
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 reduction with protons and electrons at a boron-based reaction center†
Jordan W.
Taylor
,
Alex
McSkimming
,
Laura A.
Essex
and
W. Hill
Harman
 *
*
Department of Chemistry, University of California, Riverside, California 92521, USA. E-mail: hill.harman@ucr.edu
First published on 6th August 2019
Abstract
Borohydrides are widely used reducing agents in chemical synthesis and have emerging energy applications as hydrogen storage materials and reagents for the reduction of CO2. Unfortunately, the high energy cost associated with the multistep preparation of borohydrides starting from alkali metals precludes large scale implementation of these latter uses. One potential solution to this issue is the direct synthesis of borohydrides from the protonation of reduced boron compounds. We herein report reactions of the redox series [Au(B2P2)]n (n = +1, 0, −1) (B2P2, 9,10-bis(2-(diisopropylphosphino)phenyl)-9,10-dihydroboranthrene) and their conversion into corresponding mono- and diborohydride complexes. Crucially, the monoborohydride can be accessed via protonation of [Au(B2P2)]−, a masked borane dianion equivalent accessible at relatively mild potentials (−2.05 V vs. Fc/Fc+). This species reduces CO2 to produce the corresponding formate complex. Cleavage of the formate complex can be achieved by reduction (ca. −1.7 V vs. Fc/Fc+) or by the addition of electrophiles including H+. Additionally, direct reaction of [Au(B2P2)]− with CO2 results in reductive disproportion to release CO and generate a carbonate complex. Together, these reactions constitute a synthetic cycle for CO2 reduction at a boron-based reaction center that proceeds through a B–H unit generated via protonation of a reduced borane with weak organic acids.
Introduction
Since their discovery by Schlesinger and Brown in the 1940s,1 borohydrides have become ubiquitous reducing agents in the synthesis of fine and commodity chemicals.2 More recently, borohydrides have attracted interest in energy storage applications,3 both as a dense and readily handled source of H2 and in the reduction of CO2 to fuels such as formic acid and methanol.4 The first report of the reaction of NaBH4 with CO2 dates back to 1955 when Wartik and Pearson described the solution (dimethyl ether) and solid-state reactions with mass-balance and hydrolytic analysis of the products.5 In 1967, the reaction of NaBH4 with CO2 in aqueous conditions was shown to produce sodium formate.6 The intrinsic role of the borohydride ion in CO2 reduction was later investigated by Mizuta in 2014 who showed that, in the presence of a catalytic amount of NaBH4, BH3·THF effectively reduces CO2 to trimethylboroxine.7 A year later, Cummins reported the direct reaction of NaBH4 with CO2 yields the triformatoborohydride that could be hydrolyzed to yield formic acid.8 Additional studies over the last decade have found that in the presence of a suitable catalyst, pinacol borane (HBpin) and catechol borane (HBcat) can reduce CO2 to a range of products9 including carbon monoxide10 and formaldehyde,11 formic acid,12 and methanol equivalents.13Despite their broad utility, one drawback to these reagents is the substantial energy required for their production. For example, millions of kg of NaBH4 are produced each year by the NaH reduction of B(OMe)3 in the Brown–Schlesinger process.14
| B(OCH3)3 + 4NaH → NaBH4 + 3NaOCH3 | (1) |
NaH is prepared from the reaction of metallic Na with H2 gas,14 and the reduction of NaCl to metallic Na in the Downs process. The process requires high temperature (>600 °C) and operating potentials of 8 V or more for practical reaction rates.15 As a result, the stoichiometric reduction of CO2 with NaBH4, or similarly produced B–H species,16 entails a significant energy cost.
One potential solution to this issue would involve the direct construction of B–H bonds from the protonation of reduced boron compounds accessed at potentials tolerable to common solvents. Such a process would be analogous to the generation of transition metal hydrides via the reduction and protonation of a transition metal complex (Fig. 1). Despite the difficulty in accessing doubly reduced boranes,17 there has been recent progress in the protonation of low valent boron compounds to generate B–H bonds (Fig. 2). In 2006, Nozaki et al. reported the isolation of the boryllithium compound LiB(2,6-iPrC6H4)(NCH2)2, which reacts as a boryl anion equivalent, and showed that it could be protonated with H2O to give the corresponding hydroborane.18 Bertrand et al. have prepared the carbene-stabilized borylene (CAAC)2BH (CAAC = cyclic (alkyl)(amino)carbene) that could be protonated with trifluoromethanesulfonic acid (TfOH) to yield the corresponding boronium salt.19 A related bis(oxazol-2-ylidene)-phenylborylene system reported by Kinjo also produced a B–H moiety via protonation with TfOH.20 Furthermore, Wagner synthesized a diborylmethane dianion and a doubly boron-doped dibenzo[g,p]chrysene dianion that could be protonated to yield the corresponding hydride bridged B–H–B species.21 Additionally, Szymczak et al. reported a conceptually relevant series of Mn/Cr–borazine complexes that could be reduced and then protonated to yield a bridging metalloborohydride.22 The reactivities (in particular, towards hydride transfer) of these B–H bonds formed via protonation have not been reported.
