
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTip-enhanced Raman spectroscopy for structural analysis of two-dimensional covalent monolayers synthesized on water and on Au (111)†
Li-Qing
Zheng‡
a,
Marco
Servalli‡
b,
A. Dieter
Schlüter
b and
Renato
Zenobi
 *a
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 3, 8093 Zurich, Switzerland. E-mail: zenobi@org.chem.ethz.ch
bDepartment of Materials, Institute of Polymer Chemistry, ETH Zurich, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland
First published on 24th September 2019
Abstract
A two-dimensional (2D) covalent monolayer based on [4 + 4] cycloaddition reactions between adjacent anthracene units was synthesized at an air/water interface. For structural analysis, tip-enhanced Raman spectroscopy (TERS) provides direct evidence for the covalent bonds formed between monomer molecules. For the first time, progress of the photopolymerization reaction was monitored by irradiation (λ = 385 nm) of the monomer monolayer for different times, based on averaged TER spectra extracted from maps. In addition, a 2D polymerization on a Au (111) substrate was realized, which opens up new possibilities for such chemical transformations. This work uses TERS as a minimally invasive tool to investigate how the reaction conditions affect polymerization conversion. We show that the high sensitivity and the high spatial resolution of TERS can be used to estimate the crystallinity of 2D covalent monolayers, which is a key question in polymer synthesis.
Introduction
Two dimensional (2D) polymers are one atom- or one monomer unit-thin, long-ranging periodically structured, covalent networks.1–4 They resemble a “molecular fishing net”1,5 and have attracted attention due to their wide range of potential applications in electronics,6 catalysis,6 sensing,4,7–9 and micro/nano fabrication.10 Recently, polymer monolayers were used as rewritable ‘molecular paper’ and as negative photoresists for lithographic applications.10,11 One way12,13 of synthesizing 2D polymers is through the air/water interface approach.1,6,14 This has the benefit of providing laterally ‘infinite’ monolayer sheets, but has the disadvantage that structural analysis is challenging because X-ray diffraction (XRD) cannot easily be applied. Another way, although resulting in smaller 2D polymer patches, is through synthesis on a metal surface, which is usually done under ultrahigh vacuum conditions.15–17When it comes to monolayer analysis, one is faced with the extremely small quantities of material available (often <1 μg) and the intrinsic softness and flexibility 2D polymers can exhibit, a characteristic due to their high density of nano-pores. Therefore, many routine analytical techniques such as conventional XRD and conventional NMR spectroscopy are not applicable.1 Fortunately, there are several techniques that are sensitive enough to analyze monolayers, including scanning probe microscopy (SPM),18,19 such as high-resolution atomic force microscopy (HR-AFM) and scanning tunneling microscopy (STM), selected area electron diffraction (SAED),6 X-ray photoelectron spectroscopy (XPS),20 scanning electron microscopy (SEM)6,14,20 and tip-enhanced Raman spectroscopy (TERS).14,20–22 Among these techniques, HR-AFM, STM and SAED have provided evidence for at least local crystallinity of covalent monolayers. These methods, however, fail when it comes to investigating chemical connectivity. They do not provide information on the exact nature of the newly formed covalent bonds. While XPS may provide at least some useful information in this regard, TERS gives detailed chemical information in particular if the signals obtained are assigned with the help of calculated spectra.
Raman spectroscopy provides chemical fingerprint information even of complex molecules. However, the small Raman cross-section of non-resonant molecules usually renders this method unsuitable for studying monolayers. Spatial resolution is another bottleneck.23 Gratifyingly, TERS can overcome these shortcomings. It couples SPM with plasmon-enhanced Raman spectroscopy,24–26 and therefore combines spatial resolution with very high sensitivity.27,28 Typically, a Ag or Au tip with a radius of around 20 nm is used as a probe. The intensity of the Raman signals of molecules located right under the tip increases by ca. 106−7-fold29 due to the strongly enhanced electromagnetic field generated by surface plasmon resonances (SPR).
TERS is about to become an important method for structural analysis of 2D polymers: Opilik et al. reported TERS to be a minimally-invasive tool for the analysis of 2D polymer monolayers.21 TERS was further utilized to reveal nano-defects within a 2D polymer monolayer synthesized at an air/water interface.22 The stacking configuration of interfacial monomer molecules due to π–π interactions was demonstrated by TERS imaging combined with density functional theory (DFT) calculations.30 Moreover, TERS provided direct spectroscopic evidence for the kind of crosslinks formed in a covalently bonded monolayer sheet and allowed estimating the conversion of the 2D polymerization.20 Regarding the synthesis of 2D polymers, the key questions are at what point the polymerization reaction is complete, and whether there are defects. Among the very few tools available for the characterization of covalent monolayers, TERS is capable of providing detailed chemical information on molecular monolayers with nanometer spatial resolution. Here, we show for the first time that TERS offers the opportunity to monitor the polymerization progress at an air/water interface. This is important for studying the kinetics and determining the polymerization reaction endpoint. We also show that TERS is not limited to studying 2D polymers after transfer from the air/water interface, but also allows to compare one and the same reaction performed on a metal surface. To the best of our knowledge, on-metal surface photopolymerization under ambient conditions has never been reported.
