
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-curing super-stretchable polymer/microgel complex coacervate gels without covalent bond formation†
Shanglin
Wu
 *a,
Mingning
Zhu
a,
Dongdong
Lu
a,
Amir H.
Milani
a,
Qing
Lian
a,
Lee A.
Fielding
a,
Brian R.
Saunders
*a,
Matthew J.
Derry
b,
Steven P.
Armes
b,
Daman
Adlam
c and
Judith A.
Hoyland
cd
*a,
Mingning
Zhu
a,
Dongdong
Lu
a,
Amir H.
Milani
a,
Qing
Lian
a,
Lee A.
Fielding
a,
Brian R.
Saunders
*a,
Matthew J.
Derry
b,
Steven P.
Armes
b,
Daman
Adlam
c and
Judith A.
Hoyland
cd
aSchool of Materials, University of Manchester, MSS Tower, Manchester, M13 9PL, UK. E-mail: shanglin.wu@postgrad.manchester.ac.uk; brian.saunders@manchester.ac.uk
bDepartment of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK
cDivision of Cell Matrix Biology and Regenerative Medicine, Faculty of Biology, Medicine and Health, University of Manchester, Oxford Road, Manchester, M13 9PT, UK
dNIHR Manchester Biomedical Research Centre, Central Manchester Foundation Trust, Manchester Academic Health Science Centre, Manchester, M13 9WL, UK
First published on 3rd August 2019
Abstract
Elastic physical gels are highly desirable because they can be conveniently prepared and readily shaped. Unfortunately, many elastic physical gels prepared in water require in situ free-radical polymerization during the gel formation stage. In contrast, complex coacervate gels are physical gels that can be prepared by simply mixing two pre-formed oppositely-charged polyelectrolytes. However, as far as we are aware, highly elastic complex coacervate gels have not yet been reported. Herein, we combine polyanionic microgel particles with a well-known commercially-available cationic polyelectrolyte to prepare polymer/microgel complex coacervate (PMCC) physical gels. This new family of gels requires annealing at only 37 °C and behaves like a covalent gel but does not form covalent bonds. Thermal reconfiguration of the dynamic ionic bonds transforms the shapeable pre-gel into a highly elastic gel that is super-stretchable, adhesive, self-healing, highly swellable and can be further toughened using Ca2+ as an ionic crosslinker. Our PMCC gels have excellent potential for applications as engineering gels and structural biomaterials, as well as for wound healing and water purification.
Introduction
Hydrogels can be prepared with excellent mechanical properties by judicious control of the spatial arrangement and crosslinking mechanism.1–4 They have been utilized as tissue scaffolds5–7 and injectable implants,8–10 as well as for wound healing,11 drug delivery,12 water purification,13 electronic skin,14,15 soft robotics16 and energy storage.17 Recently, hydrogels have been reported with outstanding tensile fracture stress and strain properties, including double network hydrogels,18 nanocomposite hydrogels,19 microgel-reinforced hydrogels,20 slide-ring hydrogels21 and hybrid ionically-crosslinked hydrogels.22 One common feature for such gels is that covalent bonds are formed during gel preparation, which limits their potential applications.In contrast, physical gels do not contain chemical crosslinks and hence can be prepared under mild conditions.2,23–25 Some dynamic crosslinked gels can be also formed under physiological conditions based on the Schiff based reaction.26 Thus such gels are well-suited for use as biomaterials27 and have been constructed using peptides,28 hydrogelators,29 polyelectrolytes,30 crystallization31 and polymer-particle association.32 Peptide and hydrogelator-based gels utilize reversible association and can repair damage through self-healing. Polymer-particle self-assembled gels have employed reversible attractive hydrophobic interactions between the components. However, these gel networks tend to fail at relatively low strain.32
Recently, nanocomposite33 and supramolecular34 biocompatible hydrogels that are tough and self-healing have been synthesized using free-radical polymerization accompanying host–guest interaction,34 hydrogen bonding,33,34 electrostatic interaction33 and physical entanglement.33 Physical hydrogels with such properties have also been prepared from oppositely charged polyelectrolytes.30,35–37 These polyelectrolyte complex gels utilize reversible ionic bonds for physical crosslinking and are potentially attractive biomaterials for in vivo applications because covalent crosslinking is not required. However, to avoid the strong association between the pair of oppositely charged polyelectrolytes that would normally form coacervates,38 such gel preparation requires in situ polymerization. Because of their high ionic conductivity, polyelectrolyte-based gels may be useful for nerve tissue repair and as cardiac patches.23 Complex coacervates are widely used in food science39 and involve spontaneous phase separation of an aqueous solution of oppositely-charged macromolecules into solvent-rich and solvent-depleted phases.40 Similarly, organisms such as the sandcastle worm secrete coacervated proteins that act as adhesives for mineral particles.41 The inclusion of solvophilic units within pre-formed triblock copolymers has been exploited to prepare reversible gels formed by charge-driven self-assembly.37,42 However, these and other related coacervate gels are not stretchable.43 An intriguing question is whether pre-formed oppositely-charged macromolecules can be designed such that upon mixing produce mechanically robust, stretchable, hydrogels.
Here, we investigate a binary polyelectrolyte combination of an anionic polyacid microgel (termed MG) and a cationic branched polyethyleneimine (PEI). MGs are crosslinked polymer colloids that swell when the pH approaches the particle pKa.44 PEI is well known as a vehicle for gene transfer.45 In contrast to polyelectrolyte complex gels reported in the literature,37,42,43 physical hydrogels with excellent shapeability and stretchability are prepared simply by mixing MG and PEI followed by thermal annealing under mild conditions. A series of polymer/MG complex coacervate (PMCC) gels are prepared by varying the dry weight PEI/MG mass ratio. Each gel is denoted by PEI/MG(T-MR), where T is the annealing temperature (°C) and MR corresponds to the PEI-to-MG mass ratio. The heated gels are termed T-gels. For example, PEI/MG(37-0.67) corresponds to a T-gel prepared from a pre-gel using a mass ratio of 0.67 by thermal annealing at 37 °C. We show that these new PMCC gels are shapeable, super-stretchable, self-healing, adhesive, highly swellable and can be toughened using Ca2+.
Results and discussion
We utilized dynamic ionic bonds introduced by mixing pre-formed components which could rearrange2 to form stronger bonds consisting of multiple ionic bonds between the two components (see Fig. 1A). The pH-responsive PEA–MAA–DVB particles (where EA, MAA and DVB denote ethyl acrylate, methacrylic acid and divinylbenzene, respectively) were previously used to prepare hydrogels of covalently-interlinked MGs46 which differs considerably from the current work. Herein, the MGs had a number-average diameter of 55 nm as determined from TEM studies (see Fig. S1A, ESI†) and acid titration gave a pKa of 6.5. Branched PEI is a polycationic polymer with a pKa of 8.5 for its protonated form.47 It has been used for encapsulating cells with microgels48 as well as for DNA and RNA transfer into cells.49,50 We provide evidence below that the pre-gel “heat-sets” to form an elastic T-gel that is highly stretchable. The T-gels swell strongly when the solution pH exceeds the pKa of PEI. However, unlike other polyelectrolyte complexes,42 degelation does not occur.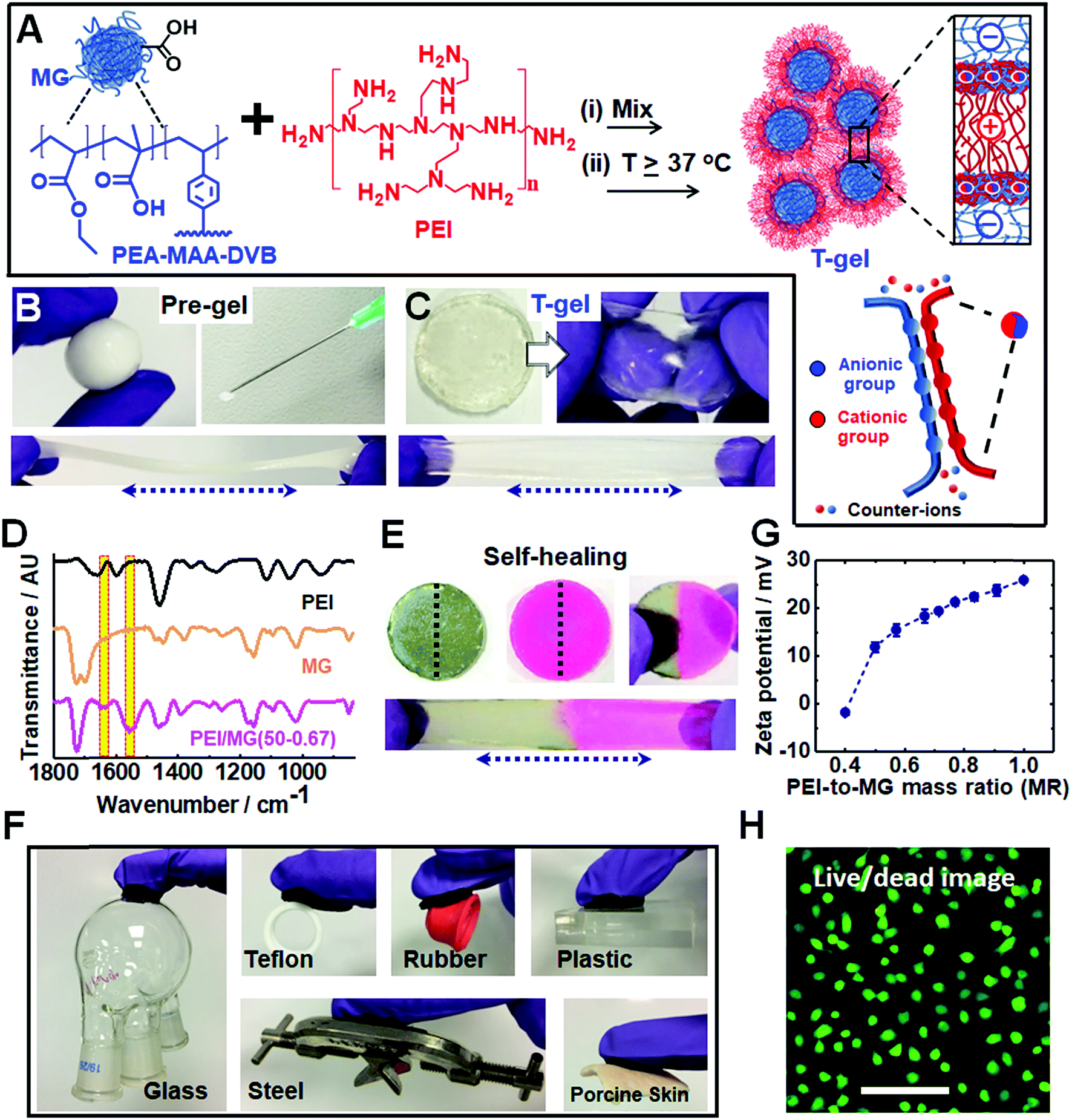 | ||
| Fig. 1 (A) Schematic representation of PEI/MG hydrogel preparation and cooperative ionic bonding. (B) Plastic flow and injectability of shapeable PEI/MG(0.67) pre-gel. (C) Stretchable behavior of a PEI/MG(50-0.67) T-gel of 19 mm diameter. (D) FTIR spectra for PEI/MG(50-0.67), MG and PEI. (E) Self-healing for the PEI/MG(50-0.67) T-gel (19 mm) after being cut (black dashed line) into two separate parts. Dyes were used to aid clarity. (F) PEI/MG(50-0.67) adheres to various materials including glass, Teflon, steel, plastic rubber and porcine skin. (G) Zeta potentials for dilute PEI/MG mixed dispersions recorded for various mass ratios (MR). (H) Live/dead assay for chondrocyte cells in the presence of PEI/MG(50-0.50) after 72 h. Scale bar 100 μm. | ||
Simply hand-mixing MGs and PEI gave a ‘putty like’ soft gel that became strongly elastic when annealed at mild temperatures. The pre-gel deformed plastically, could be molded into various shapes and was injectable (see Fig. 1B). Such pre-gels can be formed under physiologically-relevant conditions (e.g. pH 7.4 and 37 °C). The pH-responsive DVB-crosslinked MG building blocks contain a high MAA content (35.8 wt%). The size and pH-dependent properties of the MGs were characterized using potentiometric titration, dynamic light scattering (DLS) and zeta potential measurements (see Fig. S1, ESI†). These particles swell substantially from 74 nm at pH 4.7 to 502 nm at pH 11 and exhibit highly negative zeta potentials above pH 6.0. PEI had a manufacturer's molecular weight of 10 kD. Remarkably, temperature curing (at 37, 50 or 80 °C for 20 h) transformed the deformable pre-gels into highly elastic gels (compare Fig. 1C to Fig. 1B). Hence, the PMCC pre-gels could be shaped and then set in any desired shape simply by heating.
FTIR spectra were recorded for the T-gels cured at 37, 50 or 80 °C (see Fig. 1D and S2C, ESI†) and exhibit new bands at 1552 and 1637 cm−1 which are assigned to –COO− (asymmetric stretch) and –NH3+, respectively.51 These bands indicate formation of ionic crosslinks. Hydrogen bonding interactions are also indicated by the red shift in the PEI N–H stretch52 from 3450 to 3400 cm−1 (see Fig. S2, ESI†). There was no evidence for the formation of new covalent amide bonds. Hence, the PMCC gels are formed as a result of extensive ionic and hydrogen bonding.
Why do the pre-gels transform to elastic T-gels after heating? We envisage that the MGs act as nanosized crosslinkers and PEI as flexible bridges between adjacent MGs. A related morphology has been reported for covalent nanostructured hydrogels.53 Drawing upon atomic force microscopy studies of the interactions between two oppositely charged polyelectrolyte brushes,38 we propose that the PEI chains initially adsorbed in a flat conformation and then interpenetrated into the MG peripheries upon annealing (Fig. 1A). Such interpenetration increases the number of ion pairs which is driven, in turn, by the entropy increase due to mobile ion release.54 Hence, curing increases the number-density of elastically effective ionic crosslinks which increases gel modulus according to rubber elasticity theory.55
Self-healing or self-repairing gels are highly desirable because they offer the possibility of extended operational lifetimes. Fig. 1E shows a stretched PEI/MG(50-0.67) gel formed from two disk halves obtained from two different disks that were cut and then manually interfaced for 24 h at room temperature. Self-healing occurred, which suggests that dynamic bonds were present. This self-repairing behavior is attributed to the mobile PEI component.48 Tensile stress–strain measurements (Fig. S3, ESI†) showed that the recovery of the initial breaking strain (self-healing efficiency) was 92%, 64% and 20% for the PEI/MG(T-0.67) gels heated at 37, 50 and 80 °C, respectively. The less efficient self-healing observed for gels heated at 50 and 80 °C indicates more extensive ionic bond formation.
Our T-gels adhered to a remarkably wide range of materials (see Fig. 1F). For example, PEI/MG(50-0.67) adhered to plastic, rubber, steel, glass and Teflon. The gel can easily sustain the heavy glassware or steel (∼150 g) in air. Adhesive strengths of 15.2 kPa and 8.9 kPa were determined for glass and Teflon, respectively (below). The PEI/MG(50-0.67) gel also adhered tightly to a model tissue surface (porcine skin) which demonstrates its bioadhesive properties and is quantified later. The zeta potential for a dilute PEI/MG dispersion (MR = 0.67) was +18.3 mV at pH 7.3 (Fig. 1G), which accounts for the strong adhesion to negatively charged surfaces such as glass. The adhesion to Teflon is attributed to the relatively hydrophobic nature of coacervate complexes.38 Live/dead assays were measured for chondrocyte cells in the presence of PEI/MG(50-0.50) gel (Fig. 1H). Additional time points and a control are shown in Fig. S4 (ESI†). MTT assay results showed that the cell viability after 1, 2 and 3 days was 90%, 78% and 70%, respectively (see Fig. S5, ESI†). The gels had relatively low cytotoxicity. In principle, the cell viability could be increased by (1) using a PEI/MG gel with a lower MR, (2) chemically functionalising PEI56 and/or (3) chemically crosslinking PEI to prevent any free PEI chains from leaving the gel.48
The T-cured gels proved to be highly stretchable (Fig. 2A) (see also Movie S1, ESI†). The relationship between the tensile stress–strain properties and MR (Fig. 2B) was investigated (see also and Tables S1 and S2 (ESI†)). A breaking strain of 1015% was reached for PEI/MG(50-0.67). The highest Young's modulus (E = 39.0 kPa) and strength (64.0 kPa) were obtained for PEI/MG(50-0.40). As MR increased the MG particles were diluted by PEI and the average separation between MGs increased. Therefore, the PEI bridging length increased and the proportion of crosslinking centers decreased which decreased the modulus (Fig. 2C). The breaking strain passed through a maximum (Fig. 2C) because in the early stages of the MR increase the length of the bridging chains increased. However, at higher MR values fewer PEI chains were able to bridge neighboring MGs. The effect of PEI molecular weight was also investigated. Low molecular weight PEI (0.60 kD) could not form a T-cured elastic gel network (Fig. S6, ESI†); whereas 70 kD PEI formed a robust gel with E = 14.8 kPa. However, the latter gel exhibited a lower breaking strain compared to that of the PEI/MG(50-0.67) gel prepared using 10 kD PEI. The 70 kD PEI gel was not homogenous due to the high viscosity of its pre-gel.
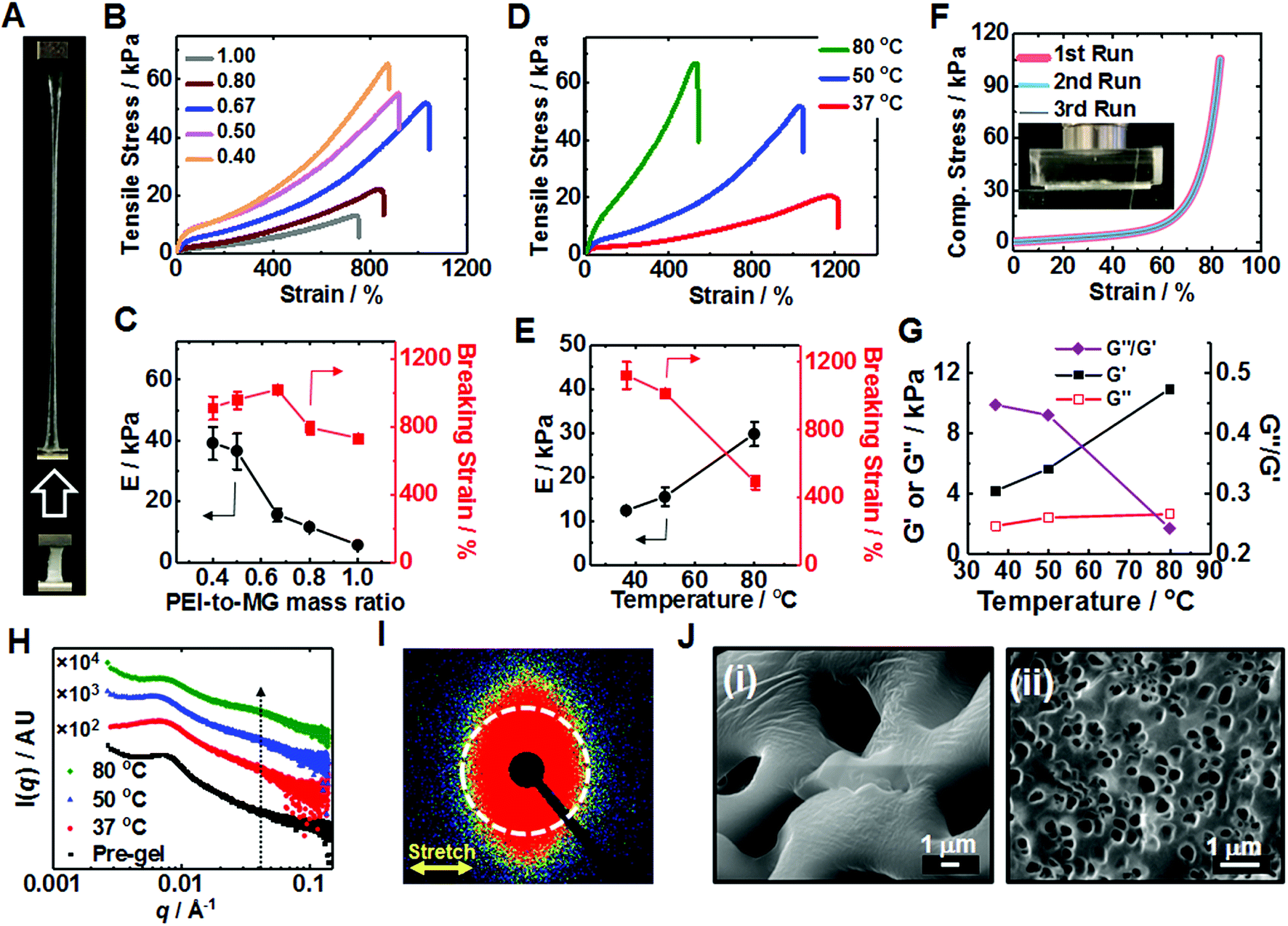 | ||
| Fig. 2 (A) Stretched PEI/MG(50-0.67) gel prepared at pH 7.3. Uniaxial tensile stress–strain data (B) and modulus and breaking strain (C) for PEI/MG(50-MR) gels prepared at 50 °C using various mass ratios. Tensile stress–strain data (D) and modulus and breaking strain data (E) for PEI/MG(T-0.67) gels annealed at various temperatures. (F) Multiple uniaxial compressive stress–strain loading cycles for a PEI/MG(50-0.67) gel. (G) Storage modulus (G′), loss modulus (G′′) and loss tangent (G′′/G′) for PEI/MG(T-0.67) gels obtained using a frequency and strain of 1 Hz and 1%, respectively. (H) 1D SAXS patterns recorded for various PEI/MG(T-0.67) gels. (I) 2D SAXS pattern for a stretched PEI/MG(50-0.67) gel. The strain (direction shown) was 155%. (J) SEM images of (i) PEI/MG(0.67) pre-gel and (ii) PEI/MG(37-0.67). | ||
We investigated the effect of annealing temperature on T-gel stretchability. Tensile measurements (Fig. 2D) showed that an PEI/MG(37-0.67) gel (cured at 37 °C) exhibited the highest breaking strain of all the gels (1122%) while an PEI/MG(80-0.67) gel possessed the highest modulus (29.8 kPa) and strength (62.5 kPa) with a relatively high breaking strain (489%). PEI/MG(50-0.67) had the highest toughness (0.194 MJ m−3) using the area under the stress–strain curve. The annealing time also affected gel mechanical properties. The PEI/MG(80-0.67) gel modulus and breaking strain increased and decreased, respectively, with increasing annealing time (Fig. S7, ESI†). We propose that thermal annealing accelerates reconfiguration and increases the number-density of ionic bonds (Fig. 1A), leading to higher moduli and lower breaking strain (Fig. 2E). PEI/MG(50-0.67) prepared using 10 kD PEI was judged to have the best overall mechanical properties. This gel also exhibited excellent compressive properties with no failure occurring at 84% strain even when repeatedly compressed (Fig. 2F). Its Young's modulus of 11.5 kPa compares well with that of muscle.57 Cyclic compression data confirmed that the residual strain was proportional to the maximum cycle strain whilst greater hysteresis was observed at higher applied strain (Fig. S8, ESI†). Frequency-sweep dynamic rheology data were measured for the PEI/MG(T-0.67) gels (see Fig. S9, ESI†). Fig. 2G shows the variation of the storage modulus (G′), loss modulus (G′′) and G′′/G′ (=tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) with cure temperature. The increase with G′ with temperature follows the same trend as that observed for the Young's modulus in Fig. 2E. The G′ values increased and G′′/G′ decreased with increasing temperature indicating that the gels became increasingly solid-like.58
δ) with cure temperature. The increase with G′ with temperature follows the same trend as that observed for the Young's modulus in Fig. 2E. The G′ values increased and G′′/G′ decreased with increasing temperature indicating that the gels became increasingly solid-like.58
Small-angle X-ray scattering (SAXS) was employed to gain further insight regarding the excellent gel stretchability. Immersion of the gels in saturated CaCl2 solution for 30 s was beneficial because it greatly increases their X-ray scattering contrast. As will be discussed below the MG particles within the PEI/MG gels partially de-swell due to added Ca2+ and the center-to-center distance decreases. However, the MGs were in a highly elastic environment prior to (and after) Ca2+ addition which would have prevented any major changes in the nearest neighbor locations in the gels occurring (e.g., such as aggregation) due to this treatment.
The structure peak observed at q = 0.0065 Å−1 for PEI/MG(50-0.67) (Fig. 2H) corresponds to a mean centre-to-centre distance between neighboring MG particles of 97 nm, which is larger than that observed for a pure 17 wt% MG dispersion (87 nm, see Fig. S10, ESI†). The presence of comparable structure peaks in these SAXS patterns supports the view that the SAXS data for the Ca2+-treated gels are representative of the as-made T-gels without Ca2+. A new shoulder emerges at q = 0.04 Å−1 after annealing which may indicate greater local order. SAXS studies of a stretched PEI/MG(50-0.67) film (Fig. 2I) revealed a distinctly different anisotropic 2D pattern. This anisotropy was confirmed by azimuthal profile analysis (Fig. S11, ESI†) and indicates directional alignment of the submicrometer MGs within the stretched gels. SEM studies showed that the pore size of freeze-dried gels was tuneable using temperature (Fig. 2J). The pre-gel had the largest pores (5.06 μm), with the mean pore size being reduced to just 0.34 μm for PEI/MG(37-0.67) (see also Fig. S12, ESI†).
The dynamic tensile stress–strain data for the PEI/MG(50-0.67) gel indicated energy dissipation, which increased linearly with maximum strain (see Fig. S13, ESI†). The maximum stress and dissipated energy rapidly became constant. The lack of change of dissipated energy with cycling implies that chains do not slide past each other. Rather, the sacrificial bonds that were broken are reformed in the absence of stress. The dynamic nature of these ionic (and hydrogen) bonds is a cause of the excellent stretchability of the T-gels.
When the solution pH is increased beyond the PEI pKa of ∼8.5 (ref. 47) the pre-gels expanded to fill the entire fluid volume, but remained a free-standing gel as judged by the tube inversion test, see Fig. 3A. Only in highly alkaline media (pH ∼ 14) did the pre-gels dissolve (Fig. S14A, ESI†). The T-gels swelled strongly to form a giant disk (Fig. 3B) at pH 9.8. Intriguingly, the T-gels did not dissolve even at pH 14 (Fig. S14B, ESI†). This is in striking contrast to the dissolution behavior reported for conventional coacervate gels.42 In the pH range from 4.7 to 8.4, the increase in mass owing to the degree of swelling (w/w%) was negligible (102–105%), see Fig. 3C. The swelling degree reached 1470% at pH 9.8 and 2704% at pH 11. Zeta potential data for dilute PEI/MG dispersions indicated that these hybrid particles (and hence most likely also the pre-gel) became negatively charged above pH 8.4 (Fig. 3D). Additionally, the MG building blocks started to swell above pH 6.1 (see Fig. S1C, ESI†). At higher pH, ionic crosslinks no longer constrained the gel and the repulsive forces between anionic carboxylate groups caused gel swelling. Nevertheless gel dissolution was prevented.
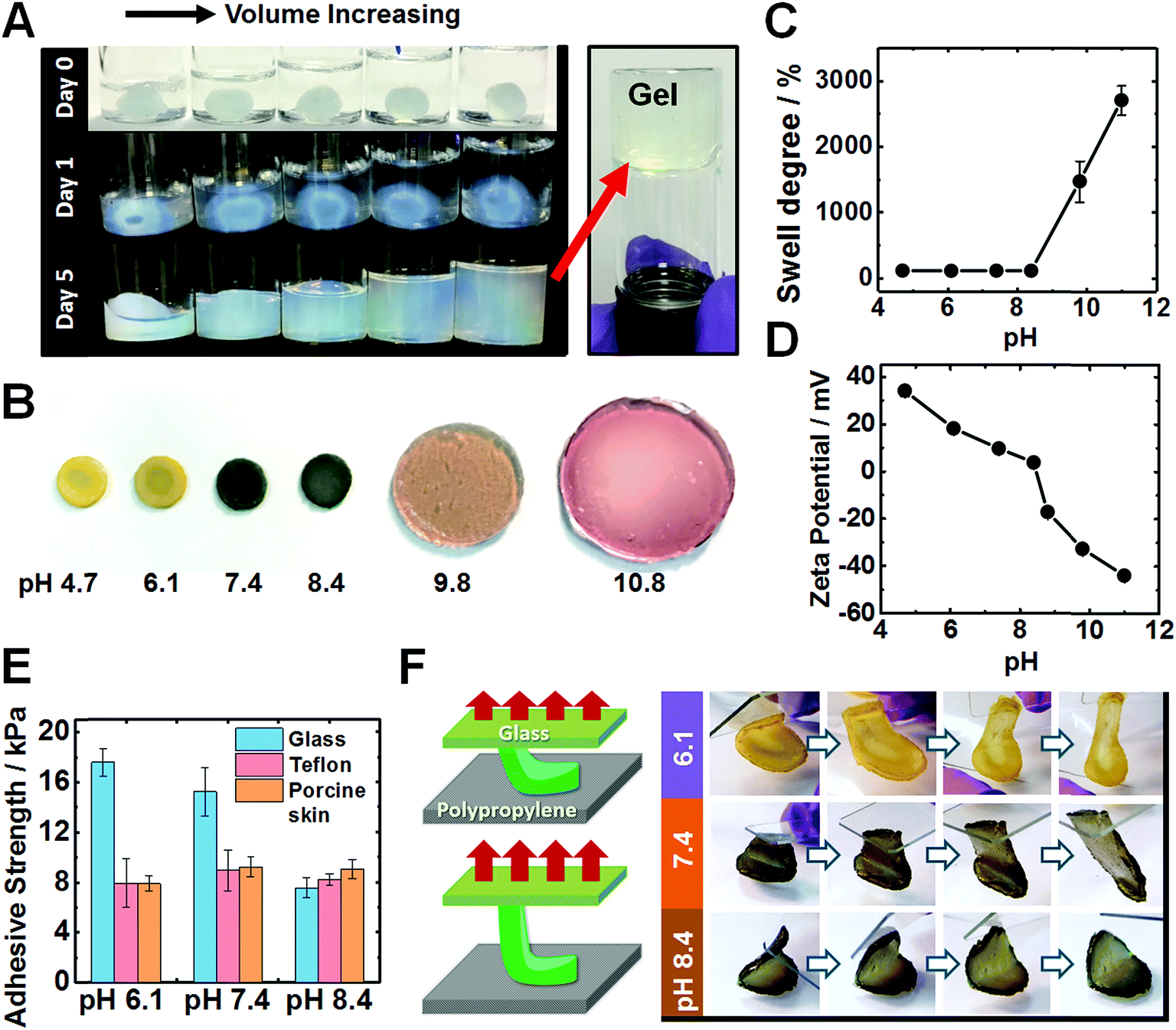 | ||
| Fig. 3 (A) Aqueous swelling behavior of an PEI/MG(0.67) pre-gel at pH 11. Such pre-gels swell to fill the available volume. (B) Swelling of an PEI/MG(50-0.67) T-gel after immersion in various buffers containing pH-indicator. (C) Degree of swelling for the PEI/MG(50-0.67) gels as a function of solution pH. (D) Zeta potential vs. pH curves for a dilute aqueous dispersion containing a mixture of PEI/MG(0.67). (E) Adhesion strengths for PEI/MG(50-0.67) gels on glass, Teflon and porcine skin measured at various solution pH values. (F) A “flip test” for PEI/MG(50-0.67) gel to identify the influence of solution pH on its adhesive strength. The corresponding schematic cartoon shows that glass was in contact with the upper surface of the gel and polypropylene was in contact with its lower surface in such adhesion tests. | ||
An interesting question concerning the swollen T-gels is why they do not re-disperse when the pH exceeds the pKa of PEI (Fig. 3B and S14B, ESI†). We attribute this behavior to persistent PEI bridging between the MGs at high pH. The mechanism for maintaining anchoring of PEI to adjacent MGs is likely to involve multiple hydrogen bonds from interpenetrating PEI and MG chains near the MG peripheries. Candidates for such hydrogen bonds are primary and secondary amine groups from PEI with MG carboxylate and ester groups.59
After immersing PEI/MG(50-0.67) gels in various buffers, their adhesion was evaluated using a lap-shear test (see Fig. 3E). The gel contact time was 10 seconds prior to each measurement. Adhesion strengths ranged from 7.5 to 17.5 kPa on both glass, Teflon and porcine skin for all pH values tested. Interestingly, weaker adhesion was observed for glass at higher pH (see Fig. 3E). PEI/MG(50-0.67) gel was placed on a smooth polypropylene film. A “flip test” confirmed a reduction in its adhesive strength from pH 6.1 to 8.4 as this gel was released from the upper glass slide (Fig. 3F). The overall surface charge of the gel was positive below pH 8.4. Hence weaker adhesion to this substrate was observed when the pH was close to neutral or when the gel acquired negative surface charge. Therefore, electrostatic attraction appears to be the dominant adhesion mechanism below pH 8.4. In contrast, the adhesive strength of porcine skin was measured as 8.9 kPa at pH 7.4, and was independent of pH (Fig. 3E). The results are suggestive of a hydrophobic interaction, which has been also reported for a poly(N-isopropylacrylamide) based complex coacervates.43
Immersion of an PEI/MG(50-0.67) gel into saturated CaCl2 solution caused a time-dependent enhancement of the modulus through ionic crosslinking (depicted in Fig. 4A). The Ca2+-toughened gels were foldable (Fig. 4B) and thin fibers could support relatively heavy loads (Fig. 4C) (see also Table S3†). The gel continuously stiffened for up to 180 min in CaCl2 (6.7 M) solution (Fig. 4D). An increase in gel modulus was accompanied by a reduction in the breaking strain, but the latter parameter remained above 70% (Fig. 4E). The modulus increased dramatically from an initial value of 0.016 MPa to 0.30 MPa and up to 34 MPa after immersion for 0.5 and 180 min, respectively. The latter modulus is a record for coacervate gels. In contrast, saturated NaCl solution did not discernibly stiffen the gel. Importantly, the T-gels could not be dissolved by soaking in such a solution for more than 3 months when heated at 50 °C confirming the irreversible formation of these remarkable physical gels. We propose that gel stiffening is the result of Ca2+ crosslinking primarily within the MGs. We characterized the change in structure using SAXS (Fig. 4F). The structure peak moved to a slightly higher q value (0.0070 Å−1) which indicates a reduction in center-to-center MG spacing to 90 nm, which is ascribed to partial MG de-swelling from Ca2+ ionic crosslinking.
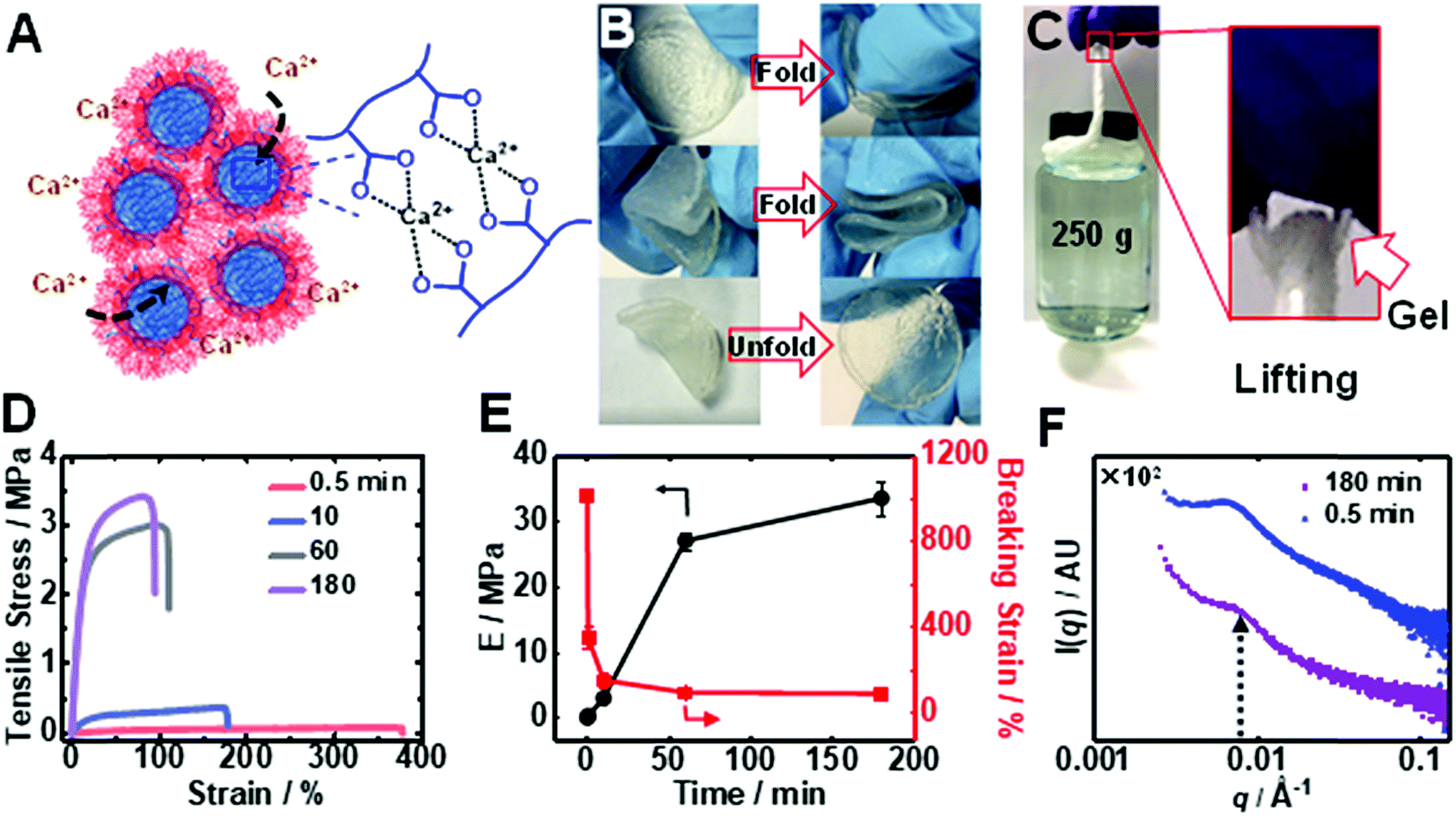 | ||
| Fig. 4 (A) Depiction of the mechanism for Ca2+-toughening of the PEI/MG T-gels. The Ca2+-toughened gel (180 min soaking time) was foldable (B) and a thin fiber supported a heavy load (C). (D) Tensile data obtained for PEI/MG(50-0.67) exposed to saturated CaCl2 solution for varying times at room temperature. (E) Variation of Young's modulus and strain-at-break with immersion time. (F) 1D SAXS patterns for the gel immersed in saturated CaCl2 solution for 0.5 and 180 min at room temperature. | ||
Conclusions
In summary, by simply mixing oppositely-charged MG and PEI we obtain soft, moldable PMCC gels that can be transformed into strong, elastic physical gels with outstanding mechanical properties under mild conditions (37 °C, neutral pH) without using any covalent crosslinking chemistry. This new family of complex coacervate gels is super-stretchable, self-healable, highly adhesive, super-swellable and dramatically toughened by addition of Ca2+. These new PMCC gels offer potential applications as engineering gels, structural biomaterials including cartilage repair, wound healing, and also for water purification membranes. All gel components are commercially available and there appears to be no barrier to the industrial scale production of PMCC gels.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by a 5 years EPSRC Established Career Fellowship awarded to BRS (EP/M002020).Notes and references
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed.
- P. Rao, T. L. Sun, L. Chen, R. Takahashi, G. Shinohara, H. Guo, D. R. King, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1801884 CrossRef PubMed.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS PubMed.
- J. P. K. Armstrong, J. L. Puetzer, A. Serio, A. G. Guex, M. Kapnisi, A. Breant, Y. Zong, V. Assal, S. C. Skaalure, O. King, T. Murty, C. Meinert, A. C. Franklin, P. G. Bassindale, M. K. Nichols, C. M. Terracciano, D. W. Hutmacher, B. W. Drinkwater, T. J. Klein, A. W. Perriman and M. M. Stevens, Adv. Mater., 2018, 30, 1802649 CrossRef PubMed.
- T. E. Brown, J. S. Silver, B. T. Worrell, I. A. Marozas, F. M. Yavitt, K. A. Günay, C. N. Bowman and K. S. Anseth, J. Am. Chem. Soc., 2018, 140, 11585–11588 CrossRef CAS PubMed.
- H.-p. Lee, L. Gu, D. J. Mooney, M. E. Levenston and O. Chaudhuri, Nat. Mater., 2017, 16, 1243 CrossRef CAS PubMed.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- M. Patenaude and T. Hoare, ACS Macro Lett., 2012, 1, 409–413 CrossRef CAS.
- A. Sinclair, M. B. O'Kelly, T. Bai, H.-C. Hung, P. Jain and S. Jiang, Adv. Mater., 2018, 30, 1803087 CrossRef PubMed.
- D. R. Griffin, W. M. Weaver, P. O. Scumpia, D. Di Carlo and T. Segura, Nat. Mater., 2015, 14, 737 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- H. Gao, Y. Sun, J. Zhou, R. Xu and H. Duan, ACS Appl. Mater. Interfaces, 2013, 5, 425–432 CrossRef CAS PubMed.
- M. L. Hammock, A. Chortos, B. C.-K. Tee, J. B.-H. Tok and Z. Bao, Adv. Mater., 2013, 25, 5997–6038 CrossRef CAS PubMed.
- A. Chortos, J. Liu and Z. Bao, Nat. Mater., 2016, 15, 937 CrossRef CAS PubMed.
- S. Bauer, S. Bauer-Gogonea, I. Graz, M. Kaltenbrunner, C. Keplinger and R. Schwödiauer, Adv. Mater., 2014, 26, 149–162 CrossRef CAS PubMed.
- Y. Shi, L. Pan, B. Liu, Y. Wang, Y. Cui, Z. Bao and G. Yu, J. Mater. Chem. A, 2014, 2, 6086–6091 RSC.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- J. Hu, T. Kurokawa, T. Nakajima, T. L. Sun, T. Suekama, Z. L. Wu, S. M. Liang and J. P. Gong, Macromolecules, 2012, 45, 9445–9451 CrossRef CAS.
- J. Araki and K. Ito, Soft Matter, 2007, 3, 1456–1473 RSC.
- X. Li, H. Wang, D. Li, S. Long, G. Zhang and Z. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 31198–31207 CrossRef CAS PubMed.
- B. Guo and P. X. Ma, Biomacromolecules, 2018, 19, 1764–1782 CrossRef CAS PubMed.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2002, 54, 13–36 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- B. Guo, J. Qu, X. Zhao and M. Zhang, Acta Biomater., 2019, 84, 180–193 CrossRef CAS PubMed.
- J. Li, X. Ni and K. W. Leong, J. Biomed. Mater. Res., Part A, 2003, 65A, 196–202 CrossRef CAS PubMed.
- R. J. Mart, R. D. Osborne, M. M. Stevens and R. V. Ulijn, Soft Matter, 2006, 2, 822–835 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722–2727 CrossRef CAS PubMed.
- F. Yokoyama, I. Masada, K. Shimamura, T. Ikawa and K. Monobe, Colloid Polym. Sci., 1986, 264, 595–601 CrossRef CAS.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef CAS PubMed.
- Z. Deng, T. Hu, Q. Lei, J. He, P. X. Ma and B. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 6796–6808 CrossRef CAS PubMed.
- Z. Deng, Y. Guo, X. Zhao, P. X. Ma and B. Guo, Chem. Mater., 2018, 30, 1729–1742 CrossRef CAS.
- W. Li, R. Feng, R. Wang, D. Li, W. Jiang, H. Liu, Z. Guo, M. J. Serpe and L. Hu, J. Mater. Chem. B, 2018, 6, 4799–4807 RSC.
- S. Ladet, L. David and A. Domard, Nature, 2008, 452, 76 CrossRef CAS PubMed.
- D. V. Krogstad, N. A. Lynd, S.-H. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2013, 46, 1512–1518 CrossRef CAS.
- J. van der Gucht, E. Spruijt, M. Lemmers and M. A. Cohen Stuart, J. Colloid Interface Sci., 2011, 361, 407–422 CrossRef CAS PubMed.
- C. Schmitt and S. L. Turgeon, Adv. Colloid Interface Sci., 2011, 167, 63–70 CrossRef CAS PubMed.
- H. Shao and R. J. Stewart, Adv. Mater., 2010, 22, 729–733 CrossRef CAS PubMed.
- H. Zhao, C. Sun, R. J. Stewart and J. H. Waite, J. Biol. Chem., 2005, 280, 42938–42944 CrossRef CAS PubMed.
- M. Lemmers, J. Sprakel, I. K. Voets, J. van der Gucht and M. A. Cohen Stuart, Angew. Chem., Int. Ed., 2010, 49, 708–711 CrossRef CAS PubMed.
- M. Dompé, F. J. Cedano-Serrano, O. Heckert, N. van den Heuvel, J. van der Gucht, Y. Tran, D. Hourdet, C. Creton and M. Kamperman, Adv. Mater., 2019, 1808179 CrossRef PubMed.
- B. R. Saunders and B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 1–25 CrossRef CAS.
- C. L. Gebhart and A. V. Kabanov, J. Controlled Release, 2001, 73, 401–416 CrossRef CAS PubMed.
- Z. Cui, W. Wang, M. Obeng, M. Chen, S. Wu, I. Kinloch and B. R. Saunders, Soft Matter, 2016, 12, 6985–6994 RSC.
- S. Choosakoonkriang, B. A. Lobo, G. S. Koe, J. G. Koe and C. R. Middaugh, J. Pharm. Sci., 2003, 92, 1710–1722 CrossRef CAS PubMed.
- S. Saxena and L. A. Lyon, Mol. Syst. Des. Eng., 2016, 1, 189–201 RSC.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS PubMed.
- M. E. Johnson, J. Shon, B. M. Guan, J. P. Patterson, N. J. Oldenhuis, A. C. Eldredge, N. C. Gianneschi and Z. Guan, Bioconjugate Chem., 2016, 27, 1784–1788 CrossRef CAS PubMed.
- C. Peniche, W. Argüelles-Monal, N. Davidenko, R. Sastre, A. Gallardo and J. San Román, Biomaterials, 1999, 20, 1869–1878 CrossRef CAS PubMed.
- H. Yuk, T. Zhang, S. Lin, G. A. Parada and X. Zhao, Nat. Mater., 2015, 15, 190 CrossRef PubMed.
- L.-W. Xia, R. Xie, X.-J. Ju, W. Wang, Q. Chen and L.-Y. Chu, Nat. Commun., 2013, 4, 2226 CrossRef PubMed.
- V. S. Rathee, H. Sidky, B. J. Sikora and J. K. Whitmer, J. Am. Chem. Soc., 2018, 140, 15319–15328 CrossRef CAS PubMed.
- L. R. G. Treloar, The physics of rubber elasticity, Oxford Univ. Press, 1975 Search PubMed.
- A. Zintchenko, A. Philipp, A. Dehshahri and E. Wagner, Bioconjugate Chem., 2008, 19, 1448–1455 CrossRef CAS PubMed.
- A. J. Engler, M. A. Griffin, S. Sen, C. G. Bönnemann, H. L. Sweeney and D. E. Discher, J. Cell Biol., 2004, 166, 877–887 CrossRef CAS PubMed.
- E. Kristo, C. G. Biliaderis and N. Tzanetakis, Food Chem., 2003, 83, 437–446 CrossRef CAS.
- C. L. Perrin and J. B. Nielson, Annu. Rev. Phys. Chem., 1997, 48, 511–544 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02555c |
| This journal is © The Royal Society of Chemistry 2019 |