
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating the surface defects of titanium oxides and consequent reactivity of Pt catalysts†
Yanan
Wang‡
a,
Sihang
Liu‡
a,
Chunlei
Pei
a,
Qiang
Fu
 b,
Zhi-Jian
Zhao
a,
Rentao
Mu
*a and
Jinlong
Gong
*a
b,
Zhi-Jian
Zhao
a,
Rentao
Mu
*a and
Jinlong
Gong
*a
aKey Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering and Technology, Tianjin University; Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China. E-mail: jlgong@tju.edu.cn; murt@tju.edu.cn
bState Key Laboratory of Catalysis, iChEM, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 27th September 2019
Abstract
In heterogeneous catalysis, it is widely believed that the surface states of catalyst supports can strongly influence the catalytic performance, because active components are generally anchored on supports. This paper describes a detailed understanding of the influence of surface defects of TiO2 supports on the catalytic properties of Pt catalysts. Pt was deposited on reduced (r-), hydroxylated (h-), and oxidized (o-) TiO2 surfaces, respectively, and the different surface states of TiO2 not only lead to differences in metal dispersion, but also distinct electronic interactions between the metal and the support. The highest reactivity for catalytic CO oxidation can be achieved over the Pt catalyst supported on reduced TiO2 with surface oxygen vacancies. The turnover frequency (TOF) of this catalyst is determined to be ∼11 times higher than that of Pt supported on oxidized TiO2. More importantly, the reactivity is seen to increase in the sequence of Pt/o-TiO2 < Pt/h-TiO2 < Pt/r-TiO2, which is well consistent with the trend of the calculated Bader charge of Pt.
Introduction
It has long been recognized that supports play an important role in heterogeneous catalysis, because supports can promote the catalytic performance and reduce the usage of noble metal catalysts.1–5 Understanding the interfacial interaction between metals and supports, studied here, is of critical importance as metal–support interactions help the dispersion of active components and sintering resistance. Additionally, a support has also been proposed as a promoter to cause changes in the electronic structure of active components and thereby alter their catalytic properties. As such, a detailed understanding of metal interactions with supports is of high importance. Previously, extensive studies showed that metal nanoparticles and single atom catalysts exhibit variable catalytic reactivity when they are deposited on different supports, such as SiO2, Al2O3, CeO2, TiO2, FeOx and many others.6–12 Surprisingly, the influence of different surface states of oxide supports on the structure and catalytic performance of metal catalysts has only been studied using model surface science systems while little is known about this factor in real supported catalysts.13–16Recently, several related studies on this issue have been published. Jia and Si et al. discovered that the surface states of iron oxide supports, hydroxylated or dehydrated, play an important role in the performance of Au/FeOx in CO oxidation.17 Similarly, Wang et al. compared the performance of Au single atoms on perfect and defective TiO2 nanosheet supports, in which the defective one shows a better performance in CO oxidation for a lower energy barrier and weaker competitive adsorption.18 Furthermore, the study of Christopher et al. provides a detailed understanding of the structure–performance relationship of atomically dispersed catalysts.19,20 They show that not only the intrinsic properties of metal atoms, but also the local coordination environment plays a pivotal role in the performance of catalysts.
Surface defects are ubiquitous in many materials and can affect their physical and chemical properties significantly. For example, Xie et al. showed that the Zn vacancies of ZnIn2S4 mediate the electron–hole separation efficiency and boost the reactivity of CO2 reduction.21 Surface science studies have shown that the surface defects of supports will affect the binding strength of metal atoms on them. For example, Thornton et al. recently unraveled the binding sites of Au atoms on a reduced TiO2 (110) substrate, and found that bridging oxygen vacancies are the preferential anchoring sites for Au atoms.22 Nevertheless, Buratto et al. discovered that the Au atoms on oxygen vacancies can be easily replaced by water molecules.23 Besenbacher et al. demonstrated that oxygen adatoms bind metal atoms stronger than oxygen vacancies.13–15
As one of the most frequently used supports in heterogeneous catalysis, reducible oxides show a diversity of point defects at surfaces, such as oxygen vacancies, hydroxyls, oxygen adatoms, etc. It has been demonstrated that these defects are fragile, environmentally sensitive and facile to transform from one to another. For example, the oxygen vacancies at the surface of many oxides can interact with water molecules and easily break them into two hydroxyl groups.24–28 Upon annealing treatment in a vacuum or a reductive atmosphere, hydroxyls will recombine into water again, forming a reduced surface with oxygen vacancies.29 Consequently, direct studies of the influence of a specific kind of surface defect on the catalytic reactivity remain a great challenge, in particular for catalysts supported on oxide nanoparticles.