We here use the macrocyclic anthraphane monomer 1 (Scheme 1A).10 Its shape-persistency combined with its amphiphilicity is likely to result in a tight face-to-face (ftf) stacked packing at the air/water interface. This packing may form spontaneously or when an appropriate surface pressure is applied. We report on the air/water interfacial behavior of monomer 1, and the structural analysis of the covalent sheet obtained via anthracene dimerization reaction when irradiating the spread monolayer at λ = 385 nm (Scheme 1B). For this, different analytical techniques were employed, including in situ Brewster angle microscopy (BAM), SEM (after transfer onto holey copper grids), AFM (after transfer onto SiO2-coated Si wafers) and finally TERS (after transfer onto Au substrates). TERS mapping (with Ag-tip) was also used to monitor and quantify the conversion of the polymerization at different irradiation time. Finally, the monolayer of monomer 1 was transferred onto a Au (111) surface prior to polymerization, followed by irradiation at 385 nm. The conversion number of the polymerization on this metal surface was compared to that of the polymerization at the air/water interface.
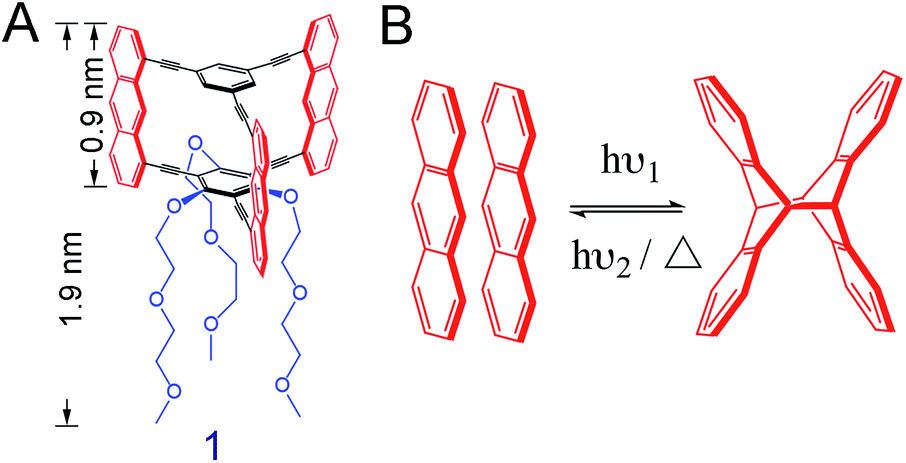 | ||
| Scheme 1 (A) Chemical structure of amphiphilic anthraphane monomer 1. The monomer has a rigid hydrocarbon cyclophane scaffold bearing three anthracene photoreactive units (red). The three diethylene glycol methyl ether (DEGME) groups (blue) render the monomer amphiphilic. (B) Reversible photochemical reaction between two anthracenes. The dimerization reaction is triggered by light irradiation and is in principle reversible. | ||
Results and discussion
Monomer structure, molecular packing model, and in situ fluorescence measurement at the air/water interface
To study the interfacial behavior, a solution of monomer 1 was spread at the air/water interface in a Langmuir–Blodgett trough, where a surface pressure (SP) vs. mean molecular area (MMA) isotherm was recorded at room temperature (Fig. S1†). In order to achieve a 2D polymer, the monomer should pack at the interface so that every anthracene unit is ftf-stacked with the anthracene units of neighboring monomers, achieving a hexagonal honeycomb-like packing. According to DFT simulations, such packing should have a MMA value of 252 Å2.10 From the isotherm, this value is reached in SP regimes ranging from 2–7 mN m−1, and we therefore aimed at performing the polymerizations and monolayer transfer around these values (see ESI†). The monolayer was then studied by in situ BAM, whereby good homogeneity was confirmed in the desired SP range (Fig. S2†). The thickness of the layer was determined by tapping mode AFM after transferring the film onto a SiO2/Si wafer by the Schäfer technique.31hAFM ≈ 1.17 nm was estimated (Fig. S3†), suggesting that the DEGME tails of monomer 1 are either stretched out on the surface or folded back towards the anthracene units, not contributing to the overall thickness of the monolayer. We then performed in situ fluorescence spectroscopy, detecting exclusively a pure anthracene excimeric emission, characterized by a broad, unstructured peak centered at around 510 nm (Fig. S4†). The absence of the typical vibronic signature of isolated anthracenes32 indicated that all the anthracene units of the monolayer were stacked ftf as in the target molecular packing and ready to be converted into cross-links upon photoirradiation. We then performed an in situ fluorescence decay experiment by irradiating the monolayer at 365 nm. As shown in Fig. S5,† the fluorescence signal decreases dramatically with irradiation time, indicating excimer consumption by photodimerization. After irradiation for 2 h, the degree of polymerization is estimated to be 93% (Fig. S6†). To have another qualitative insight into network formation, the polymerized monolayer after a 2 h irradiation was transferred onto a TEM copper grid and imaged by SEM. The irradiated film was shown to span the 20 × 20 μm2 holes of the grid in high yields (Fig. S7†). This mechanical integrity suggested that a large fraction of monomer molecules was covalently connected within the network. As control experiment, a non-irradiated monolayer was shown to not span any hole of the grid. According to bond percolation theory, the displayed mechanical integrity also suggested a high polymerization degree.20,33The irradiated monolayer was also transferred onto a SiO2/Si wafer, where it could be easily detected by color contrast. A differential interference microscopy image (Fig. S8†) showed high surface coverage and a large, homogeneous sheet with a size of several hundreds of micrometers. So far, all the analytical results hinted at the formation of a covalent monolayer. However, a quantitative analytical method that provides chemical information, such as vibrational spectroscopy, was required to confirm the nature of the 2D polymerization reaction.
