
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMild synthesis of diboryldiborenes by diboration of B–B triple bonds†
Tobias
Brückner
ab,
Rian D.
Dewhurst
 ab,
Theresa
Dellermann
ab,
Marcel
Müller
ab and
Holger
Braunschweig
*ab
ab,
Theresa
Dellermann
ab,
Marcel
Müller
ab and
Holger
Braunschweig
*ab
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 11th June 2019
Abstract
A set of diboryldiborenes are prepared by the mild, catalyst-free, room-temperature diboration of the B–B triple bonds of doubly base-stabilized diborynes. Two of the product diboryldiborenes are found to be air- and water-stable in the solid state, an effect that is attributed to their high crystallinity and extreme insolubility in a wide range of solvents.
Introduction
The propensity of polyboron species to form clusters as a way of quenching the natural electron deficiency of boron is now well documented.1 A consequence of this phenomenon is that networks of hypovalent boron atoms, bound through electron-precise (2e2c, 4e2c or 6e2c) bonds, are extremely difficult to deliberately construct.2 Thus, a chemistry based on boron chains, analogous to the ubiquitous chain chemistry of carbon, is simply nonexistent. While boranes (BR3) and diboranes(4) (B2R4)3 are now relatively well studied compounds, even some of the most simple boron analogues of organic species, such as short chains and small cyclic species, are extremely rare and suffer from difficult syntheses. Nöth's syntheses of linear tri-, tetra-, penta- and hexaboranes in 1970 and 1994,4 which are based on the reductive coupling of haloboranes, still represent some of the only rational synthetic routes to boron chain species, as exemplified in Fig. 1A. However, these synthetic routes rely on somewhat temperamental B–B coupling steps under harsh, functional-group-intolerant reductive conditions, making these reactions likely only possible with diorganylamino-substituted borane precursors. Nöth's boron chains have recently been supplemented by syntheses of B4 chains using low-valent boron precursors. In 2012 we reported the unexpected transition-metal-templated catenation of four borylene ligands into a B4 chain (Fig. 1B).5 In the absence of transition metal templation, Kinjo and coworkers found that diboration of a geminally-base-stabilized B2 species with bis(catecholato)diboron (B2cat2) provided a highly unusual B4 chain (Fig. 1C),6 while in our laboratories, the same reagent led to both 1,1- and 1,2-diborations of doubly base-stabilized diborenes (LRB![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BRL) (Fig. 1D).7
BRL) (Fig. 1D).7
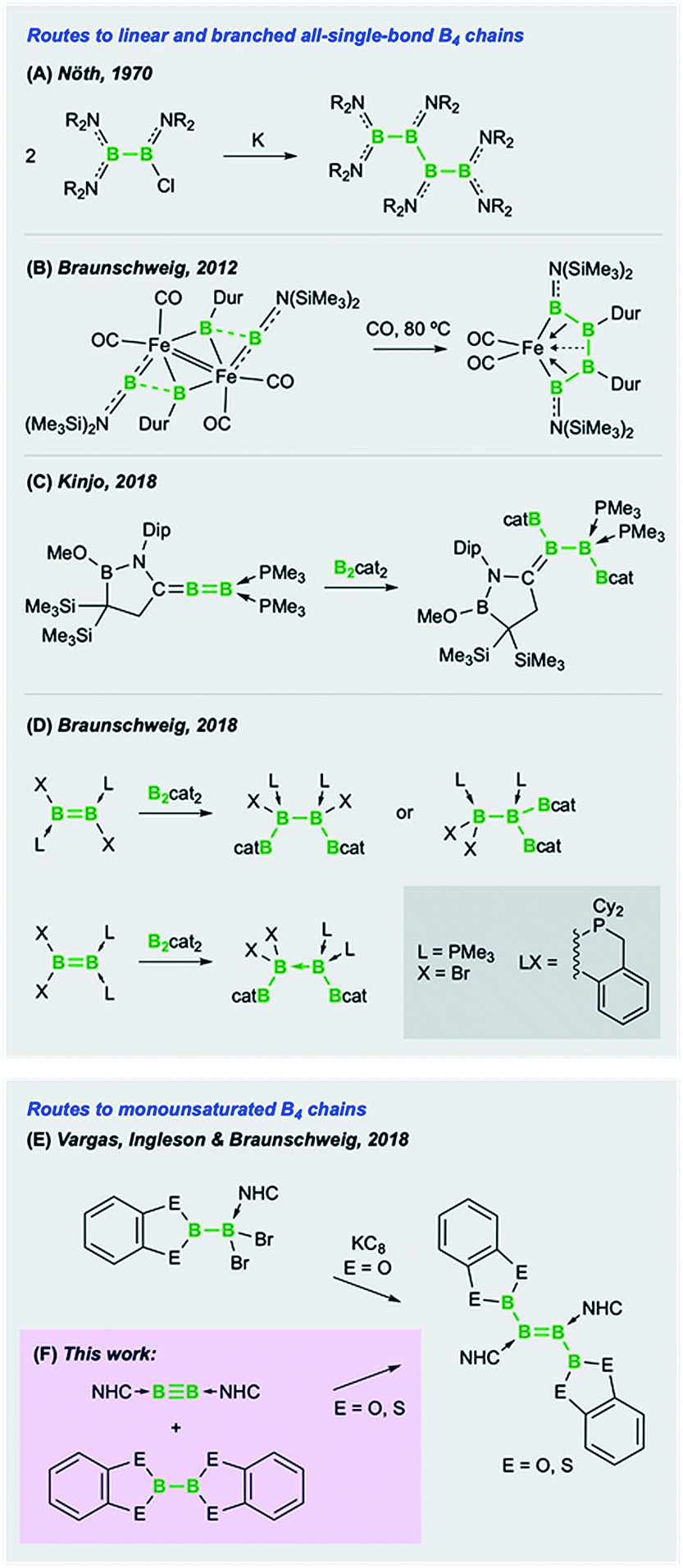 | ||
| Fig. 1 Synthetic routes to saturated (A–D) and monounsaturated (E and F) B4 chains (NHC = N-heterocyclic carbene). | ||