This paper focuses on the influence of different surface defects of TiO2 supports on the catalytic properties of Pt catalysts, because Pt/TiO2 has been extensively used in heterogeneous catalysis, photocatalytic water splitting, and electrochemistry.30–34 By finely tuning the surface states of TiO2, we prepare Pt catalysts deposited on reduced (r-), hydroxylated (h-) and oxidized (o-) surfaces, respectively. It is shown that the surface states of TiO2 not only influence the degree of metal dispersion, but also change the electronic metal–support interactions which affect the catalytic properties of Pt directly.
Results and discussion
Modulating the surface states of TiO2 supports
TiO2 supports with different surface states were prepared in a specific atmosphere. The detailed preparation process is given in Fig. S1.† First, commercial rutile TiO2 was calcined in Ar and O2 to remove contaminants, such as carbonate and moisture. To prepare a reduced surface, TiO2 was treated in an Ar atmosphere at 700 °C for 1 h.35 Upon the exposure of r-TiO2 to water vapor at 130 °C for 1 h, a hydroxylated surface can be obtained. At this temperature, the molecular water should be desorbed.36 On the other hand, oxidized TiO2 was prepared by exposing r-TiO2 to O2 at room temperature. X-ray diffraction (XRD) results (Fig. 1A and Table S1†) show the same peak position and peak shape over the different TiO2 supports, indicating that their bulk structures are the same. Additionally, the Eg peak and full width at half maximum (FWHM) in visible Raman spectra (Fig. 1B and Table S2†) do not change after the hydroxylation and oxidation of reduced TiO2, which also indicates that the bulk states of the different TiO2 samples should be the same.37,38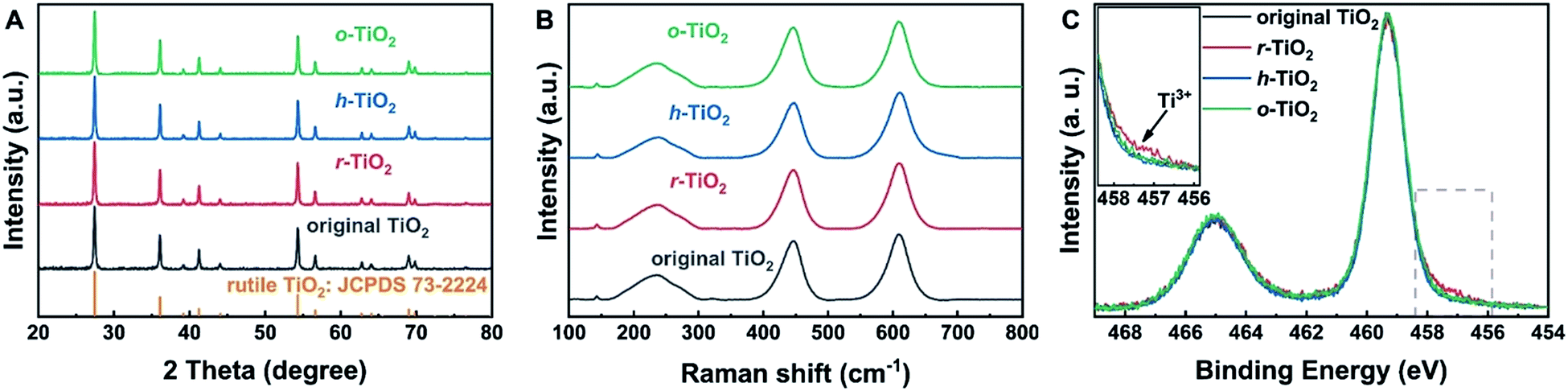 | ||
| Fig. 1 Bulk and surface properties of TiO2 supports. (A) XRD patterns of different TiO2 supports; (B) Raman spectra of different TiO2 supports; (C) XPS Ti 2p peaks of different TiO2 supports. The inset in (C) shows the Ti3+ species on reduced TiO2. | ||
To understand the surface states of TiO2, X-ray photoelectron spectroscopy (XPS) was carried out (Fig. 1C). The binding energy (BE) of XPS Ti 2p3/2 on hydroxylated and oxidized surfaces is 459.3 eV, which can be assigned to Ti4+.39,40 Over reduced TiO2, a small shoulder peak at a lower BE of 457.5 eV is observed, which indicates the presence of the Ti3+ state on the reduced surface.39,40 When r-TiO2 was exposed to water vapor, a small shoulder peak at a higher BE of the O 1s peak appears (Fig. S2A†), suggesting the formation of surface hydroxyl groups via H2O dissociation at oxygen vacancies.41 Upon the exposure of r-TiO2 to O2, the oxygen vacancies should be filled and oxygen adatoms will be generated on the top of regular Ti5c sites (Fig. S2B†). The dissociation mechanism of O2 on TiO2 was clarified in previous investigations.42–44 The disappearance of the XPS Ti3+ peak on h-TiO2 and o-TiO2 further indicates that the dissociative adsorption of water and O2 can occur at oxygen vacancies.