DFT calculations of monomer and polymer model compounds
To figure out the expected changes in the vibrational spectrum upon polymerization, calculations at the B3LYP/6-31G* level were performed21 on monomer and polymer model compounds. As model compound for monomer 1, a hydrocarbon anthraphane was chosen,34 without DEGME chains (Fig. 1A, structure a). To mimic the polymer, one, two or all three of the anthracene-units of the model monomer were dimerized with an anthracene molecule (Fig. 1A, structures b–d, respectively). The strong Raman band at 2215 cm−1 corresponding to the triple bond stretching vibrations is observed for all of the model compounds and was therefore used for normalization purposes. For better visibility of the spectral range below 1800 cm−1, it is however omitted. All Raman peaks that were used for differentiation between reacted and unreacted anthracene units are indicated with dashed lines. As shown in Fig. 1B, for the monomer model compound (black curve a) the strong Raman bands at 1382 cm−1 and 1563 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching modes of anthracene) and the band at 1447 cm−1 corresponding to anthracene vibrations,21 are absent from the Raman spectrum of the fully polymerized model (purple curve d; see Movie S1†). Moreover, the intensity of the band at 1364 cm−1 (predominantly C–C vibrations with a minor contribution from anthracene vibrations) slightly decreases after polymerization. The peak at 1224 cm−1 (C–H rocking mode of anthracene) shifts to 1190 cm−1 upon anthracene dimerization and the intensity of the peak at 1580 cm−1 (the benzene ring vibration) increases. In all the spectra of gradually polymerized model compounds, we can see a new peak appearing at 1604 cm−1 (CC stretching vibrations of polymerized anthracene ring; Movie S2†). Note that the peak at 982 cm−1 (marked with a light blue bar) appearing in both monomer and polymer model compounds is a ring breathing mode of the cyclophane's benzene units. After polymerization, the intensity of this peak remains the same since the benzene rings in the center of the structures remain unaffected. Therefore, this peak was chosen as reference for the experimental spectra presented later.
C stretching modes of anthracene) and the band at 1447 cm−1 corresponding to anthracene vibrations,21 are absent from the Raman spectrum of the fully polymerized model (purple curve d; see Movie S1†). Moreover, the intensity of the band at 1364 cm−1 (predominantly C–C vibrations with a minor contribution from anthracene vibrations) slightly decreases after polymerization. The peak at 1224 cm−1 (C–H rocking mode of anthracene) shifts to 1190 cm−1 upon anthracene dimerization and the intensity of the peak at 1580 cm−1 (the benzene ring vibration) increases. In all the spectra of gradually polymerized model compounds, we can see a new peak appearing at 1604 cm−1 (CC stretching vibrations of polymerized anthracene ring; Movie S2†). Note that the peak at 982 cm−1 (marked with a light blue bar) appearing in both monomer and polymer model compounds is a ring breathing mode of the cyclophane's benzene units. After polymerization, the intensity of this peak remains the same since the benzene rings in the center of the structures remain unaffected. Therefore, this peak was chosen as reference for the experimental spectra presented later.