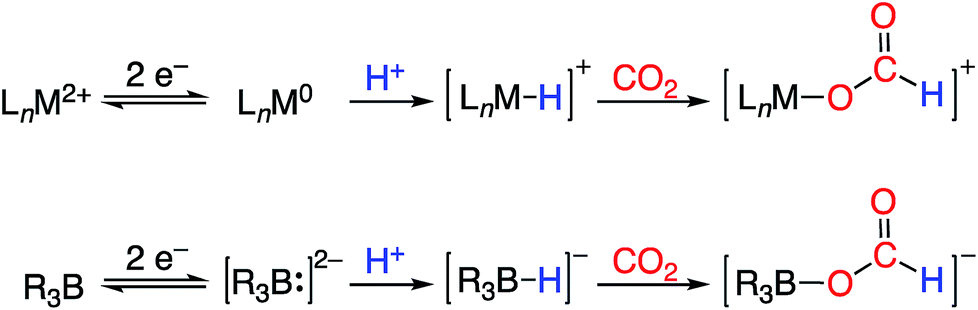 | ||
| Fig. 1 Redox-coupled hydride formation at a transition metal complex (top) and borane (bottom) as a strategy for CO2 electroreduction to formate. | ||
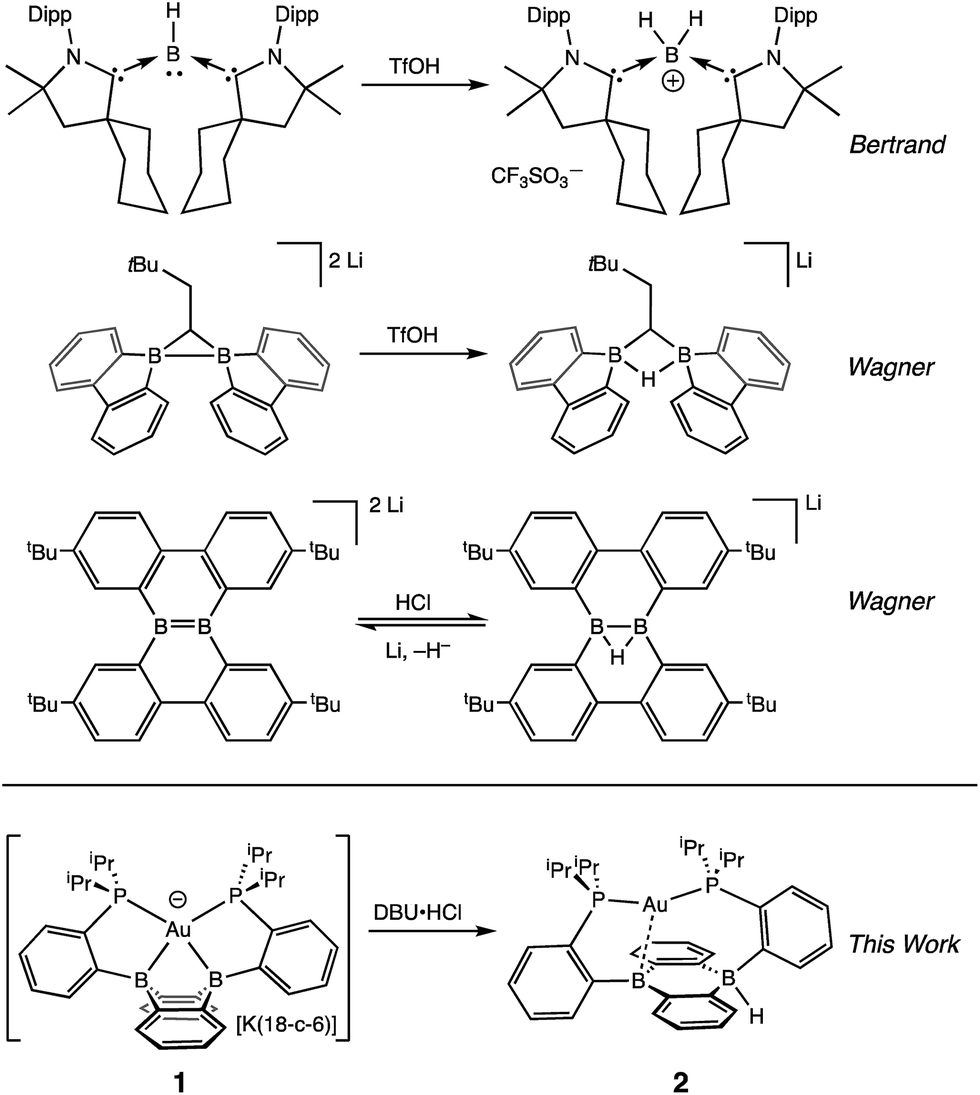 | ||
| Fig. 2 Protonation of reduced boron compounds to give tetracoordinate B–H moieties. | ||
We recently reported the synthesis of the disphosphine tethered diboraanthracene ligand B2P2 (9,10-bis(2-(diisopropylphosphino)phenyl)-9,10-dihydroboranthrene) and a series of its gold complexes in three states of charge. The anionic complex, [Au(B2P2)][K(18-c-6)] (1), which features an unprecedented boroauride moiety, could be accessed at remarkably mild potentials (−2.05 V vs. Fc/Fc+, MeCN)23 owing to the formation of a strong 3-centered, 2-electron bond between Au and the two B atoms.24 Considering that 1 can be thought of as a masked borane dianion, we wondered if protonation of this complex could provide direct access to a hydridic B–H unit (Fig. 2). We herein report the synthesis and characterization of this borohydride complex, Au(B2P2)H (2), which can be generated via direct hydride reduction of Au(B2P2)Cl (3), FLP-type dihydrogen activation, H-atom addition to the boron-centered radical complex Au(B2P2) (4) or, most notably, by protonation of 1 with mild organic acids. Borohydride 2 is sufficiently reactive to reduce CO2 to formate, and cleavage of the resulting B–OCHO bond can be achieved either by addition of an electrophile (including H+) or by one electron reduction. These results collectively represent a synthetic cycle for CO2 reduction to formate with protons and electrons, with the key finding being the generation of a hydridic borohydride unit via protonation of a reduced borane at modest potentials. This reaction sequence, along with Wagner's recent reports of CO2 hydrogenation25 and the reductive disproportionation of CO2 (ref. 26) catalyzed by DBA derivatives, illustrates the promise of redox-active boranes in the reduction of CO2, an area dominated by transition metal chemistry.27 Furthermore, given the recent application of boron-doped graphene to CO2 electroreduction,28 these molecules offer well-defined molecular models29 of fundamental chemical processes that may underlie small molecule reactivity at doped graphitic carbon surfaces.