Preparation of Pt/TiO2 catalysts
To prepare Pt catalysts on different supports, the pre-treated TiO2 was transferred into a glove box filled with Ar, and thus the surface states of pre-treated TiO2 will remain unchanged. In order to exclude the influence of O2, water and other contaminants in air during the preparation process, Pt/TiO2 catalysts were also synthesized in a glove box under the protection of an Ar atmosphere.The influence of surface states of TiO2 on the dispersion of Pt catalysts was determined by chemisorption, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and XPS measurements. The CO chemisorption and H2–O2 titration results suggest that the dispersion of Pt catalysts is highest on r-TiO2, followed by Pt/h-TiO2 and Pt/o-TiO2 (Tables 1 and S3†). From HAADF-STEM studies (Fig. 2), it was found that the Pt nanoparticles on r-TiO2 present uniform size distribution with an average diameter of 1.19 nm. In contrast, larger Pt nanoparticles are observed on h-TiO2 (1.56 nm) and o-TiO2 (1.61 nm). For the size distribution of catalysts after reaction (1.46 ± 0.36 nm, 1.46 ± 0.39 nm and 1.61 ± 0.43 nm for Pt/r-TiO2, Pt/h-TiO2 and Pt/o-TiO2, respectively), only a small variation of particle size can be observed. From XPS investigations (Table S4†), it was found that the Pt/r-TiO2 catalyst shows a stronger normalized peak area of Pt 4f/Ti 2p than Pt/h-TiO2 and Pt/o-TiO2, indicating that the Pt nanoparticles are more highly dispersed on r-TiO2. The results from H2–O2 titration, CO chemisorption, HAADF-STEM and XPS are well consistent with each other, which illustrates that the dispersion of Pt catalysts increases in the sequence of Pt/o-TiO2 < Pt/h-TiO2 < Pt/r-TiO2.
| Sample | Dispersiona | Specific rate × 100b (mol CO/(g Pt·h)) | TOFa × 100c (s−1) | TOFb × 100d (s−1) |
|---|---|---|---|---|
| a The dispersion is tested by CO chemisorption. b The specific rate is derived from the CO conversion at 80 °C. c The TOFa is calculated based on the dispersion of metal nanoparticles. d The TOFb is calculated based on the length of the perimeter between Pt nanoparticles and TiO2. | ||||
| Pt/r-TiO2 | 86% | 226 | 14.2 | 200.2 |
| Pt/h-TiO2 | 53% | 81 | 8.2 | 187.0 |
| Pt/o-TiO2 | 36% | 8 | 1.3 | 42.6 |
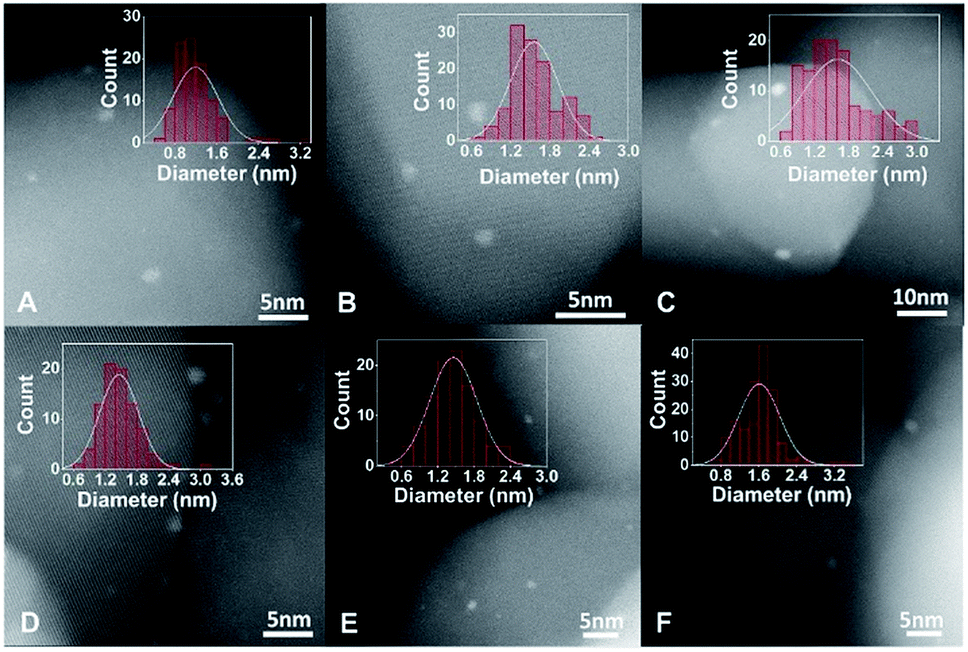 | ||
| Fig. 2 HAADF-STEM and size distribution of Pt/TiO2 catalysts. (A–C) and (D–F) are HADDF-STEM and size distribution of Pt/r-TiO2, Pt/h-TiO2 and Pt/o-TiO2 before and after reaction, respectively. | ||
Catalytic performance
