
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReply to the ‘Comment on “The chemical reactions in electrosprays of water do not always correspond to those at the pristine air–water interface”’ by A. J. Colussi and S. Enami, Chem. Sci., 2019, 10, DOI: 10.1039/c9sc00991d
Adair
Gallo
Jr.
 abc,
Andreia S. F.
Farinha
abc,
Abdul-Hamid
Emwas
ad,
Adriano
Santana
abc,
Robert J.
Nielsen
e,
William A.
Goddard
III
e and
Himanshu
Mishra
*abc
abc,
Andreia S. F.
Farinha
abc,
Abdul-Hamid
Emwas
ad,
Adriano
Santana
abc,
Robert J.
Nielsen
e,
William A.
Goddard
III
e and
Himanshu
Mishra
*abc
aKing Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: Himanshu.Mishra@Kaust.edu.sa
bWater Desalination and Reuse Center (WDRC), Thuwal 23955-6900, Saudi Arabia
cDivision of Biological and Environmental Sciences (BESE), Thuwal 23955-6900, Saudi Arabia
dCore Labs, Thuwal 23955-6900, Saudi Arabia
eMaterials and Process Simulation Center, California Institute of Technology, Pasadena, CA 91125, USA
First published on 23rd July 2019
Abstract
The air–water interface serves as a crucial site for numerous chemical and physical processes in environmental science and engineering, such as cloud chemistry, ocean-atmosphere exchange, and wastewater treatment. The development of “surface-selective” techniques for probing interfacial properties of water therefore lies at the forefront of research in chemical science. Recently, researchers have adapted electrospray ionization mass spectrometry (ESIMS) to generate microdroplets of water to investigate interfacial phenomena at thermodynamic equilibrium. In contrast, using a broad set of experimental and theoretical techniques, we found that electrosprays of water could facilitate partially hydrated (gas-phase) ions (e.g., H3O+·(H2O)2) to drive/catalyze chemical reactions that are otherwise not possible to accomplish by purely interfacial effects (e.g., enhanced water–hydrophobe surface area) (Chem. Sci., 2019, 10, 2566). Thus, techniques exploiting electrosprays of water cannot be relied upon as generalized surface-selective platforms. Here, we respond to the comments raised by Colussi & Enami (Chem. Sci., 2019, 10, DOI: 10.1039/c9sc00991d) on our paper.
Chemical and physical phenomena at the air–water interface remain a hot topic in chemical science due to their relevance in natural and industrial contexts. In our article, “The chemical reactions in electrosprays of water do not always represent those at the pristine air–water interface”, we assessed the suitability of electrospray ionization mass spectrometry (ESIMS) to investigate thermodynamic properties of the air–water interface. To do so, we used ESIMS, home-built electrosprays, proton nuclear magnetic resonance (1H-NMR), and density functional theory calculations (M06 flavor).1 Our investigation was in response to recent ESIMS-based claims of the “superacidity” of the air–water interface of pH ≤ 3–5 (ref. 2–5) water and recent reports of dramatic rate accelerations in chemical reactions in electrosprayed microdroplets – produced either by shearing water using jets of air6 or applying electrical voltage to water solutions flowing through metallic capillaries7 – leading to the reactions and mass spectrometric characterization of the reactants/products all within ∼1 ms. We wanted to understand if the role of the electrosprays leading to such fast reactions was limited only to producing large, fresh, uncontaminated water–hydrophobe interfacial areas, which is reminiscent of “on-water” chemistries in vigorously agitated oil–water emulsions,8–11 or if the electrosprays of water entailed additional mechanisms that were responsible for the dramatic accelerations. To this end, we investigated chemical reactions between isoprene and pH-adjusted water at normal temperature and pressure (NTP, 293 K, 1 atm) in water–isoprene emulsions and intersecting electrosprays of pH-adjusted water with isoprene gas. Our intention behind choosing the isoprene–water system was to benchmark our findings against previous studies with an aim to unravel underlying mechanisms. We found that:
1. Upon intersecting isoprene gas (partial pressure, p ≈ 0.6 atm at NTP) with electrosprays of water at bulk pH ≤ 4, we observed mass spectral peaks of protonated isoprene (ISO·H+) and protonated oligomers of isoprene (ISO)nH+, within ∼1 ms, as previously reported for isoprene3,5 and terpenes.4 However, we also observed the same mass spectral peaks when gaseous isoprene reacted with electrosprays of water at bulk pH > 12 and electrosprays of water comprising 10−1 to 10−8 M NaCl maintained at pH 5.6 (Fig. 2C, 3C, and S1-B3 in ref. 1). Also, the inflection points of the curves relating the total intensity of the mass spectral peaks of the oligomers and the water-pH shifted towards lower ionic strengths as the capillary voltage was increased.