Another goal of short boron chain chemistry is the synthesis of chains with partially filled π orbitals. In an attempt to prepare such species, in joint work with the groups of Vargas and Ingleson, we recently reported the reductive coupling of boryl-substituted dihaloborane Lewis adducts to form diboryldiborenes featuring significantly conjugated B4 chains with two π electrons (Fig. 1E).8 These reactions, while allowing access to these unusual compounds, were hampered by the combination of the relatively labile B–B bond in the precursor and the harsh reductive conditions required. A more convergent and potentially advantageous strategy to prepare diborylated diborenes – as well as other difunctionalized diborenes – would be the direct addition of E–E σ bonds across the B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B triple bond of diborynes (Fig. 1F).2 This strategy would eliminate the need for installing various functional groups directly into the Lewis-base-bound dihaloborane precursor (LB)BX2(ERn) (LB = Lewis base; ERn = functional group), as well as circumventing the incompatibility of this functional group with the harsh reduction conditions needed to form the BB double bond.
B triple bond of diborynes (Fig. 1F).2 This strategy would eliminate the need for installing various functional groups directly into the Lewis-base-bound dihaloborane precursor (LB)BX2(ERn) (LB = Lewis base; ERn = functional group), as well as circumventing the incompatibility of this functional group with the harsh reduction conditions needed to form the BB double bond.
The success of such a strategy would require reliable 1,2-addition chemistry of σ-bound E–E species across the BB triple bonds. While both doubly base-stabilized diborenes (LRBBRL) and diborynes (LBBL) have shown diverse reactivity with unsaturated species and elemental chalcogens,2 they have shown only limited propensity to undergo simple 1,2-addition reactions with other σ-bonded species, and often react unpredictably or not at all. In addition to the diboration reactions described above (Fig. 1D), doubly base-stabilized diborenes undergo hydroboration with the hydroborane HBCat; however, application of the more Lewis acidic borane 9-borabicyclo[3.3.1]nonane led instead to disproportionation and cluster formation.9 The only other confirmed 1,2-addition to a diborene consisted of an unexpected intramolecular C–H addition across the BB bond.10 Like diborenes, diborynes have also shown a marked reluctance to undergo conventional 1,2-additions with labile σ-bonded species. Hydrogenation of diborynes has been demonstrated with some diborynes but appears not to take place with others.11 In 2016 we reported that combination of diaryl ditellurides ArTeTeAr (Ar = Ph, 4-C6H4F) to a diboryne resulted in addition of only one “TeAr” fragment to the B2 unit and formation of an aryltelluride salt.12 However, the lighter diorganyldichalogenides were more recently found to undergo conventional 1,2-additions across diborynes, producing either diborenes or diradical products.13
We present herein convergent syntheses of monounsaturated B4 chains, doubly base-stabilized diboryldiborenes, by simple, uncatalyzed, room-temperature diboration of boron–boron triple bonds. The products feature linear chains of four sp2-hybridized boron atoms, with the outer boron atoms possessing varying degrees of coplanarity and conjugation with the central BB double bonds.