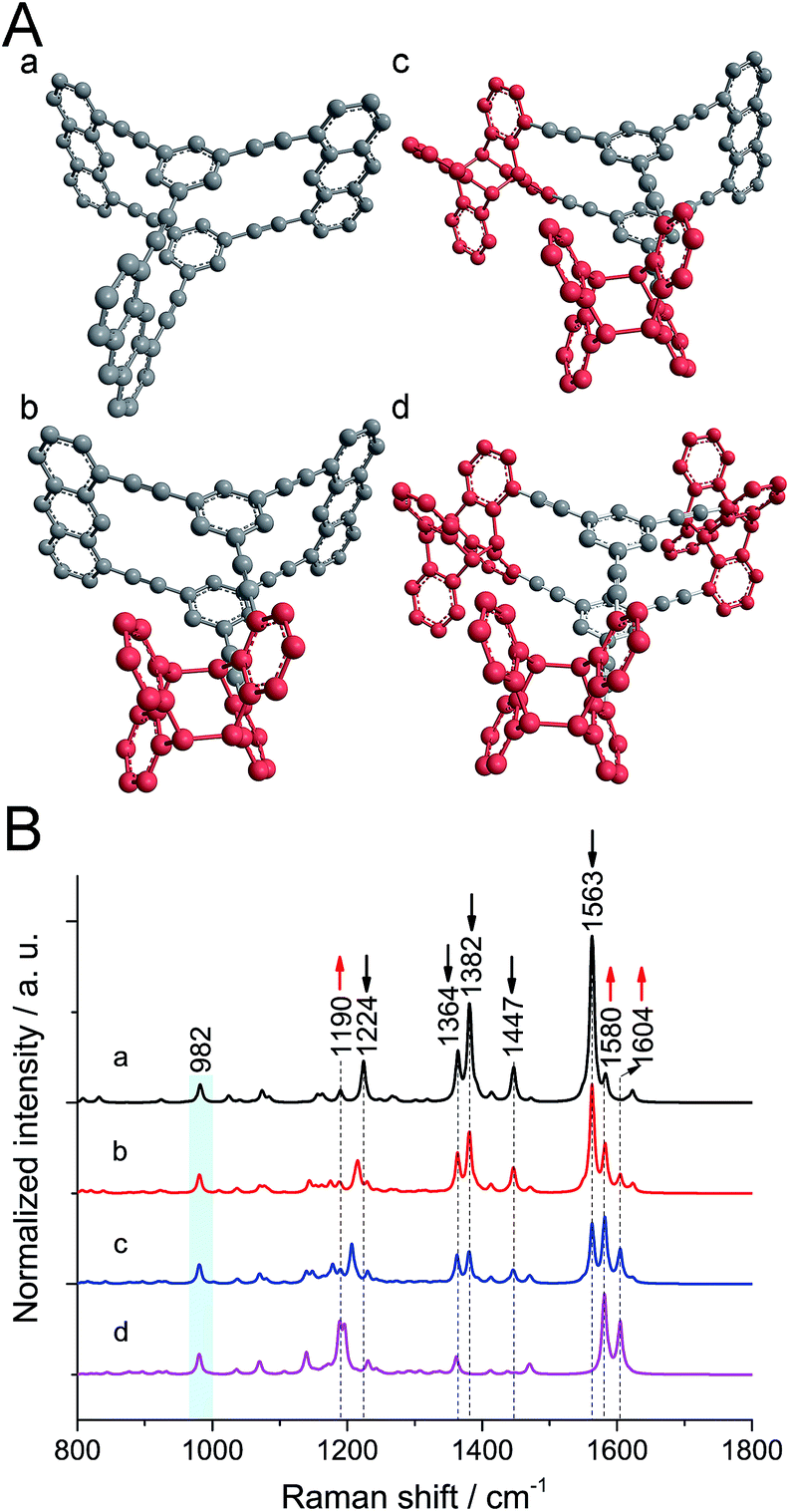 | ||
| Fig. 1 (A) Model compounds a–d used in (B). DEGME groups are neglected and polymer model compounds (b, c, d) have one to three of their anthracene moieties in dimeric form (dimerized moieties shown in red). (B) Simulated Raman spectra of monomer and polymer model compounds shown in (A). All Raman spectra were normalized to the triple bond's Raman peak at 2215 cm−1 (not shown in the spectra). | ||
Optimization of confocal Raman and TERS measurements
After completion of the calculations, confocal Raman measurements on monomer powders were conducted to check the accuracy of the calculations. The measurements required optimization due to the sensitivity of the monomer to the laser irradiation (see ESI†). The confocal Raman spectrum (Fig. S9†) was found to be in good agreement with the theoretical spectrum (curve a in Fig. 1B). Next, the TER spectrum of a monomer monolayer was recorded under optimized conditions (Fig. S10†) after transfer onto a Au substrate. It was found to be consistent with the confocal Raman. In particular, the signals typical for anthracene at 1385, 1447 and at 1560 cm−1 are present in both spectra. Noticeably, a new peak appeared at around 1620 cm−1 (Fig. S11†), which is ascribed to sample degradation driven by plasmon-induced chemistry.35 Given the sensitivity of monomer 1 during Raman measurements, TERS with its greatly enhanced sensitivity is preferred over confocal Raman spectroscopy, as it requires much less laser power and shorter acquisition times.Structural analysis of polymer monolayers using TERS
We then turned our attention to the polymerization at the air/water interface: monolayers were transferred onto Au substrates at different irradiation times to monitor the progress of the reaction. 1 × 1 μm2 TERS maps with a 100 nm pixel size were collected on non-irradiated and irradiated monomer monolayers to obtain local information as well as statistics. The samples were found to degrade when a TERS map with a 30 nm pixel size was recorded (data not shown), due to the fact that the monomer and polymer molecules are prone to photo damage (Fig. S9†) and degradation by plasmon-induced processes (Fig. S10†). This indicates that the samples are not compatible with measurements using a very high spatial resolution. This is the reason why we used a pixel size of 100 nm to analyze the samples.From the TERS map of the monomer monolayer, we determined standard deviation of the 1385 cm−1 peak position of only ±0.3 cm−1, whereas the value for the peak at 995 cm−1 is ±1.5 cm−1. The 1385 cm−1 peak has a full width at half maximum (FWHM) of 15 ± 3 cm−1. The intensity ratio between the 1385 cm−1 and 995 cm−1 peaks is 18, with a 10% variation (Fig. S12†). After 1.5 h of irradiation, the standard deviation of the peak position at 1385 cm−1 increases to ±1.5 cm−1 and the FWHM to 28 ± 11 cm−1, the position of the peak at 995 cm−1 varies by ±2 cm−1. The intensity ratio decreases to 5, with a 9% variation (Fig. S13†). After UV irradiation for 1.5 h, the peak intensity at 1385 cm−1 of the monomer monolayer drops to 73% (Fig. S14†), which is unambiguous evidence for the polymerization reaction. Furthermore, the FWHM of the 1385 cm−1 peak in the TERS maps of polymers with different irradiation times was evaluated and is shown in Table S1.