Results and discussion
Our initial synthetic approaches to a B–H containing derivative of Au(B2P2) began with direct hydride for halide substitution (Scheme 1). Addition of 1 equiv. K[sec-Bu3BH] to 3 gave borohydride 2 in 89% yield. This complex could also be prepared in good yield by exposing 3 to an excess of Et3SiH. Additionally, 2 could be synthesized via frustrated Lewis pair (FLP) H2 activation in the presence of DBU (DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene): exposure of a THF solution of equimolar 3 and DBU to 1 atm H2 resulted in the immediate formation of 2 as judged by 1H, 11B, and 31P NMR spectroscopies. This reaction is presumably mediated by the pseudo-three-coordinate boron and is consistent with a relatively weak Au–B interaction in 3. Single-crystal X-ray diffraction (XRD) studies of 2 show it to be a zwitterion in the solid state with an intact B–H bond on the DBA face opposite the Au center, analogous to the previously reported structure of zwitterion 3 (Fig. 3). The H atom was located in the electron difference map and is bound to a quasi-tetrahedral B atom (∑∠C–B–C = 340.5°). On the opposite side of the DBA linker, the Au ion is bound by both P donors in a roughly linear fashion (∠P–Au–P = 156.8°) with a single Au–B interaction (dAuB = 2.644(1) Å) to one nearly-planar B atom (∑∠C–B–C = 359.8°). The 1H NMR spectrum of 2 features a signal at 5.09 ppm for a B–H proton resonance with the expected four-line pattern for coupling to a single 11B atom (JBH = 80 Hz). The 11B NMR shows a corresponding doublet at −10.01 ppm (JBH = 80 Hz) while the 31P NMR spectrum features two doublets at 58.4 and 55.9 ppm (JPP = 255.0 Hz), consistent with inequivalent phosphine ligands on the NMR timescale. Given the precedent for H-atom transfer reactivity at boron,30 we also targeted the synthesis of 2 from the neutral radical species 4via H-atom addition. Reaction of Au(B2P2) with one equivalent of Bu3SnH produces 2 and Bu6Sn2 over 12 hours via apparent H-atom transfer as judged by NMR of reaction mixtures. | ||
| Scheme 1 Synthesis of Au(B2P2)H from H−, H˙, and H+. | ||
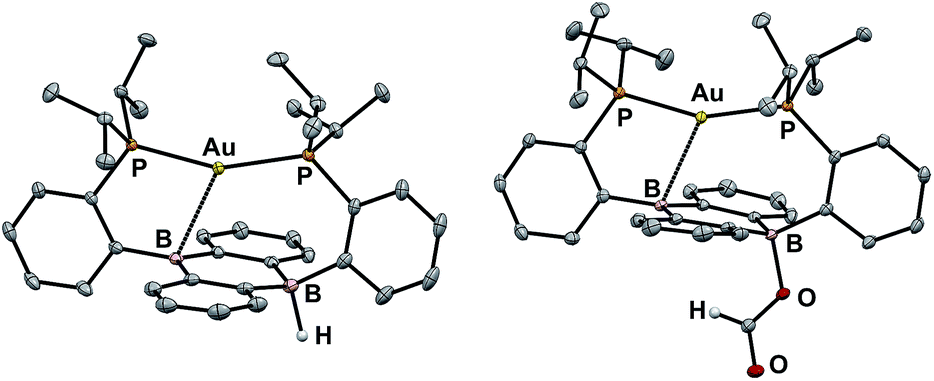 | ||
| Fig. 3 Thermal ellipsoid plots (50%) of Au(B2P2)H (2, left) and Au(B2P2)(O2CH) (5, right). Unlabeled ellipsoids correspond to carbon. Solvent molecules and most hydrogen atoms have been omitted for clarity. | ||
To probe the synthesis of 2via protonation of 1, we exposed 1 to a series of organic acids. Addition of one equivalent of solid [Ph2NH2][Cl] or [2,6-Me2pyH][Cl] to CD3CN solutions of 1 resulted in immediate effervescence and formation of dark purple reaction mixtures which were revealed by NMR analysis to contain a mixture of H2, 3, and 4. Fortunately, addition of one equivalent of DBU·HCl to a solution 1 in CD3CN gave rise to a pale-yellow solution exhibiting 1H, 11B, and 31P NMR resonances that matched authentic samples of 2 (Fig. S5–S7†) with no H2 observed. Furthermore, 2 is stable to treatment with excess DBU·HCl.
With borohydride 2 in hand, we examined its reactivity with CO2. Exposure of a C6D6 solution of 2 to 1 atm of CO2 resulted in quantitative formation of the corresponding formate complex Au(B2P2)(O2CH) (5) within seconds (Scheme 2). The solid-state structure of 5 confirms the insertion of CO2 into the B–H bond, with the resulting formate ion bound through a single O to one boron atom (Fig. 3). The zwitterionic structure of 5 is analogous to 2 and 3, with a tetrahedral boron center bound to the formate oxygen (∑∠C–B–C = 339.1°) and an intermediate length contact between Au and the non-formate bound B atom (dAuB = 2.632 Å). The 1H NMR spectrum of 5 in CDCl3 is consistent with C2v symmetry in solution, with a lone singlet in the 31P NMR spectrum at 56.1 ppm and 11B{1H} NMR displaying a broad singlet at 26.9 ppm. Similar to 3, the observed C2v symmetry in solution along with a 11B chemical shift between that of bona fide three- and four-coordinate B centers in DBA containing molecules17h,f implies rapid exchange of the formate ion between each boron site of the DBA ring, a phenomenon also observed in 3.22 The 13C{1H} NMR spectrum of 5 contains a resonance at 168.2 ppm corresponding to the formate carbon atom, and FTIR spectroscopy revealed a strong band at 1672 cm−1 for the formate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch.
O stretch.
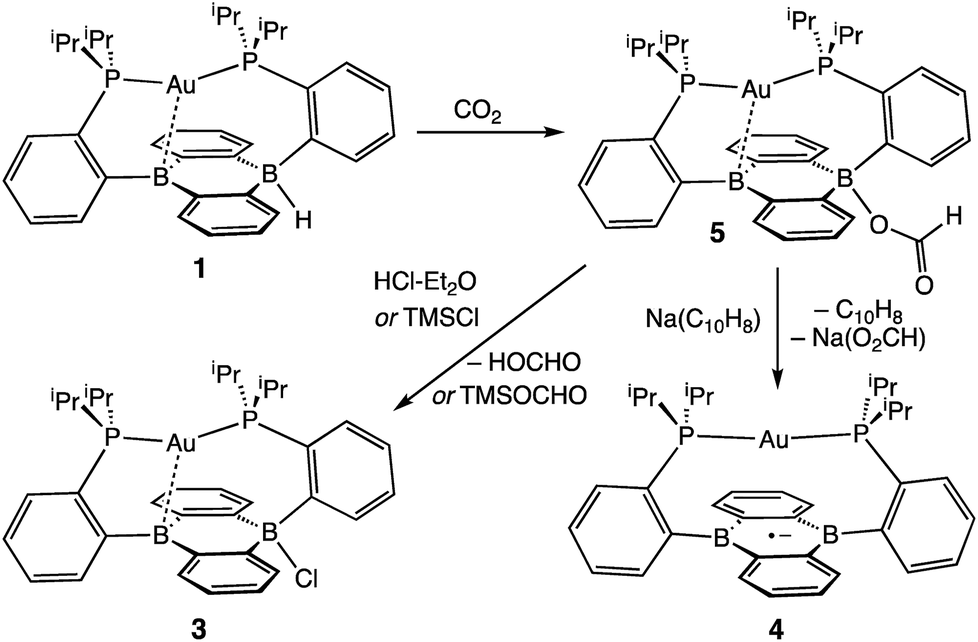 | ||
| Scheme 2 CO2 reduction to formate by Au(B2P2)H. | ||
Next, we assessed the ability of electrophiles such as TMSCl and HCl to effect B–O bond scission. Treatment of 5 with excess TMSCl resulted in the immediate formation of 3 as judged by 1H and 31P NMR along with new resonances for trimethylsilylformate (Fig. S13†), accomplishing the net hydrosilylation of CO2.31 While silyl electrophiles have strong precedent to mediate E–O bond cleavage in main group systems,32 we wondered if a suitable acid would be able to protonate the B–OCHO bond to yield formic acid. To this end, addition of [Ph2NH2][Cl] or DBU·HCl to CDCl3 solutions of 5 saw no observable reaction over the course of days at room temperature. However, addition of one equivalent of HCl·Et2O to 5 resulted in the immediate formation of the chloride complex 3 by 1H and 31P NMR spectroscopy with resonances for HCO2H observed in the 1H NMR spectrum.