Results and discussion
Combination of the doubly NHC-stabilized diboryne 1a (Fig. 2) with equimolar amounts of either B2cat2 or bis(dithiocatecholato)diboron (B2Scat2), and stirring in benzene, led to a color change to blue and the emergence of pairs of new 11B NMR spectral signals. Drying of the solution and washing with hexane provided blue solids 2a,b (Fig. 2), which displayed 11B NMR signals (2a: δ 43.0, 27.7; 2b: δ 69.1, 29.6) differing from those of the precursors 1a (δ 56), B2cat2 (δ 31), and B2Scat2 (δ 59). The upfield 11B NMR signals of 2a,b fall at the lower-field end of those of known NHC-stabilized diborenes (δ 18–30),2,13a and the pair of signals for 2a match those of the previously-reported diboryldiborenes bearing Bcat groups (δ 42–44 and 27–28).8 The observation of single 1H and 13C NMR spectral resonances for the CH3 groups of the NHC donors indicated the symmetry of the molecule in solution. High-resolution LIFDI mass spectrometry confirmed the molecular formulae of 2a,b corresponding to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 addition of the precursor diborane to diboryne 1a.
:1 addition of the precursor diborane to diboryne 1a.
 | ||
| Fig. 2 Catalyst-free diboration of diborynes. Inset photos: suspensions of diborenes 2c (top) and 2d (bottom) in water under ambient atmosphere. Dip = 2,6-diisopropyl-phenyl; Dep = 2,6-diethylphenyl. | ||
The combination of diboryne 1b, featuring unsymmetrically substituted NHC donors, with either B2cat2 or B2Scat2 led to a color change from red to brown and the precipitation of orange and red crystals, respectively (2c,d; Fig. 2). High-resolution LIFDI mass spectrometry and elemental analysis performed on these crystals again indicated 1:1 addition of the diborane to 1b. The crystals of 2c,d proved to be highly insoluble, allowing only partial characterization of 2c by solution 1H and 11B NMR spectroscopy and precluding solution NMR spectroscopy for 2d. Diborene 2c showed a broad 11B NMR signal at δ 26, but the remaining signal could not be identified due to the low concentration of the sample. Unfortunately, attempts to record solid-state MAS NMR spectra of 2d provided either no signal (without rotation) or resulted in decomposition of the sample under the pressure created by the sample rotation.
Single-crystal X-ray diffraction analysis of 2a–d provided final confirmation of the structures of the products (Fig. 3). While all four compounds show relatively long BB double bonds (2a: 1.605(3) Å; 2b: 1.617(3), 1.619(3) Å; 2c: 1.608(3) Å; 2d: 1.627(2) Å) that could be indicative of increased π conjugation with the outer boron atoms, the Bcat and BScat groups show a relatively broad range of E–B–BB torsion angles (16–54°), the larger of which indicate strong non-coplanarity and thus discount the presence of significant π conjugation in the solid state. The outer B–B bond distances of 2a–d (2a: 1.650(3) Å; 2b: 1.685(4), 1.691(4), 1.689(4), 1.687(4) Å; 2c: 1.652(2) Å; 2d: 1.664(2) Å) are in line with those of the previously-reported diboryldiborenes (1.65–1.68 Å).
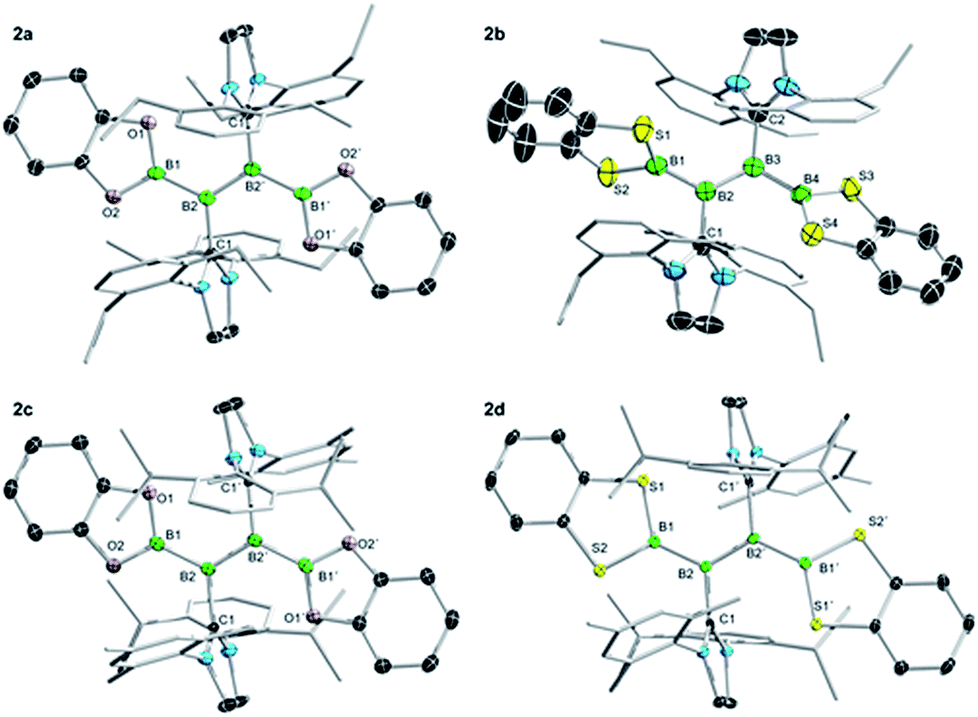 | ||
| Fig. 3 Crystallographically derived structures of 2a–d. Ellipsoids shown at the 50% probability level. Some ellipsoids and all hydrogen atoms and solvent molecules (one molecule of C6H6 in 2c) have been removed for clarity. Selected bond lengths (Å) and angles (°) for 2a: B1–B2 1.650(3), B2–B2′ 1.605(3), O1–B1–B2–B2′ 15.9(3). For 2b (two molecules in unit cell): B1–B2 1.685(4), 1.691(4), B2–B3 1.617(3), 1.619(3), B3–B4 1.689(4), 1.687(4), S1–B1–B2–B3 48.9(3), 52.9(3), S3–B4–B3–B2 53.6(3), 50.3(3). For 2c: B1–B2 1.652(2), B2–B2′ 1.608(3), O1–B1–B2–B2′ 19.4(2). For 2d: B1–B2 1.664(2), B2–B2′ 1.627(2), O1–B1–B2–B2′ 29.8(2). | ||
We were surprised to observe that while the SIDep-substituted diborenes 2a,b decompose within 15 minutes in air, diborenes 2c,d are stable for days in the solid state in ambient air and even as a suspension in water (Fig. 2, inset photos). Although the appearance of solid 2c remains unchanged, a small boronic acid signal can be observed in the 1H and 11B NMR spectra after approximately one week in D2O. These suspensions gradually decolorize within one hour upon addition of CH2Cl2, into which the diborenes are slowly solubilized and decomposed. As diborenes 2a–d are relatively similar in terms of electronics and sterics, the remarkable solid-state stability of 2c,d is likely a consequence of their high crystallinity. Once dissolved, even in miniscule amounts, the compounds quickly decompose in the presence of moisture.
The UV-vis spectra of the diboryldiborenes 2 are remarkably different in their features (see ESI†). The spectrum of the orange compound 2c (λ 451 nm) resembles that of the previously-reported yellow diboryldiborene8 [(IMe)(catB)BB(Bcat)(IMe)] (IMe = 1,3-dimethylimidazol-2-ylidene; λmax = 435 nm), both having low-wavelength features and no absorption in the longer wavelength region. However, the other three diborenes 2a,b,d have significant absorptions in the region 550–650 nm (2a: λ 422, 578 (max.) nm; 2b: λ 503, 608 (max.) nm; 2d: λ 543 (max.), 622 nm). Overall the longer wavelength absorptions of 2a–d relative to those of [(IMe)(catB)BB(Bcat)(IMe)] suggest that the more σ-donating and π-withdrawing saturated-backbone NHCs in the former lead to significant decreases in the HOMO–LUMO gaps of the molecules.
It should also be noted that, in an attempt to induce double diboration, the diborynes 1a,b were treated with two molar equivalents of the diboranes B2cat2 and B2Scat2. However, after monitoring conversion to the respective diboryldiborenes 2a,b, no further reaction was observed, even with heating (100 °C) or under photolytic conditions.
Conclusions
The diboryne diboration reactions herein provide convergent access to monounsaturated boron chains and provide a new tool in the challenging construction of electron-precise B–B bonds. Moreover, the high stability of the bulky diboryldiborene products is very encouraging. The extreme sensitivity generally shown by diborenes is the main practical impediment towards their use as “π superdonor” units in molecular electronic materials, thus the discovery of derivatives able to withstand air and water – even if only in the solid state – is a significant step forward.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this project was provided by the European Research Council (ERC) under the European Union Horizon 2020 Research and Innovation Program (Advanced Grant agreement no. 669054). The authors also thank Allychem Co., Ltd. (Dalian, China) for a generous gift of B2cat2 and Mr Dominic Prieschl for photography.Notes and references
- (a) A. Stock, Hydrides of Boron and Silicon, Cornell Univ. Press, Ithaca, NY, 1933 Search PubMed; (b) W. N. Lipscomb, Adv. Inorg. Chem., 1959, 1, 117–156 CrossRef CAS; (c) K. Wade, J. Chem. Soc., Chem. Commun., 1971, 792–793 RSC; (d) R. E. Williams, Inorg. Chem., 1971, 10, 210–214 CrossRef CAS; (e) M. A. Fox and K. Wade, Pure Appl. Chem., 2003, 75, 1315–1323 CAS; (f) E. Osorio, J. K. Olson, W. Tiznado and A. I. Boldyrev, Chem.–Eur. J., 2012, 18, 9677–9681 CrossRef CAS PubMed.
- (a) K. Nozaki, Y. Aramaki, M. Yamashita, S.-H. Ueng, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 11449–11451 CrossRef CAS PubMed; (b) H. Braunschweig and R. D. Dewhurst, Angew. Chem., Int. Ed., 2013, 52, 3574–3583 CrossRef CAS; (c) M. Arrowsmith, H. Braunschweig and T. E. Stennett, Angew. Chem., Int. Ed., 2017, 56, 96–115 CrossRef CAS PubMed.
- (a) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (b) D. G. Hall, Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, 2005 CrossRef.
- (a) K. H. Hermannsdörfer, E. Matejčikova and H. Nöth, Chem. Ber./Recl., 1970, 103, 516–527 CrossRef; (b) G. Linti, D. Loderer, H. Nöth, K. Polborn and W. Rattay, Chem. Ber., 1994, 127, 1909–1922 CrossRef CAS.
- H. Braunschweig, Q. Ye, A. Vargas, R. D. Dewhurst, K. Radacki and A. Damme, Nat. Chem., 2012, 4, 563–567 CrossRef CAS PubMed.
- W. Lu, Y. Li and R. Kinjo, Angew. Chem., Int. Ed., 2018, 57, 15691–15695 CrossRef CAS PubMed.
- T. E. Stennett, R. Bertermann and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 15896–15901 CrossRef CAS PubMed.
- A. Hermann, J. Cid, J. D. Mattock, R. D. Dewhurst, I. Krummenacher, A. Vargas, M. J. Ingleson and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 10091–10095 CrossRef CAS PubMed.
- (a) H. Braunschweig, R. D. Dewhurst, C. Hörl, A. K. Phukan, F. Pinzner and S. Ullrich, Angew. Chem., Int. Ed., 2014, 53, 3241–3244 CrossRef CAS PubMed; (b) H. Braunschweig and C. Hörl, Chem. Commun., 2014, 50, 10983–10985 RSC.
- S. R. Wang, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, V. Paprocki and L. Winner, Chem. Commun., 2017, 53, 11945–11947 RSC.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik, T. Dellermann and K. Hammond, Chem.–Eur. J., 2016, 22, 17169–17172 CrossRef CAS PubMed.
- H. Braunschweig, P. Constantinidis, T. Dellermann, W. C. Ewing, I. Fischer, M. Hess, F. R. Knight, A. Rempel, C. Schneider, S. Ullrich, A. Vargas and J. D. Woollins, Angew. Chem., Int. Ed., 2016, 55, 5606–5609 CrossRef CAS PubMed.
- (a) J. Böhnke, T. Dellermann, M. A. Celik, I. Krummenacher, R. D. Dewhurst, S. Demeshko, W. C. Ewing, K. Hammond, M. Heß, E. Bill, E. Welz, M. Röhr, R. Mitrić, B. Engels, F. Meyer and H. Braunschweig, Nat. Commun., 2018, 9, 1197 CrossRef PubMed; (b) E. Welz, J. Böhnke, R. D. Dewhurst, H. Braunschweig and B. Engels, J. Am. Chem. Soc., 2018, 140, 12580–12591 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra. CCDC 1882019–1882023. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02544h |
| This journal is © The Royal Society of Chemistry 2019 |