† The variation of the peak width is large, which could be due to the different enhancement of the TERS tip at different locations. To minimize this effect, all hundred TER spectra within a map were accumulated and then averaged to obtain statistical information. The peak of the cyclophane's benzene at 995 cm−1 was used to normalize all the averaged TER spectra (Fig. 2). After the monomer monolayer sheet was irradiated at 385 nm, the intensity of the two anthracene peaks at 1385 cm−1 and 1450 cm−1 significantly decreased. The signal at around 1600 cm−1 broadened due to spectral overlap of multiple peaks. We fitted Gaussian profiles to this region of the spectra (Fig. S15 and S16†). This showed that the intensity of the peak at 1600 cm−1 increased, while that of the peak at 1560 cm−1 decreased (Table S2†). Noticeably, the peaks at 1580 cm−1 and 1600 cm−1 merged into a single peak in the TER spectra of polymers, inevitably leading to peak broadening. The small peak at 1660 cm−1 (ν(CO)) in the TER spectra of polymers after 2 h, 5 h and 6.5 h irradiation is due to anthracene photooxidation via a [4 ± 2] cycloaddition reaction. This side reaction can be induced by prolonged UV irradiation at the interface at ambient conditions or by plasmon-driven photocatalysis. However, photooxidation is not competitive with photodimerization, due to the high concentration of the stacked anthracene at the interface.36 Since the peak at 1168 cm−1 is an indicator for the dimerization of anthracene, we further analyzed this region by applying a Gaussian fit of this peak on the averaged TER spectra of the monomer and polymer monolayers irradiated for different times (Fig. S17 and S18†): the peak intensity and peak area significantly increased in the TER spectra of polymer monolayers (Table S3†). Despite a few less than perfect fits, all these spectral changes clearly indicate the formation of a polymerized product. Notably, the intensity of the peaks at 1168 cm−1 and 1600 cm−1 gradually increases when we prolong the irradiation time from 1.5 h to 5 h, however, the intensities slightly decrease when the irradiation time is 6.5 h. We interpret this as a sign of sample degradation when the irradiation time exceeds 5 h. The fluctuations of the peak at 2215 cm−1 (Table S4†) are due to the fact that the intensity of this Raman band is strongly dependent on the molecular orientation. Even a small change in either the orientation of the molecular plane or the angle of the symmetry axis with respect to the surface can considerably influence the peak intensity.22 After transfer onto a gold substrate, the adsorption orientation of molecules is influenced by the topography of the underlying Au surface, which is different from that at the air/water interface. Moreover, the DEGME groups within monomer 1 render the adsorption orientation of molecules difficult to predict. Finally, such sheets are very unlikely to be in tight contact with the substrate. It is possible that wrinkles and/or other defects at some spots are formed upon transfer. To minimize the damage of monolayers, the transfer process was conducted at a very low rate and the substrate was left to dry overnight (see ESI†).
