconstruction strategy enabled by COF nanofibers. Furthermore, no extra steps (e.g., removing sacrificial materials from the substrates) are required as the COF nanofibers are integrated as a functional, mediation layer into the composite membranes. Therefore, our strategy is distinguished from other studies with similar construction methods.11,22
Nanofiltration performances
To illuminate the interplay between the membrane morphology and the performance, we carried out NF tests with a batch of TFC NF membranes prepared with various COF nanofiber deposition densities (Fig. 4a). The TFC NF membrane without the COF layer shows a water permeance of 12.7 L m−2 h−1 bar−1 and a Na2SO4 rejection rate of up to ∼95%. The water permeance of this membrane is higher than that of those TFC membranes fabricated with ultrafiltration substrates, because of the low mass transfer resistance of the macroporous substrate. With the COF nanofiber deposition density to 0.019 mg cm−2, however, the surface of the PES substrate cannot be fully covered (Fig. S6†), and the membrane shows a slightly enhanced water permeance of 21.6 L m−2 h−1 bar−1 without a decrease in the Na2SO4 rejection rate. With the COF nanofiber deposition density increased up to 0.038 mg cm−2, the surface of the PES substrate was completely covered by the COF layer. Under these conditions, the membrane shows the optimal NF performance with a water permeance of 31.1 L m−2 h−1 bar−1 and a Na2SO4 rejection rate of ∼95%.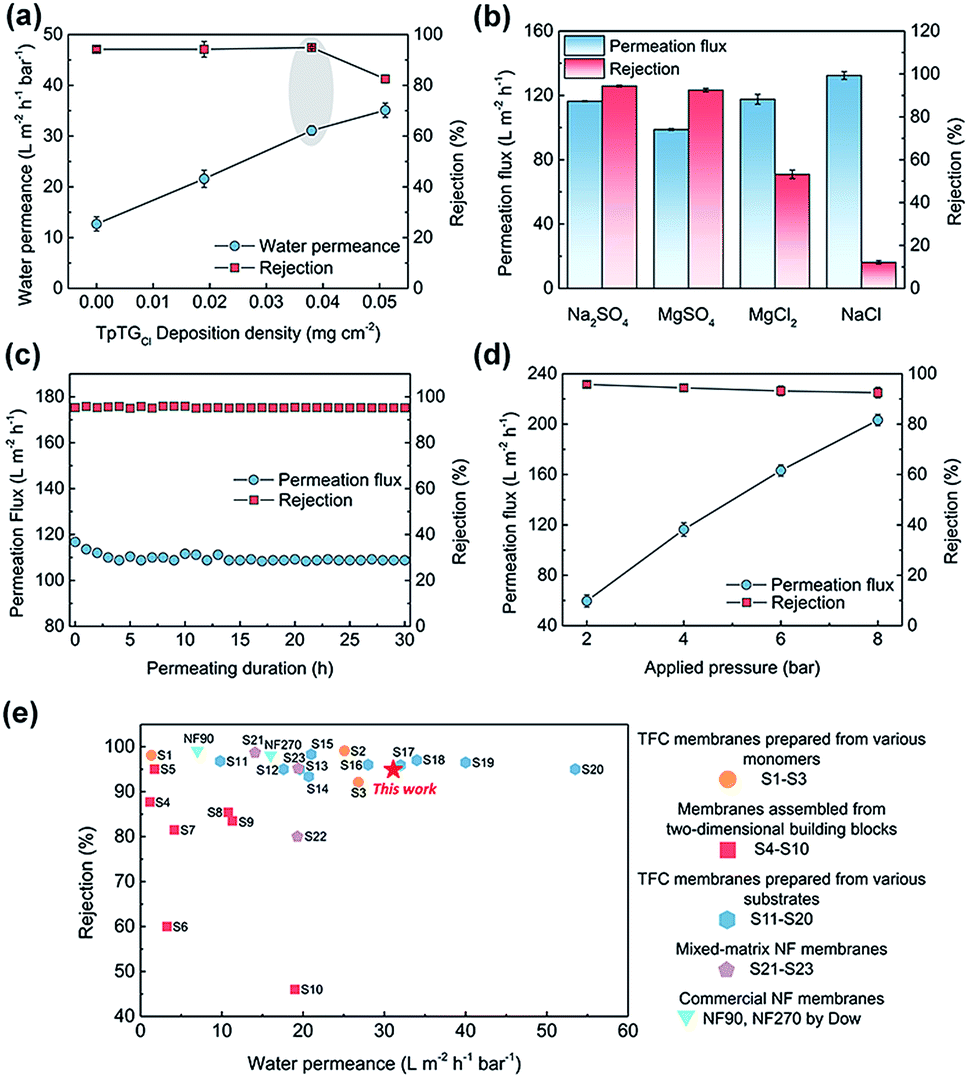 | ||
| Fig. 4 Nanofiltration performances of the PA composite membranes. (a) Effects of different TpTGCl deposition densities on water permeance and Na2SO4 rejection. (b) Effects of different inorganic salt solutions on permeation flux and rejection. (c) Variation of permeation flux and Na2SO4 rejection under a permeating duration of 30 h. (d) Variation of permeation flux and Na2SO4 rejection at different applied pressures. (e) NF performance comparison between our membrane and other membranes reported in the literature. | ||