Following successful electrophilic B–O bond cleavage in 5 to yield 3, the reductive cleavage of the B–OCHO unit was explored. Cyclic voltammetry (CV) performed on 5 revealed a quasireversible process at E1/2 = −1.74 V vs. Fc/Fc+ which gave rise to an oxidative daughter wave at ca. −1.5 V (Fig. 4). This daughter wave occurs at roughly the same potential as the oxidation of [Au(B2P2)]0/+1, consistent with the hypothesis that one electron reduction of 5 forms [5]− which then undergoes formate dissociation on the CV timescale to afford neutral 4. To unambiguously confirm the liberation of formate ion, 5 was treated with 1 equivalent of Na(C10H8) in THF. A turbid, dark-purple solution immediately formed, indicating formation of 4. The precipitated solids were collected by filtration and dissolved in D2O with 1H NMR analysis revealing formation of NaCO2H (Fig. S12†). To our knowledge, this is the only example of reductive cleavage of a B–OCHO bond to yield formate. This process is presumably driven by the accessible one-electron reduction chemistry of 5 (−1.74 V vs. Fc/Fc+) enabled by the DBA core and speaks to the utility of frontier orbital modulation by incorporation of boron into polycyclic aromatic hydrocarbon frameworks.33 Collectively, these results outline a synthetic cycle for CO2 reduction with protons and electrons mediated exclusively at boron (Fig. 5).
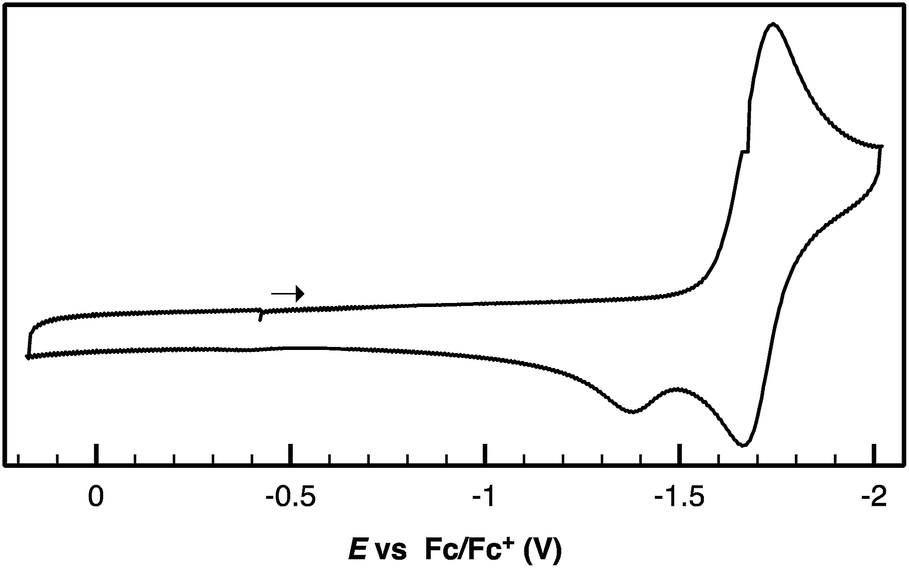 | ||
| Fig. 4 Cyclic voltammogram of Au(B2P2)(O2CH) performed in 0.1 M [nBu4N][PF6] in CH3CN at a scan rate of 100 mV s−1. | ||
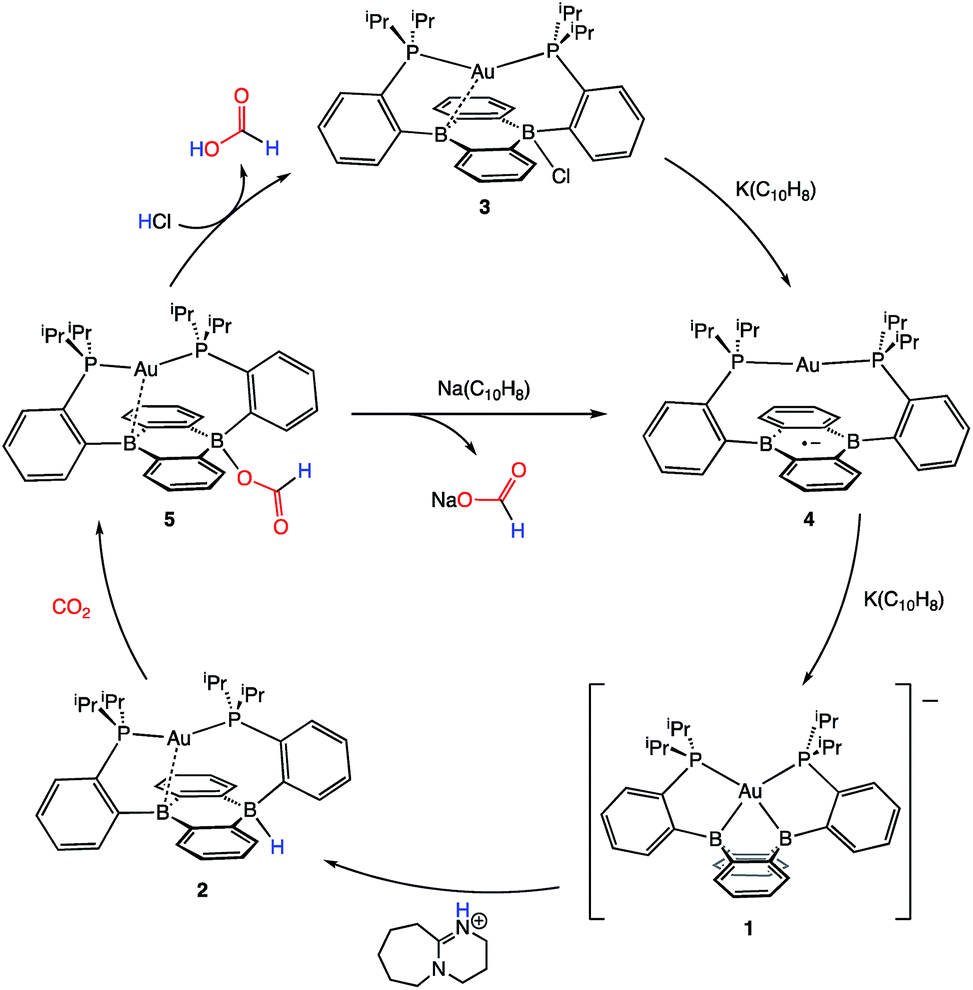 | ||
| Fig. 5 A closed synthetic cycle for the reduction of CO2 to formic acid with protons and electrons mediated by a redox active, metal-stabilized diboraanthracene platform. | ||