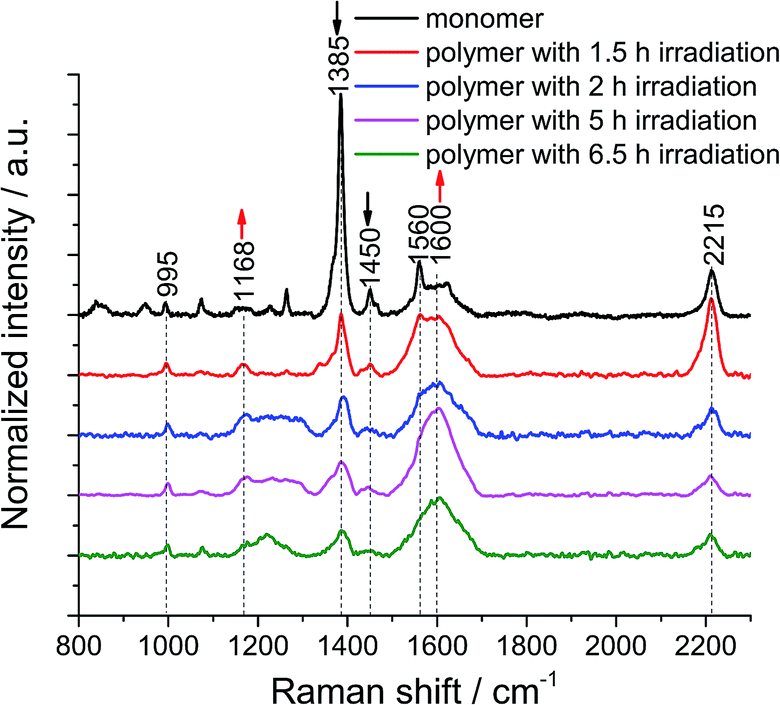 | ||
| Fig. 2 Baseline corrected average TER spectrum from a map of a monomer monolayer (black curve) and average TER spectra from TERS maps of polymer monolayers with different UV irradiation times. The size of all maps is 1 × 1 μm2 with a 100 nm pixel size. All average TER spectra were normalized to the Raman peak of the unaffected cyclophane's central benzenes at 995 cm−1. | ||
Next, the decrease of the anthracene peak at 1385 cm−1 with respect to the one at 995 cm−1 was used to estimate conversion numbers of the polymerization (Table 1). Because the peaks at 1364 cm−1 and at 1385 cm−1 overlap, peak intensities rather than peak areas were used (for details including a discussion on error bars, see ESI†). As the data in Table 1 show, conversion increases with irradiation time, and high conversion values can be obtained, which is reflected in the mechanical coherence observed by SEM. To verify the reproducibility of the conversion calculated based on one TERS map, we measured three TERS maps at different places on a monomer sample and a polymer sample after 2 h irradiation, respectively (Fig. S19†). The conversion number is calculated to be 80 ± 3% (Table S5†), which is in agreement with the value shown in Table 1. The peak at 1168 cm−1 in these TER spectra was also quantified. The peak intensity and area are confirmed to increase in the spectra of the polymer (Table S6†).
| Sample ML1 | Peak intensity ratio [1385/995 cm−1] | Conversion [%] |
|---|---|---|
| Not irradiated | 18.2 ± 1.8 | — |
| Irrad. for 1.5 h | 5.0 ± 0.5 | 73 ± 13 |
| Irrad. for 2 h | 3.3 ± 0.4 | 82 ± 13 |
| Irrad. for 5 h | 2.8 ± 0.4 | 85 ± 13 |
| Irrad. for 6.5 h | 1.5 ± 0.2 | 92 ± 14 |
Photopolymerization on Au (111) surface
Finally, the polymerization at the air/water interface was compared with the ‘same’ polymerization on a solid substrate. For this purpose, a monomer monolayer was transferred onto Au (111) and then irradiated for 2.5 h. To minimize the influence of surface roughness on the orientation of monomer molecules, a flat Au (111) substrate with a roughness of 0.39 nm was used (Fig. S20†). TERS maps before and after UV irradiation were collected using the same tip for both measurements, and are shown as waterfall plots to see the spectral changes (see Fig. 3A and B). The average peak intensity ratio in the TERS maps of monomer before and after irradiation was 14 with a 10% variation and 5.4 with a 12% variation, respectively (Fig. S21†). Occasionally, signals were weak or even absent, as indicated by horizontal blue lines. We tentatively assign these occurrences to transient loss of enhancement by the Ag tip. The averaged TER spectra from both maps show that the intensity of the characteristic Raman bands of anthracene at 1385 cm−1, 1450 cm−1 and 1560 cm−1 decreases, while the intensity of the peaks at 1600 cm−1 and 1170 cm−1 increases (Fig. 3C and S22–S24†). Although a few fits are imperfect due to low signal-to-noise ratio of the spectra, the spectral changes clearly show that the polymerization of monomer molecules induced by UV irradiation also happens on Au. Based on the same calculation method, a conversion of 62.4% ± 11% was estimated (Table S7†). In this case photooxidation was also not observed. Since monolayers of 1 were shown to polymerize with high conversion on silicon oxide substrates,10 we assume that the lower conversion observed here is specifically due to the use of gold as substrate. A tentative explanation could be that specific monomer–substrate interactions inhibit the polymerization or that the absorption of UV light from the monomer monolayer on gold is not as efficient as on water. | ||
| Fig. 3 Waterfall plot of all TER spectra from maps of a monomer monolayer on Au (111) before (A) and after UV irradiation for 2.5 h (B). The size of all maps is 1 × 1 μm2 with a 100 nm pixel size. (C) Baseline corrected average TER spectra from the above two TERS maps. These two TER spectra were normalized to the Raman peak at 995 cm−1. | ||
Conclusions
In summary, TERS and DFT calculations were used to analyze the structure of a photochemically polymerized monolayer film created from monomer 1. Upon UV-irradiation the TER spectra show a decrease by approx. 90% (within 6.5 h) of the characteristic vibrations of anthracene and the corresponding appearance of a new Raman peak at 1167 cm−1, which could unambiguously be assigned to the vibration of dimerized anthracene rings and thus indicates polymerization. For the first time, such a 2D polymerization process was monitored by TERS mapping. The cross-links formed during polymerization are clearly due to anthracene dimers, the extended 2D sheet produced is homogeneous with ≤10% spot-to-spot variation, and the final conversion is high and also verified by in situ fluorescence spectroscopy. This suggests a high degree of crystallinity in the final product, indicating that the product qualifies as a 2D polymer.Interestingly, monolayers of monomer 1 can also be polymerized on surfaces other than water: when transferred onto Au (111), photopolymerization can also be induced. This is the first case, in which a two-dimensional polymerization was brought about on a metal substrate under ambient conditions. This opens up new perspectives and widens the palette of conditions available for such polymerizations.
Notes
The original data used in this publication are made available in a curated data archive at ETH Zurich (https://www.research-collection.ethz.ch) under the DOI: 10.3929/ethz-b-000353523.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Feng Shao (ETH Zurich) for insightful discussions, and Dr Payam Payamyar and Dr Kemal Celebi for AFM and SEM measurements. L.-Q. Z. is indebted to the Chinese Scholarship Council for a Ph.D. student fellowship (scholarship # 201506190126). We thank the High Performance Computing Team at ETH Zurich for help with DFT calculations and the ERC program (grant # 741431 – 2DNanoSpec) for financial support.References
- X. Feng and A. D. Schlüter, Angew. Chem., Int. Ed., 2018, 57, 13748–13763 CrossRef CAS PubMed.
- J. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS PubMed.
- P. Payamyar, B. T. King, H. C. Öttinger and A. D. Schlüter, Chem. Commun., 2016, 52, 18–34 RSC.
- J. Sakamoto, J. van Heijst, O. Lukin and A. D. Schlüter, Angew. Chem., Int. Ed., 2009, 48, 1030–1069 CrossRef CAS PubMed.
- M. Servalli, H. C. Öttinger and A. D. Schlüter, Phys. Today, 2018, 71, 40–47 CrossRef CAS.
- H. Sahabudeen, H. Qi, B. A. Glatz, D. Tranca, R. Dong, Y. Hou, T. Zhang, C. Kuttner, T. Lehnert, G. Seifert, U. Kaiser, A. Fery, Z. Zheng and X. Feng, Nat. Commun., 2016, 7, 13461 CrossRef PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- A. C. Ferrari, et al. , Nanoscale, 2015, 7, 4598–4810 RSC.
- X. Zhuang, Y. Mai, D. Wu, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 403–427 CrossRef CAS PubMed.
- M. Servalli, K. Celebi, P. Payamyar, L.-Q. Zheng, M. Polozij, B. Lowe, A. Kuc, T. Schwarz, K. Thorwarth, A. Borgschulte, T. Heine, R. Zenobi and A. D. Schlüter, ACS Nano, 2018, 12, 11294–11306 CrossRef CAS PubMed.
- V. Müller, T. Hungerland, M. Baljozovic, T. Jung, N. D. Spencer, H. Eghlidi, P. Payamyar and A. D. Schlüter, Adv. Mater., 2017, 29, 1701220 CrossRef PubMed.
- C. Backes, T. M. Higgins, A. Kelly, C. Boland, A. Harvey, D. Hanlon and J. N. Coleman, Chem. Mater., 2017, 29, 243–255 CrossRef CAS.