On the basis of these results, the water permeance improvement can be described as a synergetic effect from the thinner thickness and increased water permeable domains of the PA layer. Without the mediation of the COF layer, the TFC membrane revealed a ∼100 nm thick PA layer with a relatively flat surface (Fig. S6†), and the water permeance was low. In the case of partial nanofiber coverage, although the controlled release occurred and created a thin PA layer with rugged structures, a thick and flat PA nanofilm still formed on the exposed regions of the PES substrate (Fig. S6†). Therefore, the elevation of water permeance was limited. As the COF nanofiber deposition density reached 0.038 mg cm−2, namely the optimal condition, an integral fibrous COF scaffold layer appeared on the PES substrate. The completely formed ultrathin PA layer with a rugged surface as well as the inherent water permeable channels inside the COF nanofibers endow the TFC membrane with the highest water permeance. Moreover, when we further increased the deposition density of the COF nanofiber to 0.051 mg cm−2, the NF performance is compromised. It is possibly due to the tiny defects within the PA layer produced by the excess loading amounts together with the rough COF layer. Nonetheless, it still shows a high water permeance of 35.1 L m−2 h−1 bar−1 with a moderate Na2SO4 rejection rate of ∼83%.
We evaluated the separation behavior of the prepared TFC NF membranes toward different inorganic salts with the same concentration of 1000 ppm (Fig. 4b). The desalination process of the NF membranes is dominated by the collaborative effect of size-sieving and Donnan exclusion.35,36 According to the steric hindrance effect, bivalent ions with a larger hydrated radius have higher rejection rates than monovalent ions with a smaller hydrated radius. Moreover, because of the negatively charged PA layer, the TFC membranes have higher rejection rates to bivalent anions than to monovalent anions. Consequently, the rejection behavior followed the order: Na2SO4 > MgSO4 > MgCl2 > NaCl. The TFC membrane exhibits a stable NF performance with a negligible permeation flux decrease under a continuous testing duration, indicating its good long-term stability (Fig. 4c). Furthermore, the TFC membrane was also applied at various transmembrane pressures for estimating its pressure resistance (Fig. 4d). There is a slight decrease in rejection rates at higher pressure caused by the concentration polarization.37,38 Similarly, a slight decrease in permeation flux occurred at high pressures, resulting from the compaction of the macroporous PES substrate. Nevertheless, this TFC membrane can withstand high pressures required for NF operations.
We compared the NF performance of the TFC membranes prepared in this work with that of other membranes reported in the literature (applied for the rejection of Na2SO4, shown in ESI, S1–S23†), including the TFC membranes prepared using various monomers and substrates, membranes assembled from two-dimensional building blocks, mixed-matrix NF membranes and commercial NF membranes. As shown in Fig. 4e, our membrane exhibits excellent NF performance with a high water permeance and a satisfactory salt rejection rate, outperforming most reported NF membranes. We are aware of the fact that the water permeance of our membrane prepared at the current stage is lower than that of a few NF membranes prepared by other methods.12,15,17,22 However, our method is a “two-in-one” strategy and requires no extra steps to remove the mediation materials, and these features are highly important for large-scale preparation and real-world applications. Moreover, the membrane performances are expected to be further improved by tuning the structure of the COF nanofiber layer and the IP parameters.
Conclusions
In summary, we have demonstrated a two-in-one strategy for the fabrication of ultra-permeable PA layers featuring ultrathin and increased water transport domains. The unique structures were enabled by a cationic COF nanofiber scaffold mediated IP process. The scaffold layers were constructed by a simple vacuum filtration of COF nanofibers on macroporous PES substrates. The nanofiber scaffold layers evolved from a relatively flat surface with randomly stacked nanofibers into a rugged surface of abundant nanofiber aggregates, which was achieved through the wetting and infiltration of aqueous solutions prior to IP. As a consequence, such COF layers led to ultrathin (∼20 nm) PA layers via controlled released of piperazine monomers. Moreover, the rugged COF layers also led to enlarged water permeable domains of PA layers. Benefiting from the synergetic effect of the ultrathin PA layer and increased water transport domains, the prepared TFC NF membranes exhibit an excellent water permeation of 31.1 L m−2 h−1 bar−1 and a Na2SO4 rejection rate of about 95%, outperforming most NF membranes prepared by other methods. This facile and highly controlled COF-nanofiber-enabled substrate reconstruction strategy provides a new opportunity for the fabrication of ultra-permeable NF membranes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Basic Research Program of China (2015CB655301), the National Science Fund for Distinguished Young Scholars (21825803), and Jiangsu Natural Science Foundation (BK20150063) is acknowledged. We also thank the Program of Excellent Innovation Teams of Jiangsu Higher Education Institutions and the Project of Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) for support.Notes and references
- J. R. Werber, C. O. Osuji and M. Elimelech, Nat. Rev. Mater., 2016, 1, 1–15 CrossRef.
- W. J. Lau, A. F. Ismail, N. Misdan and M. A. Kassim, Desalination, 2012, 287, 190–199 CrossRef CAS.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, 1138–1148 CrossRef PubMed.
- Z. Jiang, S. Karan and A. G. Livingston, Adv. Mater., 2018, 30, 1705973 CrossRef PubMed.
- F. A. Pacheco, I. Pinnau, M. Reinhard and J. O. Leckie, J. Membr. Sci., 2010, 358, 51–59 CrossRef CAS.
- J. Zhu, J. Hou, R. Zhang, S. Yuan, J. Li, M. Tian, P. Wang, Y. Zhang, A. Volodin and B. Van der Bruggen, J. Mater. Chem. A, 2018, 6, 15701–15709 RSC.
- G. M. Geise, H. B. Park, A. C. Sagle, B. D. Freeman and J. E. McGrath, J. Membr. Sci., 2011, 369, 130–138 CrossRef CAS.
- P. S. Singh, S. V. Joshi, J. J. Trivedi, C. V. Devmurari, A. P. Rao and P. K. Ghosh, J. Membr. Sci., 2006, 278, 19–25 CrossRef CAS.
- I. L. Alsvik and M.-B. Hägg, J. Membr. Sci., 2013, 428, 225–231 CrossRef CAS.
- A. K. Ghosh and E. M. V. Hoek, J. Membr. Sci., 2009, 336, 140–148 CrossRef CAS.
- S. Karan, Z. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- Y. Zhu, W. Xie, S. Gao, F. Zhang, W. Zhang, Z. Liu and J. Jin, Small, 2016, 12, 5034–5041 CrossRef CAS PubMed.
- M.-B. Wu, Y. Lv, H.-C. Yang, L.-F. Liu, X. Zhang and Z.-K. Xu, J. Membr. Sci., 2016, 515, 238–244 CrossRef CAS.
- G. Gong, P. Wang, Z. Zhou and Y. Hu, ACS Appl. Mater. Interfaces, 2019, 11, 7349–7356 CrossRef CAS PubMed.
- S. Gao, Y. Zhu, Y. Gong, Z. Wang, W. Fang and J. Jin, ACS Nano, 2019, 13, 5278–5290 CrossRef CAS PubMed.
- Z. Zhou, Y. Ying and X. Peng, Appl. Mater. Today, 2017, 8, 54–59 CrossRef.
- J.-J. Wang, H.-C. Yang, M.-B. Wu, X. Zhang and Z.-K. Xu, J. Mater. Chem. A, 2017, 5, 16289–16295 RSC.
- X. Yang, Y. Du, X. Zhang, A. He and Z.-K. Xu, Langmuir, 2017, 33, 2318–2324 CrossRef CAS PubMed.
- X. Wu, Y. Li, X. Cui, J. Wang, X. Cao, P. Zhang and L. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 10445–10453 CrossRef CAS PubMed.
- X. Zhang, Y. Lv, H.-C. Yang, Y. Du and Z.-K. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 32512–32519 CrossRef CAS PubMed.
- Z. Yang, Z.-W. Zhou, H. Guo, Z. Yao, X.-H. Ma, X. Song, S.-P. Feng and C. Y. Tang, Environ. Sci. Technol., 2018, 52, 9341–9349 CrossRef CAS PubMed.
- Z. Wang, Z. Wang, S. Lin, H. Jin, S. Gao, Y. Zhu and J. Jin, Nat. Commun., 2018, 9, 2004 CrossRef PubMed.
- Z. Tan, S. Chen, X. Peng, L. Zhang and C. Gao, Science, 2018, 360, 518–521 CrossRef CAS PubMed.
- Z. Zhai, C. Jiang, N. Zhao, W. Dong, H. Lan, M. Wang and Q. J. Niu, J. Mater. Chem. A, 2018, 6, 21207–21215 RSC.
- Z. Sun, Q. Wu, C. Ye, W. Wang, L. Zheng, F. Dong, Z. Yi, L. Xue and C. Gao, Nano Lett., 2019, 19, 2953–2959 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 1–19 Search PubMed.
- S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS PubMed.
- W. Huang, Y. Jiang, X. Li, X. Li, J. Wang, Q. Wu and X. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 8845–8849 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- F. Zhang, J. Zhang, B. Zhang, X. Tan, D. Shao, J. Shi, D. Tan, L. Liu, J. Feng, B. Han, G. Yang, L. Zheng and J. Zhang, ChemSusChem, 2018, 11, 3576–3580 CrossRef CAS PubMed.
- H. Sun and P. Wu, J. Membr. Sci., 2018, 564, 394–403 CrossRef CAS.
- Z. Zhou, Y. Hu, C. Boo, Z. Liu, J. Li, L. Deng and X. An, Environ. Sci. Technol. Lett., 2018, 5, 243–248 CrossRef CAS.
- X.-L. Wang, T. Tsuru, S.-i. Nakao and S. Kimura, J. Membr. Sci., 1997, 135, 19–32 CrossRef CAS.
- M. Dalwani, N. E. Benes, G. Bargeman, D. Stamatialis and M. Wessling, J. Membr. Sci., 2011, 372, 228–238 CrossRef CAS.
- J. Luo and Y. Wan, J. Membr. Sci., 2013, 438, 18–28 CrossRef CAS.
- J. Wang, D. S. Dlamini, A. K. Mishra, M. T. M. Pendergast, M. C. Y. Wong, B. B. Mamba, V. Freger, A. R. D. Verliefde and E. M. V. Hoek, J. Membr. Sci., 2014, 454, 516–537 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03088c |
| This journal is © The Royal Society of Chemistry 2019 |