In addition to the monoborohydride chemistry described above, we were able to generate a diborohydride species via formal sequential hydride addition to 3. Two equivalents of K[sec-Bu3BH] reacted with 3 in Et2O to give the anionic diborohydride complex [Au(B2P2)H2][K(Et2O)] (6) (Scheme 3). Single-crystal XRD reveals 6 to be a dimer in the solid state, (Fig. 6) with two [Au(B2P2)H2]− units bridged by two [K(Et2O)]+ cations via contacts between the B–H and K atoms. The [Au(B2P2)H2]− component features a linear P–Au–P moiety (∠P–Au–P = 161.9°) that sits above the DBA core with no appreciable interaction between Au and B (dAuB > 3.25 Å). The H atoms bound to both B atoms were located on the electron difference map and both B centers exhibit a pseudo-tetrahedral geometry (∑∠C–B–C = 337.5°). 1H and 11B NMR spectra feature a 4-line pattern and doublet, respectively, for the B–H moieties in 6, analogous to the monoborohydride 2 (Fig. S14†). Compound 6 is structurally related to the diborohydride species resulting from H2 activation by reduced DBA compounds reported by Wagner.34
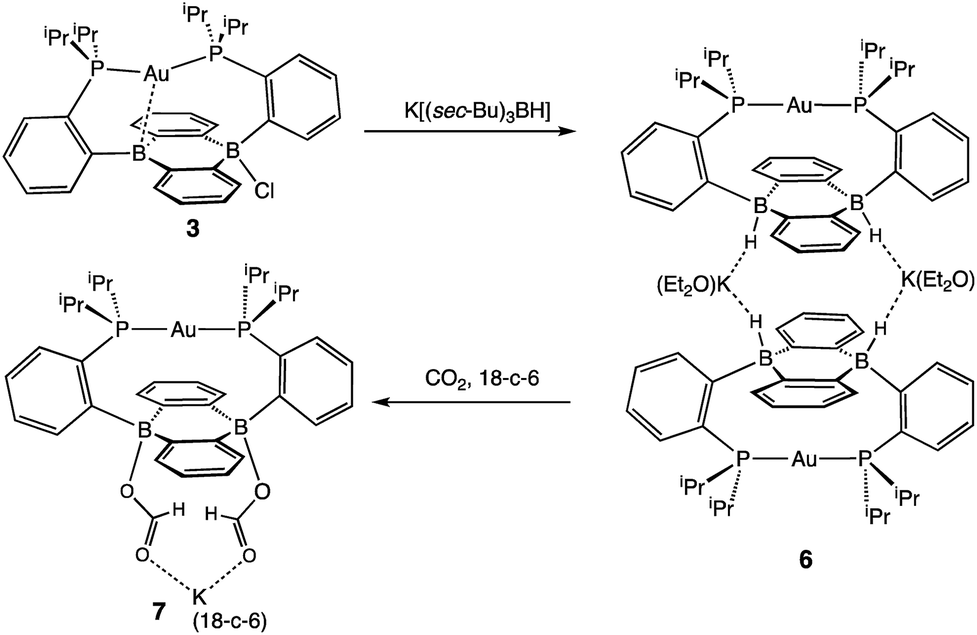 | ||
| Scheme 3 Synthesis of Diborohydride 6 and its reactivity with CO2. | ||
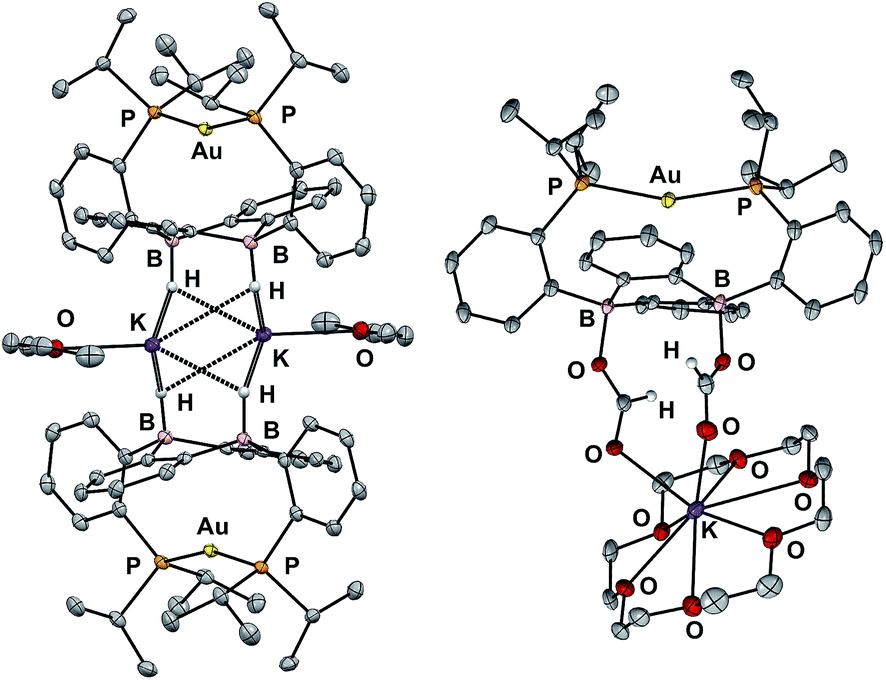 | ||
| Fig. 6 Thermal ellipsoid plots (50%) of the dimer of [Au(B2P2)H2][K(Et2O)] (6, left) and [Au(B2P2)(O2CH)2][K(18-c-6)] (7, right). Unlabeled ellipsoids correspond to carbon. Solvent molecules, a disordered crown ether component in 6, and most hydrogen atoms have been omitted for clarity. | ||
Diborohydride 6 is also reactive towards CO2 (1 atm), cleanly generating the corresponding diformate complex, which was isolated as its 18-crown-6 adduct, [Au(B2P2)(O2CH)2][K(18-c-6)] (7). Single-crystal XRD confirmed the insertion of CO2 into both B–H bonds, with B atoms puckered out of planarity to accommodate each OCHO unit. Additionally, the formate moieties in 7 were identified by 1H NMR (8.34 ppm), 13C{1H} NMR (168.2 ppm) and FTIR spectroscopy (CO, 1672 cm−1).