The reactivity to CO oxidation was comparatively studied over the different catalysts (Fig. 3A). Not surprisingly, the best reactivity is observed on Pt/r-TiO2. With this catalyst, 50% CO conversion takes place at 87.6 °C. In contrast, the Pt/h-TiO2 and Pt/o-TiO2 catalysts exhibit worse performance, in which 50% CO conversion occurs at 102.3 and 126.3 °C, respectively. It is noteworthy that a similar trend of CO oxidation reactivity can be observed over the catalysts with different concentrations of surface defects, which are prepared through different treatment times (Fig. S3 and S4†).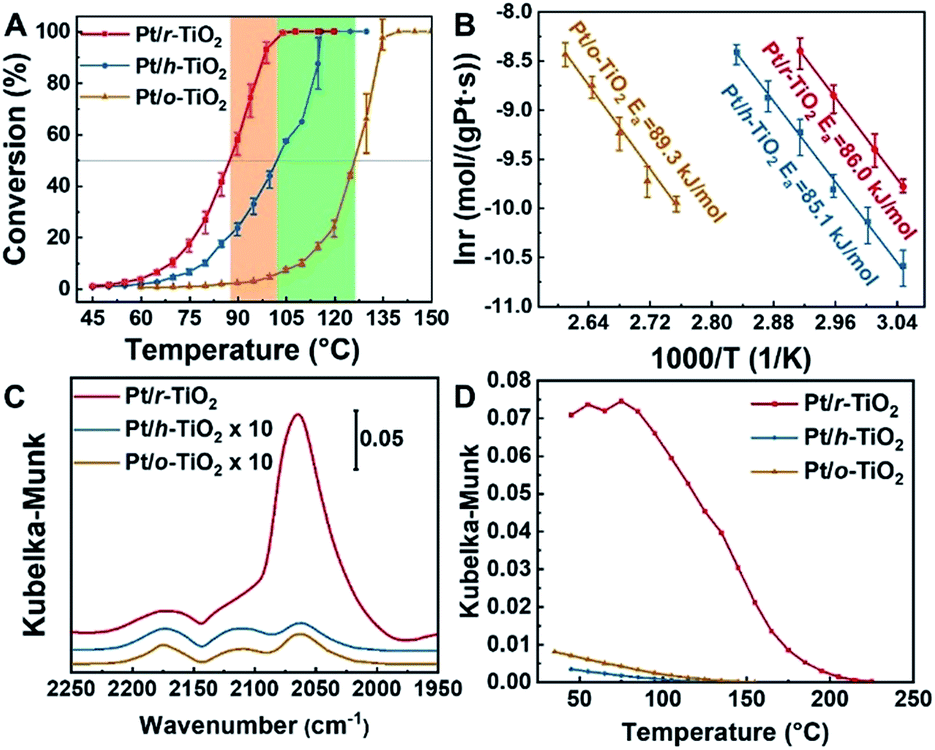 | ||
Fig. 3 Reactivity, kinetic properties and DRIFTS of CO adsorption on Pt/TiO2 catalysts. (A) Light-off curves of CO conversion on different catalysts. The CO oxidation was conducted with a gas composition of 1% CO, 20% O2 and He balanced, keeping GHSV at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ml gcat.−1 h−1. Each point is tested at a fixed temperature three times and then ramped to higher temperature. (B) Arrhenius plot of CO oxidation on different catalysts. (C) DRIFTS in CO oxidation (1% CO, 20% O2 and He balanced) at 80 °C. The spectra were acquired after 5 minutes of reaction. (D) Plot of CO adsorption amount as a function of temperature in an Ar atmosphere. CO was pre-adsorbed at room temperature. 000 ml gcat.−1 h−1. Each point is tested at a fixed temperature three times and then ramped to higher temperature. (B) Arrhenius plot of CO oxidation on different catalysts. (C) DRIFTS in CO oxidation (1% CO, 20% O2 and He balanced) at 80 °C. The spectra were acquired after 5 minutes of reaction. (D) Plot of CO adsorption amount as a function of temperature in an Ar atmosphere. CO was pre-adsorbed at room temperature. | ||
The kinetic measurement of CO oxidation was further conducted. It can be found that the specific rate of Pt/r-TiO2 to CO oxidation is ∼2.8 and ∼28 times higher than that of Pt/h-TiO2 and Pt/o-TiO2, respectively (Table 1). In addition, the turnover frequency (TOF) values based on the surface area (TOFa) and the perimeter length of Pt nanoparticles (TOFb) are shown in Tables 1 and S3.† The Pt/r-TiO2 catalyst shows a TOFa of 0.142 s−1 and a TOFb of 2.002 s−1, while the Pt/h-TiO2 and Pt/o-TiO2 catalysts exhibit much lower values. For the CO oxidation reaction occurring over noble metal catalysts, many previous studies suggested that the reaction obeys the Mars–van Krevelen (M–vK) mechanism on reducible catalysts or the Langmuir–Hinshelwood (L–H) mechanism on non-reducible catalysts.30,45–47 The reaction order test shows that the orders of CO and O2 on different catalysts are almost the same, negative for CO and near to zero for O2 (Fig. S5†), which is consistent with the M–vK mechanism. Furthermore, the comparable apparent activation energy (Ea) derived from the Arrhenius plot demonstrates that the CO oxidation on the different catalysts should follow the same reaction mechanism (Fig. 3B).
Electronic metal–support interactions
It has been generally agreed that supported Pt catalysts show low structure sensitivity for CO oxidation. For example, Iglesia and Lu et al. suggested that the catalytic properties of Pt/Al2O3 and Pt/TiO2 catalysts for CO oxidation are independent of Pt cluster size.30,47 Although theoretical calculations and surface science experiments suggested that the reaction should be highly structure sensitive,48–50 Christopher et al. showed that CO-induced structure reconstruction of Pt nanoparticles mitigates the inherent structure sensitivity.51 Therefore, the observed reactivity difference of Pt/TiO2 catalysts can be attributed to the different electronic metal–support interactions.From XPS studies of nano-sized materials, it was found that the final state effect will make BE shift to a higher position for smaller metal nanoparticles.52–54 Although the size of Pt nanoparticles on r-TiO2 is smaller than that on h-TiO2 and o-TiO2, the BE of XPS Pt 4f7/2 peaks from Pt/r-TiO2 is even ∼0.2 eV lower than that from Pt/h-TiO2 (Fig. S6, S7 and Table S4†). This indicates that Pt may accept more electrons from r-TiO2 or transfer fewer electrons to r-TiO2 compared to Pt/h-TiO2 and Pt/o-TiO2 catalysts.
Diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) of CO adsorption was also conducted to demonstrate the electronic metal–support interactions. It can be found that the better catalyst for CO oxidation shows a larger amount of CO adsorption in Kubelka–Munk units, which are assumed to be linearly related to the adsorbate coverage (Fig. 3C).4,55 Besides the larger CO adsorption amount, the Pt/r-TiO2 catalyst also presents ∼80 °C higher temperature for complete CO desorption compared to Pt/h-TiO2 and Pt/o-TiO2, which indicates that CO binds stronger with Pt/r-TiO2 (Fig. 3D). The higher CO coverage induced by electronic interactions should be detrimental to the reactivity of Pt/r-TiO2 in kinetic aspects due to the negative reaction order. However, Pt/r-TiO2 shows better reactivity than Pt/h-TiO2 and Pt/o-TiO2. Therefore, it can be drawn that electronic interactions play a dominant role thermodynamically in reactivity modulation which is demonstrated by the DFT calculations.
Theoretical calculations
Density functional theory (DFT) calculations provide further insights into the electronic interactions between metals and supports, and their influence on catalytic reactivity. Bader charge analysis shows that the average charges of Pt on r-TiO2, h-TiO2 and o-TiO2 are determined to be +0.04, +0.08 and +0.10, respectively (Fig. S8†), which agrees with XPS measurements.The CO adsorbed at the interface of Pt/r-TiO2 is found to have a binding energy of 2.08 eV, which is 0.29 and 0.21 eV higher than those for the CO adsorbed at the interface of Pt/h-TiO2 and Pt/o-TiO2, respectively. This result is well consistent with the DRIFTS results. Furthermore, the reaction barriers of adsorbed CO with the lattice oxygen of TiO2 (CO + O) were also calculated. Fig. 4 shows the energy barriers and TS geometries for CO oxidation on the different catalysts. It can be found that the energy barriers for the CO reaction are 0.89 eV, 0.96 eV and 1.04 eV on Pt/r-TiO2, Pt/h-TiO2 and Pt/o-TiO2, respectively. The barrier is consistent with Ea derived from the kinetic test (∼0.95 eV). However, compared with the reaction path on Pt/h-TiO2 and Pt/o-TiO2, the route on Pt/r-TiO2 is energetically more favorable which is downhill for further CO2 desorption and the free energy barrier of the CO oxidation process is also slightly favored. This indicates that Pt/r-TiO2 should exhibit better reactivity for CO oxidation than Pt/h-TiO2 and Pt/o-TiO2. Subsequently, the reaction barriers for CO oxidation on different Pt/TiO2 surfaces with oxygen vacancies (CO + O2 + O_vac) were calculated (Fig. S9†). It can be seen that the reaction barriers for CO + O2 + O_vac are much lower than those for CO + O over different catalysts.
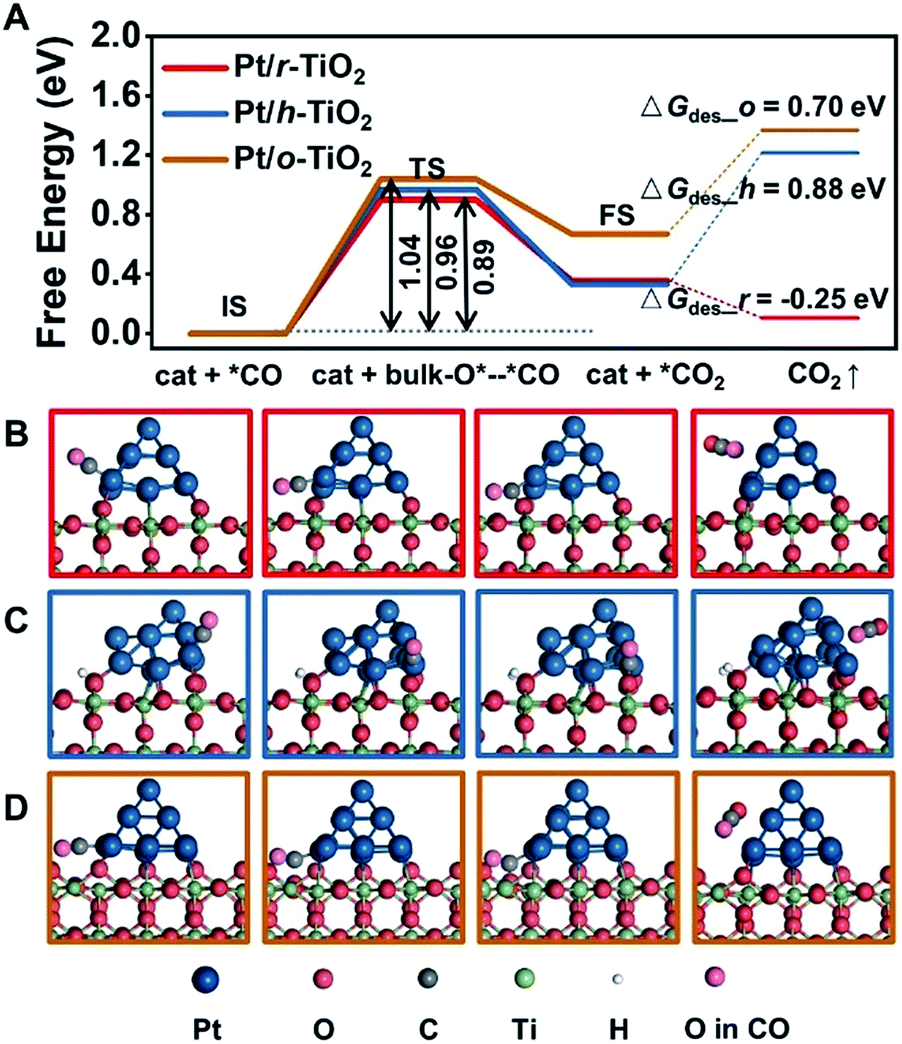 | ||
| Fig. 4 Calculated CO oxidation over Pt/TiO2 catalysts. (A) Free energy barriers for CO oxidation. (B–D) Models from left to right represent the initial state (IS), transition state (TS), final state (FS) and CO2 desorption of CO oxidation on Pt/r-TiO2, Pt/h-TiO2 and Pt/o-TiO2, respectively. | ||
Conclusions
Although it has been known that the surface states of supports should play an important role in the catalytic properties of catalysts, an understanding of the correlation between the surface states of supports and catalytic performance is still lacking, especially for real supported catalytic systems. Surface science investigations have shown that metal nanoparticles can be trapped at O adatoms and O vacancies on the TiO2 (110) surface under ultra-high vacuum conditions.13–15,22 However, in practice, the influence of air atmosphere and the structural complexity has made direct studies of real supported catalysts very difficult. In the present study, the preparation of different catalysts was carried out under the protection of an Ar atmosphere, and thus the influence of active molecules (O2, H2O, etc.) in air can be excluded, but this was often ignored before.In summary, we demonstrate how sensitively the catalytic performance of Pt catalysts depends on the surface states of TiO2 supports. Before depositing Pt catalysts, different TiO2 supports, including reduced, hydroxylated and oxidized types, were obtained via pre-treatments in different atmospheres. The highest reactivity for CO oxidation was achieved using Pt catalysts supported on r-TiO2, whereas worse performance was seen for the catalysts supported on h-TiO2 and o-TiO2. The established surface state–electronic structure–reactivity relationship clearly illustrates the important role of surface defects in catalytic performance and can be readily extended to many other metal/oxide catalytic systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (No. 2016YFB0600901), the National Science Foundation of China (No. 21525626, 91645106, and 21603159) and the Program of Introducing Talents of Discipline to Universities (No. B06006). We thank Jie Xu and Prof. Jun Luo from Tianjin University of Technology for HADDF-STEM characterization and Chao Wang from Dalian Institute of Chemical Physics for XPS characterization. We also thank Hao Li from Tianjin University, and Dr Lijun Gao and Dr Jinhu Dong from Dalian Institute of Chemical Physics for fruitful discussions.References
- M. Ahmadi, H. Mistry and B. Roldan Cuenya, J. Phys. Chem. Lett., 2016, 7, 3519–3533 CrossRef CAS PubMed.
- F. Shi, L. R. Baker, A. Hervier, G. A. Somorjai and K. Komvopoulos, Nano Lett., 2013, 13, 4469–4474 CrossRef CAS PubMed.
- N. J. Divins, I. Angurell, C. Escudero, V. Pérez-Dieste and J. Llorca, Science, 2014, 346, 620–623 CrossRef CAS PubMed.
- Y. Wang, D. Widmann and R. J. Behm, ACS Catal., 2017, 7, 2339–2345 CrossRef CAS.
- Y. Wang, D. Widmann, M. Heenemann, T. Diemant, J. Biskupek, R. Schlögl and R. J. Behm, J. Catal., 2017, 354, 46–60 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- D. A. J. M. Ligthart, R. A. van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2011, 50, 5306–5310 CrossRef CAS PubMed.
- H. Li, S. Zha, Z.-J. Zhao, H. Tian, S. Chen, Z. Gong, W. Cai, Y. Wang, Y. Cui, L. Zeng, R. Mu and J. Gong, ACS Catal., 2018, 8, 5526–5532 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Y. Lou and J. Liu, Ind. Eng. Chem. Res., 2017, 56, 6916–6925 CrossRef CAS.
- D. Matthey, J. G. Wang, S. Wendt, J. Matthiesen, R. Schaub, E. Lægsgaard, B. Hammer and F. Besenbacher, Science, 2007, 315, 1692–1696 CrossRef CAS PubMed.
- J. Ø. Hansen, E. Lira, P. Galliker, J.-G. Wang, P. T. Sprunger, Z. Li, E. Lægsgaard, S. Wendt, B. Hammer and F. Besenbacher, J. Phys. Chem. C, 2010, 114, 16964–16972 CrossRef CAS.
- F. Rieboldt, L. B. Vilhelmsen, S. Koust, J. V. Lauritsen, S. Helveg, L. Lammich, F. Besenbacher, B. Hammer and S. Wendt, J. Chem. Phys., 2014, 141, 214702 CrossRef CAS PubMed.
- S. Bonanni, K. Aït-Mansour, W. Harbich and H. Brune, J. Am. Chem. Soc., 2012, 134, 3445–3450 CrossRef CAS PubMed.
- H.-Z. Cui, Y. Guo, X. Wang, C.-J. Jia and R. Si, Catalysts, 2016, 6, 37 CrossRef.
- J. Wan, W. Chen, C. Jia, L. Zheng, J. Dong, X. Zheng, Y. Wang, W. Yan, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1705369 CrossRef PubMed.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang, A. S. Hoffman, I. Ro, G. W. Graham, S. R. Bare, G. Pacchioni, X. Pan and P. Christopher, Nat. Mater., 2019, 18, 746–751 CrossRef CAS PubMed.
- E. C. H. Sykes, Nat. Mater., 2019, 18, 663–664 CrossRef CAS PubMed.
- X. Jiao, Z. Chen, X. Li, Y. Sun, S. Gao, W. Yan, C. Wang, Q. Zhang, Y. Lin, Y. Luo and Y. Xie, J. Am. Chem. Soc., 2017, 139, 7586–7594 CrossRef CAS PubMed.
- A. Mellor, D. Humphrey, C. M. Yim, C. L. Pang, H. Idriss and G. Thornton, J. Phys. Chem. C, 2017, 121, 24721–24725 CrossRef CAS PubMed.
- X. Tong, L. Benz, S. Chrétien, H. Metiu, M. T. Bowers and S. K. Buratto, J. Phys. Chem. C, 2010, 114, 3987–3990 CrossRef CAS.
- O. Bikondoa, C. L. Pang, R. Ithnin, C. A. Muryn, H. Onishi and G. Thornton, Nat. Mater., 2006, 5, 189–192 CrossRef CAS.
- S. Wendt, J. Matthiesen, R. Schaub, E. K. Vestergaard, E. Lægsgaard, F. Besenbacher and B. Hammer, Phys. Rev. Lett., 2006, 96, 066107 CrossRef CAS PubMed.
- R. Mu, D. C. Cantu, X. Lin, V.-A. Glezakou, Z. Wang, I. Lyubinetsky, R. Rousseau and Z. Dohnálek, J. Phys. Chem. Lett., 2014, 5, 3445–3450 CrossRef CAS PubMed.
- R. Mu, D. C. Cantu, V.-A. Glezakou, I. Lyubinetsky, R. Rousseau and Z. Dohnálek, J. Phys. Chem. C, 2015, 119, 23552–23558 CrossRef CAS.
- Z.-T. Wang, Y.-G. Wang, R. Mu, Y. Yoon, A. Dahal, G. K. Schenter, V.-A. Glezakou, R. Rousseau, I. Lyubinetsky and Z. Dohnálek, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1801–1805 CrossRef CAS PubMed.
- R. Mu, Z.-j. Zhao, Z. Dohnalek and J. Gong, Chem. Soc. Rev., 2017, 46, 1785–1806 RSC.
- N. Li, Q.-Y. Chen, L.-F. Luo, W.-X. Huang, M.-F. Luo, G.-S. Hu and J.-Q. Lu, Appl. Catal., B, 2013, 142–143, 523–532 CrossRef CAS.
- G. T. K. K. Gunasooriya, E. G. Seebauer and M. Saeys, ACS Catal., 2017, 7, 1966–1970 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644–3653 CrossRef CAS.
- S. Wendt, R. Schaub, J. Matthiesen, E. K. Vestergaard, E. Wahlström, M. D. Rasmussen, P. Thostrup, L. M. Molina, E. Lægsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Surf. Sci., 2005, 598, 226–245 CrossRef CAS.
- J. Ø. Hansen, J. Matthiesen, E. Lira, L. Lammich and S. Wendt, Surf. Sci., 2017, 666, 113–122 CrossRef CAS.
- J. C. Parker and R. W. Siegel, Appl. Phys. Lett., 1990, 57, 943–945 CrossRef CAS.
- H. Zhang, J. Cai, Y. Wang, M. Wu, M. Meng, Y. Tian, X. Li, J. Zhang, L. Zheng, Z. Jiang and J. Gong, Appl. Catal., B, 2018, 220, 126–136 CrossRef CAS.
- A. N. Shultz, W. Jang, W. M. Hetherington, D. R. Baer, L.-Q. Wang and M. H. Engelhard, Surf. Sci., 1995, 339, 114–124 CrossRef CAS.
- F. Guillemot, M. C. Porté, C. Labrugère and C. Baquey, J. Colloid Interface Sci., 2002, 255, 75–78 CrossRef CAS PubMed.
- J. Balajka, M. A. Hines, W. J. I. DeBenedetti, M. Komora, J. Pavelec, M. Schmid and U. Diebold, Science, 2018, 361, 786–789 CrossRef CAS PubMed.
- Y. Du, Z. Dohnálek and I. Lyubinetsky, J. Phys. Chem. C, 2008, 112, 2649–2653 CrossRef CAS.
- Z.-T. Wang, Y. Du, Z. Dohnálek and I. Lyubinetsky, J. Phys. Chem. Lett., 2010, 1, 3524–3529 CrossRef CAS.
- E. Lira, J. Ø. Hansen, P. Huo, R. Bechstein, P. Galliker, E. Lægsgaard, B. Hammer, S. Wendt and F. Besenbacher, Surf. Sci., 2010, 604, 1945–1960 CrossRef CAS.
- S. T. Daniells, A. R. Overweg, M. Makkee and J. A. Moulijn, J. Catal., 2005, 230, 52–65 CrossRef CAS.
- M. Kotobuki, R. Leppelt, D. A. Hansgen, D. Widmann and R. J. Behm, J. Catal., 2009, 264, 67–76 CrossRef CAS.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS PubMed.
- T. Jiang, D. J. Mowbray, S. Dobrin, H. Falsig, B. Hvolbæk, T. Bligaard and J. K. Nørskov, J. Phys. Chem. C, 2009, 113, 10548–10553 CrossRef CAS.
- S. Bonanni, K. Aït-Mansour, W. Harbich and H. Brune, J. Am. Chem. Soc., 2014, 136, 8702–8707 CrossRef CAS PubMed.
- O. Balmes, G. Prevot, X. Torrelles, E. Lundgren and S. Ferrer, ACS Catal., 2016, 6, 1285–1291 CrossRef CAS.
- M. J. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS PubMed.
- D. P. Anderson, J. F. Alvino, A. Gentleman, H. A. Qahtani, L. Thomsen, M. I. J. Polson, G. F. Metha, V. B. Golovko and G. G. Andersson, Phys. Chem. Chem. Phys., 2013, 15, 3917–3929 RSC.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS PubMed.
- T. Armaroli, T. Bécue and S. Gautier, Oil Gas Sci. Technol., 2004, 59, 215–237 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and theoretical details, supporting figures and tables. See DOI: 10.1039/c9sc03119g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |