DOI:
10.1039/C9SC02598G
(Edge Article)
Chem. Sci., 2019,
10, 10097-10105
Electrostatically PEGylated DNA enables salt-free hybridization in water†
Received
28th May 2019
, Accepted 11th September 2019
First published on 12th September 2019
Abstract
Chemically modified nucleic acids have long served as a very important class of bio-hybrid structures. In particular, the modification with PEG has advanced the scope and performance of oligonucleotides in materials science, catalysis and therapeutics. Most of the applications involving pristine or modified DNA rely on the potential of DNA to form a double-stranded structure. However, a substantial requirement for metal-cations to achieve hybridization has restricted the range of applications. To extend the applicability of DNA in salt-free or low ionic strength aqueous medium, we introduce noncovalent DNA–PEG constructs that allow canonical base-pairing between individually PEGylated complementary strands resulting in a double-stranded structure in salt-free aqueous medium. This method relies on grafting of amino-terminated PEG polymers electrostatically onto the backbone of DNA, which results in the formation of a PEG-envelope. The specific charge interaction of PEG molecules with DNA, absolute absence of metal ions within the PEGylated DNA molecules and formation of a double helix that is significantly more stable than the duplex in an ionic buffer have been unequivocally demonstrated using multiple independent characterization techniques.
Introduction
Ever since the structural elucidation of deoxyribose nucleic acid (DNA) in 1953,1 this biomacromolecule has gained a considerable amount of interest from researchers across many disciplines and it is regarded as one of the most promising materials for programmed and predictable structure formation.2–4 The combination of minuscule size, self-recognition properties and the ability to undergo self-assembly has led to the widespread use of DNA for the fabrication of several functional DNA-based systems5,6 and has significantly contributed towards the advancement of various supramolecular DNA assemblies.7,8 The variety of such assemblies was extended by fabricating hybrid structures of DNA with a wide range of materials including organic molecules,9,10 polymers,11,12 nanoparticles13–16 and metal complexes17,18 enabling their implementation in diverse fields, ranging from templated synthesis,19,20 catalysis21,22 to (opto)-electronics,23–25 magnetics26 and biomimetics.27 Moreover such hybrids expanded the scope of diagnostic,28,29 biomedical,30–32 and therapeutic systems.33
All the above mentioned pristine DNA assemblies and the hybrid architectures of DNA depend on the presence of metal ions to achieve the fundamental purine–pyrimidine base-pairing of adenine (A) with thymine (T) and guanine (G) with cytosine (C).1 The highly charged polyanionic nature of nucleic acids requires nonspecifically bound cations, such as sodium or magnesium ions, to neutralize the negative charges on the polymer backbone. This screening of charges allows to overcome the repulsive coulombic forces between the phosphates, so that the nucleic acid molecules can fold into its compact native structures.34–36 A report highlights that the majority of experimental measurements regarding the study of the helix's thermal stability are performed in presence of Na+ salts, at a concentration of 1 M.37 Although the specified salt concentration is not an obvious requirement for obtaining thermodynamically stable double helices38 the above mentioned reference clearly highlights the need of a reasonably high salt concentration for duplex stability. Moreover, the presence of metal-cations to obtain a stable duplex has restricted some applications and is often not desirable.39 Magnesium ions can trigger undesirable enzymatic activity.40 Furthermore, they may affect the brightness of fluorescent dye molecules41 and may precipitate inorganic nanoparticles.42,43 Recent reports dealing with divalent cation-free assembly of DNA objects,44 deposition of salt-free DNA origami nanostructures,45 study on the stability of DNA origami at low salt conditions,46 and the finding of dynamic behavior of DNA nanostructures in a metal-ion-free buffer47 highlight the urge for obtaining salt-free DNA hybridization. Here, we introduce a noncovalent approach for the PEGylation of oligonucleotides that allows obtaining electrically neutral DNA–PEG supramolecular constructs. The resulting DNA architectures are capable of forming a double-stranded structure through Watson–Crick base-pairing between complementary constructs in aqueous milieu in complete absence of metal-cations (Fig. 1). The fabrication of the constructs relies on noncovalent functionalization of the DNA backbone, by grafting positively charged polyethylene glycol (PEG) chains onto the polyanionic DNA, with the help of electrostatic interactions.48 Although electrostatically conjugated DNA–PEG assemblies obtained by employing naturally occurring double-stranded DNA (dsDNA) molecules were reported earlier49,50 and the double helical structure is proven to remain intact in PEG-decorated DNA melts50 none such assemblies were reported to allow salt-free hybridization of specific short complementary DNA strands in aqueous phase neither was pristine DNA hybridization in absence of metal-cations demonstrated. Moreover, we found unexpected stabilization of the double helix through the noncovalent PEG envelope in absence of metal ions.
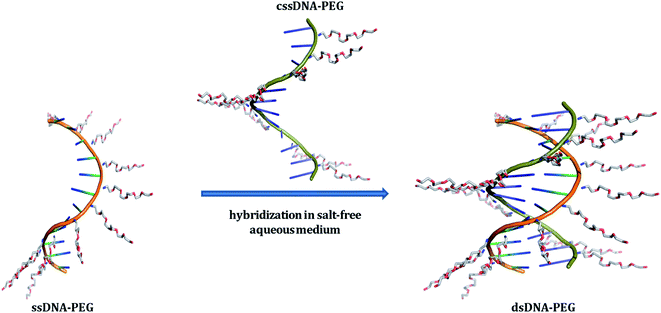 |
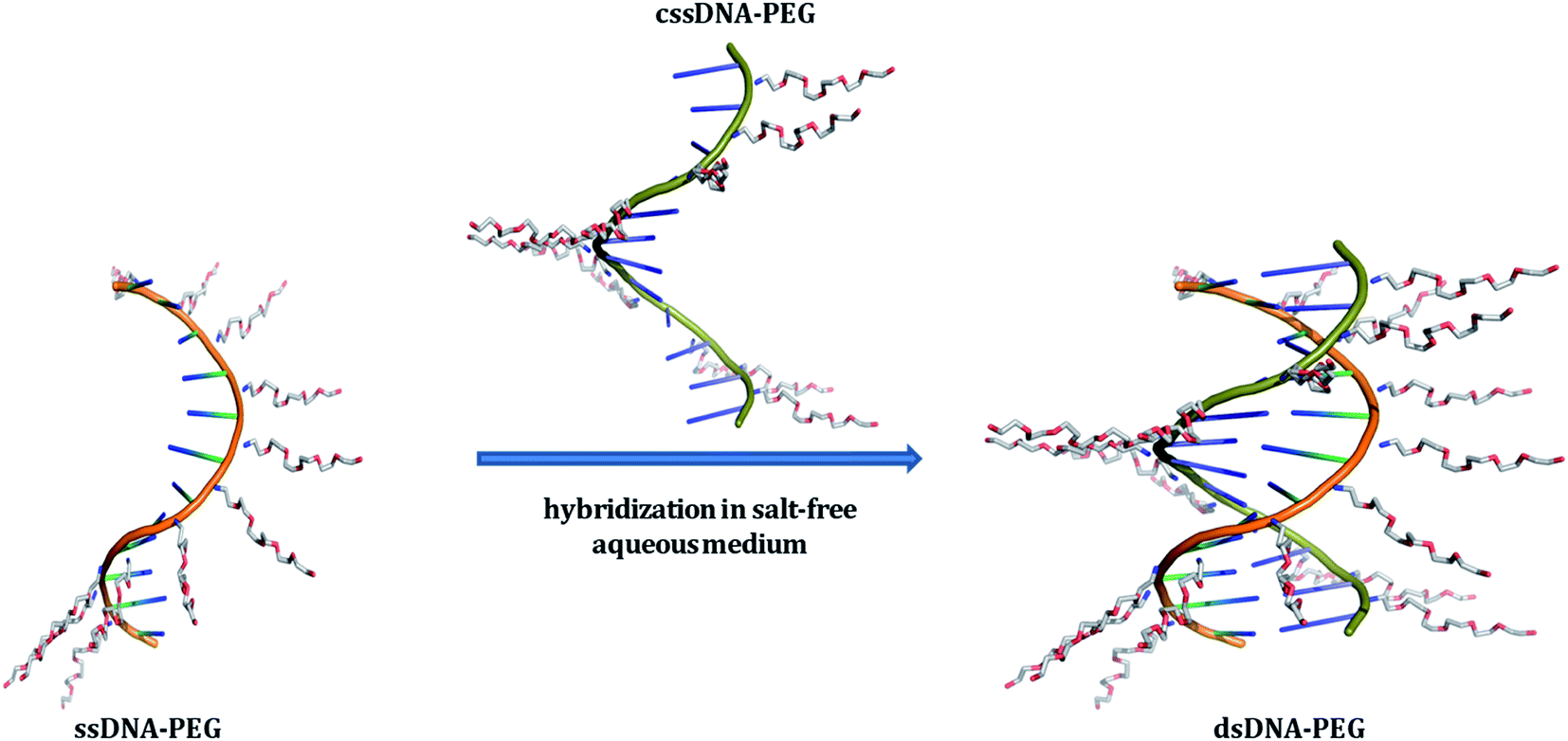
| | Fig. 1 Schematic representation of single-stranded DNA (ssDNA) and its complementary strand (cssDNA), both exhibiting a PEG shell, undergoing hybridization in salt-free aqueous medium. The DNA backbones of ssDNA–PEG and cssDNA–PEG have been shown in orange and green colors, respectively. The cationic PEG molecules attached to the negatively charged phosphates of DNA strands are shown in blue (for nitrogen), grey (for carbon) and red (for oxygen). PyMOL Molecular Graphics System, Version 1.8.2.1 Schrödinger, LLC has been used for preparing the above schematic. | |
Results and discussion
Noncovalent strategy for PEGylation of oligonucleotides
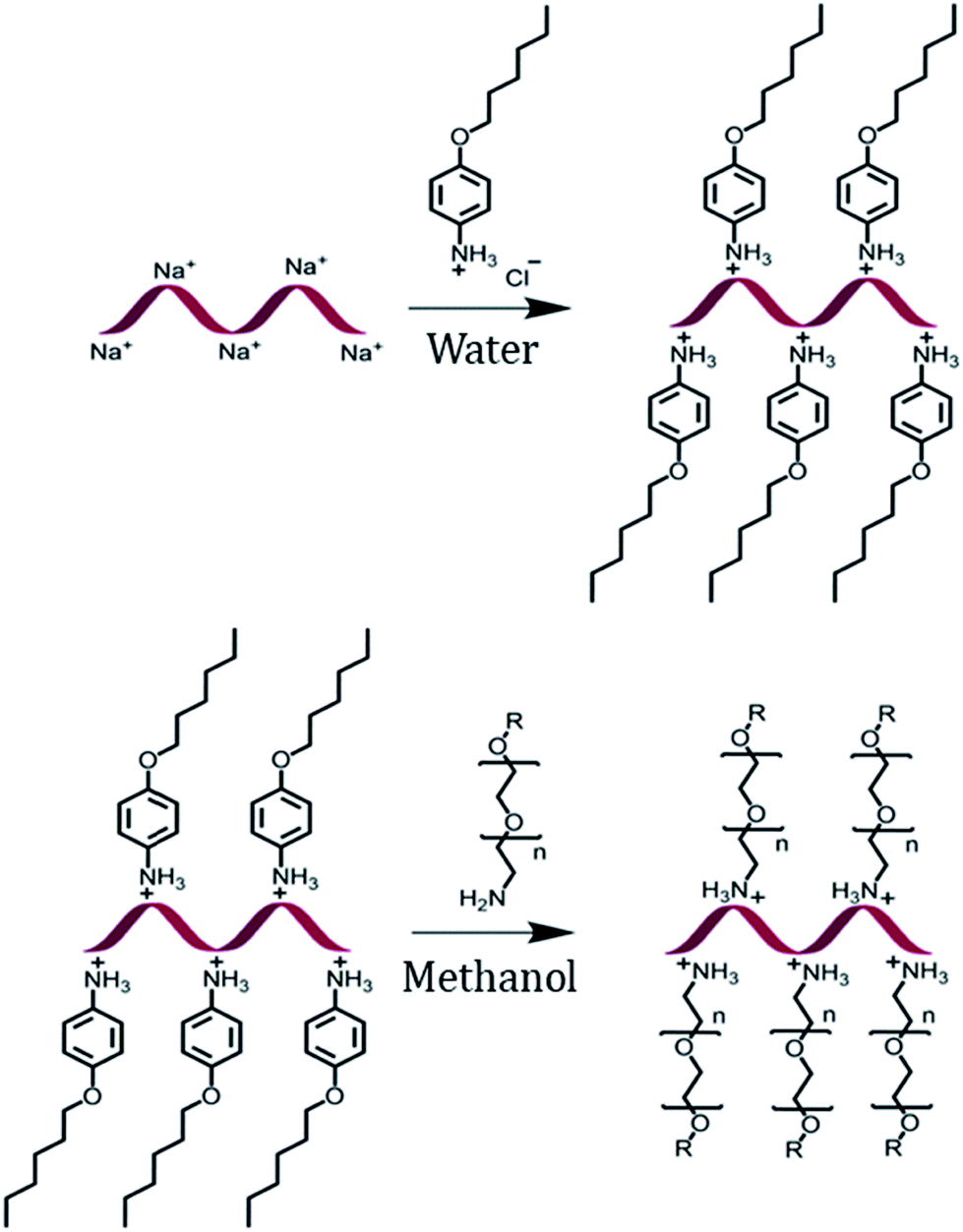
The encapsulation of DNA with a PEG shell was achieved by the method of anion exchange as described by Chen, et al. for small molecules.48 The detailed procedure to obtain a DNA–PEG complex is described in the ESI.† In brief, the ssDNA was precipitated from aqueous medium by electrostatic complexation with 4-(hexyloxy)anilinium (ANI) (Scheme 1). The DNA–ANI complex was freeze-dried and resuspended in methanol. Excess amount of methoxyPEG with a terminal primary amine group (mPEG-Amine), dissolved in methanol was added to the DNA–ANI solution. After complete exchange of ANI molecules by cationic amino PEGs, the complex was washed to remove the unbound PEG and free ANI molecules. Finally, the DNA–PEG complex was obtained after freeze-drying.
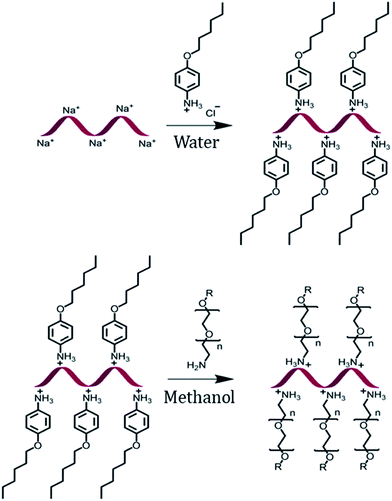 |
| | Scheme 1 Schematic representation of the formation of a DNA–PEG complex. | |
Characterization of PEG-grafted DNA
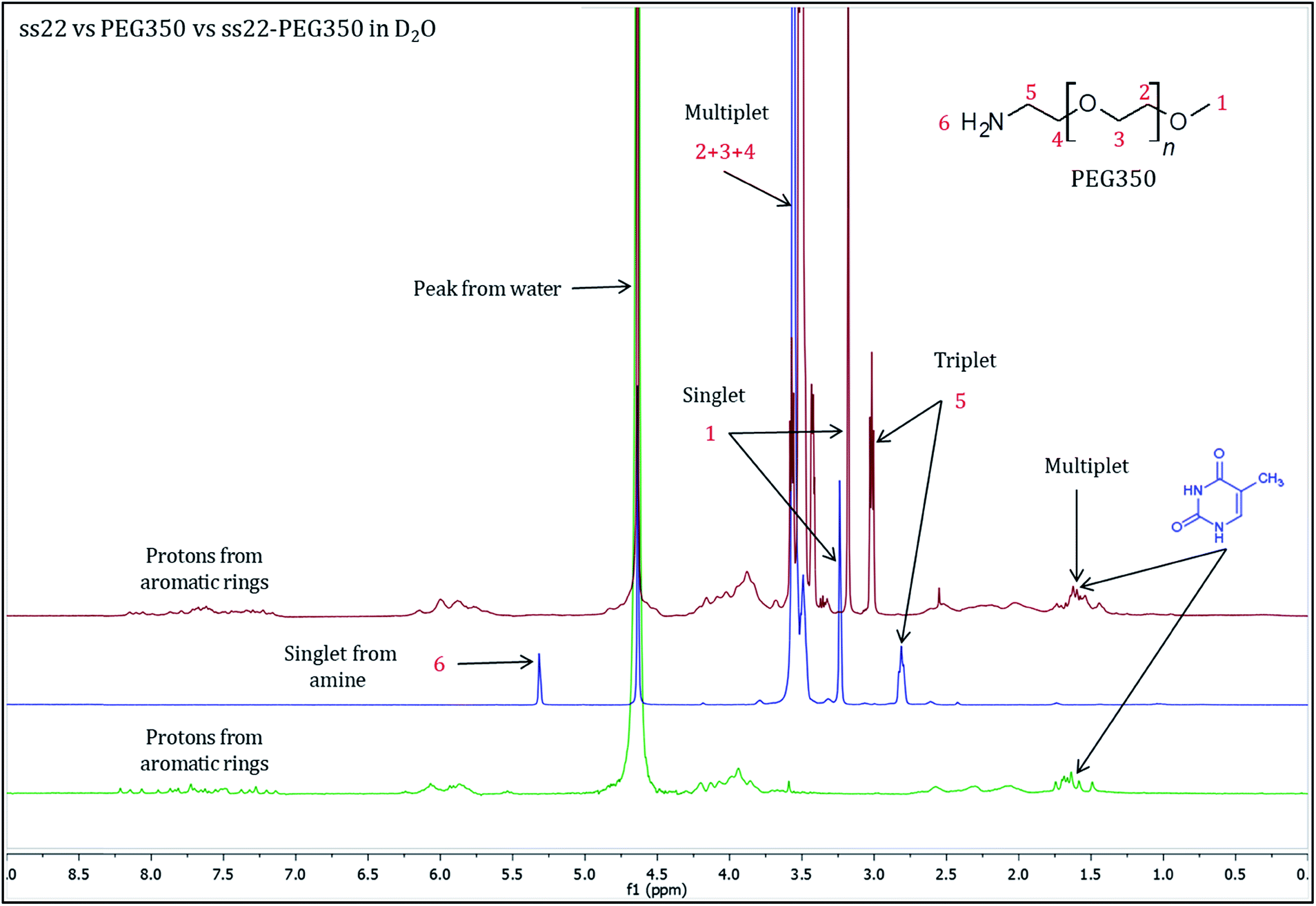
To validate the complex formation, 1H NMR spectra of pristine DNA and mPEG-Amine were compared to the spectrum of the DNA–PEG complex. The charge-specific grafting of PEG molecules onto the DNA backbone was verified with four DNA sequences (ss22, css22, ss14 and css14) and by attaching mPEG-Amine of different molecular weights (PEG350, PEG750 and PEG2000). ss22 is complementary to css22 and ss14 is a complementary sequence to css14. The PEGylation attains completion irrespective of the lengths, base compositions of the oligonucleotide sequences used and is independent of the molecular weights of the investigated PEG polymers (Fig. S4 and S7–S19†). For illustration, 1H NMR spectra of ss22 and PEG350 were compared to the spectrum of ss22–PEG350 complex (Fig. 2). The signals from the methyl groups of thymine bases (in the range of 1.5–2.0 ppm)51,52 in ss22–PEG350 complex appear in a position similar to the uncomplexed ss22, which indicates the presence of intact DNA in the complex. A shift of the α-methylene peak (position 5) by 0.2 ppm (approx.) in ss22–PEG350 complex towards lower field in comparison to the free PEG350 substantiates the charged nature of the PEGs encasing the DNA molecules. A clear triplet originating from α-methylene group is present in the spectrum of free PEG350, whereas a sharp but unresolved peak in the DNA–PEG complex is seen. We ascribe the absence of a well resolved triplet to the restricted mobility of the PEG amino terminus after complexation with DNA. The presence of a sharp instead of a broad peak directs towards a non-aggregated state of the DNA–PEG complex.48 The number of protons corresponding to the terminal methyl group of PEG350 was found to be 63 in ss22–PEG350 (Fig. S7†) whereas the same group in free PEG350 showed only 3 protons (Fig. S3†). The ratio obtained by dividing the number of protons related to the specified methyl group in ss22–PEG350 complex by the ones in free PEG350 allows the precise assessment of the grafting stoichiometry of the complex. The number of PEG chains grafted onto the backbone of ss22 was determined to be 21, this is equivalent to the total number of negative charges of ss22. This establishes the electrostatic conjugation of one PEG polymer to each negatively charged phosphate of the DNA. The specific charge interaction was further validated by comparing the signal of the α-methylene group adjacent to the terminal amine in PEG350 with the corresponding signal in ss22–PEG350 complex. Further details dealing with the step-wise realization of the PEGylation process by 1H NMR are described in ESI (Fig. S5 and S6†).
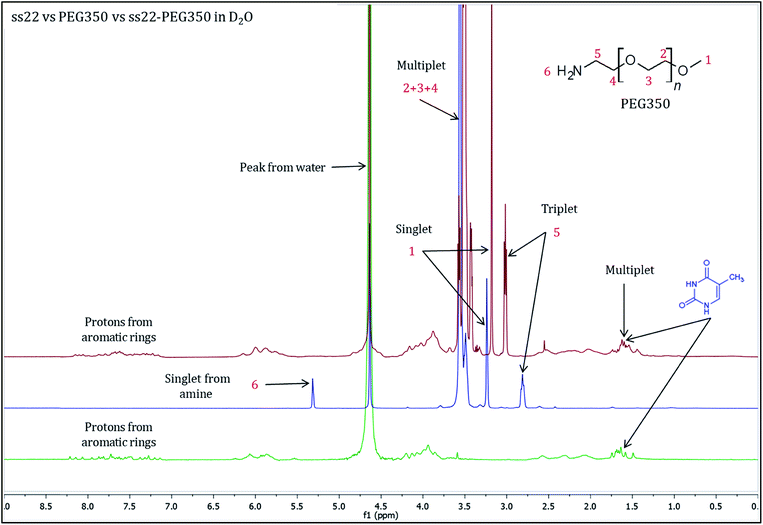 |
| | Fig. 2
1H NMR spectra of ss22 (green), PEG350 (blue) and ss22–PEG350 complex (red) in D2O. | |
To further confirm the non-aggregated state of DNA–PEG complexes, ultraviolet-visible (UV-vis) spectra of ss22–PEG350 in ultrapure water at low concentrations (0.5–8 μM) were recorded. A distinct DNA absorbance peak at 260 nm was obtained for all the concentrations and a linear correlation is observed in the plot of absorbance at 260 nm vs. concentration of ss22–PEG350 (Fig. S20†). Inductively coupled plasma optical emission spectrometry (ICP-OES) was used to determine the concentrations of Na+, K+ and Mg2+ in the DNA–PEG complex solutions (Fig. S21†). These experiments were further verified by inductively coupled plasma mass spectrometry (ICP-MS) measurements. The data (Tables S1 and S2†) reveal the complete absence of metal ions in the PEGylated form of DNA. All the spectroscopic experiments emphasize the encasing of the DNA molecules by a PEG shell.
Salt-free hybridization of PEGylated DNA
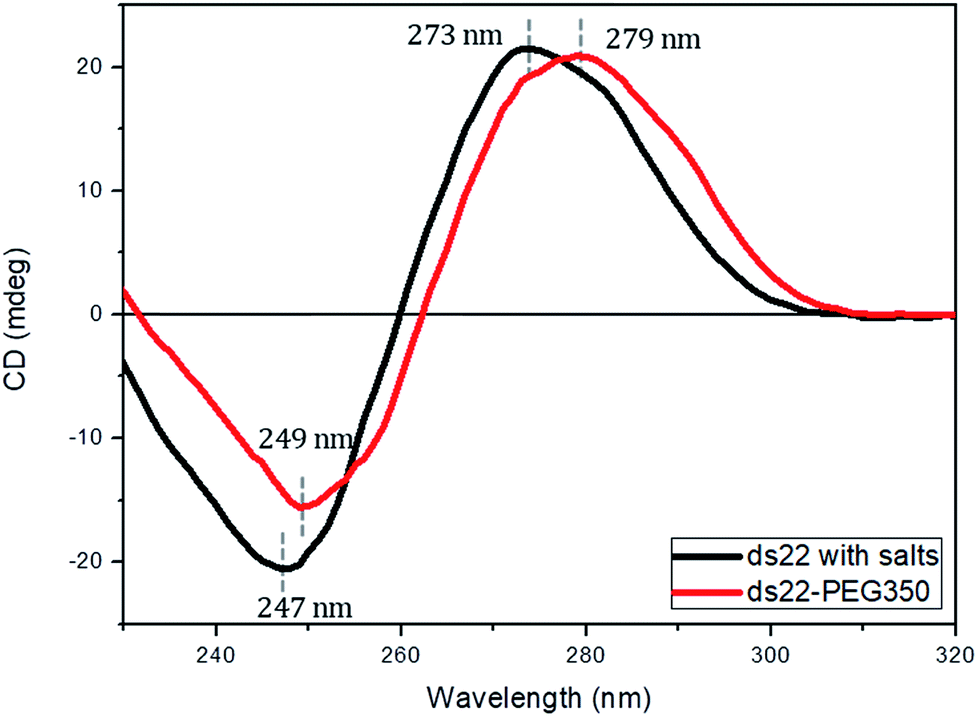
Subsequent to the characterization of the DNA–PEG complexes, two individually PEGylated complementary strands were allowed to form a hybridized structure. The detailed protocol regarding the formation of dsDNA is mentioned in ESI.† Circular dichroism (CD) spectroscopy was used for mapping the conformational properties of DNA–PEG complexes. A positive band at 279 nm due to the base stacking and a negative band at 249 nm due to the right-handed helicity are observed for ds22–PEG350 (Fig. 3), confirming the intact canonical B-form of the PEGylated DNA duplex.53–55 Slight shifts in the positive and negative bands by 6 nm and 2 nm, respectively, to longer wavelengths and a small decrease in intensity compared to ds22 with salts in water can be attributed to minor changes in the interactions between the bases due to complexation with cationic PEG molecules.56,57 In order to investigate the significance of tethered PEGs to maintain the intact B-form of DNA duplex in the complete absence of metal ions, control experiments under practically achievable “no salt” and at “no added salt” conditions were performed. ssDNA samples (ss22 and css22) were dialyzed against ultrapure water (pH 4.5–5.0) to ensure the replacement of most of the metal counterions on the backbones of the oligonucleotides by H+ ions and thereby achieving the stated “no salt” condition. The “no added salt” expression describes the condition in which a DNA sample contains the minimum number of metal-cations necessary to maintain its overall neutrality. Further details regarding the dialysis of ssDNA samples and concentrations of the investigated metal ions under the studied conditions are mentioned in ESI.† The CD spectrum of ds22 at no added salt seems to overlap with the ds22–PEG350 bands but the CD bands of ds22 obtained by annealing two individually dialyzed complementary ssDNA molecules shows a drastic decrease in the intensities of the peaks, in addition to the shifts in the peak positions towards the long wavelength region (Fig. S22†). The overlap of bands of ds22 at no added salt with ds22–PEG350 emphasizes the necessity for the complete neutralization of the DNA backbone, either by metal-cations or by PEG molecules to obtain an intact canonical B-form of the double helix. The significant decrease in the intensities of positive and negative bands of the ds22 obtained from dialyzed strands implies a weak stacking of bases in absence of salts, necessary for the electrostatic component of base-stacking interaction58 and confirms the disruption of right-handed helicity, respectively. This finding is further substantiated by comparison with CD spectra of ssDNA. Noticeable differences in the CD spectra of PEGylated ssDNA and dsDNA are observed (Fig. S23d†). The observed differences are similar to the CD spectra of DNA obtained in presence of salts (Fig. S23a†) and at no added salt condition (Fig. S23b†). However, in the dialyzed sample a weaker positive Cotton effect was observed for the mixture of the complementary strands (Fig. S23c†) compared to the other samples (Fig. S23a, b and d†). The little difference in CD spectrum of ds22 obtained from dialyzed strands in comparison to ss22 can be ascribed to the partial hybridization induced by neutralization of the participating ssDNA molecules by H+ ions. The formation of dsDNA under salt-free condition was further established employing DNA-decorated with higher molecular weight PEGs (PEG750 and PEG2000) (Fig. S24†). No considerable changes in the positive and negative bands of ds22–PEG750 and ds22–PEG2000 in comparison to ds22–PEG350 were observed (Fig. S25†). This indicates both base-stacking and right-handed helicity in the PEGylated double helices are independent of the molecular weight of the PEG shell encasing the DNA.
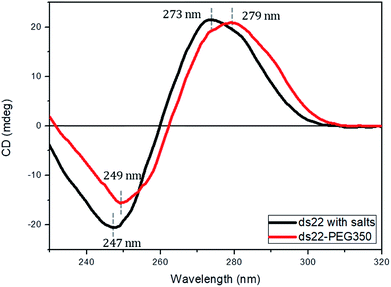 |
| | Fig. 3 CD spectra of 5 μM ds22 with salts and ds22–PEG350 complex without salt in aqueous medium. | |
Solution small-angle X-ray scattering profiles of DNA–PEG complexes
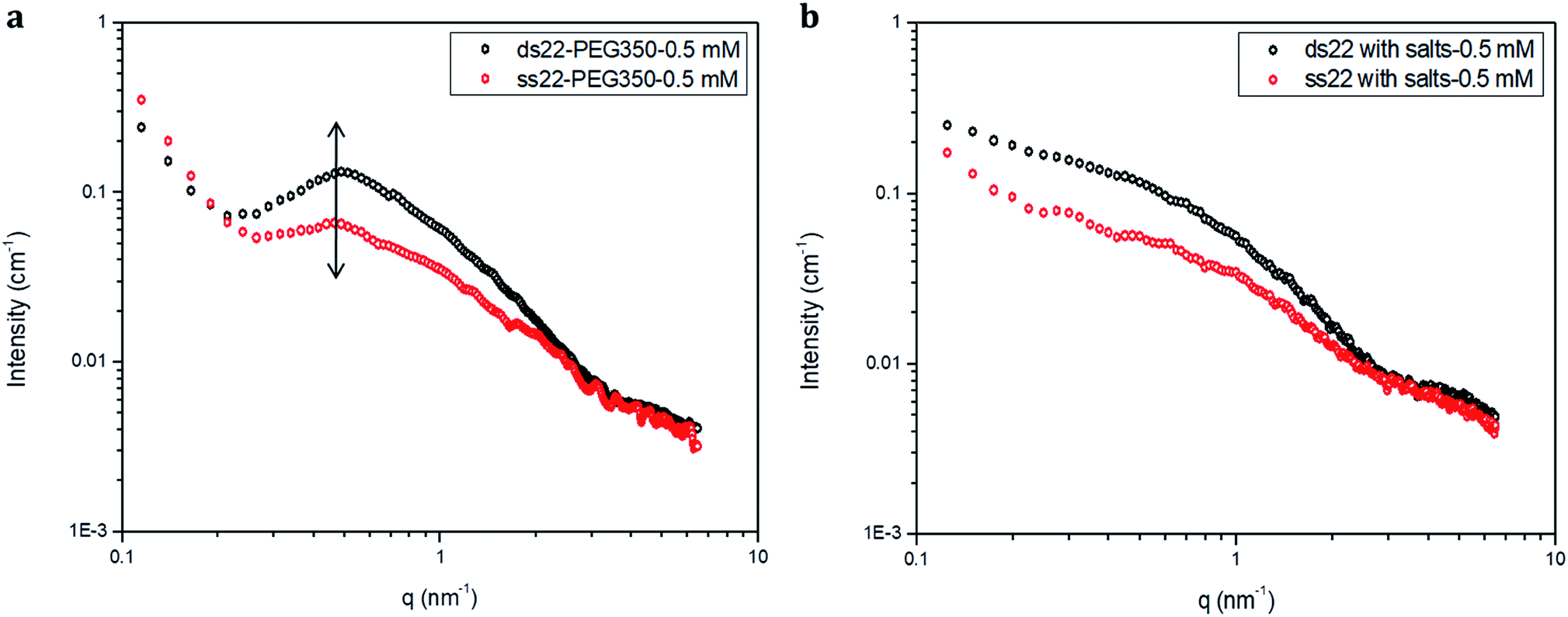
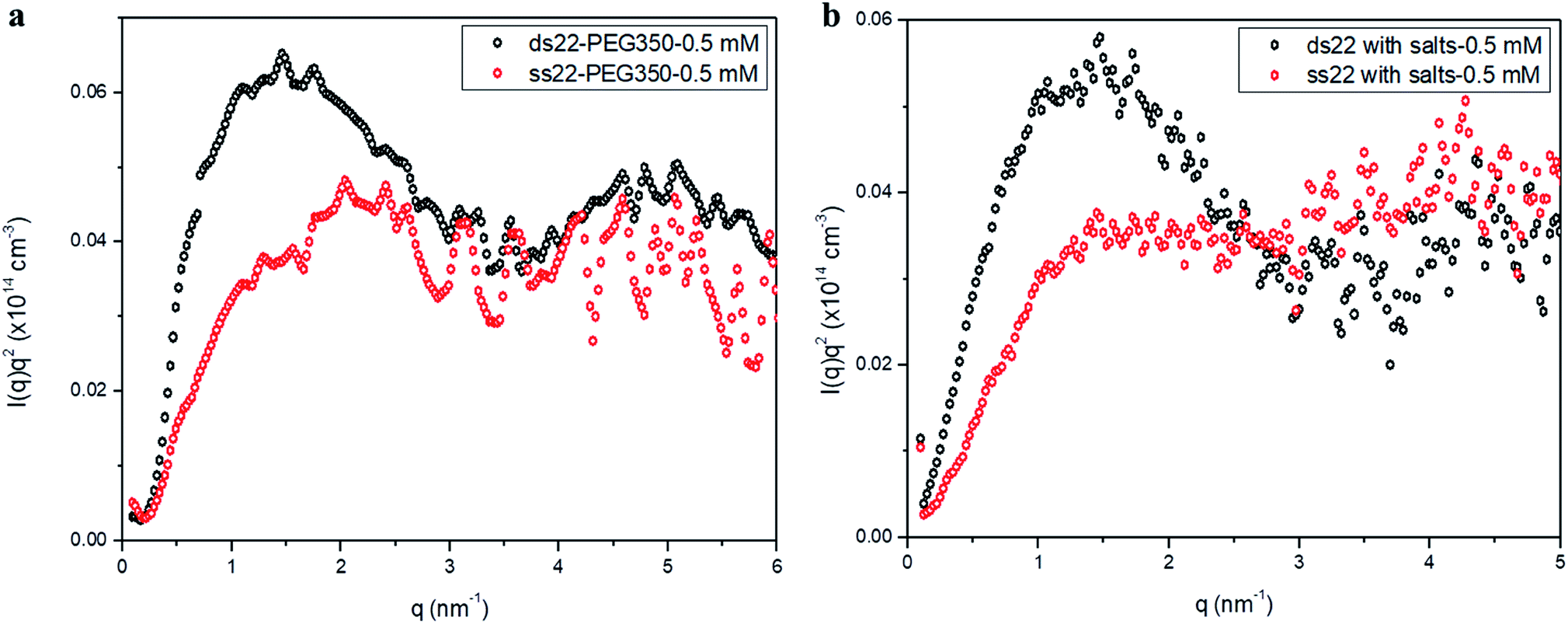
Furthermore, solution small-angle X-ray scattering (SAXS) was employed to investigate the structures of DNA–PEG complexes in ultrapure water. To differentiate the hybridized structure from the single-stranded ones in case of PEGylated DNA, SAXS profiles at two different concentrations (0.5 mM and 1.5 mM) were obtained. In both cases, the SAXS intensities of ds22–PEG350 and ss22–PEG350 differ significantly from q < 2 nm−1 with the ds22–PEG350 showing systematically higher intensity (Fig. 4a and S26†). The SAXS intensity, in general, is defined as I(q) = (Δρ)2N/VP(q)S(q) where Δρ2 is the electron density difference between the scattering objects (i.e., studied DNA systems) and the surrounding media (i.e., ultrapure water and ions), N/V is the scattering object concentration (i.e., numerical density, related to the object concentration), P(q) is the so-called form factor related to the shape and size of the DNA system and S(q) is the so-called structure factor, which is related to the interactions between the macromolecules.59 In the case of PEGylated oligonucleotides, devoid of salts, for both ds22–PEG350 and ss22–PEG350, the scattering intensities feature a pronounced peak resulting from the repulsive interactions contributing towards the structure factor. The position of this peak, qmax, is related to the average distance between the interacting DNA–PEG complexes by Bragg's law (dmax = 2π/qmax). The perfect match of the peak positions for both, single-stranded and hybridized DNA–PEG complexes at a particular concentration substantiates the presence of same number of particles per volume (N/V factor mentioned above) in the complex solutions, as the position of such peaks is reported to depend on the concentration of polyions in salt-free and at very low ionic strength aqueous solutions.60–63 Remarkable shifts in the peak positions with increase in the concentrations of ss22–PEG350 (Fig. S27a†) and ds22–PEG350 (Fig. S27b†) towards larger q regions are observed. The peak positions are found to be independent of the structure (double helix or flexible coil) of the investigated complexes but highly depend on the concentrations (density of scatterers) in the solutions. These observations, exclude the possibility of the presence of partially unhybridized PEGylated DNA molecules in the double helices obtained from the individually PEGylated strands in salt-free aqueous medium. Further comparison with the SAXS patterns of ss22 and ds22 in presence of high concentrations of salts (Fig. 4b) and under “low salt” (Fig. S28a†) and “no added salt” (Fig. S28b†) conditions validate the formation of an intact duplex with PEGylated DNA strands in a solution devoid of metal-cations. Overlapping intensities and different peak positions obtained for the same concentration of ss22 and ds22 measured could potentially be attributed to the presence of unhybridized ssDNA at “no added salt” condition. In the presence of salts, complete screening of the intermolecular forces (i.e., electrostatic interactions) occurs and both ss22 and ds22 do not show an interaction peak.62 However, similar to the case of PEGylated oligonucleotides, the ds22 scattering intensity is higher than the ss22 one. The deviation in the shape and intensity of the scattering curves of the hybridized structures from the single-stranded ones could be attributed to a different molecular shape of the duplex with respect to the coil-like structure of the ssDNA. Indeed the duplex structure possesses higher rigidity, compactness and electron density in comparison to the single strand structure. The difference in shape between duplex and the corresponding single strand is already evident from the log–log plots in Fig. 4. This difference can be easily appreciated using the Kratky plot (Fig. 5). While the Kratky plot of the DNA duplex shows a broad peak indicative of a compact shape; the one for the single strand does not show the peak and the curve is typical for unfolded, more extended structures.64 Further details dealing with the structural characterization and intermolecular correlations of PEGylated DNA are beyond the scope of the present study and will be discussed in a separate article.
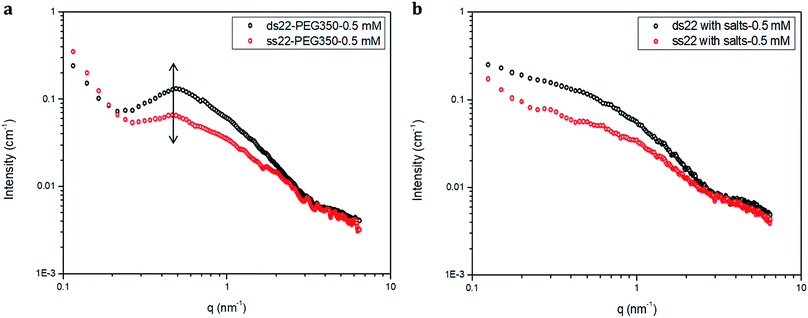 |
| | Fig. 4 SAXS profiles of (a) PEGylated DNA without salt and (b) non-PEGylated DNA with high concentrations of salts in aqueous medium. | |
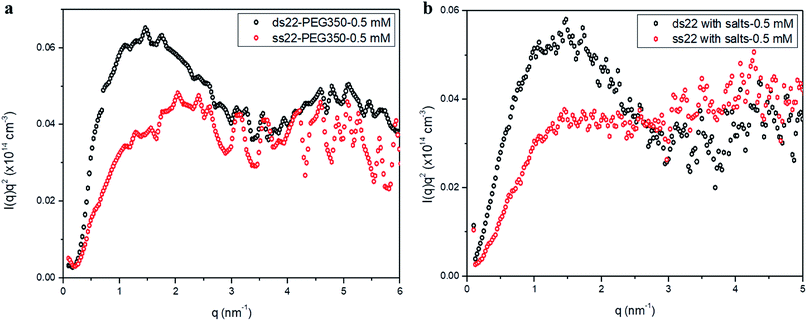 |
| | Fig. 5 Kratky plots of (a) PEGylated DNA without salt and (b) non-PEGylated DNA with high concentrations of salts in aqueous medium. | |
Stability of PEGylated DNA
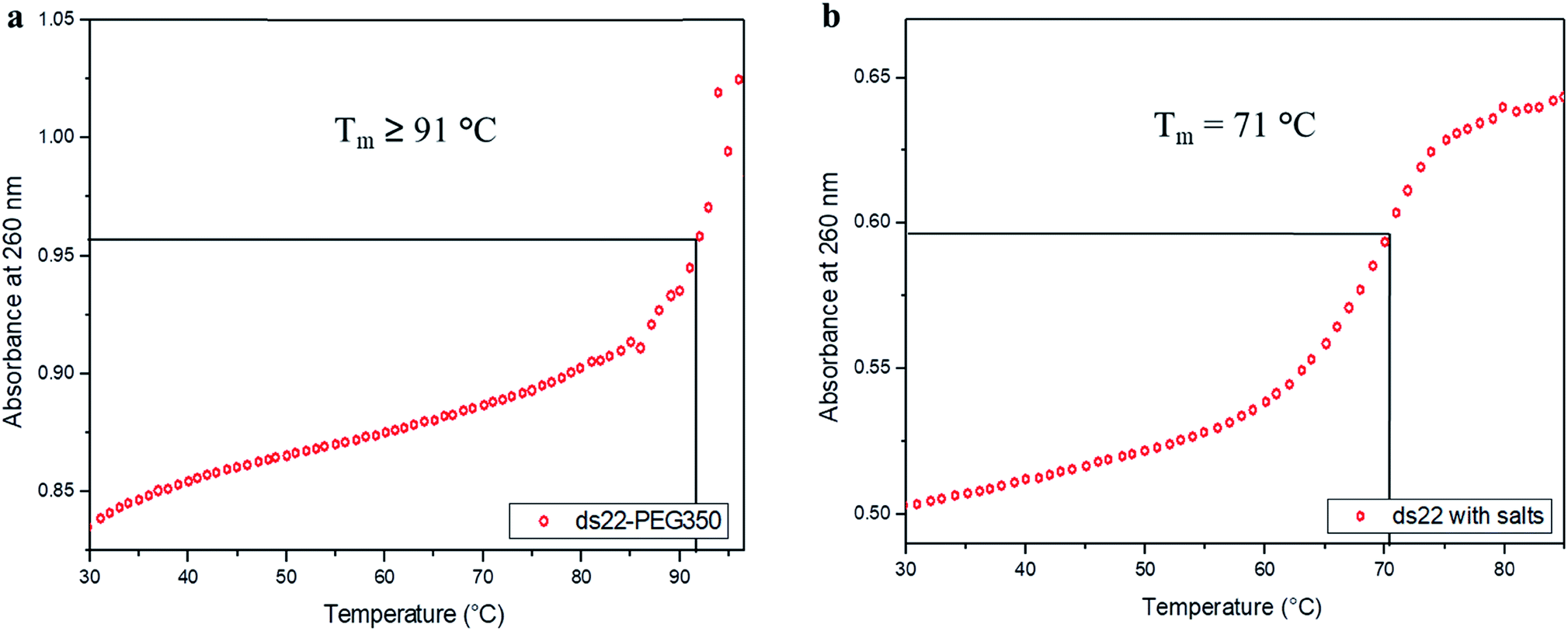
In the next step, the stability of the PEG-grafted DNA was investigated. Melting temperature (Tm) is one of the key features associated with the stability of DNA molecules. It provides a direct evidence for the structural transition occurring due to the application of heat.65 The melting curve of ds22–PEG350 without salt in ultrapure water consists of a sigmoidal curve with a pronounced hyperchromicity indicating a clear transition from hybridized state at lower temperature to unhybridized molecules at higher temperature (Fig. 6a) analogous to the thermal denaturation profile of ds22 in presence of salts (Fig. 6b). No increase in the absorbance values was observed for ss22–PEG350 and ss22 for the studied temperature range (Fig. S29†). Moreover, an increase of ≥20 °C in Tm of ds22–PEG350 in comparison to the Tm of ds22 was found. Subsequently, the effect of the molecular weight of the PEG shell on the thermal stability of the DNA duplex was studied by recording the melting curves of ds22–PEG750 (Fig. S30a†) and ds22–PEG2000 (Fig. S30b†). An increase in absorbance at 260 nm with increasing temperature, in contrary to the melting profiles of ss22–PEG750 (Fig. S31a†) and ss22–PEG2000 (Fig. S31b†), was observed. This substantiates the hybridized state of DNA duplex-enveloped with PEG750 and PEG2000. The absence of the final plateau phase for ds22–PEG750 and ds22–PEG2000 within the investigated temperature range direct towards the enhancement in the stability of the double-stranded structure with increase in the molecular weights of the PEG chains. The dramatic increase of the Tm of the PEGylated DNA duplexes is indicative of higher thermal stability due to electrostatic PEGylation. The increase in the thermostability could be an effect of intrinsic crowding associated with the PEGylated strands,66 alteration in the base-pairing kinetics,67 the overall brush-type architecture of the PEGylated DNA molecules68 or a combination of them. Other factors, that might play a substantial role in increasing the melting temperature of PEG-coated dsDNA include the excluded volume effect of PEG that can increase the effective concentration of the oligonucleotide leading to more favorable binding, osmotic pressure, DNA condensation or change in dielectric property of the reaction medium.69,70 To elucidate the driving force responsible for the increase in the melting temperature of electrostatically PEGylated DNA duplex further investigations will be performed.
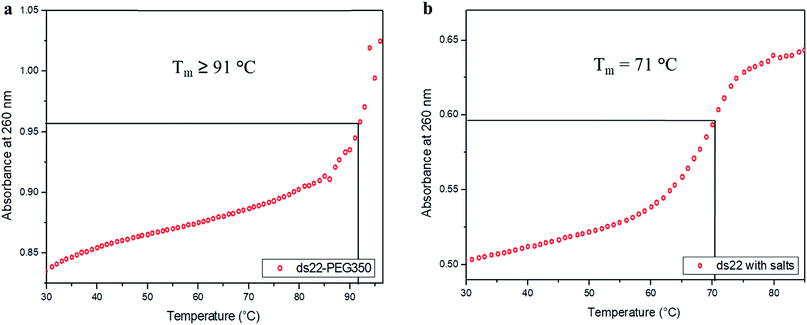 |
| | Fig. 6 Melting curves of (a) ds22–PEG350 without salt and (b) ds22 with salts in aqueous medium. | |
Effect of salt on DNA–PEG complexes
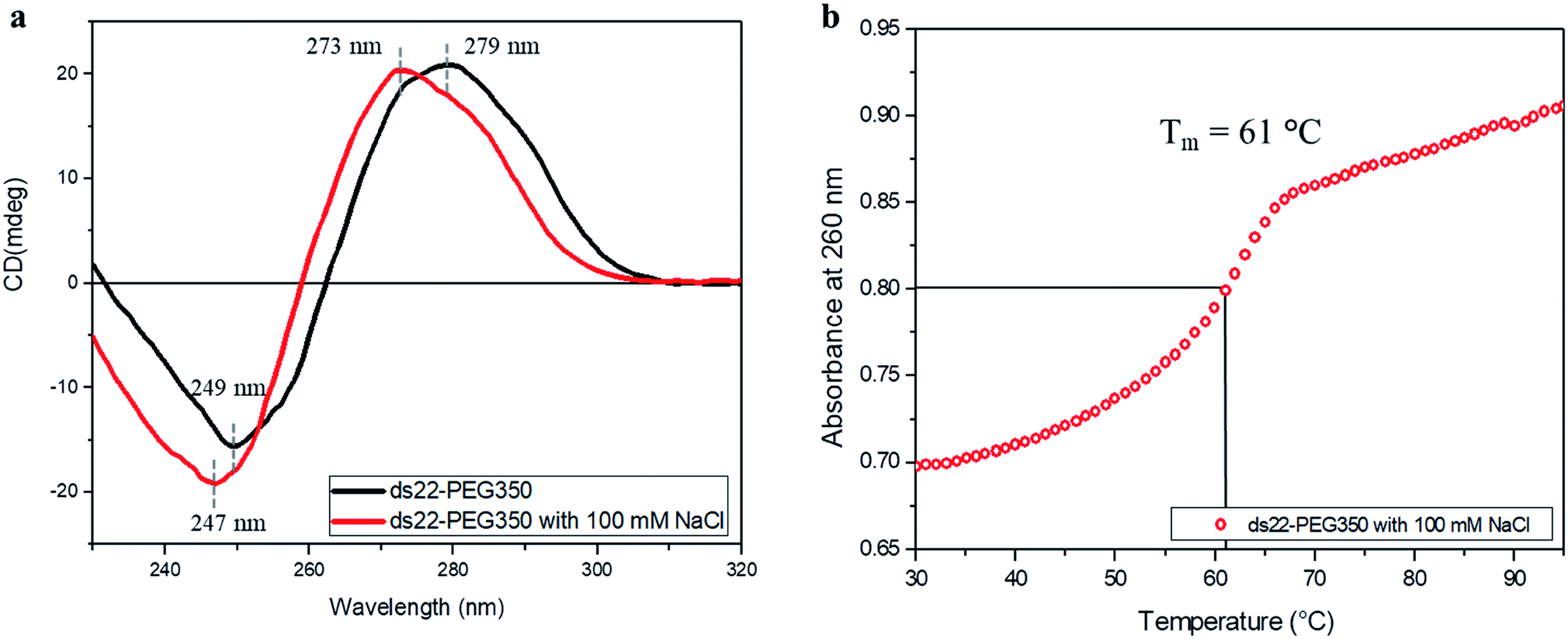
Subsequent to establishing the noncovalent PEGylation of DNA and demonstration of salt-free hybridization achieved using PEG-grafted DNA, the effect of salt on these complexes was investigated. Two separately PEGylated complementary strands were allowed to undergo hybridization in ultrapure water containing NaCl. The conformation and thermostability of the resulting structure were studied. A positive band at 273 nm and a negative band at 247 nm were observed in the CD spectrum of ds22–PEG350 with 100 mM NaCl (Fig. 7a). The obtained bands and their intensities are different from the CD spectrum of ds22–PEG350 acquired under salt-free condition but are similar to the spectrum of ds22 with salts (Fig. 3). The observed similarities between the CD spectral features of ds22–PEG350 with 100 mM NaCl and ds22 with salts point towards the restoration of base–base interactions between the complementary strands. This potentially results from the substitution of the PEG chains from the DNA backbone by metal-cations. The replaceable nature of the PEGs is established further by comparing the melting temperatures of the PEGylated duplex in presence and absence of salt. A decrease in the Tm of ds22–PEG350 recorded in presence of NaCl (Fig. 7b) by ∼30 °C in comparison to ds22–PEG350 obtained without salt (Fig. 6a) clearly demonstrates the destabilizing effect of salt on PEG-grafted DNA double helix. The difference in the Tm of ds22–PEG350 with 100 mM NaCl (Fig. 7b) in comparison to ds22 with salts (Fig. 6b) can be ascribed to the difference in salt composition of the reaction media.37,38 All the above observations pertaining to the effect of salt on the PEG-enveloped DNA duplex were further validated by recording the CD spectra (Fig. S32†) and melting temperatures (Fig. S33†) of ds22–PEG750 and ds22–PEG2000 in presence of salt. Similar CD bands (Fig. 7a and S32†) and same melting temperatures (Fig. 7b and S33†) of ds22–PEG350, ds22–PEG750 and ds22–PEG2000 in presence of 100 mM NaCl explicitly demonstrate the displacement of PEGs in solution. A detailed study dealing with salt-induced step-wise dePEGylation of the designed supramolecular complexes is beyond the scope of the present study and will be discussed in a separate article.
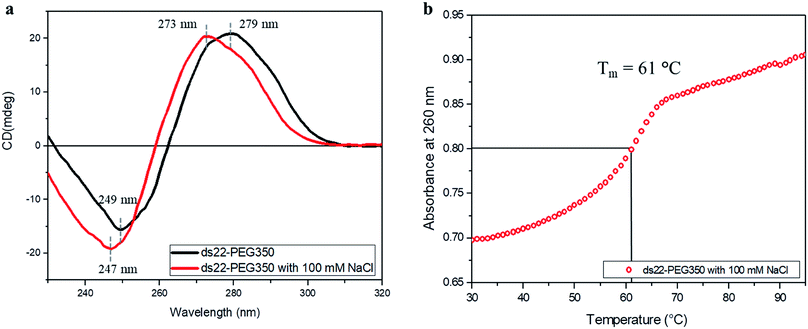 |
| | Fig. 7 (a) CD spectra of 5 μM ds22–PEG350 complex without salt and with 100 mM NaCl in ultrapure water and (b) thermal denaturation profile of ds22–PEG350 with 100 mM NaCl in ultrapure water. | |
Conclusions
In summary, we have developed a robust and facile strategy for the noncovalent PEGylation of oligonucleotides. The method allows charge-specific grafting of hydrophilic PEG molecules onto the DNA backbone, unlike previously reported chemical modification to obtain DNA–PEG conjugates.21,71–74 The noncovalent approach for PEGylation of oligonucleotides described here can potentially be considered as a third generation PEGylation approach in relation to the existing methods.75,76 The process renders metal-free DNA molecules. The PEGylated DNA strands are highly soluble in water and enable salt-free hybridization of DNA with markedly enhanced thermostability of the resulting duplex. The charge-specific interaction of PEG molecules with DNA, complete absence of metal ions in the PEGylated DNA molecules and formation of the hybridized structure have been demonstrated using various independent characterization techniques. In addition, the replaceable nature of the PEGs encasing the DNA, in presence of considerably high salt concentration was shown. We believe that this approach broadens the scope for functional DNA systems including DNA nanostructures for applications in salt-free or low ionic strength aqueous medium.77 Our PEGylation strategy could possibly be explored towards the fabrication of DNA-based supramolecular catalytic cores to achieve metal-free catalysis, as reported in case of organocatalysts.78,79 The extensive PEGylation and overall brush-type architecture of the DNA–PEG complexes might be suitable to protect oligonucleotides in biopharmaceutical contexts. Further research addressing the morphology, brush thickness, persistence length of the new architectures80,81 and a thorough study dealing with cation induced dePEGylation82 is a prerequisite for biological applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Prof. B. L. Feringa and his group members for allowing us to use the CD spectropolarimeter. The authors are grateful to J. van der Velde and J. IJmker for their contributions to the ICP-OES and ICP-MS measurements, respectively. The assistance of Nicola Pontillo in designing the schematic of DNA hybridization using PyMOL is gratefully acknowledged. G. C. is thankful to the Erasmus Mundus Programme for economic assistance through the NAMASTE scholarship. The study was financially supported by the European Union (European Research Council Advanced Grant SUPRABIOTICS No. 694610).
References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS PubMed.
- D. Yang, M. R. Hartman, T. L. Derrien, S. Hamada, D. An, K. G. Yancey, R. Cheng, M. Ma and D. Luo, Acc. Chem. Res., 2014, 47, 1902–1911 CrossRef CAS PubMed.
- Z. Li, J. Wang, Y. Li, X. Liu and Q. Yuan, Mater. Chem. Front., 2018, 2, 423–436 RSC.
- F. C. Simmel and W. U. Dittmer, Small, 2005, 1, 284–299 CrossRef CAS PubMed.
- M. K. Beissenhirtz and I. Willner, Org. Biomol. Chem., 2006, 4, 3392–3401 RSC.
- C. K. McLaughlin, G. D. Hamblin and H. F. Sleiman, Chem. Soc. Rev., 2011, 40, 5647–5656 RSC.
- Y. Shao, H. Jia, T. Cao and D. Liu, Acc. Chem. Res., 2017, 50, 659–668 CrossRef CAS.
- J. Shi and D. E. Bergstrom, Angew. Chem., Int. Ed., 1997, 36, 111–113 CrossRef CAS.
- B. Datta and G. B. Schuster, J. Am. Chem. Soc., 2008, 130, 2965–2973 CrossRef CAS.
- K. J. Watson, S. J. Park, J. H. Im, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2001, 123, 5592–5593 CrossRef CAS PubMed.
- M. Kwak and A. Herrmann, Angew. Chem., Int. Ed., 2010, 49, 8574–8587 CrossRef CAS PubMed.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- C. J. Ackerson, M. T. Sykes and R. D. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13383–13385 CrossRef PubMed.
- D. S. Seferos, A. E. Prigodich, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 308–311 CrossRef CAS.
- W. Cheng, M. J. Campolongo, J. J. Cha, S. J. Tan, C. C. Umbach, D. A. Muller and D. Luo, Nat. Mater., 2009, 8, 519–525 CrossRef CAS PubMed.
- K. M. Stewart and L. W. McLaughlin, J. Am. Chem. Soc., 2004, 126, 2050–2057 CrossRef CAS PubMed.
- K. M. Stewart, J. Rojo and L. W. McLaughlin, Angew. Chem., Int. Ed., 2004, 43, 5808–5811 CrossRef CAS PubMed.
- F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Angew. Chem., Int. Ed., 2010, 49, 7948–7951 CrossRef CAS PubMed.
- H. Abe, N. Abe, A. Shibata, K. Ito, Y. Tanaka, M. Ito, H. Saneyoshi, S. Shuto and Y. Ito, Angew. Chem., Int. Ed., 2012, 51, 6475–6479 CrossRef CAS PubMed.
- S. S. Sankar, K. Sangeetha, K. Karthick, S. Anantharaj, S. R. Ede and S. Kundu, New J. Chem., 2018, 42, 15784–15792 RSC.
- H. Nishioka, X. Liang, T. Kato and H. Asanuma, Angew. Chem., Int. Ed., 2012, 51, 1165–1168 CrossRef CAS PubMed.
- N. Lu, H. Pei, Z. Ge, C. R. Simmons, H. Yan and C. Fan, J. Am. Chem. Soc., 2012, 134, 13148–13151 CrossRef CAS PubMed.
- M. Kwak, J. Gao, D. K. Prusty, A. J. Musser, V. A. Markov, N. Tombros, M. C. A. Stuart, W. R. Browne, E. J. Boekema, G. ten Brinke, H. T. Jonkman, B. J. van Wees, M. A. Loi and A. Herrmann, Angew. Chem., Int. Ed., 2011, 50, 3206–3210 CrossRef CAS PubMed.
- G. H. Clever, S. J. Reitmeier, T. Carell and O. Schiemann, Angew. Chem., Int. Ed., 2010, 49, 4927–4929 CrossRef CAS PubMed.
- K. Börjesson, J. Tumpane, T. Ljungdahl, L. M. Wilhelmsson, B. Nordén, T. Brown, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2009, 131, 2831–2839 CrossRef PubMed.
- J. M. Gibbs, S. J. Park, D. R. Anderson, K. J. Watson, C. A. Mirkin and S. T. Nguyen, J. Am. Chem. Soc., 2005, 127, 1170–1178 CrossRef CAS PubMed.
- D. K. Prusty and A. Herrmann, J. Am. Chem. Soc., 2010, 132, 12197–12199 CrossRef CAS PubMed.
- Y. Wu, K. Sefah, H. Liu, R. Wang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5–10 CrossRef CAS PubMed.
- T. Schnitzler and A. Herrmann, Acc. Chem. Res., 2012, 45, 1419–1430 CrossRef CAS PubMed.
- C. C. Wang, S. M. Wu, H. W. Li and H. T. Chang, ChemBioChem, 2016, 17, 1052–1062 CrossRef CAS PubMed.
- T. Chen, C. S. Wu, E. Jimenez, Z. Zhu, J. G. Dajac, M. You, D. Han, X. Zhang and W. Tan, Angew. Chem., Int. Ed., 2013, 52, 2012–2016 CrossRef CAS PubMed.
- V. A. Bloomfield, Biopolymers, 1997, 44, 269–282 CrossRef CAS PubMed.
- A. M. Pyle, J. Biol. Inorg Chem., 2002, 7, 679–690 CrossRef CAS PubMed.
- S. A. Woodson, Curr. Opin. Chem. Biol., 2005, 9, 104–109 CrossRef CAS PubMed.
- Z. J. Tan and S. J. Chen, Biophys. J., 2006, 90, 1175–1190 CrossRef CAS PubMed.
- J. SantaLucia, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1460–1465 CrossRef CAS PubMed.
- K. Liu, J. Varghese, J. Y. Gerasimov, A. O. Polyakov, M. Shuai, J. Su, D. Chen, W. Zajaczkowski, A. Marcozzi, W. Pisula, B. Noheda, T. T. M. Palstra, N. A. Clark and A. Herrmann, Nat. Commun., 2016, 7, 11476 CrossRef CAS PubMed.
- W. Yang, J. Y. Lee and M. Nowotny, Mol. Cell, 2006, 22, 5–13 CrossRef CAS PubMed.
- J. P. Kring and J. N. Williams, J. Biol. Chem., 1955, 212, 751–756 CAS.
- J. Sharma, R. Chhabra, Y. Liu, Y. Ke and H. Yan, Angew. Chem., Int. Ed., 2006, 45, 730–735 CrossRef CAS PubMed.
- J. D. Carter and T. H. LaBean, ACS Nano, 2011, 5, 2200–2205 CrossRef CAS PubMed.
- T. G. Martin and H. Dietz, Nat. Commun., 2012, 3, 1103 CrossRef PubMed.
- V. Linko, B. Shen, K. Tapio, J. J. Toppari, M. A. Kostiainen and S. Tuukkanen, Sci. Rep., 2015, 5, 15634 CrossRef CAS PubMed.
- N. P. Agarwal, M. Matthies, F. N. Gür, K. Osada and T. L. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 5460–5464 CrossRef CAS PubMed.
- Y. Li, L. Song, B. Wang, J. He, Y. Li, Z. Deng and C. Mao, Angew. Chem., Int. Ed., 2018, 57, 6892–6895 CrossRef CAS PubMed.
- W. Chen, J. Y. Gerasimov, P. Zhao, K. Liu and A. Herrmann, J. Am. Chem. Soc., 2015, 137, 12884–12889 CrossRef CAS PubMed.
- S. Katayose and K. Kataoka, Bioconjugate Chem., 1997, 8, 702–707 CrossRef CAS PubMed.
- A. M. Leone, S. C. Weatherly, M. E. Williams, H. H. Thorp and R. W. Murray, J. Am. Chem. Soc., 2001, 123, 218–222 CrossRef CAS PubMed.
- S. S. Wijmenga, M. Kruithof and C. W. Hilbers, J. Biomol. NMR, 1997, 10, 337–350 CrossRef CAS PubMed.
- R. L. Benoit and M. Frechette, Can. J. Chem., 1986, 64, 2348–2352 CrossRef CAS.
- J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlickova, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- J. Wang and X. Yang, Spectrochim. Acta, Part A, 2009, 74, 421–426 CrossRef PubMed.
- Y. Chen, K. Ma, T. Hu, B. Jiang, B. Xu, W. Tian, J. Z. Sun and W. Zhang, Nanoscale, 2015, 7, 8939–8945 RSC.
- C. S. Braun, G. S. Jas, S. Choosakoonkriang, G. S. Koe, J. G. Smith and C. R. Middaugh, Biophys. J., 2003, 84, 1114–1123 CrossRef CAS PubMed.
- C. R. Middaugh and J. D. Ramsey, Anal. Chem., 2007, 79, 7240–7248 CrossRef PubMed.
- P. Yakovchuk, E. Protozanova and M. D. F. Kamenetskii, Nucleic Acids Res., 2006, 34, 564–574 CrossRef CAS PubMed.
-
O. Glatter and O. Kratky, Small-angle X-ray scattering, Academic Press, London, 1982 Search PubMed.
- A. Patkowski, E. Gulari and B. Chu, J. Chem. Phys., 1980, 73, 4178–4184 CrossRef CAS.
- M. H. J. Koch, Z. Sayers, P. Sicre and D. Svergun, Macromolecules, 1995, 28, 4904–4907 CrossRef CAS.
- L. Skibinska, J. Gapinski, H. Liu, A. Patkowski, E. W. Fischer and R. Pecora, J. Chem. Phys., 1999, 110, 1794–1800 CrossRef CAS.
- W. Häußler, A. Wilk, J. Gapinski and A. Patkowski, J. Chem. Phys., 2002, 117, 413–426 CrossRef.
- L. Pollack, Annu. Rev. Biophys., 2011, 40, 225–242 CrossRef CAS.
- A. T. Ansevin, D. L. Vizard, B. W. Brown and J. McConathy, Biopolymers, 1976, 15, 153–174 CrossRef CAS PubMed.
- C. H. Spink and J. B. Chaires, J. Am. Chem. Soc., 1995, 117, 12887–12888 CrossRef CAS.
- X. B. Gu, S. Nakano and N. Sugimoto, Chem. Commun., 2007, 2750–2752 RSC.
- F. Jia, X. Lu, X. Tan, D. Wang, X. Cao and K. Zhang, Angew. Chem., Int. Ed., 2017, 56, 1239–1243 CrossRef CAS PubMed.
- D. Miyoshi and N. Sugimoto, Biochimie, 2008, 90, 1040–1051 CrossRef CAS PubMed.
- S. Nakano, D. Miyoshi and N. Sugimoto, Chem. Rev., 2014, 114, 2733–2758 CrossRef CAS PubMed.
- A. Jäschke, J. P. Fürste, D. Cech and V. A. Erdmann, Tetrahedron Lett., 1993, 34, 301–304 CrossRef.
- A. Jäschke, J. P. Fürste, E. Nordhoff, F. Hillenkamp, D. Cech and V. A. Erdmann, Nucleic Acids Res., 1994, 22, 4810–4817 CrossRef PubMed.
- G. M. Bonora, E. Ivanova, V. Zarytova, B. Burcovich and F. M. Veronese, Bioconjugate Chem., 1997, 8, 793–797 CrossRef CAS PubMed.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- X. Lu and K. Zhang, Nano Res., 2018, 11, 5519–5534 CrossRef CAS PubMed.
- N. Ponnuswamy, M. M. C. Bastings, B. Nathwani, J. H. Ryu, L. Y. T. Chou, M. Vinther, W. A. Li, F. M. Anastassacos, D. J. Mooney and W. M. Shih, Nat. Commun., 2017, 8, 15654 CrossRef CAS PubMed.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- F. G. Fontaine and É. Rochette, Acc. Chem. Res., 2018, 51, 454–464 CrossRef CAS PubMed.
- M. S. Rocha, I. M. Storm, R. F. Bazoni, É. B. Ramos, A. Hernandez-Garcia, M. A. Cohen Stuart, F. Leermakers and R. de Vries, Macromolecules, 2018, 51, 204–212 CrossRef CAS PubMed.
- F. Jia, D. Wang, X. Lu, X. Tan, Y. Wang, H. Lu and K. Zhang, Nano Lett., 2018, 18, 7378–7382 CrossRef CAS PubMed.
- L. Kong, F. Campbell and A. Kros, Nanoscale Horiz., 2019, 4, 378–387 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Comprehensive description of the materials, method to obtain a DNA–PEG complex, characterization of PEG-grafted DNA (1H NMR spectra, UV-vis spectra, ICP-OES and MS data) and characterization of PEGylated DNA duplex (CD spectra, SAXS profiles and melting curves). See DOI: 10.1039/c9sc02598g |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Konstantin
Balinin
abc,
Giuseppe
Portale
ab,
Konstantin
Balinin
abc,
Giuseppe
Portale