- R. Dong, T. Zhang and X. Feng, Chem. Rev., 2018, 118, 6189–6235 CrossRef CAS PubMed.
- W. Dai, F. Shao, J. Szczerbínski, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 55, 213–217 ( Angew. Chem. , 2016 , 128 , 221–225 ) CrossRef CAS PubMed.
- S. Clair, M. Abel and L. Porte, Chem. Commun., 2014, 50, 9627–9635 RSC.
- J. M. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. L. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Nat. Chem., 2012, 4, 215–220 CrossRef CAS PubMed.
- D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. T. Song, M. Lackinger, A. D. Schlüter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450–3453 CrossRef CAS PubMed.
- V. Müller, A. Hinaut, M. Moradi, M. Baljozovic, T. A. Jung, P. Shahgaldian, H. Möhwald, G. Hofer, M. Kröger, B. T. King, E. Meyer, T. Glatzel and A. D. Schlüter, Angew. Chem., Int. Ed., 2018, 57, 10584–10588 ( Angew. Chem. , 2018 , 130 , 10744–10748 ) CrossRef PubMed.
- V. Müller, F. Shao, M. Baljozovic, M. Moradi, Y. Zhang, T. Jung, W. B. Thompson, B. T. King, R. Zenobi and A. D. Schlüter, Angew. Chem., Int. Ed., 2017, 56, 15262–15266 ( Angew. Chem. , 2017 , 129 , 15464–15468 ) CrossRef PubMed.
- L. Opilik, P. Payamyar, J. Szczerbínski, A. P. Schütz, M. Servalli, T. Hungerland, A. D. Schlüter and R. Zenobi, ACS Nano, 2015, 9, 4252–4259 CrossRef CAS PubMed.
- F. Shao, W. Dai, W. Zhang, A. D. Schlüter and R. Zenobi, ACS Nano, 2018, 12, 5021–5029 CrossRef CAS PubMed.
- X. Wang, S.-C. Huang, T.-X. Huang, H.-S. Su, J.-H. Zhong, Z.-C. Zeng, M.-H. Li and B. Ren, Chem. Soc. Rev., 2017, 46, 4020–4041 RSC.
- S.-Y. Ding, J. Yi, J.-F. Li, B. Ren, D.-Y. Wu, R. Panneerselvam and Z.-Q. Tian, Nat. Rev. Mater., 2016, 1, 16021 CrossRef CAS.
- M. Richard-Lacroix, Y. Zhang, Z. Dong and V. Deckert, Chem. Soc. Rev., 2017, 46, 3922–3944 RSC.
- E. A. Pozzi, G. Goubert, N. Chiang, N. Jiang, C. T. Chapman, M. O. McAnally, A.-I. Henry, T. Seideman, G. C. Schatz, M. C. Hersam and R. P. Van Duyne, Chem. Rev., 2017, 117, 4961–4982 CrossRef CAS PubMed.
- J.-H. Zhong, X. Jin, L. Meng, X. Wang, H.-S. Su, Z.-L. Yang, C. T. Williams and B. Ren, Nat. Nanotechnol., 2017, 12, 132–136 CrossRef CAS PubMed.
- L.-Q. Zheng, X. Wang, F. Shao, M. Hegner and R. Zenobi, Angew. Chem., Int. Ed., 2018, 130, 1037–1041 ( Angew. Chem. , 2018 , 130 , 1037–1041 ) CrossRef.
- B. Pettinger, P. Schambach, C. J. Villagómez and N. Scott, Annu. Rev. Phys. Chem., 2012, 63, 379–399 CrossRef CAS PubMed.
- F. Shao, V. Müller, Y. Zhang, A. D. Schlüter and R. Zenobi, Angew. Chem., Int. Ed., 2017, 56, 9361–9366 CrossRef CAS PubMed.
- G. G. Roberts, Formats and Editions and Langmuir Blodgett Films, Plenum Press, New York, 1990 Search PubMed.
- T. Hayashi, N. Mataga, Y. Sakata, S. Misumi, M. Morita and J. Tanaka, J. Am. Chem. Soc., 1976, 98, 5910–5913 CrossRef CAS.
- S. Feng and P. N. Sen, Phys. Rev. Lett., 1984, 52, 216–219 CrossRef.
- M. Servalli, N. Trapp, M. Wörle and F.-G. Klärner, J. Org. Chem., 2016, 81, 2572–2580 CrossRef CAS PubMed.
- J. Szczerbiński, L. Gyr, J. Kaeslin and R. Zenobi, Nano Lett., 2018, 18, 6740–6749 CrossRef PubMed.
- P. Payamyar, L. Kaja, C. Ruiz-Vargas and A. D. Schlüter, et al. , Adv. Mater., 2014, 26, 2052–2058 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03296g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |