
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultralong room-temperature phosphorescence of a solid-state supramolecule between phenylmethylpyridinium and cucurbit[6]uril†
Zhi-Yuan
Zhang
 and
Yu
Liu
*
and
Yu
Liu
*
Department of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: yuliu@nankai.edu.cn
First published on 1st July 2019
Abstract
Long-lived organic room-temperature phosphorescence (RTP) has received great attention because of its various potential applications. Herein, we report a persistent RTP of a solid-state supramolecule between a cucurbit[6]uril (CB[6]) host and a heavy-atom-free phenylmethylpyridinium guest. Significantly, the long-lived phosphorescence completely depends on the host–guest complexation, revealing that the non-phosphorescent guest exhibits a 2.62 s ultralong lifetime after being complexed by CB[6] under ambient conditions. The ultralong RTP is because of tight encapsulation of CB[6], which boosts intersystem crossing, suppresses nonradiative relaxation and possibly shields quenchers. Moreover, several phosphorescent complexes possessing different lifetimes are prepared and successfully applied in triple lifetime-encoding for data encryption and anti-counterfeiting. This strategy provides a new insight for realizing purely organic RTP with ultralong lifetime and expands its application in the field of information protection.
Introduction
Organic room-temperature phosphorescence (RTP) with persistent luminescence has received enormous attention because of its various applications in biological imaging,1,2 light-emitting devices,3 information storage and encryption,4,5 and so on. However, achieving ultralong RTP (generally from seconds to hours) is extremely difficult due to two main issues: (i) the intrinsically weak spin–orbit coupling of purely organic molecules makes it hard to achieve efficient intersystem crossing (ISC); (ii) a long triplet state (T1) lifetime means that T1 will suffer tremendously from quenching by a very low concentration of impurities even in crystals.6 Several methods have been reported to actualize long-lived RTP, such as forming charge-separated states,7 crystallization,8–17 polymerization,18–21 embedding into suitable matrices,22–24 and so on.25–27 Recently, Huang and co-workers achieved lifetimes of 1.35 s and 1.91 s by reasonable crystal design.28,29 Fukushima and Nakai et al. found that the lifetime of arylboronic esters in crystals could reach 1.85 s due to an appropriate molecular packing.30 Zhao and co-workers reported ultralong RTP with a lifetime of 0.75 s by embedding phosphors into polymers.31 Tian and co-workers developed purely organic amorphous polymers with a lifetime of 0.537 s by utilizing hydrogen-bonding networks between polymeric chains, and prepared cyclodextrin based phosphors.32–34 Wu and co-workers reported self-assembled nanoparticles with RTP for cell imaging.35 Besides, theoretical descriptors were also utilized to assist in the molecular design of efficient and long-lived RTP.36More recently, we found that the phosphorescence quantum yield of bromophenyl-methyl-pyridinium chloride (PYCl) could reach 81.2% after being complexed by cucurbit[6]uril (CB[6]).37 In general, the introduction of heavy atoms facilitates ISC and thus contributes to high phosphorescence quantum yield, but the heavy atoms will result in a shorter lifetime (normally below a millisecond) because of the acceleration of radiative and non-radiative decay rates of the triplet state.6 The high efficiency and short lifetime (τ = 5.40 ms) of the complex (PYCl/CB[6]) in our recent work is a typical case.37 Therefore, removing a heavy atom (Br) from PYCl will slow down radiative and non-radiative decay of the triplet state and probably achieve long-lived RTP. It is reasonable to realize this proposal by a solid-state supramolecular strategy because the aforementioned issues in achieving ultralong RTP will no longer exist: firstly, the encapsulation of CB[6] promotes ISC of the guest and thus boosts the production of triplet states, which will (partially) offset the absence of the heavy atom; secondly, the extreme restriction of the guest by CB[6] will greatly suppress molecular motions (vibrations, rotations, and inter-collisions) and prolong the lifetime of phosphorescence; thirdly, CB[6] will act as a fine shell to prevent quenchers (oxygen, impurities and so on) from attacking triplet states. To verify our hypothesis, phenylmethylpyridinium chloride (PBC) was designed and synthesized. Furthermore, a series of derivatives were prepared to explore the generalizability of the solid-state supramolecular strategy and uncover the relationship between the structure of guests and the phosphorescence properties of complexes (Scheme 1 and Fig. S1–S14†).
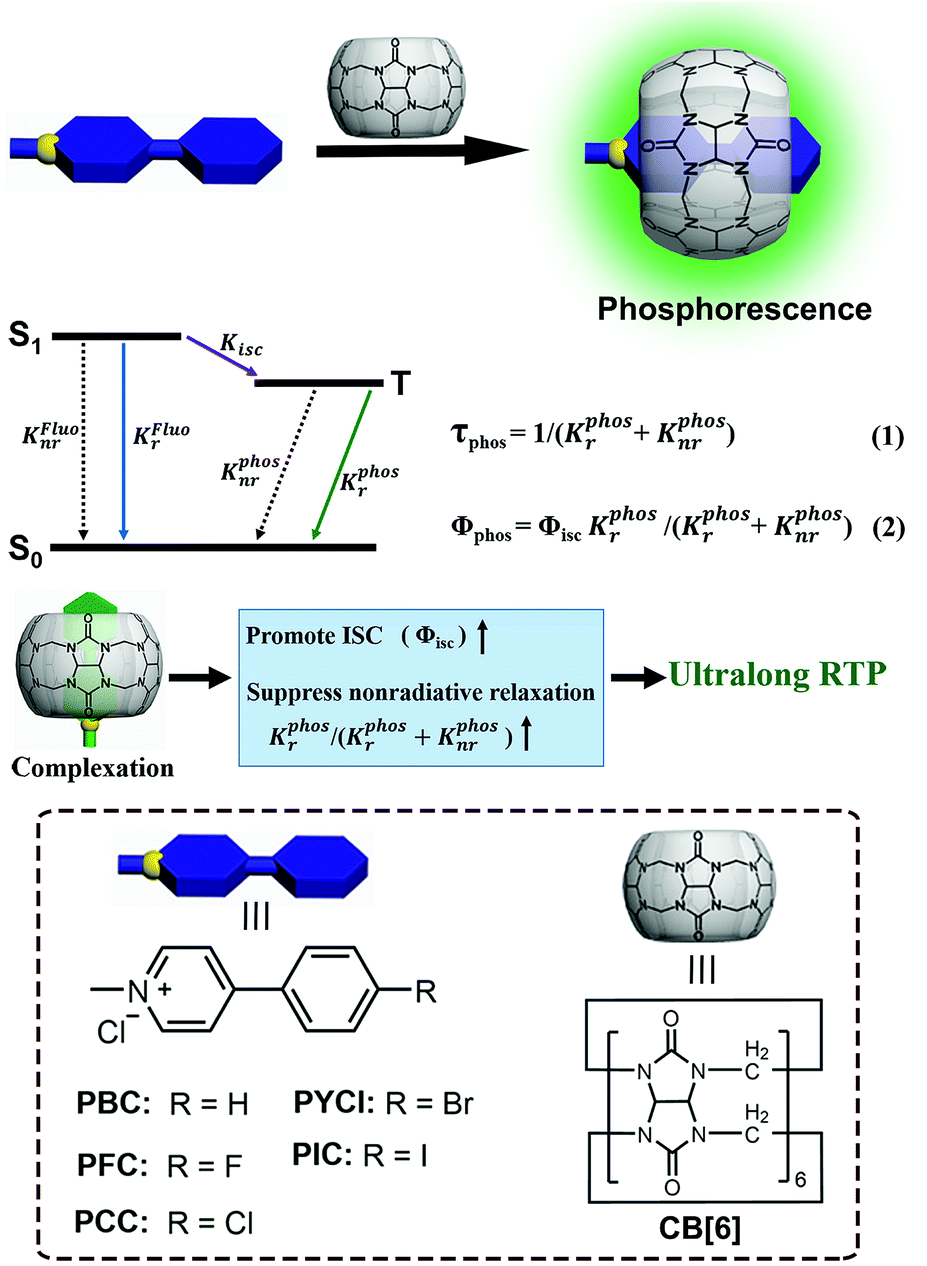 | ||
| Scheme 1 Schematic illustration of the solid-state supramolecular strategy and Jablonski diagram for radiative and non-radiative processes. (KFluor is the radiative rate constant of the lowest excited singlet state S1; KFluonr is the nonradiative rate constant of the lowest excited singlet S1; Kisc is the intersystem crossing rate constant from the excited singlet state to the triplet state; KPhosr is the radiative rate constant of the lowest excited triplet state T1; KPhosnr is the nonradiative rate constant of the lowest excited triplet state T1; τPhos is the lifetime of the excited triplet state; ΦPhos is the quantum yield of phosphorescence; and Φisc is the quantum yield of intersystem crossing from the excited singlet state to the triplet state; inset: the molecular structures of the guests and host). | ||
Experimental
Materials and methods
All chemicals were commercially available unless noted otherwise. NMR spectroscopy was performed on a Bruker AV400 spectrometer. XRD patterns obtained at Rigaku SmartLab 3 kW. High-resolution mass spectrometry (HR-MS) was performed on a Q-TOF LC-MS in ESI/MALDI mode. Photoluminescence spectra and lifetimes were measured on an FLS900 and FLS1000. Phosphorescence quantum efficiencies were measured on a HAMAMATSU C9920-02.Synthesis of PX
Compound PBC was synthesized according to the literature.38 4-Phenylpyridine (1.09 g, 5.85 mmol) and methyl iodide (2 mL, 30 mmol) were dissolved in anhydrous ethanol (50 mL) and then heated under reflux for 24 h. After being cooled to room temperature, the reaction mixture was concentrated and the resulting pale gold precipitate was washed with dichloromethane and then dried under vacuum to yield a pale gold solid (1.32 g, 76%). After dispersing the solid in 30 mL acetonitrile, saturated aqueous NH4PF6 was added until all of the solid dissolved. After evaporating the acetonitrile and filtering, a white solid was obtained. After dissolving the solid in CH3CN, tetrabutylammonium chloride was added until no solid appeared any more. PBC (white solid) was obtained after filtering and washing with CH3CN (10 mL × 3). 1H NMR (400 MHz, DMSO) δ 9.12 (d, J = 6.2 Hz, 2H), 8.53 (d, J = 6.3 Hz, 2H), 8.09 (d, J = 5.6 Hz, 2H), 7.64 (d, J = 5.8 Hz, 3H), 4.38 (s, 3H); 13C NMR (101 MHz, DMSO) δ 154.16, 145.68, 133.54, 131.99, 129.64, 128.05, 124.06, 46.96.Compound PFC was synthesized according to the literature.38 Cesium carbonate (0.98 g, 3.0 mmol) and tetrakis(triphenylphosphine)palladium (0.23 g, 0.20 mmol) were added to a stirred solution of 4-bromopyridine hydrochloride (0.49 g, 2.5 mmol) and 4-fluorobenzeneboronic acid (0.42 g, 3.0 mmol) in dioxane/methanol (125 mL). The mixture was heated under reflux for 24 h and then concentrated. The slurry was triturated with ethyl acetate (40 mL) and the organic phase was washed with saturated aqueous sodium bicarbonate (30 mL × 3), water (30 mL × 2), and brine (100 mL), and then dried over sodium sulfate. After removal of the solvent by evaporation, the resulting residue was subjected to column chromatography (petroleum ether/ethyl acetate 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give a white solid. Then the solid reacted with methyl iodide and was purified in a similar manner to PBC. After ion exchange, PFC was obtained as a white solid. 1H NMR (400 MHz, D2O) δ 8.69 (d, J = 6.5 Hz, 2H), 8.16 (d, J = 6.5 Hz, 2H), 7.88 (dd, J = 8.6, 5.3 Hz, 2H), 7.28 (t, J = 8.7 Hz, 2H), 4.32 (s, 3H); 13C NMR (101 MHz, D2O) δ 166.16, 163.66, 154.96, 144.81, 130.35, 124.44, 116.75, 47.17; HRMS (m/z): [M–Cl]+ calcd for C12H11NF+, 188.0875; found, 188.0874.
:1) to give a white solid. Then the solid reacted with methyl iodide and was purified in a similar manner to PBC. After ion exchange, PFC was obtained as a white solid. 1H NMR (400 MHz, D2O) δ 8.69 (d, J = 6.5 Hz, 2H), 8.16 (d, J = 6.5 Hz, 2H), 7.88 (dd, J = 8.6, 5.3 Hz, 2H), 7.28 (t, J = 8.7 Hz, 2H), 4.32 (s, 3H); 13C NMR (101 MHz, D2O) δ 166.16, 163.66, 154.96, 144.81, 130.35, 124.44, 116.75, 47.17; HRMS (m/z): [M–Cl]+ calcd for C12H11NF+, 188.0875; found, 188.0874.
Compound PCC was prepared as a white powder according to a procedure similar to that described for compound PBC: 1H NMR (400 MHz, DMSO) δ 9.10 (d, J = 6.8 Hz, 2H), 8.54 (d, J = 6.9 Hz, 2H), 8.13 (d, J = 8.6 Hz, 2H), 7.72 (d, J = 8.6 Hz, 2H), 4.36 (s, 3H); 13C NMR (101 MHz, DMSO) δ 152.92, 145.77, 137.12, 132.42, 129.96, 129.69, 124.13, 47.08.
Compound PIC was prepared as a pale yellow powder according to a procedure similar to that described for compound PFC: 1H NMR (400 MHz, D2O) δ 8.70 (d, J = 6.5 Hz, 2H), 8.15 (d, J = 6.5 Hz, 2H), 7.89 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 4.33 (s, 3H); 13C NMR (101 MHz, D2O) δ 154.96, 144.92, 138.72, 133.03, 129.22, 124.38, 98.87, 47.33; HRMS (m/z): [M–Cl]+ calcd for C12H11NI+, 295.9936; found, 295.9934.
Compound PEC: 1H NMR (400 MHz, DMSO) δ 8.93 (d, J = 6.8 Hz, 2H), 8.26 (d, J = 6.8 Hz, 2H), 8.05 (d, J = 16.4 Hz, 1H), 7.76 (d, J = 6.8 Hz, 2H), 7.59–7.42 (m, 4H), 4.28 (s, 3H); 13C NMR (101 MHz, DMSO) δ 152.43, 145.20, 140.58, 135.18, 130.39, 129.14, 128.12, 123.59, 123.36, 46.89.
Compound PC: 1H NMR (400 MHz, DMSO) δ 9.13 (d, J = 5.8 Hz, 2H), 8.59 (t, J = 7.8 Hz, 1H), 8.15 (t, J = 7.1 Hz, 2H), 4.41 (s, 3H); 13C NMR (101 MHz, DMSO) δ 145.66, 145.05, 127.67, 47.81.
Results and discussion
Using this strategy, we successfully realized ultralong RTP. The chromophore PBC itself emitted only blue fluorescence (λmax = 441 nm) with a lifetime (τ) of 2.25 ns (Fig. S15†). Notably, after forming the PBC/CB[6] complex by a grinding method,37 the solid emitted a delayed luminescence (λmax = 510 nm) with an ultralong phosphorescence of 2.62 s, along with fluorescence at λmax = 427 nm (τ = 8.40 ns) (Fig. 1a, b and S16†). And the afterglow could last more than 9 s after turning off the UV lamp (Fig. 1c and Video 1†). This excellent long luminescence verified our hypothesis that the removal of the heavy atom could prolong the lifetime of the complex and demonstrated that the complexation of CB[6] could “turn on” the phosphorescence of PBC. Furthermore, the decent phosphorescence quantum yield (9.7%) indicated that the encapsulation of CB[6] promoted ISC of the guest even without the assistance of the heavy atom (Table 1 and Fig. S17b†). Indeed, the intersystem crossing rate (Kisc) was as high as 1.2 × 107 s−1. Considering that there was no heavy atom to promote ISC, the result, which was only one magnitude smaller than that of PYCl/CB[6] (2.2 × 108 s−1), was satisfactory. Significantly, the radiative decay rate of phosphorescence (KPhosr) was very low (3.7 × 10−2 s−1), about 4000-fold lower than that of PYCl/CB[6] (150 s−1), revealing that the absence of the heavy atom would slow down the decay of T1 and favor long lifetimes. Normally, a long T1 lifetime means that T1 will suffer tremendously from quenching by a very low concentration of impurities because of triplet energy migration, so that extremely pure samples and complete exclusion of oxygen are needed in order to observe ultralong RTP.6 However, these rigorous conditions were no longer necessary in our case; the PBC/CB[6] powder showed quite robust persistent RTP even upon being exposed to ambient conditions. Two reasons were proposed for this property: (i) CB[6] played the role of a tight shell to prevent quenchers (oxygen, impurities and so on) from attacking the chromophoric core (PBC); (ii) every PBC molecule possessed an individual micro-circumstance provided by CB[6]. The large outer diameter (14.4 Å)39 and high rigidity of CB[6] made each complex an independent phosphor unit. Possible defects (empty CB[6] or free PBC) would not produce serious triplet energy migration and therefore had little effect on the whole phosphorescence. They jointly stabilized the triplet state and guaranteed the robust persistent RTP.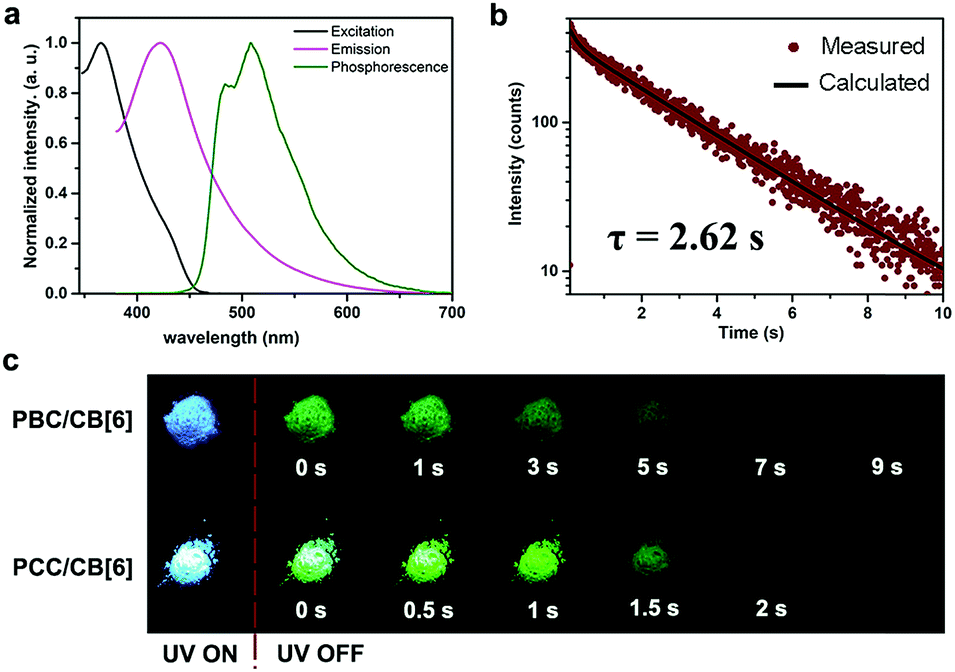 | ||
| Fig. 1 Photophysical properties of PX/CB[6]. (a) Excitation (black), photoluminescence (magenta) and phosphorescence spectra (olive) of PBC/CB[6] in the solid state; (b) time resolved PL decay of PBC/CB[6] at 510 nm in the solid state at room temperature; (c) luminescence photographs of PBC/CB[6] and PCC/CB[6] powder under 365 nm UV irradiation and at different time intervals after removal of the ultraviolet lamp. | ||
| Compound | EX (nm) | λ Fluo (nm) | λ Phos (nm) | τ Fluo (ns) | τ Phos (ms) | Φ Fluo (%) | Φ Phos (%) | K Fluor (s−1) | K Fluonr (s−1) | K isc (s−1) | K Phosr (s−1) | K Phosnr (s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The radiative decay rate constant of fluorescence KFluor = ΦFluo/τFluo. b The nonradiative decay rate constant of fluorescence KFluonr = (1 − ΦFluo − ΦPhos)/τFluo. c The intersystem crossing rate constant Kisc = ΦPhos/τFluo. d The radiative decay rate constant of phosphorescence KPhosr = ΦPhos/τPhos. e The nonradiative decay rate constant of phosphorescence KPhosnr = (1 − ΦPhos)/τPhos. f Not detected. g Not calculated. | ||||||||||||
| PBC/CB[6] | 366 | 427 | 510 | 8.40 | 2620 | 17.3 | 9.7 | 2.1 × 107 | 8.7 × 107 | 1.2 × 107 | 3.7 × 10−2 | 0.34 |
| PFC/CB[6] | 330 | 434 | 520 | 5.61 | 0.0095 | 27.7 | 1.9 | 4.9 × 107 | 1.2 × 108 | 1.4 × 107 | 2.0 × 103 | 1.0 × 105 |
| PCC/CB[6] | 370 | 420 | 500 | 2.52 | 275 | 41.8 | 26.7 | 1.6 × 108 | 1.2 × 108 | 1.1 × 108 | 0.97 | 2.7 |
| PYCl/CB[6] | 334 | 388 | 500 | 3.62 | 5.40 | 3.7 | 81.2 | 1.0 × 107 | 4.2 × 107 | 2.2 × 108 | 150 | 34.8 |
| PIC/CB[6] | 407 | 477 | 575 | 1.53 | 1.40 | 2.6 | 3.9 | 1.7 × 107 | 6.1 × 108 | 2.5 × 107 | 27.9 | 6.9 × 102 |
| PEC/CB[6] | 429 | 494 | —f | 7.52 | —f | 4.34 | —f | 5.8 × 106 | 1.3 × 108 | —g | —g | —g |
| PC/CB[6] | 250 | 494 | —f | 23.14 | —f | 2.60 | —f | 1.1 × 106 | 4.2 × 107 | —g | —g | —g |
The complex was further characterized by 1H NMR, which revealed that methyl protons Ha and the aromatic protons Hb, and He,f of PBC underwent enormous upfield shifts of 0.15, 0.29 and 0.26 ppm, respectively (Fig. 2a). These complexation-induced shifts indicated that PBC was deeply encapsulated into the cavity of CB[6]. High resolution mass spectrometry also proved the formation of the PBC/CB[6] complex (Fig. 2b). The intense peak of [M–Cl]+ (m/z 1166.3912) fitted well with the calculated value (1166.3914). The powder X-ray diffraction (XRD) pattern of the PBC/CB[6] complex revealed its ordered array like that of PYCl/CB[6] (Fig. 2c). The XRD peaks with d-spacings of 5.58 and 3.84 Å were indexed to diffractions from the (220) and (330) planes, respectively. The d-spacings of 7.00, 4.44, 3.51, and 3.27 Å were assigned to (020), (400), (040), and (240) planes (Fig. 2c). Taking into account the extremely similar structures of PBC and PYCl, this result was reasonable. To verify if the arrangement of the complex influences the lifetime, we further ground the complex powder. The XRD patterns revealed that many peaks became weaker and some even disappeared, indicating the loss of order (Fig. S18a†). But the nearly identical lifetimes (2.61 s and 2.62 s) confirmed that the change in the microstructure had little effect on the phosphorescence of the complex, which further proved the aforementioned proposal that each complex was an individual phosphor and the phosphorescence originated from complexation (Fig. S18b†).
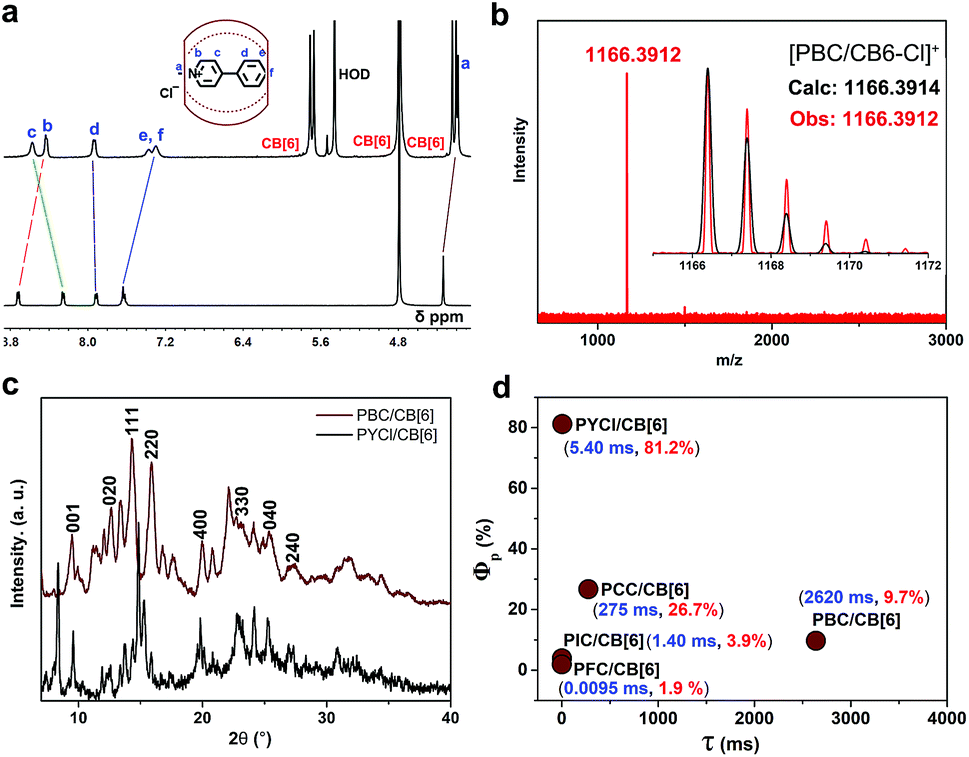 | ||
| Fig. 2 (a) 1H NMR spectra of PBC/CB[6] and PBC; (b) high resolution mass spectrometry of PBC/CB[6]; (c) XRD patterns of PBC/CB[6] (wine) and PYCl/CB[6] (black); (d) phosphorescence quantum yield (Φp) and lifetime (τ) of PX/CB[6]. | ||
We then synthesized guests with halogen atoms (F, Cl and I) possessing different coupling abilities to explore the relationship between the structure of the guests and the phosphorescence properties of the complexes. Besides, methyl-pyridinium chloride (PC) and styryl-methyl-pyridinium chloride (PEC) were also synthesized to verify whether the structure of phenylmethylpyridinium was necessary for generating phosphorescence (Scheme 1). Interestingly, the PFC/CB[6], PCC/CB[6] and PIC/CB[6] complexes emitted phosphorescence peaks at 520 nm (τ = 0.0095 ms), 500 nm (τ = 275 ms) and 575 nm (τ = 1.40 ms), accompanied by strong fluorescence emission peaks at 434 nm (τ = 5.61 ns), 420 nm (τ = 2.52 ns) and 477 nm (τ = 1.53 ns) in the solid state (Table 1 and Fig. S19–S21†). In contrast, guests only showed fluorescence peaks at 443 nm (τ = 10.62 ns) for PFC, 434 nm (τ = 3.49 ns) for PCC and 505 nm (τ = 1.20 ns) for PIC (Fig. S22†). These results proved the universality of the solid-state supramolecular strategy and indicated that the phosphorescence properties of the complex could be tuned by substituents. When the aromatic moieties were separated (PEC) or broken (PC), no phosphorescence occurred in the monomer (PEC and PC) or the complex (PEC/CB[6] and PC/CB[6]) (Fig. S23 and S24†). This means that the structure of (substituted) phenylmethylpyridinium was indispensable for generating phosphorescence of the complex in these cases. The high resolution mass spectra showed an intense peak of [PX/CB6-Cl]+, which proved the complexation between these guests and CB[6] (Fig. S25†). Moreover, enormous upfield shifts of methyl protons and aromatic protons of guests further confirmed that these guests were deeply complexed by CB[6] (Fig. S26–S30†).
On the basis of the measured quantum yields and lifetimes of these complexes, the radiative and nonradiative decay rate constants could be calculated following the standard methods (Table 1).28,40–42 Eqn (1) and (2) in Scheme 1 indicated that small KPhosr + KPhosnr was beneficial for achieving long lifetime, while high ΦPhos was the comprehensive result of efficient ISC (high Φisc) and high efficiency of phosphorescence (KPhosr/(KPhosr + KPhosnr)). The intersystem crossing rate constant Kisc of complexes ranged between 1.2 × 107 and 2.2 × 108 s−1. The fast ISC of S1 → Tn (n ≥ 1) (1.1 × 108 s−1 for PCC/CB[6] and 2.2 × 108 s−1 for PYCl/CB[6]) produced a sufficient population of T1 and therefore provided one of the essential conditions for efficient phosphorescence. The slow nonradiative decay (KPhosnr = 2.7 s−1 for PCC/CB[6] and 34.8 s−1 for PYCl/CB[6]) of phosphorescence provided another one, resulting in high phosphorescence quantum yield (81.2% for PYCl/CB[6] and 26.7% for PCC/CB[6]). For PBC/CB[6], the extremely low value of KPhosr + KPhosnr (0.377 s−1) made T1 deactivation a very slow process and therefore resulted in ultralong lifetime. The KPhosr + KPhosnr of PCC/CB[6] was also small enough (3.67 s−1) to produce an afterglow (Fig. 1c and Video 2†). Moreover, there was no positive correlation between the phosphorescence quantum yield (ΦPhos) of the complexes and the atomic number of the substituted halogen (Table 1 and Fig. S31–S33†). A possible explanation was proposed: according to eqn (2) in Scheme 1, the increase of atomic number (from F to I) would improve Φisc and KPhosr, which was beneficial for achieving high ΦPhos.6 But KPhosnr would be improved too and ΦPhos would decrease. Therefore, KPhosr/(KPhosr + KPhosnr) would increase or decrease, making ΦPhos either higher or lower.
Information encryption and anti-counterfeiting are of great significance and have received enormous attention in this information era.43–49 On the basis of distinct lifetime and robust phosphorescence properties of these complexes, a triple encoding model was fabricated. As illustrated in Fig. 3 (dotted box), the digital pattern was painted with different complexes on flexible paper, among which PYCl/CB[6] was used to label the black part, the dark gray part was coated with PCC/CB[6], and the light gray part was painted with PBC/CB[6]. In daylight, only a colorless pattern was obtained. When a UV lamp with 365 nm excitation was turned on, an intense green “1054” appeared because of the high quantum yield of PYCl/CB[6] (Fig. 3 and Video 3†). On switching the UV lamp off, the relatively short lifetime of PYCl/CB[6] (5.40 ms) made its phosphorescence disappear immediately, while long-lived PCC/CB[6] (275 ms) and PBC/CB[6] (2620 ms) contributed to the moderate green “6849”. Later, the phosphorescence of PCC/CB[6] became invisible, and the pattern that remained was “5213”. Therefore, we achieved triple lifetime-encoding for information encryption and anti-counterfeiting by subtle utilization of the diverse lifetimes of phosphorescent complexes.
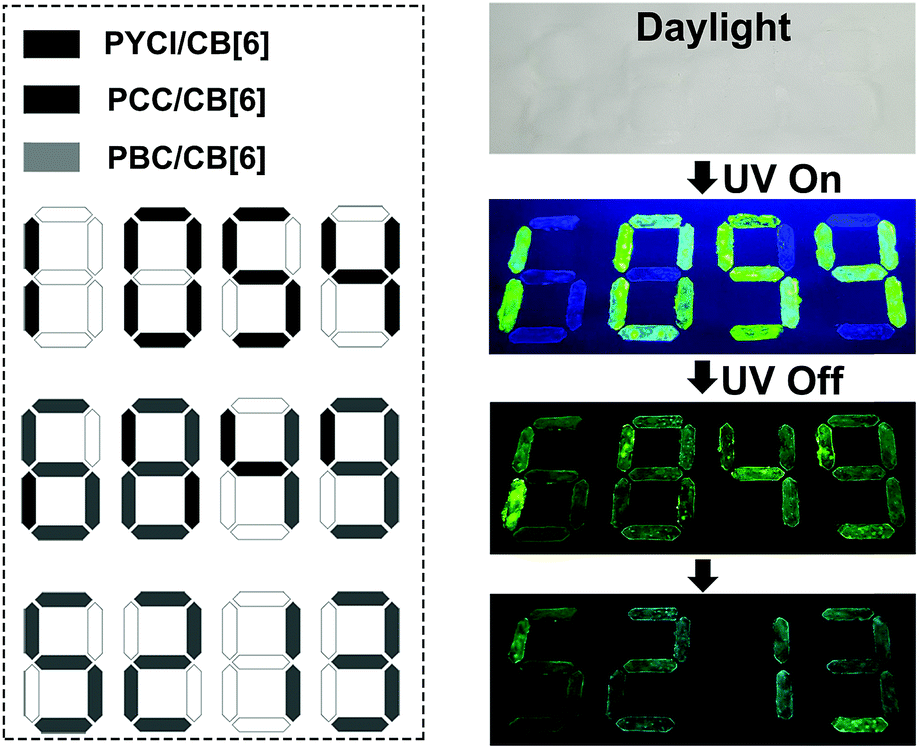 | ||
| Fig. 3 Schematic illustration of lifetime-encoding for security applications using PYCl/CB[6], PCC/CB[6] and PBC/CB[6]. | ||
Conclusion
In conclusion, we present a concise and universal solid-state supramolecular strategy to realize purely organic heavy-atom-free ultralong RTP based on CB[6] and phenylmethylpyridinium. The strategy involving the enhancement of ISC and suppression of nonradiative decay through the complexation of CB[6] can remarkably promote RTP of phenylmethylpyridinium and its derivatives under ambient conditions. Significantly, PBC/CB[6] powder exhibits a phosphorescence lifetime of 2.62 s and a decent efficiency of 9.7%. Moreover, the structure of phenylmethylpyridinium (PBC, PFC, PCC, PYCl and PIC) is vital for the generation of phosphorescence. Notably, PBC/CB[6], PCC/CB[6] and PYCl/CB[6] were successfully applied in triple lifetime-encoding by utilizing their different lifetimes, showing great potential for application in information encryption and anti-counterfeiting. This strategy possesses the advantages of facile preparation, universality and robust RTP without rigorous conditions such as low temperature, crystallization and deoxygenation. The result will help to understand purely organic ultralong RTP and promote the generation of better (longer lifetime, higher efficiency and more robust) RTP materials that may stimulate the development of data encryption, organic devices, bio-imaging, and so on.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSFC (21432004, 21672113, 21772099, and 21861132001) for financial support.Notes and references
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508–2524 RSC.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747 CrossRef CAS PubMed.
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- S. Cai, H. Shi, J. Li, L. Gu, Y. Ni, Z. Cheng, S. Wang, W.-w. Xiong, L. Li, Z. An and W. Huang, Adv. Mater., 2017, 29, 1701244 CrossRef PubMed.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384 CrossRef CAS PubMed.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- T. Zhang, Z. Zhao, H. Ma, Y. Zhang and W. Z. Yuan, Chem.–Asian J., 2019, 14, 884–889 CrossRef CAS PubMed.
- Z. Mao, Z. Yang, Z. Fan, E. Ubba, W. Li, Y. Li, J. Zhao, Z. Yang, M. P. Aldred and Z. Chi, Chem. Sci., 2019, 10, 179–184 RSC.
- G. Chen, H. Feng, F. Feng, P. Xu, J. Xu, S. Pan and Z. Qian, J. Phys. Chem. Lett., 2018, 9, 6305–6311 CrossRef CAS PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205 CrossRef CAS PubMed.
- Z. He, H. Gao, S. Zhang, S. Zheng, Y. Wang, Z. Zhao, D. Ding, B. Yang, Y. Zhang and W. Z. Yuan, Adv. Mater., 2019, 31, 1807222 CrossRef PubMed.
- T. Ogoshi, H. Tsuchida, T. Kakuta, T.-a. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- S. M. Parke, E. Hupf, G. K. Matharu, I. de Aguiar, L. Xu, H. Yu, M. P. Boone, G. L. C. de Souza, R. McDonald, M. J. Ferguson, G. He, A. Brown and E. Rivard, Angew. Chem., Int. Ed., 2018, 57, 14841–14846 CrossRef CAS PubMed.
- H. Mieno, R. Kabe, N. Notsuka, M. D. Allendorf and C. Adachi, Adv. Opt. Mater., 2016, 4, 1015–1021 CrossRef CAS.
- H. Wu, W. Chi, Z. Chen, G. Liu, L. Gu, A. K. Bindra, G. Yang, X. Liu and Y. Zhao, Adv. Funct. Mater., 2019, 29, 1807243 Search PubMed.
- Q. Li, M. Zhou, M. Yang, Q. Yang, Z. Zhang and J. Shi, Nat. Commun., 2018, 9, 734 CrossRef PubMed.
- S. Hirata, K. Totani, T. Yamashita, C. Adachi and M. Vacha, Nat. Mater., 2014, 13, 938 CrossRef CAS PubMed.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Adv. Mater., 2018, 30, 1800783 CrossRef PubMed.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685 CrossRef CAS PubMed.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728–2733 CrossRef CAS PubMed.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- X. Ma, C. Xu, J. Wang and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 10854–10858 CrossRef CAS PubMed.
- D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed.
- Y. Zhang, T.-Y. Zhou, K.-D. Zhang, J.-L. Dai, Y.-Y. Zhu and X. Zhao, Chem.–Asian J., 2014, 9, 1530–1534 CrossRef CAS PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- C. C. Byeon, M. M. McKerns, W. Sun, T. M. Nordlund, C. M. Lawson and G. M. Gray, Appl. Phys. Lett., 2004, 84, 5174–5176 CrossRef CAS.
- S.-J. Yoon, J. H. Kim, K. S. Kim, J. W. Chung, B. Heinrich, F. Mathevet, P. Kim, B. Donnio, A.-J. Attias, D. Kim and S. Y. Park, Adv. Funct. Mater., 2012, 22, 61–69 CrossRef CAS.
- P. C. Y. Chow, S. Albert-Seifried, S. Gélinas and R. H. Friend, Adv. Mater., 2014, 26, 4851–4854 CrossRef CAS PubMed.
- Y. Zhou, S.-T. Han, X. Chen, F. Wang, Y.-B. Tang and V. A. L. Roy, Nat. Commun., 2014, 5, 4720 CrossRef CAS PubMed.
- J. Xue, Z.-K. Zhou, Z. Wei, R. Su, J. Lai, J. Li, C. Li, T. Zhang and X.-H. Wang, Nat. Commun., 2015, 6, 8906 CrossRef CAS PubMed.
- Q. Huang, X. Mei, Z. Xie, D. Wu, S. Yang, W. Gong, Z. Chi, Z. Lin and Q. Ling, J. Mater. Chem. C, 2019, 7, 2530–2534 RSC.
- Z. Wang, C.-Y. Zhu, S.-Y. Yin, Z.-W. Wei, J.-H. Zhang, Y.-N. Fan, J.-J. Jiang, M. Pan and C.-Y. Su, Angew. Chem., Int. Ed., 2019, 58, 3481–3485 CrossRef CAS PubMed.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Chem. Mater., 2017, 29, 4866–4873 CrossRef CAS.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef PubMed.
- Q. Zhou, Z. Wang, X. Dou, Y. Wang, S. Liu, Y. Zhang and W. Z. Yuan, Mater. Chem. Front., 2019, 3, 257–264 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02633a |
| This journal is © The Royal Society of Chemistry 2019 |