Having probed the [Au(B2P2)] system for CO2 reduction chemistry from its mono- and dihydrides, we additionally explored the direct reaction of CO2 with 1. Wagner et al. recently reported that 9,10-dilithio-9,10-diborataanthracene reacts with CO2 to yield CO3 and CO via reductive disproportionation.25 Reaction of 1 with 1 atm of CO2 in C6D6 resulted in the immediate formation of a colorless solution with no detectable precipitate. A single crystal suitable for XRD studies was obtained from CO2 saturated reaction mixtures and revealed the product to be the carbonate adduct, [Au(B2P2)CO3][K(18-c-6)] (8, Fig. 7) (Scheme 4). The asymmetric unit of 8 contains two crystallographically distinct molecules which are chemically equivalent in gross terms but exhibit minor differences in their geometries, most notably in their B–O distances, which range from 1.550(6) Å to 1.578(8) Å. This spread of B–O distances is consistent with a soft [CO3]2− binding potential, in agreement with Wagner's results.25 Solution NMR spectroscopy of 8 is consistent with C2v symmetry and the [CO3]2− unit was identified by 13C{1H} NMR (168.0 ppm) and FTIR (1592 cm−1) spectroscopies with 13CO2 experiments confirming these assignments (see ESI†).
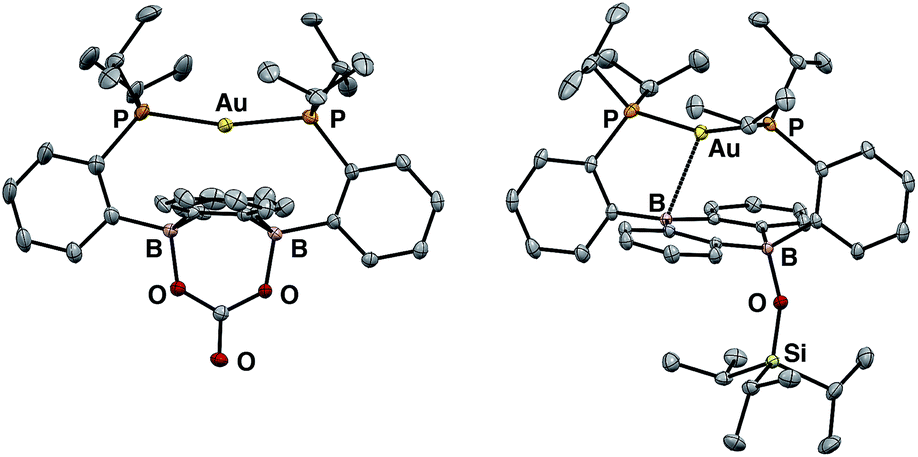 | ||
| Fig. 7 Thermal ellipsoid plots (50%) of the anion [Au(B2P2)(CO3)][K(18-c-6)] (8, left) and Au(B2P2)(OSiiPr3) (9, right). Unlabeled ellipsoids correspond to carbon. Solvent molecules, disordered components, and hydrogen atoms have been omitted for clarity. | ||
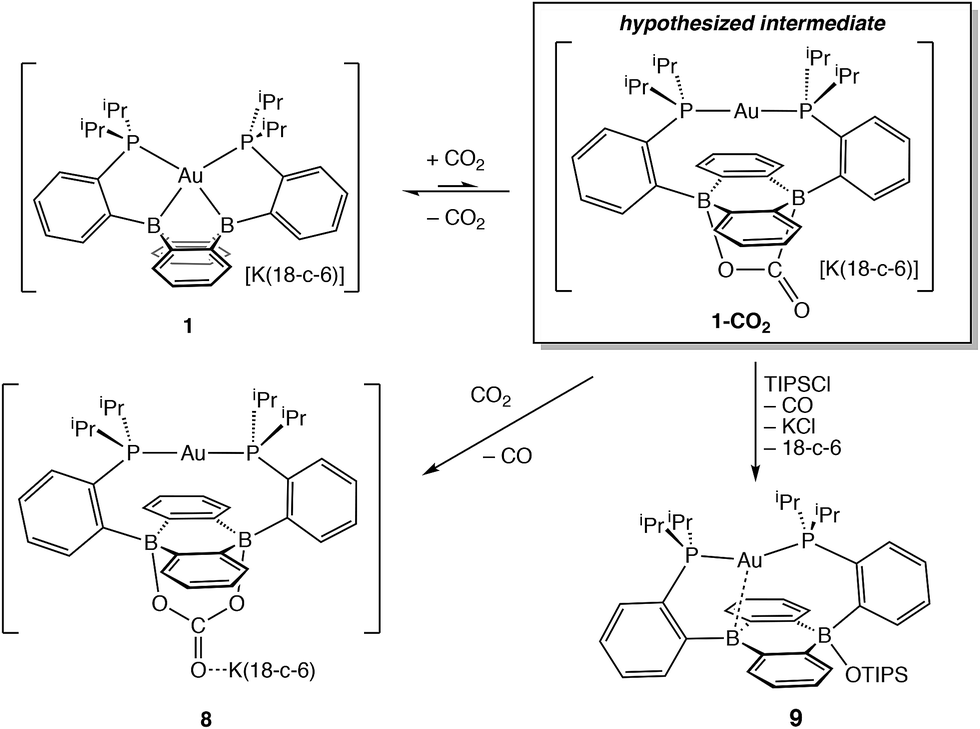 | ||
| Scheme 4 Reduction of CO2 to CO by [Au(B2P2)]−. | ||
In order to probe the mechanism of the formation of 8 from 1 and CO2, we explored the reaction by NMR spectroscopy at low temperature. Recent reports on reduced boron-containing heterocycles,21 offer extensive precedent for the cycloaddition of CO2 across the boron atoms, and we sought to probe whether a similar CO2 adduct, [Au(B2P2)CO2]− (1–CO2), might be an intermediate in the formation of 8. Addition of a single equivalent of CO2 to a frozen solution of the anion 1 in d8-toluene followed by insertion into a precooled (−50 °C) NMR spectrometer and subsequent variable temperature NMR monitoring resulted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of starting material 1 and the carbonate adduct 8. When the same experiment was carried out with 13CO2 enhanced intensity was observed corresponding to the Au(B2P2)13CO3 resonance at 168.2 ppm along with free 13CO at 184.5 ppm. These results are consistent with endergonic binding of CO2 to the DBA core followed by rapid reaction with a second equivalent of CO2 to release CO and form 8, analogous to the mechanism established by Wagner in a related system.25 As we were unable to observe the putative CO2 adduct [Au(B2P2)CO2]−, we attempted to trap it by other chemical means during the reaction of 1 with CO2. Solutions of 1 are stable in the presence of triisopropylsilylchloride (TIPSCl); however, addition of 1 atm CO2 to a thawing C6D6 solution of 1 and TIPSiCl resulted in the immediate formation of a pale-yellow solution. Single-crystal XRD studies revealed the product to be the zwitterion Au(B2P2)(OSiiPr3) (9), which contains a triisopropylsiloxide anion bound to one B atom (Fig. 7). The 31P NMR spectrum contains a set of doublets at 56.0 (JP–P = 240 Hz) and 52.5 (JP–P = 240 Hz) ppm while the 11B NMR spectrum featured a broad resonance at 51.5 and a sharper singlet at 2.99 ppm. Two crystallographically distinct molecules of 9 are present in the asymmetric unit although their geometries are essentially identical (Fig. S44†). The formation of 9 occurs with CO loss as judged by 13CO2 labeling experiments. Exposure of 9 to additional CO2 (1 atm) did not lead to any further reaction, ruling out additional reaction of the –OTIPS moiety with CO2 as has been previously observed for metal-siloxide units.35
:1 mixture of starting material 1 and the carbonate adduct 8. When the same experiment was carried out with 13CO2 enhanced intensity was observed corresponding to the Au(B2P2)13CO3 resonance at 168.2 ppm along with free 13CO at 184.5 ppm. These results are consistent with endergonic binding of CO2 to the DBA core followed by rapid reaction with a second equivalent of CO2 to release CO and form 8, analogous to the mechanism established by Wagner in a related system.25 As we were unable to observe the putative CO2 adduct [Au(B2P2)CO2]−, we attempted to trap it by other chemical means during the reaction of 1 with CO2. Solutions of 1 are stable in the presence of triisopropylsilylchloride (TIPSCl); however, addition of 1 atm CO2 to a thawing C6D6 solution of 1 and TIPSiCl resulted in the immediate formation of a pale-yellow solution. Single-crystal XRD studies revealed the product to be the zwitterion Au(B2P2)(OSiiPr3) (9), which contains a triisopropylsiloxide anion bound to one B atom (Fig. 7). The 31P NMR spectrum contains a set of doublets at 56.0 (JP–P = 240 Hz) and 52.5 (JP–P = 240 Hz) ppm while the 11B NMR spectrum featured a broad resonance at 51.5 and a sharper singlet at 2.99 ppm. Two crystallographically distinct molecules of 9 are present in the asymmetric unit although their geometries are essentially identical (Fig. S44†). The formation of 9 occurs with CO loss as judged by 13CO2 labeling experiments. Exposure of 9 to additional CO2 (1 atm) did not lead to any further reaction, ruling out additional reaction of the –OTIPS moiety with CO2 as has been previously observed for metal-siloxide units.35
Conclusions
In conclusion, we have synthesized a reactive borohydride complex, 2, via formal hydride addition, dihydrogen activation, radical H-atom addition, and, most crucially, reduction/protonation. The borohydride unit rapidly inserts CO2 to afford the formate complex 5. Both CV and synthetic studies confirm that 5 can be reduced at E1/2 = −1.79 V vs. Fc/Fc+ and undergo formate loss to give the neutral radical 4 and formate via reductive B–O cleavage. The cleavage of the B–O bond in 5 can also be achieved via addition of electrophiles including TMSCl or HCl·Et2O, to give 3 and TMSOCHO or formic acid, respectively. Additionally, direct reaction of the anion 1 with 1 atm of CO2 results in the extrusion of CO and the formation of carbonate complex 8via the reductive disproportionation of CO2. Collectively, these reactions outline a strategy for the reduction of CO2 with protons and electrons via reactive borohydride species generated at redox-active, conjugated boranes, and efforts to extend this chemistry to electrocatalytic systems are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation (CHE-1752876) and the American Chemical Society Petroleum Research Fund (57314). NMR spectra were collected on instruments funded by an NSF MRI award (CHE-162673) and an Army Research Office instrumentation grant (W911NF-16-1-0523). W. H. H. is a member of the University of California, Riverside Center for Catalysis. Dr Fook Tham and Dr Charlene Tsay are acknowledged for X-ray crystallographic analysis.Notes and references
- H. I. Schlesinger and H. C. Brown, J. Am. Chem. Soc., 1940, 62, 3429 CrossRef CAS.
- (a) H. C. Brown and S. Krishnamurthy, Tetrahedron, 1979, 35, 567 CrossRef CAS; (b) H. C. Brown, E. J. Mead and B. C. Subba Rao, J. Am. Chem. Soc., 1955, 77, 6209 CrossRef CAS.
- (a) J. Ma, N. A. Choudhury and Y. Sahai, Renewable Sustainable Energy Rev., 2010, 14, 183 CrossRef CAS; (b) M. Paskevicius, L. H. Jepsen, P. Schouwink, R. Cerny, D. Ravnsbaek, Y. Filinchuk, M. Dornheim, F. Besenbacher and T. R. Jensen, Chem. Soc. Rev., 2017, 46, 1565 RSC.
- S. Bontemps, Coord. Chem. Rev., 2016, 308, 117 CrossRef CAS.
- (a) T. Wartik and R. K. Pearson, J. Am. Chem. Soc., 1955, 77, 1075 CrossRef CAS; (b) T. Wartik and R. K. Pearson, J. Inorg. Nucl. Chem., 1958, 7, 404 CrossRef CAS.
- F. Eisenberg Jr and A. H. Bolden, Carbohydr. Res., 1967, 5, 349–350 CrossRef.
- K. Fujiwara, S. Yasuda and T. Mizuta, Organometallics, 2014, 33, 6692 CrossRef CAS.
- I. Knopf and C. Cummins, Organometallics, 2015, 34, 1601 CrossRef CAS.
- S. Bontemps, Coord. Chem. Rev., 2016, 308, 117–130 CrossRef CAS.
- (a) D. S. Laitar, P. Muller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196 CrossRef CAS PubMed; (b) H. Zhao, Z. Lin and T. B. Marder, J. Am. Chem. Soc., 2006, 128, 15637 CrossRef CAS PubMed.
- S. Bontemps, L. Vendler and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 124, 1703 CrossRef.
- (a) M.-A. Courtemanche, M.-A. Legare, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708 CrossRef CAS PubMed; (b) R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459 CrossRef CAS; (c) H.-W. Suh, L. M. Guard and N. Hazari, Chem. Sci., 2014, 5, 3859 RSC.
- (a) S. Chakraborty, J. Zhang, J. Krause and H. Guan, J. Am. Chem. Soc., 2010, 132, 8872 CrossRef CAS; (b) M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343 CrossRef CAS; (c) C. Das Neves Gomes, E. Biondiaux, P. Thuery and T. Cantat, Chem.–Eur. J., 2014, 20, 7098 CrossRef CAS PubMed; (d) M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. KociokKohn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC; (e) J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS.
- U. Wietelmann, M. Felderhoff and P. Rittmeyer, Hydrides, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2016 Search PubMed.
- A. Klemm, G. Hartmann and L. Lange, Sodium and Sodium Alloys, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2000 Search PubMed.
- Virtually all B–H reagents are ultimately derived from alkali metal hydrides or NaBH4. See ref. 14.
- (a) H. Asakawa, K.-H. Lee, K. Furukawa, Z. Lin and M. Yamashita, Chem.–Eur. J., 2015, 21, 4267 CrossRef CAS; (b) C.-W. Chiu and F. P. Gabbai, Organometallics, 2008, 27, 1657 CrossRef CAS; (c) C.-W. Chiu, Y. Kim and F. P. Gabbai, J. Am. Chem. Soc., 2009, 131, 60 CrossRef CAS; (d) J. Edwin, M. Bochmann, M. C. Boehm, D. E. Brennan, W. E. Geiger, C. Krueger, J. Pebler, H. Pritzkow and W. Siebert, J. Am. Chem. Soc., 1983, 105, 2582 CrossRef CAS; (e) G. E. Herberich, B. Buller, B. Hessner and W. Oschmann, J. Organomet. Chem., 1980, 195, 253 CrossRef CAS; (f) E. Januszewski, M. Bolte, H.-W. Lerner, M. Holthausen and M. Wagner, Dalton Trans., 2013, 42(38), 13826 RSC; (g) R. J. Kwaan, J. C. Harlan and J. R. Norton, Organometallics, 2001, 20, 3818 CrossRef CAS; (h) A. Lorbach, A. Hubner and M. Wagner, Dalton Trans., 2012, 41, 6048 RSC; (i) P. Muller, B. Gangnus, H. Pritzkow, H. Schulz, M. Stephan and W. Siebert, J. Organomet. Chem., 1995, 487, 235 CrossRef.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113 CrossRef CAS.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610 CrossRef CAS.
- L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280 CrossRef CAS PubMed.
- (a) A. Hübner, T. Kaese, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2015, 137, 3705 CrossRef PubMed; (b) T. Kaese, H. Budy, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2017, 56, 7546 CrossRef CAS PubMed.
- (a) T. J. Carter, J. W. Kampf and N. K. Szymczak, Angew. Chem., Int. Ed., 2012, 51, 13168 CrossRef CAS PubMed; (b) T. J. Carter, J. Y. Wang and N. K. Szymczak, Organometallics, 2014, 33, 1540 CrossRef CAS; (c) T. J. Carter, Z. M. Heiden and N. K. Szymczak, Chem. Sci., 2015, 6, 7258 RSC.
- J. W. Taylor, A. McSkimming, M.-E. Moret and W. H. Harman, Angew. Chem., Int. Ed., 2017, 56, 10413 CrossRef CAS PubMed.
- Wagner has recently shown that C-halogenation can render DBA derivatives significantly easier to reduce: S. Brend'amour, J. Gilmer, M. Bolte, H.-W. Lerner and M. Wagner, Chem.–Eur. J., 2018, 24, 16910–16918 CrossRef PubMed.
- E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2019, 141, 6082 CrossRef CAS PubMed.
- E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2018, 57, 16491 CrossRef CAS PubMed.
- (a) E. Benson, C. Kubiak, A. Sathrum and J. Smieja, Chem. Soc. Rev., 2009, 38, 89 RSC; (b) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed; (c) J. Qiao, Y. Liu, F. Hong and J. A. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC; (d) C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423 RSC.
- (a) N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061 RSC; (b) T. B. Tai and M. T. Nguyen, Chemistry, 2013, 19, 2942 CrossRef CAS PubMed.
- (a) J. W. Taylor, A. McSkimming, C. F. Guzman and W. H. Harman, J. Am. Chem. Soc., 2017, 139, 11032 CrossRef CAS; (b) D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef CAS.
- (a) Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346 CrossRef CAS; (b) P.-Y. Feng, Y.-H. Liu, T.-S. Lin, S.-M. Peng and C.-W. Chiu, Angew. Chem., Int. Ed., 2014, 53, 6237 CrossRef CAS PubMed; (c) Y. Su, Y. Li, R. Ganguly and R. Kinjo, Chem. Sci., 2017, 8, 7419 RSC; (d) B. Wang, Y. Li, R. Ganguly, R. D. Webster and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 7826 CrossRef CAS.
- F. J. Fernández-Alvarez, A. M. Aitani and L. A. Oro, Catal. Sci. Technol., 2014, 4, 611–624 RSC.
- A. Schnurr, H. Vitze, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 6012 CrossRef CAS.
- (a) C. A. Jaska, D. J. H. Emslie, M. J. D. Bosdet, W. E. Piers, T. S. Sorensen and M. Parvez, J. Am. Chem. Soc., 2006, 128, 10885 CrossRef CAS; (b) Z. Zhang, E. S. Penev and B. I. Yakobson, Chem. Soc. Rev., 2017, 46, 6746 RSC; (c) V. M. Hertz, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 8800 CrossRef CAS PubMed; (d) B. Su and R. Kinjo, Synthesis, 2017, 49, 2985 CrossRef CAS.
- E. von Grotthuss, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef CAS PubMed.
- (a) W. Sattler and G. Parkin, J. Am. Chem. Soc., 2011, 133, 9708 CrossRef CAS PubMed; (b) A. N. Kornev, T. A. Chesnokova, E. V. Zhezlova, L. N. Zakharov, G. K. Fukin, Y. A. Kursky, G. A. Domrachev and P. D. Lickiss, J. Organomet. Chem., 1999, 587, 113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental details and characterization data. CCDC 1921501–1921506. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02792k |
| This journal is © The Royal Society of Chemistry 2019 |