2. To investigate the effects of water–hydrophobe interfacial area alone (purely interfacial effects) on the chemical reactions, we combined isoprene with pH-adjusted water, 1 ≤ pH ≤ 13, in the volumetric ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:3 (isoprene:water:air) and agitated the emulsions at 1200 rpm for 6, 60, and 360 minutes. These experiments facilitated the formation of both isoprene–water (liquid–liquid) and isoprene–water (gas–liquid) interfaces. We analyzed the organic phases of the emulsions comprising 1 ≤ pH ≤ 4 water by 1H-NMR and ESIMS and found no evidence for oligomers, (ISO)n·H+ (n ≥ 2), using 1H-NMR, whereas the ESIMS spectra were similar to those obtained from gas–liquid collisions, i.e., those containing oligomers (Fig. 1A and C). Curiously, if we condensed the same organic phase that produced no 1H-NMR signal for the oligomers after electrospraying and reanalyzed with 1H-NMR, we found signals for oligomers (Fig. 1C, and 4A1 in ref. 1).
:6:3 (isoprene:water:air) and agitated the emulsions at 1200 rpm for 6, 60, and 360 minutes. These experiments facilitated the formation of both isoprene–water (liquid–liquid) and isoprene–water (gas–liquid) interfaces. We analyzed the organic phases of the emulsions comprising 1 ≤ pH ≤ 4 water by 1H-NMR and ESIMS and found no evidence for oligomers, (ISO)n·H+ (n ≥ 2), using 1H-NMR, whereas the ESIMS spectra were similar to those obtained from gas–liquid collisions, i.e., those containing oligomers (Fig. 1A and C). Curiously, if we condensed the same organic phase that produced no 1H-NMR signal for the oligomers after electrospraying and reanalyzed with 1H-NMR, we found signals for oligomers (Fig. 1C, and 4A1 in ref. 1).
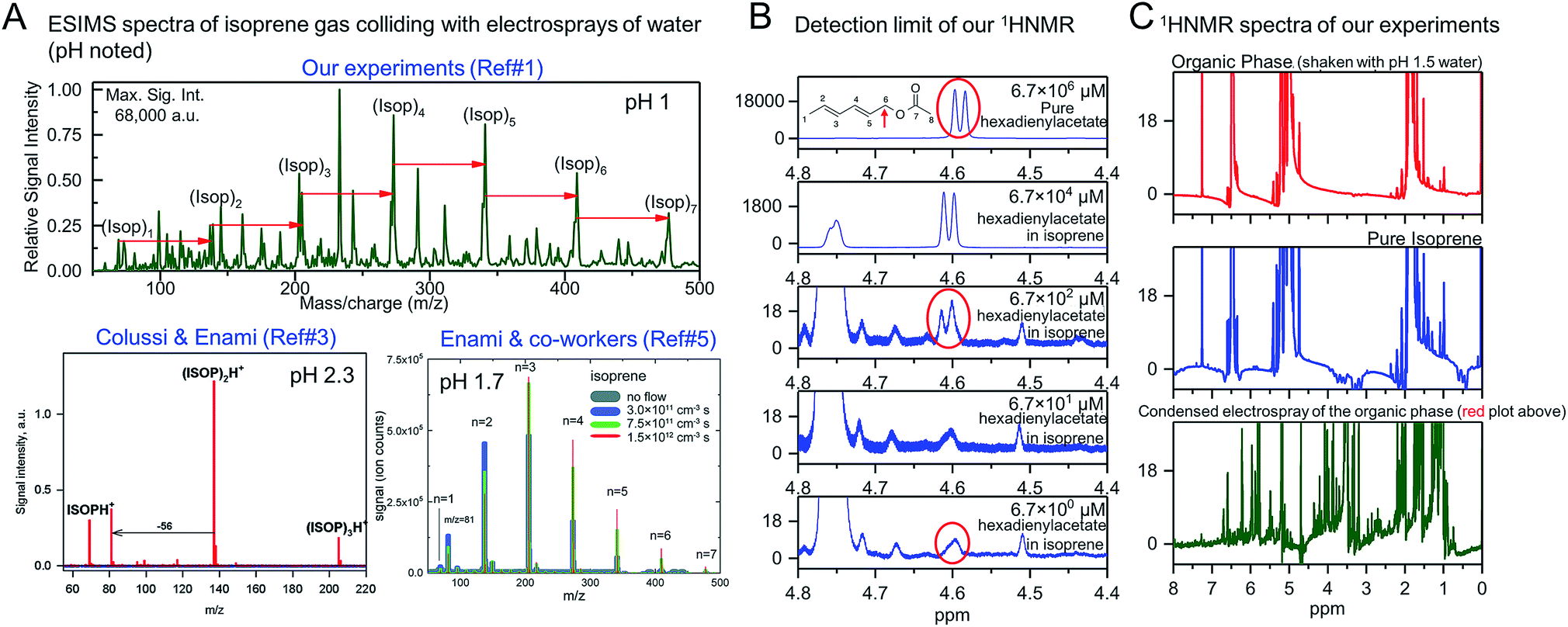 | ||
| Fig. 1 (A) Similar ESIMS spectra obtained when electrosprays of mildly acidic water (1 ≤ pH ≤ 4) are collided with gaseous isoprene in our experiments1 and those of Colussi & co-workers'3 and Enami & co-workers'.5 All the plots demonstrate significantly higher spectral peaks of oligomers ((ISO)n·H+, n ≥ 1) than isoprene ((ISO)·H+) indicating high reaction yields (figures adapted with permission from ref. 3, Copyright 2012 American Chemical Society; and from ref. 5 with permission from the PCCP Owner Societies). (B) Our 1H-NMR data demonstrate that we can detect 2,4-hexadienylacetate, our surrogate for ISO oligomers, at concentrations as low as 6.7 μM even in non-deuterated solutions such as isoprene. (C) The lack of the 1H-NMR spectral peaks for the oligomers in the organic phase (in the range 3–4 ppm) after agitation with pH 1.5 water for 6 hours (red plot) demonstrates that the reaction yields, if any, were below the 10 μM range. This appears to be in contradiction with the significantly high yields observed in ESIMS experiments in (A). Taken together, our ESIMS and 1H-NMR results, along with our quantum calculations, prove that the cationic oligomerization of isoprene takes place exclusively in electrosprays by partially-hydrated hydronium ions, such as H3O+·(H2O)2, which are unavailable at both oil–water and air–water interfaces at NTP conditions. | ||
3. Next, we used density functional theory calculations at the M06/6-311+G*/6-311++G** level to calculate kinetic barriers impeding protonation and oligomerization of ISO molecules on (H2O)2·H3O+ and (H2O)35·H3O+ water clusters as surrogates for gas-phase and interfacial reactions, respectively (Fig. 5 and 6 in ref. 1). Our calculations revealed that the kinetic barriers were consistent with the time-scale of our ESI experiments (∼1 ms) for extremely reactive (H2O)2·H3O+ species reacting with isoprene (gas-phase reactions). On the other hand, for larger (H2O)36·H+ clusters the kinetic barriers were insurmountable under NTP conditions. We note that (H2O)35·H3O+ corresponds to pH −0.2 water, which contains more than three orders of magnitude higher concentration of H3O+ ions than, for instance, in pH ≈ 4 water whose aerial interface exhibits “superacidity” according to Colussi & Enami.2
Taken together, our experiments and calculations bring into question the claims of the superacidity of water at pH ≤ 4 and demonstrate that the chemical reactions in/on electrosprays of water may not always represent those at the air–water interface under NTP.
Colussi & Enami have challenged our findings.12 Here, by raising five questions that we sought to answer in our work, we thoroughly address their points, which are:
i. “Gas-phase ion thermodynamic data alone (i.e., without recourse to ab initio calculations or molecular dynamics simulations) show that partially hydrated H3O+ ions can protonate gas-phase isoprene”.
ii. The significant mutual solubility of isoprene and water leads to “in-water” rather than “on-water” reactions.
iii. Aqueous electrosprays generated by applying voltage to metallic capillaries in our ESIMS experiments are different from theirs that utilized pneumatic nebulization for electrospraying.5 Thus, the chemical reactions investigated by those two systems cannot be compared with each other.
iv. Our 1H-NMR resolution does not have the requisite detection limit to measure oligomers, if any, formed in vigorously shaking liquid isoprene with pH-adjusted water.
v. Acidities of the microdroplets are same as those of the injected aqueous solutions during their residence inside the reaction chamber, and this is reflected by the fact that strong and weak bases show similar titration curves.
1. What is the significance of our ab initio calculations beyond the available thermochemical data? We strongly rely on gas-phase thermochemistry data in interpreting our experimental results and those of Colussi & Enami. For instance, given the proton affinities of isoprene (197 kcal mol−1) and water (166.5 kcal mol−1) under NTP,13 it is straightforward to expect that the gas-phase chemical reaction of ISO(g) + H3O+(g) → ISO·H+ + H2O(g) would proceed downhill with ΔG0 = −31.5 kcal mol−1, which our ab initio calculations also capture within reasonable accuracy (ΔG0 = −31 kcal mol−1) (Fig. S7 in ref. 1). However, as we start adding water molecules one by one to H3O+ leading to H3O+·(H2O)m (m ≥ 1), the kinetics of the proton transfer reaction of ISO(g) + H3O+·(H2O)m (m ≥ 1)(g) → ISO·H+·(H2O)m+1 (m ≥ 1)(g) becomes impossible to predict based on the thermodynamic data alone; reliable theoretical and computational tools are crucial to make such predictions. For instance, the central hypothesis underlying the putative superacidity of pH ≤ 4 water as stated by Colussi & Enami12 is that “partially hydrated hydroniums, however, may conceivably exist at the sharp air–water interface, where the concentration of condensed water drops from 55.5 M to zero in ∼1 nm, i.e., within a couple of molecular diameters”. To be more specific about hydration, Enami and co-workers5 have recently stated that “partially hydrated hydronium species, H3O+·(H2O)m (m < 5) form at the topmost layers of the surface of pH ≤ 4–5 water, which transfers H+ ions to species whose proton affinity is larger than that of gaseous water”. We bring to the reader's attention that the pH of bulk water with a proton concentration equivalent to H3O+·(H2O)m (m = 0–4) is approximately pH < −1, while H3O+·(H2O)55,554 is representative of bulk pH 3 water. Essentially, Enami & Colussi are claiming that the surface of pH ≤ 4 water is the same as H3O+·(H2O)m (m < 5) when, for instance, probed by gas-phase molecules such as isoprene. But such a scenario would necessitate that the H3O+·(H2O)m (m < 5) species appear at the air–water interface and continue to be pulled towards the air until they break all the hydrogen bonds with the rest of the water. This distinction is crucial because our DFT calculations show that even in reactions of isoprene with H3O+·(H2O)35, protonation and oligomerization are impeded by the kinetic barriers of magnitude ΔG‡ = 25.5 kcal mol−1 and ΔG‡ = 40.2 kcal mol−1 (Fig. S7 in ref. 1), rendering those reactions impossible at NTP within ∼1 ms.1 Colussi & Enami suggest that the sharp hydration gradients at the air–water interface are responsible for the appearance of the partially hydrated hydronium ions, such as represented by H3O+·(H2O)m (m < 5),5 and they cited an article by Pratt and co-workers14 to support their speculation. However, we were unable to find any connection between their claim and this reference or any other theoretical model/calculation to our knowledge. To summarize, based on the thermochemical data and our quantum mechanical calculations, protonation and oligomerization of isoprene is impossible at the air–water interface under NTP within ∼1 ms, and the observation of oligomers in electrosprays of water guarantees that the ESIMS process facilitates interactions of isoprene with gas-phase water clusters with extra hydronium ions, such as H3O+·(H2O)2.
2. Based on the mutual solubility of isoprene and water, if the cationic polymerization of isoprene took place in our vigorously agitated emulsions, which scenario would it correspond to: “on water” or “in water”? We commend Colussi & Enami for seeking clarification on the effects of mutual solubility of isoprene and water in our emulsions. They claim that,12 “These studies indicate that, given the significant mutual solubilities of water and liquid isoprene (see below), the interfacial boundary between the two liquids is diffuse. In consequence, isoprene reactions in water emulsions take place “in water” rather than “on water”. Therefore, the fundamental reason why isoprene oligomers were not formed in Gallo et al.'s “in water” experiments was the absence of the partially hydrated hydroniums thermodynamically required to protonate isoprene (and initiate its oligomerization)”. We respectfully disagree. In the section above, we have explained why the cationic polymerization of isoprene at NTP is impossible at the air–water interface (i.e. due to purely interfacial effects or “on-water”) within ∼1 ms. Next, we note that the cationic oligomerization of alkenes (e.g., C3–C8) takes place in 60–70% sulfuric acid solutions15 that are orders of magnitude more acidic than our 1 ≤ pH ≤ 4 solutions, obviating the scope of “in-water” reactions. Thus, the cationic oligomerization of isoprene is neither possible “on-water” nor “in-water” within ∼1 ms when the bulk pH of water ranges within 1–4.
Next, for the sake of semantics alone, we discuss whether the isoprene–water emulsions correspond to “on-water” or “in-water” category based on their mutual solubility. In this context, we draw the reader's attention to Table 1 in a recent study by Butler & co-workers.11 They investigated the Huisgen cycloaddition of phthalazinium-2-dicyanomethanide (melting point 253 °C and solubility in water ≤ 5 μM at 37 °C) with a series of organic molecules in oil–water emulsions as a function of their solubility in water in the 0.58–1.2 × 10−4 M range. Based on the ratio of the endo:exo products in their comprehensive study, where the exo products got enhanced when the reaction took place “on-water”, Butler & coworkers found that the “on-water” reactions took place when the solubility of the organic reactants in water were below 75 mM.11 The solubility of isoprene in water under our experimental conditions is ≤15 mM,1 which is also comparable with the solubilities of several reactants in the well-known report by Sharpless & co-workers8 that coined the term “on-water”. Thus, even from the point of view of semantics, our isoprene–water emulsions pertain to the “on-water” scenario, albeit devoid of chemical reactions.
Lastly, based on the mutual solubilities of water and isoprene, the variation in the concentration of water at the water–isoprene (liquid-phase) system is from 55.5 M to ∼15 mM, whereas that for the air–water interface at thermodynamic equilibrium with the vapor phase at NTP is from 55.5 M to 1.3 mM.16 Thus, we disagree that those two molecular interfaces are dramatically different from each other – one being “diffuse” while the other being “sharp”.
3. Do our ESIMS experiments reproduce their ESIMS experiments? In a recent article, Enami & co-workers aptly noted that, “our method is close to sonic spray ionization mass spectrometry”,5 citing the seminal work of Hirabayashi & co-workers.17 Pneumatic nebulization typically employs fast-flowing jets of neutral gases, such as N2, in a coaxial setting to shear liquids to produce a mist comprising positively and negatively charged droplets along with electrically neutral droplets; when the speed of the nebulizing gas reaches the speed of sound, the method is known as the “sonic” spray ionization (SSI).17,18 The electrically charged droplets can be directed for mass spectrometric detection by applying electric fields.17,18 Similarly to ESI, wherein higher capillary voltage produces higher charging (positive or negative), in pneumatic nebulization, higher gas pressure/velocity produces higher charging (positive and negative).18 The subsequent electrohydrodynamics of the charged droplets leading to the ejection of ions to the gas-phase has been extensively investigated.17–21 In fact, the mass spectra obtained from commercially available pneumatic nebulization setups can be tuned to be quite similar to those obtained by ESI, nanoESI, kilovolt paper sprays and even zero-volt paper sprays, for instance, by adjusting the nebulizing gas pressure, the MS inlet voltage, and the temperature and flow-rate of the drying gas, among other parameters.18,20,22 For instance, Cooks & co-workers demonstrated that the mass spectra are similar using ESI and SSI with a variety of protonated amino acid clusters by spraying diluted solutions of 1:1 water–methanol mixtures.18 Based on the extant experimental literature listing ESIMS operational conditions and using either ESI or pneumatic nebulization and yielding similar mass spectra and on the fact that, using our ESIMS,1,3 we are able to obtain the same (ISO)n·H+ (n ≥ 1) mass spectral peaks as a function of the bulk water pH as Colussi & Enami obtained (Fig. 1A), we are confident that our investigation of chemical reactions of isoprene in aqueous electrosprays is directly comparable to theirs.
In this context, we also remark on a recent report claiming the phosphorylation of common sugars, including glucose, fructose, and ribose, in aqueous microdroplets produced by pneumatic nebulization.6 The authors speculated that the observed chemical reactions were driven by interfacial effects at the edge of the droplets.6 However, their interpretation has been challenged by Jacobs et al., who reproduced the same mass spectra solely through gas-phase reactions realized by (i) ESI of aqueous solutions comprising sugars and phosphoric acid and (ii) atomizing and vaporizing the reactants first (together or through separate streams, and thus eliminating interfacial reactions) followed by MS detection.23 These findings highlight the ambiguities in current understanding of the mechanisms underlying the dramatic rate accelerations in chemical reactions in electrosprays of water conducted at ∼1 ms time-scales. In this scenario and the isoprene–water system, it is clear that gas-phase reactions during electrospraying and/or downstream mass spectrometric detection might crucially influence the outcome of the reactions and purely interfacial effects cannot be guaranteed to be the causation. That is why our investigations combined a broad spectrum of complementary experimental and theoretical/computational platforms to minimize interpretational errors.24
4. What is the significance of our proton nuclear magnetic resonance (1H-NMR) results? Colussi & Enami12 state that, “there is no reason to expect that the extent of isoprene oligomerization would be the same on gas–water and liquid–water interfaces12”. We note that while preparing our emulsions, we added isoprene to pH ≤ 4 water in glass-vials with air comprising the headspace, leading to a volumetric ratio of isoprene, water, and air of 1:6:3. On vigorous agitation, those emulsions facilitated continuous production of fresh isoprene–water (liquid–liquid) and isoprene–water (gas–liquid) interfaces. Thus, if purely interfacial effects (“on-water”) were responsible for the protonation and oligomerization of isoprene on mildly acidic water, due to the superacidity of the air–water interface at pH ≤ 4 as claimed by Colussi & Enami,2–5,12 then there is no reason why we should not have observed these effects despite using pH 1 water (three orders of magnitude higher proton concentration than pH 4 water) and after 360 minutes (>seven orders of magnitude longer duration that the duration of ESIMS experiments of ∼1 ms), and using the 1H-NMR with a detection limit of 10 μM (Fig. 1B). This means that we do not see a reaction yield of even 0.0001% even after 360 min, whereas ESIMS experiments demonstrate significantly higher yields, i.e. in terms of mass spectral intensities, ∑[(ISO)n·H+ (n ≥ 1)] ≫ [(ISO)·H+] (Fig. 1A). However, if we take the organic phases of those emulsions and electrospray them and then condense the vapor and reanalyze by 1H-NMR, we can observe signals for oligomers in similar trends as Colussi & Enami did (Fig. 1C, and 4 in ref. 1). These experiments unambiguously establish that the chemical reactions took place exclusively in the electrosprays (more discussion below). The fact that we could not see the oligomers in our emulsions by 1H-NMR in the first place was because no reactions took place therein; our model calculations attest to these findings.
Out of curiosity, we repeated 1H-NMR studies using nondeuterated isoprene (99% purity from Sigma Aldrich) as the solvent and 2,4-hexadienylacetate as a surrogate of an isoprene oligomer, recognizing that this could be a reason for doubting the sensitivity of our 1H-NMR analysis that is generally carried out in deuterated solvents. To this end, we prepared fresh isoprene solutions and spiked them with 2,4-hexadienylacetate in the range 6.7–6.7 × 106 μM and tracked the doublet peak (J = 6.7 Hz) due to the resonance of H6a and H6b (around 4.6 ppm; see the inset in top panel in Fig. 1B) using our Bruker 700 AVANCE III spectrometer equipped with a Bruker CP TCI multinuclear CryoProbe. We could clearly observe the doublet peak at 670 μM, and as a broad singlet at 6.7 μM (molar fraction: 0.000067%), which confirms the detection limit of our technique.
5. The fate of charged microdroplets in reaction chambers and beyond, and the significance of pH ≤ 4: for mass spectrometric detection, the formation of ions is paramount.25 Since our report establishes that the oligomerization of isoprene entails partially hydrated hydronium ions, the emerging picture is that oligomerization should take place wherever the microdroplets give rise to protonated water clusters of sufficient activity. Whether those gas-phase clusters are produced inside the reaction chamber and/or if it happens inside the mass spectrometer might vary from one ESIMS to another.2–7,23,26–29 It is conceivable that during gas–liquid collision experiments, organic reactant(s) adsorb onto the water surface (Fig. S4 in ref. 1), and even partially dissolve in the droplets, and during their passage for the mass spectrometric detection, chemical reactions take place in the gas-phase.2–7,23,26–28
What should we infer from the wide range of molecules that interact with electrosprays of pH-adjusted water and exhibit similar titration curves due to proton transfers and/or proton-catalyzed reactions?3,12,30–33 Interestingly, the proton affinities of the gases in the gas–liquid collision experiments studied by Colussi & co-workers were always higher than that of water.2–4 This observation suggests that they, in fact, investigated chemical reactions of those reactants with protonated water clusters, just like the one we unraveled in our own work.1 Regarding the dependence of mass spectra on pH, both our experiments and those of others18 reveal that with increasing ionic strengths (adjusted by pH or salts), the formation of electrosprays by pneumatic nebulization18 or by electrical voltage34 becomes easier and the ion counts scale proportionately. This observation could be tested by adding salts and bases to water and repeating the gas–liquid collision experiments, as we did (e.g., Fig. 3C in ref. 1). Given that, we note that there might not always be a one-to-one correspondence in terms of the mass spectral ion counts and the pH, ionic strengths, and pH–pKa relationships due to a host of reasons, including the ion-specific effects in liquid and gas phases,35 electrolytic effects (in ESI), and gas-phase chemistries36 during electrospray ionization mass spectrometry.
To summarize, we caution against the use of electrosprays, produced by electrical voltage or pneumatic nebulization, coupled with mass spectrometry as a bona fide surface-specific platform for investigating rates of chemical reactions at the air–water interface, such as in the context of atmospheric chemistry and environmental science. We advocate combining complementary experimental and theoretical/computational platforms towards testing hypotheses and minimizing interpretational ambiguities and blind-spots.24 We warmly thank Colussi & Enami for their interest in our work and hope that this discussion will prove to be useful to the wider chemical science audience.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by funding from King Abdullah University of Science and Technology (#OSR-2016-CRG5-2992). The co-authors thank Dr Mahmoud Ibrahim (KAUST) for his assistance with the 1H-NMR experiments, Professor Richard Saykally and Professor Evan Williams (University of California Berkeley) for fruitful discussions, and Dr Virginia Unkefer (KAUST) for her assistance in editing the manuscript.References
- A. Gallo, A. S. F. Farinha, M. Dinis, A.-H. Emwas, A. Santana, R. J. Nielsen, W. A. Goddard and H. Mishra, The chemical reactions in electrosprays of water do not always correspond to those at the pristine air–water interface, Chem. Sci., 2019, 10(9), 2566–2577 RSC.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Superacid chemistry on mildly acidic water, J. Phys. Chem. Lett., 2010, 1(24), 3488–3493 CrossRef CAS.
- S. Enami, H. Mishra, M. R. Hoffmann and A. J. Colussi, Protonation and Oligomerization of Gaseous Isoprene on Mildly Acidic Surfaces: Implications for Atmospheric Chemistry, J. Phys. Chem. A, 2012, 116(24), 6027–6032 CrossRef CAS.
- S. Enami, M. R. Hoffmann and A. J. Colussi, Dry deposition of biogenic terpenes via cationic oligomerization on environmental aqueous surfaces, J. Phys. Chem. Lett., 2012, 3, 3102–3108 CrossRef CAS PubMed.
- S. Ishizuka, T. Fujii, A. Matsugi, Y. Sakamoto, T. Hama and S. Enami, Controlling factors of oligomerization at the water surface: why is isoprene such a unique VOC?, Phys. Chem. Chem. Phys., 2018, 20(22), 15400–15410 RSC.
- I. Nam, H. G. Nam and R. N. Zare, Abiotic synthesis of purine and pyrimidine ribonucleosides in aqueous microdroplets, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(1), 36 CrossRef CAS PubMed.
- X. Yan, R. M. Bain and R. G. Cooks, Organic Reactions in Microdroplets: Reaction Acceleration Revealed by Mass Spectrometry, Angew. Chem., Int. Ed., 2016, 55(42), 12960–12972 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, “On water”: Unique reactivity of organic compounds in aqueous suspension, Angew. Chem., Int. Ed., 2005, 44(21), 3275–3279 CrossRef CAS.
- Y. Jung and R. A. Marcus, Protruding interfacial OH groups and ‘on-water’ heterogeneous catalysis, J. Phys.: Condens. Matter, 2010, 22(28), 284117 CrossRef PubMed.
- L. L. Thomas, J. Tirado-Rives and W. L. Jorgensen, Quantum Mechanical/Molecular Mechanical Modeling Finds Diels-Alder Reactions Are Accelerated Less on the Surface of Water Than in Water, J. Am. Chem. Soc., 2010, 132(9), 3097–3104 CrossRef CAS PubMed.
- R. N. Butler, A. G. Coyne, W. J. Cunningham and E. M. Moloney, Water and Organic Synthesis: A Focus on the In-Water and On-Water Border. Reversal of the In-Water Breslow Hydrophobic Enhancement of the Normal endo-Effect on Crossing to On-Water Conditions for Huisgen Cycloadditions with Increasingly Insoluble Organic Liquid and Solid 2π-Dipolarophiles, J. Org. Chem., 2013, 78(7), 3276–3291 CrossRef CAS.
- A. J. Colussi and S. Enami, Comment on “The chemical reactions in electrosprays of water do not always correspond to those at the pristine air–water interface”, Chem. Sci., 2019, 10 10.1039/c9sc00991d.
- E. P. L. Hunter and S. G. Lias, Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update, J. Phys. Chem. Ref. Data, 1998, 27(3), 413–656 CrossRef CAS.
- M. A. Wilson, A. Pohorille and L. R. Pratt, Molecular dynamics of the water liquid-vapor interface, J. Phys. Chem., 1987, 91(19), 4873–4878 CrossRef CAS PubMed.
- V. B. Kazansky, Solvation effects in catalytic transformations of olefins in sulfuric acid, Catal. Rev., 2001, 43(3), 199–232 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 1999, vol. 79 Search PubMed.
- A. Hirabayashi, M. Sakairi and H. Koizumi, Sonic spray mass-spectrometry, Anal. Chem., 1995, 67(17), 2878–2882 CrossRef CAS PubMed.
- Z. Takats, S. C. Nanita, R. G. Cooks, G. Schlosser and K. Vekey, Amino Acid Clusters Formed by Sonic Spray Ionization, Anal. Chem., 2003, 75(6), 1514–1523 CrossRef CAS PubMed.
- W. D. Luedtke, U. Landman, Y. H. Chiu, D. J. Levandier, R. A. Dressler, S. Sok and M. S. Gordon, Nanojets, electrospray, and ion field evaporation: Molecular dynamics simulations and laboratory experiments, J. Phys. Chem. A, 2008, 112(40), 9628–9649 CrossRef CAS PubMed.
- A. Özdemir, J.-L. Lin, Y. S. Wang and C.-H. Chen, A deeper look into sonic spray ionization, RSC Adv., 2014, 4(106), 61290–61297 RSC.
- Y.-G. Lyu, H. Bai, W.-T. Li, J.-K. Yang, Y.-J. He and Q. Ma, Progress of Sonic-Spray Ionization Mass Spectrometry and Its Applications, Chin. J. Anal. Chem., 2019, 47(1), 1–12 Search PubMed.
- M. Wleklinski, Y. Li, S. Bag, D. Sarkar, R. Narayanan, T. Pradeep and R. G. Cooks, Zero Volt Paper Spray Ionization and Its Mechanism, Anal. Chem., 2015, 87(13), 6786–6793 CrossRef CAS PubMed.
- M. I. Jacobs, R. D. Davis, R. J. Rapf and K. R. Wilson, Studying Chemistry in Micro-compartments by Separating Droplet Generation from Ionization, J. Am. Soc. Mass Spectrom., 2019, 30(2), 339–343 CrossRef CAS PubMed.
- J. R. Platt, Strong Inference - Certain Systematic Methods of Scientific Thinking May Produce Much More Rapid Progress Than Others, Science, 1964, 146(364), 7 Search PubMed.
- P. Kebarle and U. H. Verkerk, Electrospray: From Ions in Solution to Ions in the Gas Phase, What We Know Now, Mass Spectrom. Rev., 2009, 28(6), 898–917 CrossRef CAS PubMed.
- S. Banerjee, E. Gnanamani, X. Yan and R. N. Zare, Can all bulk-phase reactions be accelerated in microdroplets?, Analyst, 2017, 142(9), 1399–1402 RSC.
- A. J. Ingram, C. L. Boeser and R. N. Zare, Going beyond electrospray: mass spectrometric studies of chemical reactions in and on liquids, Chem. Sci., 2016, 7(1), 39–55 RSC.
- K. Matsuoka, Y. Sakamoto, T. Hama, Y. Kajii and S. Enami, Reactive Uptake of Gaseous Sesquiterpenes on Aqueous Surfaces, J. Phys. Chem. A, 2017, 121(4), 810–818 CrossRef CAS PubMed.
- Z. Xia and E. R. Williams, Effect of droplet lifetime on where ions are formed in electrospray ionization, Analyst, 2019, 144(1), 237–248 RSC.
- H. Mishra, S. Enami, R. J. Nielsen, L. A. Stewart, M. R. Hoffmann, W. A. Goddard and A. J. Colussi, Bronsted basicity of the air-water interface, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(46), 18679–18683 CrossRef CAS PubMed.
- H. Mishra, S. Enami, R. J. Nielsen, M. R. Hoffmann, W. A. Goddard and A. J. Colussi, Anions dramatically enhance proton transfer through aqueous interfaces, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(26), 10228–10232 CrossRef CAS PubMed.
- H. Mishra, R. J. Nielsen, S. Enami, M. R. Hoffmann, A. J. Colussi and W. A. Goddard, Quantum chemical insights into the dissociation of nitric acid on the surface of aqueous electrolytes, Int. J. Quantum Chem., 2013, 113(4), 413–417 CrossRef CAS.
- A. J. Colussi and S. Enami, Detecting Intermediates and Products of Fast Heterogeneous Reactions on Liquid Surfaces via, Online Mass Spectrom., 2019, 10(2), 47 Search PubMed.
- Y. Wang, M. K. Tan, D. B. Go and H.-C. Chang, Electrospray cone-jet breakup and droplet production for electrolyte solutions, Europhys. Lett., 2012, 99(6), 64003 CrossRef.
- J. Liigand, A. Laaniste and A. Kruve, pH Effects on Electrospray Ionization Efficiency, J. Am. Soc. Mass Spectrom., 2017, 28(3), 461–469 CrossRef CAS PubMed.
- S. L. Zhou and K. D. Cook, Protonation in electrospray mass spectrometry: Wrong-way-round or right-way-round?, J. Am. Soc. Mass Spectrom., 2000, 11(11), 961–966 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |